C C C h h h a ap a p pt t te e er r r 1 1 1 緒論
1 1 1 . . . 1 1 1 樣品背景與特性
據歷史學家研究,人類自新石器時代之後即利用陶瓷來製造器具。那何謂陶 瓷呢?「陶瓷」二字即英文的"pottery and porcelain"由陶及瓷組成,就中國字而 言,放在窯內燒的東西就是陶,質地更密實燒成更高溫的就是瓷。陶瓷器(Ceramic ware)為陶器與瓷器的概括名稱。Ceramics 是由希臘語演變而來,是指利用黏土 燒成而得的容器,或稱其原料者。現在不同國家擁有不同的定義:英、蘇、歐洲 等地的定義:由無機材料所組成的製品,先成形,再經加熱而硬化;美國和日本 的定義:凡以無機非金屬物質為原料,製造或使用中處於高溫度之製品或材料。
陶瓷並不會像金屬一樣在溫度的劇烈改變下而產生物質的變化,所以陶瓷具有耐 高溫、穩定性高且耐腐蝕的性質,這些的特性都是陶瓷所擁有的獨特性質,但現 在除了一般聯想到的磁磚、陶瓷器皿、浴缸、或是景德鎮美麗的陶瓷藝術品,其 實這些日常所用的陶瓷,通稱為『傳統陶瓷』,不過這類陶瓷並不是陶瓷研究的 全部。由於材料在工程應用上的條件日趨嚴苛,且隨著科技的突飛猛進,許多具 有特殊功能的陶瓷產品陸續誕生,使得原以傳統工藝與藝術為主的傳統陶瓷漸漸 演進成了今日科學且高科技的『精密陶瓷』( Fine Ceramics)。
所謂精密陶瓷,是以精製的高純度無機材料為原料,利用各種化學或物理方 法精確控制組成及均勻度,再以乾式壓製、鑄漿、或射出成形等方法成形後,經 過精確的反應及燒結步驟,使其顯微結構與物性與化性達到一定標準,再經過加 工製成具有卓越機能的成品。它具有堅硬、耐磨、耐壓、耐高熱、耐酸、耐鹼等 持久的特性,並具有相當優異的光、電磁、熱功能以及生物相容性。依據運用的 範圍可將精密陶瓷分成三大類:(1)電子陶瓷,(2)結構陶瓷,(3)生醫陶瓷。本文 最主要要討論的是電子陶瓷中的介電陶瓷。
最早在1939 年Rychtmyer[1]利用圓柱型的介電材料,將其彎曲至頭尾相接成 一圓環狀,可以將波週而復始的置於其中,這可以視為一個共振器或天線,可以 輻射出共振頻率並被其他的儀器所接收。這是最早介電材料在微波上的應用。在 之後的20 年左右,因為共振腔的難尋覓與對於如何測量製造出來的共振器等困
個人通訊的今日,有無窮的潛力。
好的微波通訊材料所需要的條件有三種:高介電係數、低介電損失與穩定的溫度
係數[5-7]。以下將會介紹為何需要這三種條件。
z 一個微波共振器需要有高的介電係數,因為微波元件的體積大小與其介電係 數開根號成反比。當微波以平面波進入介電物質時,為了要達成共振條件,
需滿足邊界條件(boundary condition),此時如果兩電路兩端為開路或短路,
,...
