Experimental and theoretical study of the electronic structure of Fe
3Al, Fe
2VAl, and Fe
2VGa
L.-S. Hsu
Department of Physics, National Chang-Hua University of Education, Chang-Hua 50058, Taiwan, Republic of China
Y.-K. Wang and G. Y. Guo
Department of Physics, National Taiwan University, Taipei 106, Taiwan, Republic of China
C. S. Lue
Department of Physics, National Cheng Kung University, Tainan 70101, Taiwan, Republic of China
共Received 9 July 2002; revised 12 September 2002; published 18 November 2002兲The electronic structures of Fe
3Al, Fe
2VAl, and Fe
2VGa are studied by x-ray absorption near-edge spec- troscopy
共XANES兲 at the Fe and V K edges. The experimental XANES spectra are compared with thoseobtained from first-principles electronic-structure calculations. The experimental XANES features for these intermetallic compounds reflect the Fe- and V-p unoccupied partial density of states. The magnetic moments and the density of states at the Fermi energy at different atomic sites and spins are calculated and compared with experimental and previous theoretical values.
DOI: 10.1103/PhysRevB.66.205203 PACS number
共s兲: 78.70.Dm, 78.20.Ls, 75.50.CcI. INTRODUCTION
Intermetallic compound Fe
3Al crystallizes in a face- centered cubic 共fcc兲 structure (D03) 共see Fig. 1兲. There are three Fe atoms per unit cell belonging to two different types.
The first type consists of one Fe atom 关denoted by Fe共I兲兴 in the unit cell. It is surrounded by eight Fe atoms in an octa- hedral coordination. The second type consists of two Fe at- oms 关denoted by Fe共II兲兴 in the unit cell. They are surrounded by four Al and four Fe atoms in a tetrahedral coordination.
Fe
3Al has a number of unusual electronic and magnetic properties that make it attractive for high-temperature appli- cations. It is a ferromagnetic metal with T
c⫽713 K and spin magnetic moment of 2.2 B and 1.45
B for the Fe 共I兲 and Fe 共II兲 atoms, respectively.
1Its magnetic property is coupled strongly to its crystalline structure. When Fe 共I兲 is replaced by V, the resultant structure is a Heusler-type compound (Fe
2VAl) with the cubic L2
1lattice 共see Fig. 1兲. Although the Fe
3Ga phase does not exist, the Heusler-type compound Fe
2VGa also crystallizes in the L2
1structure. Interestingly, both Fe
2VAl and Fe
2VGa show the absence of ferromagnetic ordering.
While Fe
3Al has been classified as a ferromagnetic metal, semiconductinglike behavior was observed in Fe
2VAl and Fe
2VGa, as evidenced by their negative temperature coeffi- cient of electrical resistivity.
2,3Several theoretical calculations
4 – 8predicted the presence of a pseudogap in Fe
2VAl, due to the hybridization effects. The pseudogap for- mation arises from an indirect band overlap at the Fermi energy (E
F), and thus Fe
2VAl is characterized as a semi- metal. Results of nuclear magnetic resonance 共NMR兲 measurements
9,10are in good agreement with these calcula- tions. An optical-conductivity measurement on Fe
2VAl fur- ther confirmed the existence of a pseudogap at E
F.
11Band- structure calculations also indicated that Fe
2VGa is a semimetal,
7,8and a recent NMR study on this material has revealed a small density of states 共DOS兲 at EF within the
pseudogap,
12consistent with the semimetallic characteristics of this compound.
In this article, x-ray absorption near-edge spectra 共XANES兲 at the Fe and V K edges of Fe3Al, Fe
2VAl, and Fe
2VGa were measured and were compared with theoretical XANES spectra. Such analysis is especially important for establishing a relationship between the experimental XANES spectra and the unoccupied part of the local DOS and obtain- ing quantitative information about hole numbers and charge transfer in these intermetallic compounds. The total and site- and spin-decomposed magnetic moments and DOS’s at E
F 关n(E
F) 兴 of these three intermetallic compounds are cal- culated and compared with corresponding experimental and theoretical values. This article is organized as follows. In Sec. II, the experimental and the theoretical methods are de- scribed. In Sec. III, the results and discussion are presented.
The conclusions of this work are contained in Sec. IV.
