以混合 0-1 變數線性規劃方法
尋找基因轉錄因子結合位點
研究生:傅昶瑞
指導教授:黎漢林
國立交通大學資訊管理研究所
摘要
基因轉錄因子結合位點 (Transcription factor binding site, TFBS) 的搜尋在基 因組的功能性分析上扮演關鍵性的角色。在眾多搜尋方法之中,以共同序列 (consensus sequence)為基礎的窮舉法相對最為準確,但其指數成長的運算量卻讓 這類方法無法搜尋較長的序列。藉由預先給定的樣版來輔助搜尋能顯著地減少運 算量,只是這類資訊較難以取得,也因此目前TFBS 的搜尋大多仍仰賴準確率低 但速度較快的啟發式方法。 為了能有效準確搜尋TFBS,本研究發展一套混合 0-1 線性規劃法來求解三 種不同類型的TFBS 搜尋問題。這包括固定樣版 TFBS 搜尋,模糊樣版 TFBS 搜 尋,以及無樣版TFBS 搜尋。本方法的優點包括:(1)以共同序列為基礎的設計, (2)可得到全域最佳解,以及(3)可套用結構性的限制以加速運算並提高準確率。 而在無樣版 TFBS 搜尋中,本方法更可以成功找到因結構鬆散而難以發現的 TFBS。本研究以多個範例來針對三種不同類型的 TFBS 搜尋問題進行一系列實 驗,也都在可接受的時間下成功找到了實際存在的TFBS。 關鍵詞:DNA-蛋白質交互作用,基因調控,轉錄因子結合位點,線性規劃,整 數規劃
A mixed 0-1 linear programming approach for
finding transcription factor binding sites
Student: James Changjui Fu
Advisor:Hanlin Li
Institute of Information Management
National Chiao Tung University
ABSTRACT
The discrimination of transcription factor binding sites (TFBS) in multiple DNA sequences is an essential work for function analysis of gene expression. Enumeration methods that search all possible patterns have best precision among all current algorithms but require an exponential computational time and have difficulties to search for longer patterns. A predefined shared pattern can notably prunes the searching space but such information is often unavailable. Finding unframed TFBS today still relies on heuristic approaches which compromise to accuracy.
To effectively find TFBS, this study develops a mixed 0-1 linear programming approach to solve a series of problems for issues including fixed-pattern TFBS finding, ambiguous spacer TFBS finding and pattern-free TFBS finding. The proposed method has the following advantages over current methods: (1) A pattern-driven instead of sample-driven (or sequence-driven) design; (2) A global optimal solution is promised; (3) Structural features of motifs are embeddable to help facilitate search process. And with pattern-free approaches we can successfully determine TFBS within dispersed spacers. We apply several experiments on every kind of TFBS finding programs and in these examples the real TFBS are successfully determined in an acceptable computational time.
Keywords: DNA-protein interaction, gene regulation, transcription factor binding site,
誌謝
本研究的完成,首先要感謝我的指導教授黎漢林老師。沒有他當
初的提點與指導,我不會有今日豐碩的研究成果。另外我要特別感謝
我的口試委員游伯龍老師、曾國雄老師、陳茂生老師、唐傳義老師、
林妙聰老師、以及盧錦隆老師,在我撰寫博士論文期間,他們所給予
的寶貴建議與協助,使我的博士論文能夠有更為嚴謹完善的內容。
我更要感謝我最親愛的父母以及哥哥,他們對我不變的信心以及
鼓勵,是我完成學業最大的原動力。還有我的好友晏安,總能讓我在
意想不到的地方,得到更深一層的領悟與靈感。在博士研究的期間有
許多快樂美好的回憶,感謝在這段時間陪伴我的諸位同學朋友,讓我
的生活過得充實且多采多姿。深深地感謝大家,希望未來我們能在更
大的舞台上相聚。
CONTENTS
摘要 i Abstract ii 誌謝 iii Contents iv Table vi Figure viiChapter 1 Introduction
1
1.1 DNA-binding Motifs and Their Binding Sites 1
1.2 TFBS finding problem 7
1.3 Formulation of Pattern-driven TFBS Finding 12
Chapter 2 Propositions
19
2.1 Relaxation of Binary Indicator z 19
2.2 Disaggregated Nonlinear Formulation 20
2.3 Replacement of Mixed 0-1 Product Terms 22
Chapter 3 Model 1: Fixed-pattern TFBS Finding
25
3.1 Mixed 0-1 linear program for Fixed-pattern TFBS Finding 25
3.2 Structural Constraints 27
3.3 Suboptimal Consensus 28
3.4 General Exception Constraints 29
3.5 Experimental Results 30
Chapter 4 Model 2: Ambiguous-spacer TFBS Finding
34
4.1 Problem of Ambiguous-spacer TFBS Finding 34
4.2 Mixed 0-1 linear program for Ambiguous-spacer TFBS Finding 36
4.3 Experimental Results 38