2 ,3 2 , 2
λ λ
λ = λ
= n
L ,L 為電磁波在介質中傳輸長度,λ 為微波進入介電物質 時的波長。
e jkx E
E −
= 0 , k = k'-jk" =ω µε = Re
{
ω µε}
−jIm{
ω µε}
} Re{
1 2
µε λ π
k = f
=
⇒ (1.1.1)
又 真空中光速 0
c = λ f = µε1 ,當μr~1,
L f
C r r
∝
=
=
⇒
} Re{
} Re{
0
ε ε
λ λ ,
} Re{
1 r
L∝ ε
⇒ (1.1.2)
當元件的介電常數越高時,樣品所需的體積也就越小。在微波電路中,對於介電 常數的要求大約在10~100 左右。
z 共振有兩個主要的特性,共振頻率與品質因子(Quantity Factor)。品質因子是 與共振電路電磁波儲存容量及他的能量消耗有關之參數。它的定義為元件儲 存的能量除以單位週期消耗的能量。當品質因子越高,表示能量損失越少,
介電損失越低。介電損失tanδ,又稱損失正切(loss tangent),也是代表單位 週期所消耗的能量。介電損失與品質因子互成反比,即tanδ=1/Q[3]。
~ C
I
R
t
ej
V t
V( )= 0 ω I
~ 介電物質
Z 1
R 1 θ
δ ωC
圖 1-1 (a)微波電路簡圖;(b)微波電路等效電路;(c)阻抗示意圖
一個微波共振電路如圖1-1(a),換成等效電路如圖 1-1(b),可視為為一 RC 電路。
此等效電路之電流為
t t j
t j
j j CVe
R e e V
I t
I ω
ω 0 ω ω 0
) 0
( = = + (1.1.3)
又 Z
I0 =V0 1 1 Z R j Cω
⇒ = + (1.1.4)
Z:為電路總阻抗,為一複數形式,如圖 1-1(c)
如果介電物質是理想電容,此時 R→∞,θ=π/2、δ=0,在這個狀況下此電路 無能量散失。但是因為介電物質的金屬性、介電性與考慮輻射因素,R≠∞,θ
<π/2,此時能量會隨時間函數散失,散失能量平均功率為 δ
θ sin 1 cos
2
2
0 ∝ ∝ =
= R R
Power V (1.1.5)
當R→∞,cosθ=sinδ→0,此時能量散失也趨近於零。
對一個好的、具有低介電損失的材料,sinδ ≈tanδ 。tanδ即所謂的損失正切,
又被稱為消散係數(dissipation factor)。這個係數是微波電路中很重要的物理量。
通常tanδ被要求在 1/5000~1/10000 之內。
z 因介電性質都是跟溫度有關的函數,當溫度係數趨近於零的時候,表示物質 不受溫度改變影響,能可維持原來的介電性質。對一介電共振器而言,溫度 係數有兩種,一為共振頻率溫度係數-τf,另一為介電常數溫度係數τε, 兩者定義分別為:
⎟⎠
⎜ ⎞
⎝
⎛
∆
= ∆
T f
f f
τ 1 , ⎟
⎠
⎜ ⎞
⎝
⎛
∆
= ∆ T ε
τε ε1 (1.1.6)
這兩者的關係式可以表示為,
) (
2τf αT
τε ≈− + (1.7)
αT為熱膨脹係數。所要求的溫度係數需要在 T f 0 1~3ppm/oC 2 = < ±
+τ τ
α ε 的範
圍內[3]。
介電材料在微波傳播上的應用,主要依靠此三種性質在微波中的優越性,所 以材料學者不停的研究介電材料中可以擁有更高的介電性質、更低的介電損失與 接近於零的溫度係數。本文中研究的鈣鈦礦陶瓷就是因為他在微波中擁有非常低 的介電損失而聞名。
1 1 1 . . . 2 2 2 樣品結構與光學特性
自二十世紀發現ABO 鈣鈦礦(perovskite)結構之後,因它的優越的微波
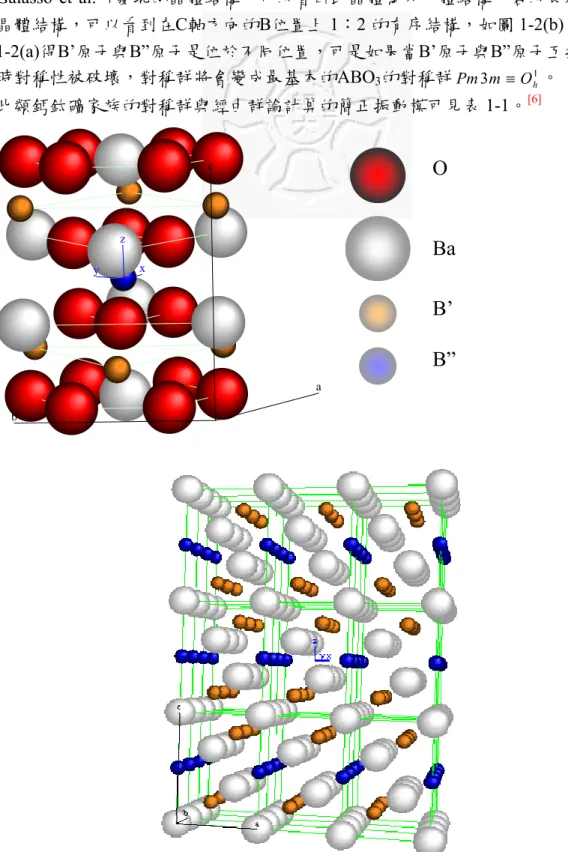
Galasso et al.所發現的晶體結構,可以看出此晶體為六方體結構,若以長程來看 晶體結構,可以看到在C軸方向的B位置上 1:2 的有序結構,如圖 1-2(b)。由圖 1-2(a)得B’原子與B”原子是位於不同位置,可是如果當B’原子與B”原子互換,此 時對稱性被破壞,對稱群將會變成最基本的ABO3的對稱群Pm3m≡O1h。
此類鈣鈦礦家族的對稱群與經由群論計算的簡正振動模可見表1-1。[6]
a b
c
y x z
O
Ba
B’
B”