FIG. 1. Unit cell of four interpenetrating fcc sublattices showing the four types of site at Al
共Ga兲 共000兲, B 共12,
12,
12兲, C 共14,
14,
14兲, andD
共34,
34,
34兲. Structures: D03Fe
3Al, B
⫽Fe共I兲, C⫽D⫽Fe共II兲;L2
1Fe
2VAl, and Fe
2VGa, B
⫽V, C⫽D⫽Fe.II. METHODS A. Experimental methods
The Fe
3Al polycrystalline sample was prepared and char- acterized as described previously.
13The Fe
2VAl and Fe
2VGa polycrystals were prepared by mixing appropriate amounts of high-purity elemental metals in a water-cooled copper cru- cible and then melting several times in an Ar arc furnace.
The resulting Fe
2VAl ingot was annealed in a vacuum-sealed (10
⫺5Torr) quartz tube at 1000 °C for two days, and at 400 °C for 12 h. The Fe
2VGa ingot was annealed at 600 °C and then 400 °C for two days. They were then furnace- cooled to room temperature. The same preparation technique has been used in other studies of these two materials,
14,15which are known to form in a single-phase L2
1Heusler-type structure. X-ray diffraction patterns were taken with the Cu K ␣ radiation on powdered substances, and the expected L2
1structure was observed for Fe
2VAl and Fe
2VGa. The lattice constants were determined to be 5.76 and 5.77 Å for Fe
2VAl and Fe
2VGa, respectively. These values are essentially the same as those reported in the literature.
2,3Wavelength disper- sive spectroscopy showed that these two compounds are uni- form in composition. No signs of a second phase were seen in this analysis.
The XANES experiments were carried out on beam lines 17C and 15B at the Synchrotron Radiation Research Center, Taiwan. A Si 共111兲 double-crystal monochromator was used and the typical energy resolution was about 1 eV. The XANES spectra were collected by recording the total yield of secondary electrons from the sample surfaces at room temperature. Standard 5- m-thick Fe and V foils were used as references for energy calibration. Their XANES data were also used as reference spectra. The photon flux was obtained simultaneously by measuring the current of a Au mesh lo- cated near the exit slit of the monochromator and used for normalization of the XANES spectra.
B. Theoretical methods
In order to calculate the theoretical XANES spectra and other physical parameters of Fe
3Al, Fe
2VAl, and Fe
2VGa, we use the highly accurate all-electron full-potential linear augmented-plane-wave 共FLAPW兲 method.16 The calcula- tions are based on the first-principles density-functional theory with the generalized gradient approximation 共GGA兲
to the exchange-correlation potential.
17We begin the calcu- lations by determining the total energies at several values of the lattice constants near the experimental ones. These ener- gies are then fitted to the Murnaghan equation of state
18to obtain the theoretical equilibrium lattice constants and bulk moduli. The theoretical and experimental lattice constants of these three compounds are shown in the fourth column in Table I. We note that previous authors
4used local spin- density approximation 共LSDA兲 to calculate the electronic structures of Fe
3Al and Fe
2VAl. We thus also used LSDA to calculate the theoretical equilibrium lattice constants of Fe
2VAl 共5.595 Å兲 and Fe2VGa 共5.99 Å兲. Obviously, these two values differ more from the respective experimental val- ues than those calculated with the GGA method 共see Table I兲.
Thus, GGA seems more accurate than LSDA in calculating the electronic structures of these compounds, and we will use it in all the calculations. The muffin-tin radius of 1.27 and 1.22 Å is used for Fe 共V兲 and Al共Ga兲 atoms, respectively. The wave functions, the charge densities, and the potentials are expanded in terms of the spherical harmonics inside the muffin-tin spheres. The cutoff angular momentum (l
max) is ten for the wave functions and six for the charge densities and the potentials. The Brillouin zone 共BZ兲 integration is carried out by using the improved tetrahedron method.
19The number of the augmented plane waves included is 96 per atom, i.e., R
mtK
max⫽9.
16The number of the k points in the irreducible BZ wedge 共IBZW兲 used in the self-consistent cal- culations is 47. To calculate the DOS curves and the theoret- ical XANES spectra, 286 k points in the IBZW were used.
We have checked the convergence of the calculated eigen- values with respect to the number of augmented plane waves used and the number of k points used.