Chapter 5 Model 3: Pattern-free TFBS Finding
41
5.1 Problem of Pattern-free TFBS Finding 42
5.2 Formulation and Linearization 43
5.3 Structural Constraints 47
5.4 Suboptimal Solutions and Exception Rules 55
5.5 Complexity Analysis of Searching Space 56
5.6 Experimental Results 58
5.7 Software Package: “Global Site Seer v2” 61
Chapter 6 Discussion
62
6.1 Features of Proposed Methods 62
6.2 Issues in Approach Design 63
6.3 Benchmark Results 64
Chapter 7 Concluding Remarks
66
Appendix
68
TABLE
Table 1 IUPAC code for nucleic acids 8
Table 2 The binary coding for each four bases 5
Table 3 Illustrative table of base comparison 5
Table 4 FNR binding sites found by program (P3) 40
Table 5 Binary base codes for pattern-free consensus 44
Table 6 A Prediction of daf-19 binding sites 60
Table 7 A Prediction of lin-32 binding sites 60
FIGURE
Figure 1 Gene expression. 2
Figure 2 Zinc-containing modules 3 Figure 3 Homeodomain-DNA complex 4 Figure 4 Dimerized DNA binding domain 5 Figure 5 PWM and IC representation 9
Figure 6 Site extraction for fixed-pattern TFBS finding 14
Figure 7 A mixed 0-1 nonlinear program for finding CRP-binding sites 17
Figure 8 Experiments for fixed-pattern TFBS finding 32
Figure 9 Site extraction for ambiguous-spacer TFBS finding 35
Figure 10 Experiments of ambiguous-spacer TFBS finding 39
Figure 11 Site extraction for pattern-free TFBS finding 43
Figure 12 Extraction of original and inv/comp candidate sets 49 Figure 13 λ repressor, an example of dimerized binding protein 51
Figure 14 Two candidate sets for even-spacer and odd-spacer dyad motifs 52
Figure 15 Zif268 zinc finger regulator, an example of series-type regulator 55
Chapter 1 Introduction
For past two decades, biologists have sequenced more and more complete genome sets of various species. To extract all the secrets of life from these huge data, procedures of how genes work in organism are continuously researched and discussed. Gene transcription, a primary gateway to gene function, is controlled by a complex regulatory mechanism in which many specific regulatory proteins bind to local regions of gene upstream, called transcription factor binding sites (TFBS), to control the gene expression. Therefore, the discrimination of TFBS from DNA sequences therefore becomes an essential work for genome function analysis.
1.1
DNA-binding Motifs and Their Binding Sites
Before the discussion about TFBS, we need to know the mechanism of gene regulatory. DNA transcription is the very first stage of gene expression. The complexes of Rribonucleic acid (RNA) polymerases and general transcription factors transcribe all kinds of genes at a basal level—like an idling engine—to remain the minimum operation. In fact, the transcription of active genes generally rises far above this basal level. To provide the needed extra boost in transcription, additional
gene-specific transcription factors (TF) play the critical role to control the throttle.
These transcription factors, also called regulators or activators, are like a set of keys capable of unlocking or locking the transcription. They bind to specific locations—like many particular keyholes—to stimulate or inhibit RNA polymerase to transcribe a gene. The activation of a gene relies on presence of all required enhancers and absence of all inhibitors (or at a low safe level).