圖 1-2 (a) A(B’1/3B”2/3)O3單位晶格結構;(b) A(B’1/3B”2/3)O3的長程有序結構
A(B’1/3B”2/3)O3單位晶格有15 顆原子,所以有 45 個自由度,經過群論計算,
可以得出有42 個A(B’1/3B”2/3)O3的簡正振動模,如表1-1。A(B’1/3B”2/3)O3屬於拉 曼的振動模共有九個,紅外線的部分有16 個,剩下的為聲學聲子與不被激發的 振動模。由此可看出,要研究晶格振動最好的利器就是拉曼與紅外線。利用拉曼
與紅外線可以得出物體內部的晶格結構,並且得出物體的內部晶格結構如何影響 巨觀的介電性質。
表格 1-1 ABO3經群論計算的簡正振動模
Compound ABO3 A(B’1/2B”1/2)O3 (A’1/2A”1/2) BO3 (A’1/2A”1/2) (B’1/2B”1/2)O3
A(B’1/3B”2/3)O3
Space group
3m O1h
Pm ≡ Fm3m≡Oh5 Fm3m≡Oh5 F4−3m≡Td2 P3−m1≡D33d
A1g(O) A1(O) 4A1g(A,B”,O) Eg(O)
Raman -
2F2g(A,O)
F2g(B)
E(O) 5Eg(A,B”,O)
9Eu(A,B’,B”,O) infrared 3F1u(A,B,O) 4F1u(A,B’,B”,O) 5F1u(A’,A”,B’B
”,O)
-
7A2u(A,B’,B”,O) Raman and
IR
- - - 6F2(A’,A”,B’,
B”,O)
-
Silent F2u(O) F1g(O)+F2u(O) 3F2u(O) 2F1(O) A2g(O)+2A1u(O)
Acoustic F1u F1u F1u F2 A2u+Eu
1 1 1 . . . 3 3 3 拉曼散射
當光進入物質中,使物質受到光電場影響而產生電偶極,受光電場影響而震 盪的電偶極會發射出電磁波,而傳播到各個方向,此電磁波並非同相所以不能互 相形成建設性干涉,此種稱為雷利散射(Rayleigh scattering)。在非吸收介質中,
物體的缺陷越多,雷利散射效應越強。伴隨雷利散射產生的還有拉曼散射(Raman Scattering),此為與物體中晶格振動的聲子結合,產生了高於原始光頻率的光 (anti-Stokes Effect)或低於原始光頻率的光(Stokes Effect)出現,如圖 1-3。這個現 象與物質本身的電性、物性、化性有相當大的關係[7]。
雷利散射 拉曼散射 拉曼散射 Rayleigh
scattering
stokes effect
antistokes effect
振動能階
電子基態
圖 1-3 雷利散射與拉曼散射的能階圖
晶格經熱振動或光激發之後產生聲子,但只有某些晶體結構所產生的振動可 以被光學激發,並被儀器所偵測到。也就是說聲子對晶格的對稱性相當敏感,尤 其對於鐵電性物質的鈣鈦礦結構更是如此。研究鐵電性晶體的方法之一就是利用 拉曼散射,因為鐵電性晶體隨著溫度變化時會有相變產生,而光譜也因此產生變 化[8]。但是A(B’1/3B”2/3)O3沒有鐵電性,只有順電性。拉曼光譜是一種非破壞性與 非侵入型的方法,不用破壞樣品本身即可探知內部結構,並對於微觀的結構非常 敏感,尤其是無結晶系統中結構的辨別,微觀拉曼測量可以對樣品的異質性做出 結構的判定、異質性。對於陶瓷這種異於晶體的雜亂度,拉曼散射比其他的測量 方法更能得出晶體結構的參數。因為拉曼散射光譜擁有非常敏感的參數Æ頻率、
強度、波形與寬度。這些參數對於能夠真正的反應出結構的改變,不管是溫度、
氣壓、電場或是應力的的改變。
高頻振動頻率改變,對應到晶格變化,屬於短程的變化。例如四面體或八面 體的鍵結強度變強,其聲子頻率也隨之變高。低頻頻率變化就是屬於全體的運動 變化,像是電偶極強度等。
拉曼的相對強度可以反應出晶體內部振動模的變化,當特定振動模強度相對 於其他振動模強度變弱時,表示晶體內部與特定振動模有關的原子位置有所改 變,即此原子並不在理想晶格中的位置,所以無法產生特定振動模的聲子,相對 強度也因此變弱。
波形與波的寬度則是與物體結構與外界干擾有關。波的寬度就是波的半高 寬,此物理量與聲子的生命期相關。
本文中樣品拉曼訊號的取得是在室溫中利用DILOR XY-800 光譜儀,以三個