III. RESULTS AND DISCUSSIONS
The calculated total energy of the ferromagnetic Fe
3Al phase is lower than that of the paramagnetic one by 1.13 eV per formula unit. Thus, the XANES spectrum obtained from the spin-polarized calculation of Fe
3Al is used to compare with the experimental spectrum. On the other hand, since Fe
2VAl and Fe
2VGa are nonmagnetic,
4the theoretical XANES spectra of the paramagnetic state are used for com- paring with the experimental XANES spectra. We did per- form the spin-polarized calculations of Fe
2VAl and Fe
2VGa, TABLE I. Crystal structure, lattice constant a, total density of states at the Fermi energy n(E
F), and bulk
modulus for Fe
3Al, Fe
2VAl, and Fe
2VGa. ‘‘Para.,’’ ‘‘ferro.,’’ and ‘‘exp.’’ represent theoretical paramagnetic, theoretical ferromagnetic, and experimental values, respectively.
Material Crystal structure a
共Å兲n(E
F)
共states/eV cell兲B
共Mbar兲Fe
3Al D0
3para. 5.645 11.73 2.04
ferro. 5.761 2.78 1.67
exp. 5.789 3.09
Fe
2VAl L2
1para. 5.712 0.18 2.12
exp. 5.76
Fe
2VGa L2
1para. 5.726 0.28 2.14
exp. 5.77
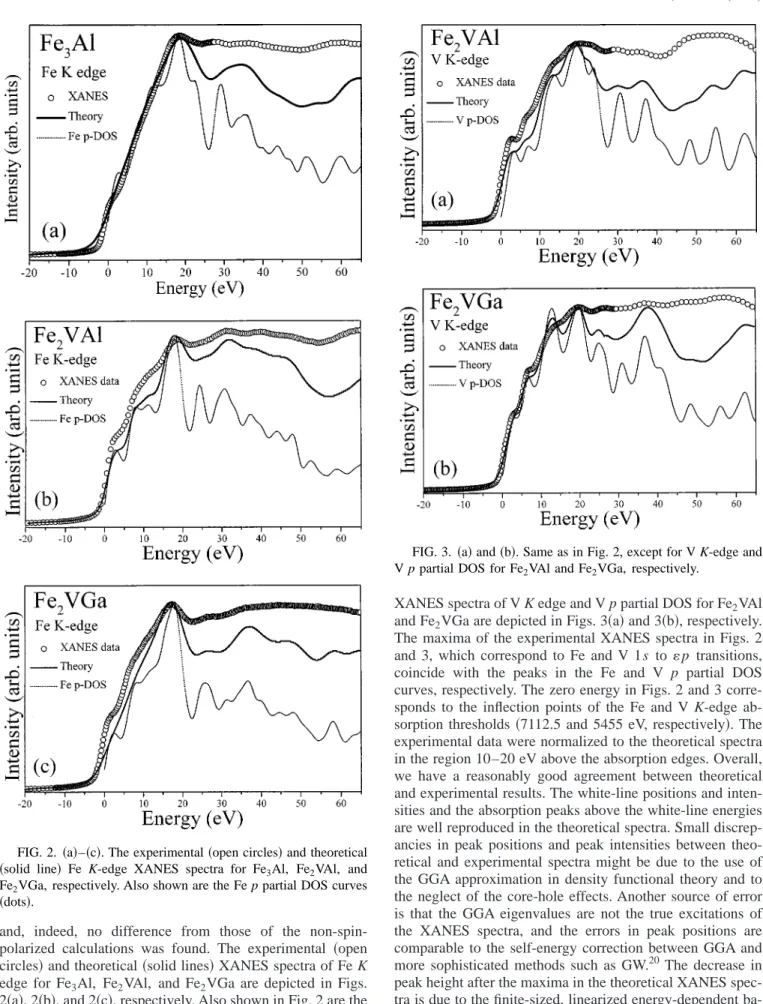
and, indeed, no difference from those of the non-spin- polarized calculations was found. The experimental 共open circles 兲 and theoretical 共solid lines兲 XANES spectra of Fe K edge for Fe
3Al, Fe
2VAl, and Fe
2VGa are depicted in Figs.
2 共a兲, 2共b兲, and 2共c兲, respectively. Also shown in Fig. 2 are the Fe p partial DOS 共dots兲 convoluted by the experimental reso- lution of 1 eV for these compounds. The corresponding
XANES spectra of V K edge and V p partial DOS for Fe
2VAl and Fe
2VGa are depicted in Figs. 3 共a兲 and 3共b兲, respectively.