Activators have at least two functional domains: a DNA-binding domain and a transcription-activation domain. Many also have a dimerization domain that allows
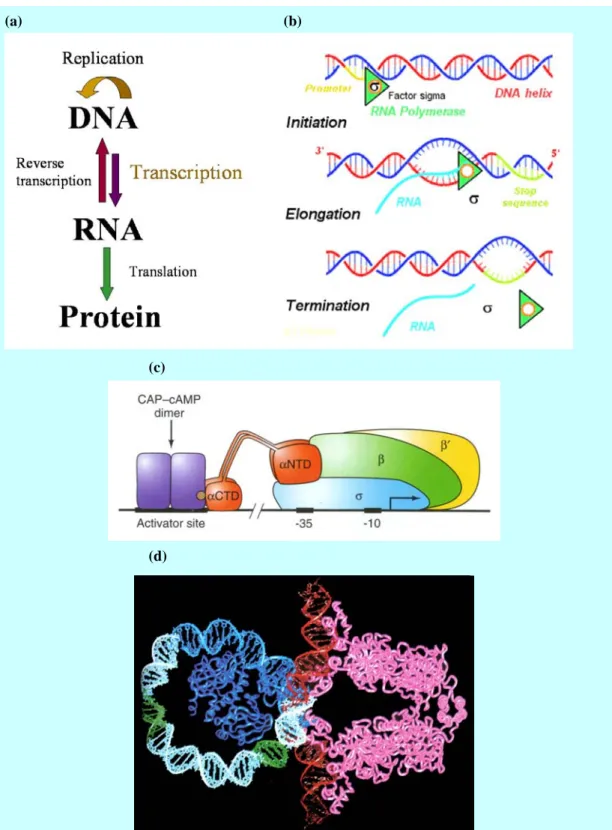
(a) (b)
(c)
(d)
Figure 1 Gene expression. (a) Central dogma; (b) Transcription of a gene in prokaryotes; (c) The complex of DNA strand, RNA polymerase, general transcription factors and CAP-cAMP (CRP) dimer—a gene specific transcription factor; (d) Computer graphic of lac repressor (pink) and CRP dimer (blue) binding to DNA.
the activators to bind to each others, forming homodimer (two identical monomers bound together), heterodimers (two different monomers bound together), or even higher multimers such as tetramers [Weaver, 2002]. Each DNA-binding domain, the most part we concern about, has a DNA-binding motif, which is the part of the domain that has a characteristic specialized for specific DNA binding. Most DNA-binding motifs fall into the following classes:
1. Zinc-containing modules. These modules use one or more zinc ions to create a
proper shape to bind to DNA and include at least three kinds of modules. The most often seen is zinc fingers, which is a chain of two or more zinc finger (a)
(b)
Figure 2 Zinc-containing modules: (a) Zinc fingers (Zif268), consisted by a series of zinc finger which contains a zinc ion; (b) The GAL4 protein, a dimerized motif which contains two zinc ions in each monomer.
monomers. Some zinc containing motifs also have dimerization domain containing two identical monomers, e.g. the GAL4 motif.
2. Homeodomains (HDs). These resemble in structure and function the
helix-turn-helix DNA-binding domains such as the λ phage repressor. The mutation of their gene may cause severe deformation. Most homeodomain proteins have weak DNA-binding specificity and rely on other proteins to help them bind specifically and efficiently to their DNA targets.
3. bZIP and bHLH motifs. Most DNA-binding motifs are of this type. They have a
highly basic DNA-binding motif linked to one or both of the protein dimerization motifs known as leucine zippers and helix-loop-helix (HLH) motifs. This kind of motifs have very strong DNA-binding specificity.
These three classes cover a large majority of DNA-binding motifs but certainly the list is not exhaustive. There are still other kinds of DNA-binding motifs not falling into any of these categories.