光柵分光,並以利用液態氮降溫的CCD偵測器所接收。使用 10 mW的氬(Ar+)離 子雷射,波長為514.5-nm作為激發光源。所得出的拉曼光譜具有 0.5 cm-1的解析 度。
1 1 1 . . . 4 4 4 X 光繞射
X 光量測原理是根據布拉格定律(Bragg Law)2dsinθ =nλ,其中 d 為晶格面間距,
2θ為散射角,λ 為 X 光射線波長。當入射光與散射光產生之波程差(path difference) 為2dsinθ,是波長λ 的整數倍時,會產生建設性干涉,就在 X 光射線譜圖上有 繞射峰產生。偵測器(detector)的同步移動,可得到各繞射峰強度的分布譜圖。實 驗在同步輻射中心與工研院所完成。
本實驗利用H. M. Rietveld 在 1969 年所提出的Rietveld 結構精算法[9],來分析X 光繞射實驗所得的譜圖,進而獲得精細的晶體結構與參數。GSAS結構精算軟體,
是美國Los Alamos 國家實驗室的Allen C. Larson 和Robert B. Von Dreele 依照 Rietveld 精算法,而發展撰寫的結構分析軟體[10]。
GSAS 結構精算軟體,將背景函數(background function)、尺度因子(scale factor)、
晶格函數(lattice parameters)、儀器的零點位置(zero point)、波形函數(profile function)等各項係數設計為可調整的參數,再加上原子的位置、熱擾動因子和各 相所佔的比例等,經逐步擬合後推算出一合於實驗譜圖的晶體結構模型。
首先,參照先前諸多關於A(B’1/3B”2/3)O3之對稱群與原子位置作為精算的基礎,
代入已輸入原始X 光繞射資料的GSAS 精算軟體中,再固定所有參數,針對背 景函數及尺度因子作計算,尺度因子大小決定整個模型的比例,而經計算後就有 一基本的繞射譜圖背景。逐步調整參數值,縮小擬合差χ2而漸趨於穩定,就可 開始精算晶格常數及儀器零點位置,相當於先前的步驟,計算模型所預測會出現 繞射峰位置會因晶格常數及儀器零點位置的修正而有所不同,反覆計算可使更趨 近於實驗值。再將這些參數固定,針對波形函數作計算,當χ2值逐漸縮小與晶 格常數趨近於穩定後,此時所計算的初步模型已可大致符合實驗值,再計算原子 位置、熱擾動因子,經過一連串的反覆計算後,各項決定譜圖的參數也逼近擬合 值,整個譜圖最終由Rietveld 結構精算法所運用的最小平方法逼近,直至每一個 參數都得到最佳擬合參數值。Rietveld 程式精算基本流程列於圖 1-4。
粗糙晶格模型 最小平方誤差法
精確晶格模型 計算鍵長鍵角 實驗數據
最初晶格模型 熱模型因子
佔用因子
背景值校正 晶格常數 波形常數
計算所得數據 Electron density
map
圖 1-4 GSAS 程式分析流程
1 1 1 . . . 5 5 5 延伸 X 光吸收精細結構
X 光吸收精細結構「X-ray absorption fine structure」(XAFS)在近十年內有長 足的發展,現已發展成與實驗數據有高吻合度之理論。此理論研究的理論基礎是 利用超過吸收邊界(absorption edge)的 X 光吸收係數震盪來觀測晶格中各種不同 種類的吸收原子與其他原子間的細微結構、電子組態和熱擾動性質。由於上述的 這些理由致使XAFS 成為研究物質微觀的一個重要利器。以下將會探討美國華盛 頓大學物理系所開發的模擬系統Æ延伸 X 光吸收精細結構(EXAFS) 和 X 光吸 收近邊緣結構(XANES)這兩種理論模擬來引申。
X 光吸收近邊緣結構 x-ray absorption near-edge structure ( XANES ) 可反應 吸收原子的電子性質,例如:氧化價數及 d-軌域的電子填滿率,並可分辨吸收 原子所處之晶位對稱性。XANES 的適用範圍大約在由吸收邊緣(absorption edge) 開始算起到 30 eV 左右,這主要是由附近原子內電子與電子之間擾動的效應產 生,此部分的理論較為複雜,必須利用多重散射(multiple-scattering)的模型來解 釋。