The maxima of the experimental XANES spectra in Figs. 2 and 3, which correspond to Fe and V 1s to p transitions, coincide with the peaks in the Fe and V p partial DOS curves, respectively. The zero energy in Figs. 2 and 3 corre- sponds to the inflection points of the Fe and V K-edge ab- sorption thresholds 共7112.5 and 5455 eV, respectively兲. The experimental data were normalized to the theoretical spectra in the region 10–20 eV above the absorption edges. Overall, we have a reasonably good agreement between theoretical and experimental results. The white-line positions and inten- sities and the absorption peaks above the white-line energies are well reproduced in the theoretical spectra. Small discrep- ancies in peak positions and peak intensities between theo- retical and experimental spectra might be due to the use of the GGA approximation in density functional theory and to the neglect of the core-hole effects. Another source of error is that the GGA eigenvalues are not the true excitations of the XANES spectra, and the errors in peak positions are comparable to the self-energy correction between GGA and more sophisticated methods such as GW.
20The decrease in peak height after the maxima in the theoretical XANES spec- tra is due to the finite-sized, linearized energy-dependent ba- sis sets in the muffin-tin spheres used in the present calcula- tions. As for the source of the discrepancy in peak width, the FIG. 2.
共a兲–共c兲. The experimental 共open circles兲 and theoretical共solid line兲 Fe K-edge XANES spectra for Fe3
Al, Fe
2VAl, and Fe
2VGa, respectively. Also shown are the Fe p partial DOS curves
共dots兲.FIG. 3.
共a兲 and 共b兲. Same as in Fig. 2, except for V K-edge andV p partial DOS for Fe
2VAl and Fe
2VGa, respectively.
hot-electron lifetime is strongly energy dependent so that it mostly damps peaks about 20 eV above the absorption edge.
The calculated and experimental total n(E
F) values for Fe
3Al, Fe
2VAl, and Fe
2VGa are listed in the fifth column in Table I, and the site- and spin-decomposed n(E
F) values are listed in Table II. Also included in Table II are those values for Fe metal. The total n(E
F) value for ferromagnetic Fe
3Al is 2.78 states/eV cell, which yields the electron-phonon mass enhancement factor of 0.11 when compared with the experi- mental value 共3.09 states/eV cell兲21 of DO
3Fe
3Al obtained from specific-heat measurement. We note that the experi- mental n(E
F) value for Fe
3Al could increase by a maximum of 20% from the above-sited value when the sample was heat treated.
21 This is because various heat treatment of Fe
3Al changes the local atomic environment of the Fe atoms to form different phases.
22 The ferromagnetic n(E
F) value for Fe
3Al in Table I is essentially the same as that 共2.75 states/eV cell 兲4 obtained from a previous self-consistent cal- culation and is much smaller than the earlier non-self- consistently calculated value 共5.79 states/eV cell兲.23 From Table II, one can see that the spin-up and spin-down n(E
F) values of Fe 共I兲 atoms in Fe3Al are close to those of the Fe metal, but the n(E
F) value of the spin-down Fe 共II兲 atoms in Fe
3Al is considerably larger than n(E
F) values of the Fe metal. This implies that the contribution to the total n(E
F) value for Fe
3Al is dominated by the spin-down Fe 共II兲 3d electrons, which was confirmed by earlier electrical- resistivity study.
24 The calculated n(E
F) values for Fe
2VAl and Fe
2VGa are 0.18 and 0.28 states/eV cell, respectively, and the major contribution to the higher n(E
F) value for Fe
2VGa comes from that of the V sites in this compound, as can be seen in Table II. The V partial d-spin contributions to the n(E
F) values for Fe
2VAl 共Ref. 10兲 and Fe2VGa 共Ref. 12兲 are 0.023 and 0.085 states/eV cell, respectively, as derived from the NMR studies on these two compounds. Our theo- retical values 共0.062 and 0.128 states/eV cell for Fe2VAl and Fe
2VGa, respectively 兲 are larger than these two experimental ones. On the other hand, our calculated total n(E
F) value for Fe
2VAl is essentially the same as that calculated previously 共0.1 states/eV cell兲.6We note that the theoretical and NMR- derived n(E
F) values for Fe
2VAl and Fe
2VGa are consider- ably larger than the experimental ones obtained from specific-heat measurements.