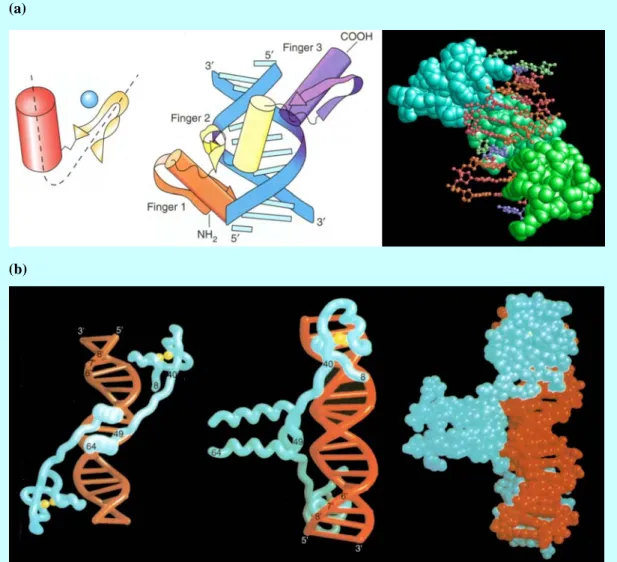
(a) (b)
Figure 3 Homeodomain-DNA complex in fruit fly Drosophila--an example of mono-type interaction. (a) Schematic representation; (b) A deformation caused by mutations in genes of these regulators: Antennapedia. It grows legs where antennae would normally be.
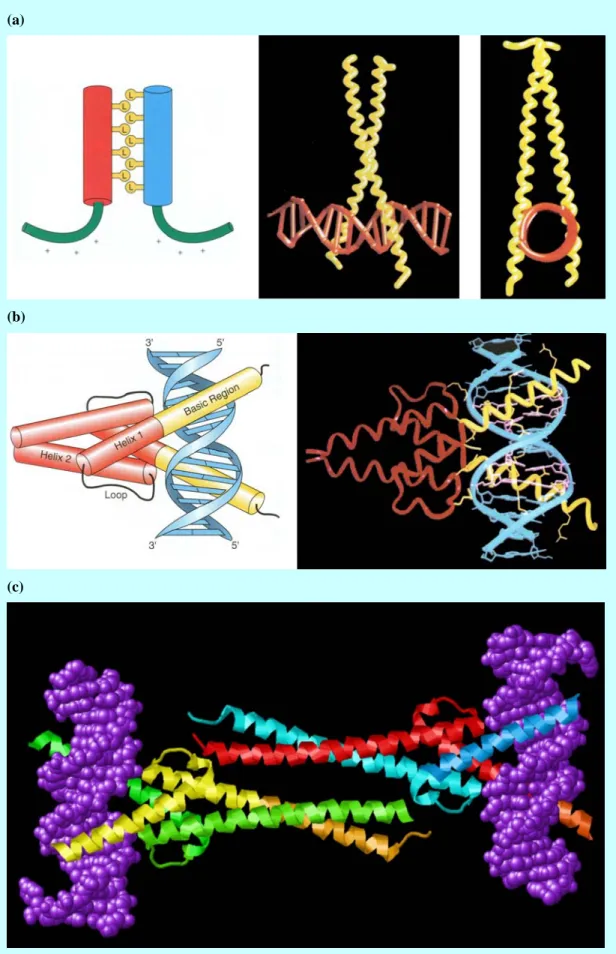
(a)
(b)
(c)
Figure 4 Dimerized DNA binding domain: (a) Leucine zipper (bZIP) complex. From left to right: dimerization of leucine zipper and two computer graph illustrating binding domain; (b) Two schematic diagrams of Helix-loop-helix (bHLH) complex; (c) Max-Myc heterotetramer.
Transcription factor binding site is a short region within a particular nucleotide sequence for a specific activator to bind. Because of various domains of DNA-protein interaction, TFBS linked to different kinds of DNA-binding motifs has particular characteristics for binding. Most TFBS can be categorized into three types:
(1) Mono-type TFBS. This kind of TFBS is for binding a monomer. DNA binding
domains like homeodomains have their binding sites of this type. Most mono-type TFBS are relatively weak signals and difficult to determined. In fact, their binding motifs usually require other auxiliary protein-protein interaction domains or DNA-protein binding domains to help their binding.
(2) Dyad-type TFBS. Dimerized regulators bind to this kind of TFBS. Dyad-type
TFBS is the most often seen type and generally not longer than 22 bases. It consists of two symmetrical half binding sites with a fixed number of in-between spacers. As a result this kind of TFBS has very strong binding specificity to regulators and relatively easy to determined.
(3) Series-type TFBS. The binding sites of chain-like regulatory protein like
zinc-fingers are of this type. This kind of TFBS contains several adjacent short units of the same size. For example of zinc fingers Zif268, it has binding site consisting of three units each of which is three-base long.
These types of TFBS have different features that make the specificity for recognition. These features can be regarded as logical rules that might be helpful for TFBS determination.