延伸 X 光吸收精細結構 ( EXAFS ) 則提供了吸收原子周圍之局部結構,其中包 括了周圍各配位層的原子種類、個數、與中心吸收原子間的平均距離及其排列的 雜亂程度等。EXAFS 的範圍大約是超過吸收邊緣(absorption edge)30 eV 再延伸 至1000 eV。EXAFS 相對於 XANES 是較為簡單,且整套的理論也已發展完成。
其發生的機制是在吸收原子處於一個四周均為凝結(condensed)的狀態。這樣情況 下才會出現EXAFS。本文將使用 FEFF8 來擬和數據並研究晶體微觀結構與微波 介電性質作關連。
9000 10000 11000
XANE 與 EXAFS 的分界可見圖 1-5
0.0 0.5 1.0
Photon energy (eV)
X-ray Absorption Coefficient
XANES
EXAFS
30eV
圖 1-5 EXAFS 與 XANES 的光譜分界
111...555..1.11
EXAFS 原理說明
當原子內層電子吸收 X 光時,會變成自由的光電子,此時光電子將會以物 質波的方式傳播。如果吸收原子周圍並沒有任何其他原子,此波將會傳播出去,
並不會造成如圖1-6 有物質波互相干涉的影響。但對於一晶體來說,當物質波傳 播出去時,將會遇到其他的原子阻擋將物質波反彈,此反彈的波與原本的波產生 干涉,而經過互相干涉的波會反應在吸收譜的震盪光譜上,當回向散射的相位 (phase)與傳播出去的光電子物質波的相位是相同時,此時會產生建設性干涉,光 譜會看見高峰,若為兩者相位相差π,則產生破壞性干涉,光譜上形成凹陷處。
見圖1-5。
圖 1-6 光電子以物質波的傳播並與周圍原子作用
Final state
Backscattering wave Outgoing wave
x-ray
Absorbing atom Core state
Scattering atom
圖 1-7 X 光吸收後光電子波狀態示意圖
圖1-6 中的實線為光電子的物質波,虛線為光電子被附近原子所散射的物質 波,而最後的狀態為這兩種物質波所形成的干涉,而這其中也必須符合選擇規 則。圖1-7 說明了被激發的光電子物質波與被周圍原子阻擋而往回傳播的物質波 (backscattering wave)互相的干涉,最後成一末態(final state),而我們所觀測結果 即為末態所產生的吸收率。在 EXAFS 範圍中所受到最大的影響是一階單重散 射;利用Broglie 的關係與能量守恆算出光電子的物質波及動能,如下式:
0 2
2
h E
m p p
h = −
= ν
λ , (1.5.1)
E0為吸收邊緣的能量
通常光子的能量範圍最好的在 40 keV;由於在這個範圍光子會被電子完全
吸收,光子的能量轉變成光電子脫離原子的位能井所需的能量;此時光電子具有 動能大約15 eV 以上;由於附近原子所造成的位能井大約是 3 eV,這個能量相對 於光電子的動能 15 eV 可以視為一種微擾項,這方便於我們計算,且光電子所留 下的內層電洞能態與電子的初始能態均可以忽略外部原子所造成位能井的影 響;這也就是為什麼我們利用X 光激發出深層電子進行 EXAFS。
XAFS 的 χ 光譜定義為利用吸收係數邏輯歸一的常數,而吸收譜超過吸收邊 緣的震盪部分如下式:
( ) [ ( ) ( ) ]
) ( 0
0 0
E E E E
µ µ χ µ
∆
= −
(1.5.2)
E0為吸收邊緣能量值,通常是定義為吸收光譜微分極大值,µ0(E)為超過吸收 邊緣後吸收原子不受周圍原子干涉之吸收光譜值,∆µ0(E0)為吸收原子因吸收了X 光所造成的躍遷高度函數(step function)的高度差值[11-12],如圖 1-8 表示。圖 1-8 為標準銅金屬的吸收譜。
9000 9900 10800
0.0 0.4 0.8
eV
µ(Ε)
Cu data µ0(E)
∆µ0(Ε0)
圖 1-8 銅的 K 層吸收譜
在目前的EXAFS 的研究工作中,主要的形式是由 Kroning 短程理論,光電子
X 光的吸收遵守費米黃金定則,躍遷率 W 為
( )
Efi z eE f
W π 0 2 2ρ 2
2 ⎟
⎠
⎜ ⎞
⎝
= ⎛
= (1.5.3)
ρ(Ef)為末狀態數的密度,所以X光的單位體積吸收功率 WNa dt
du ==ω ,u為X光被 吸收的單位體積能量。Na為單位體積吸收原子的個數。