14
obtained from a previous self-consistent cal- culation and is much smaller than the earlier non-self- consistently calculated value 共5.79 states/eV cell兲.23 From Table II, one can see that the spin-up and spin-down n(E
F) values of Fe 共I兲 atoms in Fe3Al are close to those of the Fe metal, but the n(E
F) value of the spin-down Fe 共II兲 atoms in Fe
3Al is considerably larger than n(E
F) values of the Fe metal. This implies that the contribution to the total n(E
F) value for Fe
3Al is dominated by the spin-down Fe 共II兲 3d electrons, which was confirmed by earlier electrical- resistivity study.
24 The calculated n(E
F) values for Fe
2VAl and Fe
2VGa are 0.18 and 0.28 states/eV cell, respectively, and the major contribution to the higher n(E
F) value for Fe
2VGa comes from that of the V sites in this compound, as can be seen in Table II. The V partial d-spin contributions to the n(E
F) values for Fe
2VAl 共Ref. 10兲 and Fe2VGa 共Ref. 12兲 are 0.023 and 0.085 states/eV cell, respectively, as derived from the NMR studies on these two compounds. Our theo- retical values 共0.062 and 0.128 states/eV cell for Fe2VAl and Fe
2VGa, respectively 兲 are larger than these two experimental ones. On the other hand, our calculated total n(E
F) value for Fe
2VAl is essentially the same as that calculated previously 共0.1 states/eV cell兲.6We note that the theoretical and NMR- derived n(E
F) values for Fe
2VAl and Fe
2VGa are consider- ably larger than the experimental ones obtained from specific-heat measurements.
14
Al are close to those of the Fe metal, but the n(E
F) value of the spin-down Fe 共II兲 atoms in Fe
3Al is considerably larger than n(E
F) values of the Fe metal. This implies that the contribution to the total n(E
F) value for Fe
3Al is dominated by the spin-down Fe 共II兲 3d electrons, which was confirmed by earlier electrical- resistivity study.
24The calculated n(E
F) values for Fe
2VAl and Fe
2VGa are 0.18 and 0.28 states/eV cell, respectively, and the major contribution to the higher n(E
F) value for Fe
2VGa comes from that of the V sites in this compound, as can be seen in Table II. The V partial d-spin contributions to the n(E
F) values for Fe
2VAl 共Ref. 10兲 and Fe2VGa 共Ref. 12兲 are 0.023 and 0.085 states/eV cell, respectively, as derived from the NMR studies on these two compounds. Our theo- retical values 共0.062 and 0.128 states/eV cell for Fe2VAl and Fe
2VGa, respectively 兲 are larger than these two experimental ones. On the other hand, our calculated total n(E
F) value for Fe
2VAl is essentially the same as that calculated previously 共0.1 states/eV cell兲.6We note that the theoretical and NMR- derived n(E
F) values for Fe
2VAl and Fe
2VGa are consider- ably larger than the experimental ones obtained from specific-heat measurements.
14
VAl and Fe
2VGa, respectively 兲 are larger than these two experimental ones. On the other hand, our calculated total n(E
F) value for Fe
2VAl is essentially the same as that calculated previously 共0.1 states/eV cell兲.6We note that the theoretical and NMR- derived n(E
F) values for Fe
2VAl and Fe
2VGa are consider- ably larger than the experimental ones obtained from specific-heat measurements.
14
The calculated paramagnetic and ferromagnetic values of
the bulk modulus 共B兲 for Fe3Al are 2.04 and 1.67 Mbar, respectively, and are listed in the last column in Table I. The previously reported theoretical B values 关1.59 共Ref. 22兲 and 1.83 共Ref. 25兲 Mbar兴 for Fe3Al are close to our ferromag- netic B value. The calculated B values for Fe, Fe
2VAl, and Fe
2VGa are 1.85, 2.12, and 2.14 Mbar, respectively. To our knowledge, there are no experimental B values reported for Fe
3Al, Fe
2VAl, and Fe
2VGa.
Al are close to our ferromag- netic B value. The calculated B values for Fe, Fe
2VAl, and Fe
2VGa are 1.85, 2.12, and 2.14 Mbar, respectively. To our knowledge, there are no experimental B values reported for Fe
3Al, Fe
2VAl, and Fe
2VGa.