1.2
TFBS finding problem
To find TFBS, one has a collection of sequences that are known to contain binding sites for a common factor, but neither the positions of the sites nor the specificity of the factor are known. Besides that, TFBS are usually with some degree of ambiguity. These make TFBS finding a difficult and challenging problem. Experimental methods like DNA microarray (DeRisi et al., 1997; Lockhart et al., 1996) and SAGE (Velculescu et al., 2000) are capable to precisely elucidate TFBS. However, they are too laborious and time consuming to analyze enormous genome data. More and more computer based methods like enumeration methods, probability models and heuristics have been developed to find these conserved signals. In this section we discuss current computer-based (say in silico) approaches and their limitations.
Site Representation
Most transcription factor binding sites have variability on their component bases. With this ambiguity regulatory system can take advantage of level control on the gene expression. This makes the representation of DNA binding sites more complicated. How to precisely describe this variability depends on what kind of methods is applied in searching TFBS. Generally TFBS searching methods can be classified into two categories: pattern-driven approaches and sequence-driven (also called
alignment-driven) approaches. Pattern-driven approaches search for a consensus sequence which best fits all site occurrences. And the representation of this consensus
sequence includes simple DNA sequence and IUPAC (acronym of: International
Union of Pure and Applied Chemistry) code sequence. Sequence-driven approaches
information content (IC).
The simplest TFBS representation is merely a DNA sequence consisted only by A, T, G and C. Although incapable of describing base variability, this expression is still useful in pattern-driven enumeration methods. This is because flexible representation like IUPAC code will lead to enormous searching space in enumeration methods.
IUPAC is a degenerate naming rule consisting of 16 alphabets which describe various combinations of nucleic acids codes, shown in Table 1. Any kind of ambiguities in nucleic acids has a corresponding code and so IUPAC code can be used to completely describe a TFBS consensus. An obvious defect of IUPAC code is that it fails to describe the base preference level at each position. Position weight matrix (PWM) is designed for more precisely describing base variability.
In PWM the significance of a particular TFBS consensus is given by a measure of statistical surprise from multiple aligned short sequences. It calculates log
Table 1 IUPAC code for nucleic acids IUPAC code Description
A Adenine T Thymine C Cytosine G Guanine U Uracil R A, G (purine) Y T, C (pyrimidine) K G, T (keto) M A, C (amino) S G, C (strong) W A, T (weak) B T, G, C D A, T, G H A, T, C V A, G, C N A, T, G, C (any)
likelihood ratio (LLR) of four nucleic acids at each position as: b i b p f i b LLR , 2 log ) , ( = (1)
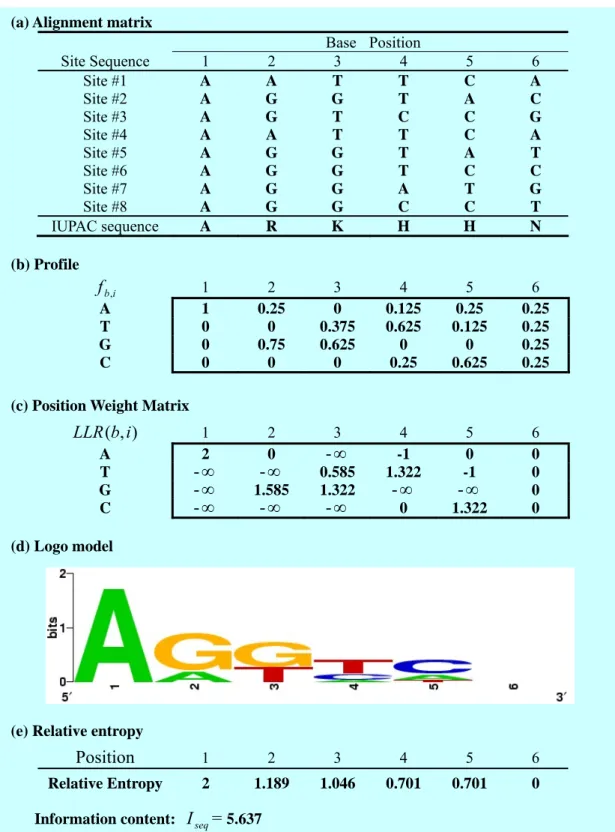
(a) Alignment matrix
Base Position Site Sequence 1 2 3 4 5 6 Site #1 A A T T C A Site #2 A G G T A C Site #3 A G T C C G Site #4 A A T T C A Site #5 A G G T A T Site #6 A G G T C C Site #7 A G G A T G Site #8 A G G C C T IUPAC sequence A R K H H N (b) Profile i b f , 1 2 3 4 5 6 A 1 0.25 0 0.125 0.25 0.25 T 0 0 0.375 0.625 0.125 0.25 G 0 0.75 0.625 0 0 0.25 C 0 0 0 0.25 0.625 0.25 (c) Position Weight Matrix
) , ( ib LLR 1 2 3 4 5 6 A 2 0 -∞ -1 0 0 T -∞ -∞ 0.585 1.322 -1 0 G -∞ 1.585 1.322 -∞ -∞ 0 C -∞ -∞ -∞ 0 1.322 0 (d) Logo model
(e) Relative entropy
Position 1 2 3 4 5 6
Relative Entropy 2 1.189 1.046 0.701 0.701 0 Information content: I = seq 5.637