(
fa
a E f zi E
N e dt WN
du ρ
ω π π
ω 2 2 02 2
4 8
−
=
= =
)
(1.5.4)又吸收率μ定義為 u dx
du =−µ ,
cdt du du =dx
c 為 X 光(電磁波)的速度,u 為巨觀電磁波能量密度 π 8
2
E0
u = 。
(
fa f zi E
c N
e ρ
ω
µ =4π2 2 2
)
(1.5.5)光電子的初狀態和末狀態是利用Hankel function來作球形近似,波函數可寫成
j ikr
r e j
的形式,當入射波與反射波互相干涉之後,產生了一正弦波的關係,而且需
要加上一相位,這是因為此時的位能已產生改變,並非一定值[11-12]。除了主要的 正弦波振盪外,還有幾個主要的因素會影響吸收率。
第一為光電子的生命期。由於光電子會受到之前遺留下的電洞與周遭的原子電子 影響,通常是有一定的生命週期,而之前利用Fermi Golden rule 並未考慮電子在 末狀態的生命期,所以在理論中加了一平均自由路徑 λ (mean free path),即
。 (1.5.6)
) / 2
( λ
χ ∝e − r
第二個因素是因為在室溫下,因晶體的熱振動或是晶體結構本身的不整齊,
使得同層的原子距離改變,導致反射波非同相位,這項因素又稱Debye-Waller factor,此項與溫度有關,在EXAFS中以愛因斯坦模型做基礎,為一高斯分佈圖
形。即χ ∝e(−2k2σ2)[11-13]。 (1.5.7)
第三個因素為當X-ray 激發原子時,還有一個效應我們必須考慮,當 X-ray 激發 原子時,把電子激發至高能態,此時內層會留下一個電洞,這個帶正電的電洞會 對帶正電的原子核產生排斥力,這會使得對於被激發至高能態電子的庫倫吸引力 減弱,反過來說原子核對電子所產生的位能井深度減少,而這個被激發至高能態 的電子就有可能因為這個原因變成光電子。這種並非因為X-ray 激發變成光電子 的自由電子稱為passive electrons。由於原來由原子核所產生的位能井,因為電洞 的效應,使得位能井減弱,讓前後兩能態作歸一的動作就不再等於1。如下式
1 ' 2
2
0 =
∏
≤i
i
i p
p
S (1.5.8)
S20通常大概是在0.7~0.8 左右[13-15]。
而最後加上單重散射近似,並將(1.5.6)式、(1.5.7)式、(1.5.8)式之因素考慮進去。
並將即為EXAFS 的主導公式。
2
2 2
) ( / 2 2
2
0 ( ) sin[2 2 ( )]
)
( j j R k k
j j
j kR k e e
kR k N f S
k δ j λ σ
χ =
∑
+ − − (1.5.9))
j(k δ
末狀態相位的改變 與吸收原子至鄰近的散射原子距離有關,而其形成
干涉性條紋的振幅大小 f(k)則與散射原子的種類及數量有關,因為原子的大小 與質量會影響散射回來的能量分布[17]。
EXAFS 的準確度大概離吸收原子 5 埃左右,屬於短程量測,而短程散射路徑也是對 吸收率的影響較大。
111...555..2.22
實驗裝置
在本文中所進行的 X 光吸收譜是在國家同步輻射中心(NSRRC)所量測,我 們所用的光束線 BL17C Wiggler 實驗站上進行。實驗站以 Si(111)雙晶體分光儀 (double crystal monochromator, DCM)分光,使用同步輻射的優點除了強度大於一 般X 光、頻寬大、具有高偏振性、準質性良好(Extremely high collimation)、而且 為脈波,具有時間結構,可做時間解析光譜,最後是提供光源穩定性高,不隨著 時間變化。
下圖所示為典型 X 光吸收光譜術之實驗站配置,同步輻射光束經雙晶體單 光器(兩片平行的 Si (111)晶體)選取單光後進入輻射屏蔽屋,首先將遇到一組 X-Y 狹縫,其水平及垂直開口大小係以手轉螺絲配合一刻度尺調節,至於狹縫位置則 可利用兩個步進馬達( hs1h 及 hs1v)分別進行水平方向及垂直方向的移動。
圖 1-9 同步輻射 X 光吸收實驗站設置
是以一氣體游離腔( Io )測量,目前BL17C1 光束線提供兩 種長
法,穿透法(Transmission Mode)、螢光法 (Fluo
樣品