In Table II, the calculated site-projected and total mag- netic moments ( M
s) are listed in the first row for Fe and Fe
3Al. For Fe
3Al, the M
svalue is 2.51
B/atom for the Fe atoms on sites with eight Fe nearest neighbors 关Fe共I兲 atoms兴, 1.96
B/atom for the other Fe atoms on sites with four Fe and four Al nearest neighbors 关Fe共II兲 atoms兴, and
⫺0.12 B/atom for the Al atoms. These values are close to those reported previously by other theorists 共2.4, 1.8, and
⫺0.13 B/atom, respectively 兲
25 and experimentalists 关2.2 and 1.45
B/atom, respectively, for Fe 共I兲 and Fe共II兲 atoms兴.1
Earlier experimentalists,
26 using neutron-diffraction mea- surement, reported similar M
s values for Fe
3Al at the Fe 共I兲 (2.18
B/atom) and Fe 共II兲 (1.50
B/atom) atomic sites.
However, the M
svalues 共2.16 and 1.26
B/atom, respec- tively 兲 obtained from a non-self-consistent calculation23 are smaller than those obtained from present self-consistent cal- culation. We should mention that, in Table II, the Fe 共I兲 atoms in Fe
3Al possess nearly the same M
s value as Fe atoms in bcc Fe metal, which also have eight Fe nearest neighbors.
This observation was confirmed by neutron-diffraction study.
27On the other hand, the difference between the M
svalues of the Fe 共II兲 atoms in Fe3Al and of the Fe atoms in Fe metal is attributed to the increase of DOS near the top of the spin-down Fe 3d band in Fe
3Al. Thus, the Fe moment in Fe and Fe
3Al is only a function of the number of nearest Fe neighbors. The total M
s value for Fe
3Al is 6.10
B/atom, which is close to the experimental values of 5.61
B/atom.
28
The number of Fe d electrons calculated for Fe
3Al is 6.25, which is close to the value 共6.59兲 reported previously.29The number of Fe d electrons for Fe metal calculated in this work is 6.15. The number of Fe d electrons calculated for Fe
2VAl and Fe
2VGa is 6.25 and 6.26, respectively; while that for V d electrons is 2.98 and 2.99, respectively. The constancy in Fe d electrons between Fe metal and these three intermetallic compounds is also observed in a lot of other metallic alloys.
29
The number of Fe d electrons for Fe metal calculated in this work is 6.15. The number of Fe d electrons calculated for Fe
2VAl and Fe
2VGa is 6.25 and 6.26, respectively; while that for V d electrons is 2.98 and 2.99, respectively. The constancy in Fe d electrons between Fe metal and these three intermetallic compounds is also observed in a lot of other metallic alloys.
29TABLE II. Magnetic moment M
s(
B/atom) and spin-up and spin-down density of states at the Fermi energy n(E
F) ↑ and n(E
F) ↓
共states/eV cell兲, respectively, at different atomic sites for Fe, Fe3Al, Fe
2VAl, and Fe
2VGa.
Material Fe
共I兲Fe
共II兲V Al
共Ga兲interstitial total
Fe M
s2.45
⫺0.072.38
n(E
F) ↑ 0.82 0.02 0.84
n(E
F) ↓ 0.23 0.03 0.26
Fe
3Al M
s2.51 1.96
⫺0.12 ⫺0.216.10
n(E
F) ↑ 0.20 0.36 0.03 0.07 0.66
n(E
F) ↓ 0.07 1.83 0.06 0.16 2.12
Fe
2VAl n(E
F) 0.096 0.062 0.007 0.017 0.18
Fe
2VGa n(E
F) 0.119 0.128 0.008 0.024 0.28
IV. CONCLUSIONS
The electronic structures of Fe
3Al, Fe
2VAl, and Fe
2VGa are studied jointly by XANES measurements at the Fe and V K edges and by theoretical analysis using FLAPW method. A reasonably good agreement between theory and experiment is found. The experimental XANES features for these com- pounds reflect the Fe- and V-p unoccupied density of states.
The magnetic moments and the density of states at the Fermi
energy at different atomic sites and spins are calculated and compared with experimental and other theoretical values.
ACKNOWLEDGMENTS
The authors thank J.-F. Lee and H. H. Hong for their help in data taking. This work was partially supported by grants from the National Science Council, Taiwan, Republic of China.