Figure 5 PWM and IC representation: (a) aligned site sequences and their consensus as an IUPAC sequence; (b) the profile of these sites; (c) Position Weight Matrix; (d) Logo display of site sequences; (e) Information content of site sequences.
where i is the position within the site, b∈
{
A,T,G,C}
refers to each of the possible bases, fb,i is the observed frequency of each base at that position and p is the bfrequency of base b in the whole genome. The maximum LLR among each position
are summed up as the significance of a given set of sites.
Information content (IC, Schneider et al., 1986), which is also known as the Kullback-Leibler distance, is the sum of all relative entropies of four types of bases in all positions defined as below:
∑∑
= i b b i b i b seq p f f I , 2 , log (2)For pattern-driven methods, the criterion for best conserved consensus sequence is to find the one with maximum matches among all site occurrences. For sequence-driven methods, the criterion for identifying the best alignment of potential sites is to choose the one with highest information content I . seq
Site Discovering
Pattern recognition approaches can also be categorized into two classes: pattern-driven approaches and sequence-driven approaches. As previously mentioned, pattern-driven approaches search for consensus sequence which best fits all site occurrences. For consensus-based TFBS finding (Stormo, 2000), pattern-driven algorithms that test all 4mm-wide possible consensus sequences promise an optimal
solution but are very time consuming and impractical for large m. (Pesole et al., 1992;
Tompa, 1999) Many heuristics are developed to prune the huge searching space including testing only the substrings in the sequences (Li et al., 1999; Gelfand et al., 2000), specifying a shared pattern to restrict the locations of mismatches (Brazma et al., 1998; Califano, 2000; Sinha and Tompa, 2003; Régnier and Denise, 2004; Li and
Fu, 2005) and clustering (Buhler and Tompa, 2002; Pevzner and Sze, 2000; Liang et al., 2004). In addition to the exact enumeration methods, efficient data structure like suffix tree with fixed mismatches (Pavesi et al., 2001; 2004) can search for patterns of longer length. This kind of approaches is not exact enumeration algorithm and takes advantages of searching time polynomial to pattern length and exponential to the number of tolerant mutations.
Sequence-driven methods are designed based on probabilistic modeling. The challenge of sequence-driven approaches is to find the location of the sites and representative PWM using only the sequence data, without any assumptions on the statistical distributions of patterns in the sequences. The criterion for the best alignment is the one with maximum IC. Current methods include a greedy algorithm that builds up an entire alignment of sites by adding in new ones in each iteration (Stormo and Hartzell, 1989; Hertz et al., 1990) and expectation maximization (EM) that iteratively substitute the location of sites by expected locations (Lawrence and Reilly, 1990) and its variant, Gibbs sampling (Lawrence et al., 1993) as a type of Markov chain Monte Carlo (MCMC) algorithm. EM algorithm is also implemented in the MEME program (Bailey and Elkan, 1995) which allows for the simultaneous identification of multiple patterns. Other implements of sequence-driven approaches include CONSENSUS (Hertz and Stormo, 1999), AlignACE (Hughes et al., 2000), ANN-spec (Workman and Stormo, 2000), BioProspector (Liu et al., 2001), MotifSampler (Thijs et al., 2001), GLAM (Frith et al., 2004), The Improbizer (Ao et al., 2004), QuickScore (Régnier and Denise, 2004), SesiMCMC (Favorov et al., 2004) and TFBSfinder (Tsai et al., 2006).