氣體離子真空腔
圖 1-10 X 光吸收儀器架設
入射光的強度經常
度( 15cm及 30 cm) 之游離腔,內部充填氣體的選擇標準是令其對入射光束 具有0.1 之吸收度,游離腔內兩片平行金屬板相距 1 cm,其間施以 300 V之電壓,
當氣體吸收X光而被游離時,所產生的電子將被正電壓之極板收集,造成一微弱 電流,再經一電流放大器( Keithley, Model 428 )轉換成電壓訊號,而後經電壓-頻 率轉換器( Nova, Model N101 VTF )得一序列脈衝訊號,最後再以一計數器( 3610 Hex Counter )累加而成強度訊號。
要求得EXAFS的光譜有三種方
rescence Mode)、電子產率法(Electron Yield Mode),因陶瓷材料通常燒結成 塊材,所以無法用穿透法,通常使用的是螢光法。[16]
Filter It Ir
I0
參考標準樣品
If
散射狹縫
θ Φ
Incident light Sample
Detector
圖 1-11 EXAFS 螢光法示意圖
⎟ ⎟
⎠
⎞
⎜ ⎜
⎝
⎛ − +
= ∆Ω
⎥⎦⎢ ⎤
⎣
⎡ +
− E E t
f tot tot
x f
f tot tot
E e E
I E
I
φµ θ µ
φ µ θ
µ
µ π
ε
sin) ( sin
) (
0
1
sin ) ( sin
) (
) (
4
(1.5.10)其中ε:螢光效率,△Ω:偵測器的立體角,Ef:反射光能量,E:入射光能量 µx(E):因其他原子所受影響的吸收率(所求)
µtot:總吸收率 µtot(E) = µx(E) + µother(E) θ:入射角(入射 X-ray 與樣品表面之間) Φ:反射角(反射 X-ray 與樣品表面之間)
t:樣品厚度,I0:入射光強度,If:反射光強度
當樣品很厚時,此時µt<<1,
φ µ θ
µ µ
π ε
sin ) ( sin
) (
) (
0 4
f tot tot
x
f E E
I E I
+
= ∆Ω (1.5.11)
並且吸收原子的濃度不多的時候,µx<<µtot,If ~ I0
µ
x(E)這兩個條件可使螢光法得到最好的量測,但是當吸收原子的濃度增高時,即 µ ~µ 時,我們就不能忽略另外一項µ 的影響,這種現象稱為自我吸收效應,
射,入射光與晶格振動產生的聲子交互作用,使得散射光與原始入射光能量產生 差異,藉以瞭解晶格內部結構與有序程度。延伸X 光吸收精細結構(EXAFS)則是 產生了光電子,光電子以物質波的形式與周圍原子產生干涉,藉以瞭解周圍原子 種類、配位數與原子間的距離等物理量。X 光繞射則是利用布拉格繞射來瞭解晶 體的對稱群與結構。各種不同的光學方法最主要的就是希望能以非破壞性的量測 方式來探測物質中內部的結構。我們將要利用此三種光學技術,來探測出物體的 微觀結構,並且與巨觀的介電性質作連結。
1 1 1 . . . 7 7 7 參考資料
[1]R. D. Richtmyer, Dielectric Resonators, J. Appl. Phys., 10(1939), 391 [2]Terrell A. Vanderah, Talking Ceramics, Science, 298 (2002), pp. 1182 - 1184
[3]Ebbe Nyfors, “CYLINDRICAL MICROWAVE RESONATOR SENSORS FOR MEASURING MATERIALS UNDER FLOW”, PhD thesis, Department of Electrical and Communications Engineering, Helsinki University of Technology, Finland, 2000 [4] F. Galasso and J. Pyle, “Ordering in Compounds of the A(B’0.33Ta0.67)O3 Type”
Inorg. Chem., 2 (1963), 482.
[5]Hiroshi Tamura, Lattice vibrations of Ba(Zn1/3Ta2/3)O3 crystal with ordered perovskite structure, Jpn. J. Appl. Phys. 25 (1986) ,787.