1
T. J. Burch, K. Raj, P. Jena, J. I. Budnick, V. Niculescu, and W. B.
Muir, Phys. Rev. B 19, 2933
共1979兲.2
Y. Nishino, M. Kato, S. Asano, K. Soda, M. Hayasaki, and U.
Mizutani, Phys. Rev. Lett. 79, 1909
共1997兲.3
K. Endo, H. Matsuda, K. Ooiwa, M. Iijima, K. Ito, T. Goto, and A. Ono, J. Phys. Soc. Jpn. 66, 1257
共1997兲; K. Endo, H. Mat-suda, K. Ooiwa, M. Iijima, T. Goto, K. Sato, and I. Umehara, J.
Magn. Magn. Mater. 177– 181, 1437
共1998兲.4
G. Y. Guo, G. A. Botton, and Y. Nishino, J. Phys.: Condens.
Matter 10, L119
共1998兲.5
D. J. Singh and I. I. Mazin, Phys. Rev. B 57, 14 352
共1998兲.6
R. Weht and W. E. Pickett, Phys. Rev. B 58, 6855
共1998兲.7
M. Weinert and R. E. Watson, Phys. Rev. B 58, 9732
共1998兲.8
A. Bansil, S. Kaprzyk, P. E. Mijnarends, and J. Tobola, Phys. Rev.
B 60, 13 396
共1999兲.9
C.-S. Lue and J. H. Ross, Jr., Phys. Rev. B 58, 9763
共1998兲.10
C. S. Lue and J. H. Ross, Jr., Phys. Rev. B 61, 9863
共2000兲.11
H. Okamura, J. Kawahara, T. Nanba, S. Kimura, K. Soda, U.
Mizutani, Y. Nishino, M. Kato, I. Shimoyama, H. Miura, K.
Fukui, K. Nakagawa, H. Nakagawa, and T. Kinoshita, Phys.
Rev. Lett. 84, 3674
共2000兲.12
C.-S. Lue and J. H. Ross, Jr., Phys. Rev. B 63, 054420
共2001兲.13
L.-S. Hsu, Y.-K. Wang, and G. Y. Guo, Nucl. Instrum. Methods Phys. Res. B
共to be published兲.14
C. S. Lue, J. H. Ross, Jr., C. F. Chang, and H. D. Yang, Phys. Rev.
B 60, R13 941
共1999兲.15
C. S. Lue, J. H. Ross, Jr., K. D. D. Rathnayaka, D. G. Naugle, S.
Y. Wu, and W.-H. Li, J. Phys.: Condens. Matter 13, 1585
共2001兲.16
P. Blaha, K. Schwarz, G. K. H. Madsen, D. Kvasnicka, and J.
Luitz,
WIEN2K, An Augmented Plane Wave
⫹Local Orbitals Pro-gram for Calculating Crystal Properties, Techn. University Wien, Austria, 2001.
17
J. P. Perdew, K. Burke, and M. Ernzerhof, Phys. Rev. Lett. 77, 3865
共1996兲.18
F. D. Murnaghan, Proc. Natl. Acad. Sci. U.S.A. 3, 244
共1944兲.19
P. E. Blochl, O. Jepsen, and O. K. Andersen, Phys. Rev. B 49, 16 223
共1994兲.20
P. Horsch, W. von der Linden, and W.-D. Lukas, Solid State Com- mun. 62, 359
共1987兲.21
H. Okamoto and P. A. Beck, Monatsh. Chem. 103, 907
共1972兲.22
R. Kuentzler, J. Phys.
共Paris兲 44, 1167 共1983兲.23
S. Ishida, J. Ishida, S. Asano, and J. Yamashita, J. Phys. Soc. Jpn.
41, 1570
共1976兲.24
W. B. Muir, J. I. Budnick, and K. Raj, Phys. Rev. B 25, 726
共1982兲.25
B. I. Min, T. Oguchi, H. J. F. Jansen, and A. J. Freeman, J. Magn.
Magn. Mater. 54– 57, 1091
共1986兲.26
S. J. Pickart and R. Nathans, Phys. Rev. 123, 1163
共1961兲.27
R. Nathans, M. T. Pigott, and C. G. Shull, J. Phys. Chem. Solids 6, 38
共1958兲.28
T. Wakiyama, J. Phys. Soc. Jpn. 32, 1222
共1972兲.29