[6]I. G. Siny, R. S. Katiyar, Cation arrangement in the complex perovskites and vibration spectra, J. Raman spectroscopy, 29 (1998), pp385-390.
[7]G Lucazeau, L Avello, “Raman spectroscopy in solid state physics and materical sciene. Theory. Techniques and applications”, Analusis, 23(1995), pp.301-311
[8]R. Loudon, “The Raman effect in crystals”, ADVANCE IN PHYSICS, 50 (2001), pp. 813-864
[9]H. M. Rietveld, “A profile refinement method for nuclear and magnetic structures”, J. Appl. Cryst., 2(1969), pp.65-71
[10]Larson, A. C. and Von Dreele, R. B., GSAS-General Structure Analysis System., Los Alamos National Laboratory, Los Alamos, New Mexico., 1994, LAUR 86-748.
[11]Koningsberger, D. C., and Prins, R., X-ray absorption principles, applications, techniques of EXAFS, SEXAFS and XANES, A Wiley-Interscience Publication.
[12]Lipkin, Harry J., Phase uncertainty and loss of interference in a simple model for mesoscopic Aharonov-Bohm experiments, Phys. Rev. A, 1990, 42, 49–54.
[13]Stern, Edward A., Theory of the Extended X-ray-Absorption Fine Structure, Phys.
Rev. B, 1974, 10, 3027.
[14]Newville, M., Ravel, B., Haskel, D., Rehr, J. J., Stern, E. A., and Yacoby, Y., Analysis of multiple-scattering XAFS data using theoretical standards, Physica B, 1995, 208-209, 154-156.
[15]Ravel, B., Practical introduction to multiple scattering theory, Journal of Alloys and Compounds, 2005, 401, 118-126.
[16]Pfalzer, P., Urbach, J. P., Elimination of self-absorption in fluorescence hard-X-ray absorption spectra, Phys. Rev. B, 1999, 60, 9335-9339.
[17]Wende, H, Recent advances in x-ray absorption spectroscopy, Rep. Prog. Phys., 2004, 67, 2105-2181.
[18]Azaroff, Leonid V., Theory of extended fine structure of X-ray absorption edges, Rev. Mod. Phys., 1963, 35, 1012-1021.