In most current TFBS finding methods, all the letters in the consensus sequence are treated as independent variables. Because only some bases in binding region are
reactive to the transcription factor, solving this problem by calculating scores of all bases may involve noise from bases inducting no interactions. Beside that, the assumption of independent and identically distributed bases in background is too strong. Even with a probability calculated from the sequence data, the contribution to the accuracy is still limited. Another type of heuristics include testing only the substrings in sequences and constructing data structures like a suffix tree or a graph to extract overrepresented signals. This kind of methods compromises to a possible situation of weeding out the exact consensus when all the motifs in sequences are somehow ambiguous.
Most current methods also have obstacles to involve specific TFBS features like inverted palindrome or direct repeats. By limitations from original concepts, statistical models like EM or HMM need a much more complex design to embed the structural features. In some tree-based enumeration methods it is even impossible to utilize these structural features.
1.3
Formulation of Pattern-driven TFBS Finding
In this study a pattern-driven approach utilizing mixed 0-1 linear program is proposed. A pattern-driven concept of discovering TFBS is to find the consensus which has maximum matches among all proposed sites from multiple sequences. This is a mixed 0-1 optimization problem and can be formulated as a mixed 0-1 nonlinear program. We start by formulating a fixed-pattern TFBS finding problem as a mixed 0-1 nonlinear program. In many cases a predefined shared-pattern is available from some preprocesses. This shared pattern provides information about positions of reactive bases in the binding sites and makes a TFBS finding problem relatively easier
to solve.
Representations of fixed-pattern TFBS finding
To find TFBS of a specific regulation, a set of DNA sequences upstream genes known co-regulated by the same factor is firstly prepared for analysis. A prerequisite condition is that this DNA sequence set shall be prepared having at least one
occurrence per sequence (OOPS). Namely, there exists at least one similar TFBS in each sequence. A pattern-driven TFBS finding problem is defined as:
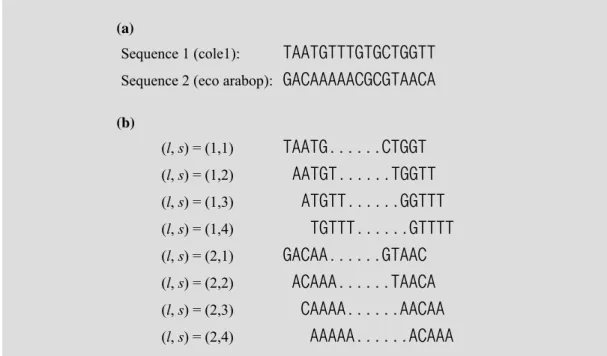
To find best conserved consensus sequence among the given sequence set, the first step is to generate a set of candidate sites from sequence data. We use the example of CRP-binding sites among DNA sequences of Escherichia coli (Stormo et
al., 1989), shown in Appendix, to illustrate the formulation. According to the predefined shared pattern, candidate sites are extracted from each starting position of each sequence, as shown in Figure 6, and indexed by (l, s) where l is the sequence
index and s is the start position. Denote dl,s,i ∈
{
A,T,G,C}
as the thi base present
in candidate site from (l, s) position.
With the pattern-driven concept, denote the consensus sequence to find as a series of binary variables. Every reactive base (i.e., the notation ‘N’ in the shared
Given
(i) A sequence set containing L sequences,
(ii) A shared pattern “NNNNN******NNNNN” in which ‘N’ and ‘*’ represent reactive and inactive bases respectively. To find the best conserved consensus sequence
x1x2x3x4x5∗∗∗∗∗∗x6x7x8x9x10,
pattern) in a consensus sequence is represented by two binary variables, u and v for four different nucleotides A, T, C and G, and indexed by its relative position, i.
Obviously the example of CRP-binding sites needs 20 binary variables to represent the consensus sequence. The binary codes for four nucleotide types are defined in Table.2. Each feasible consensus sequence with a vector of (u, v) pairs is scored by
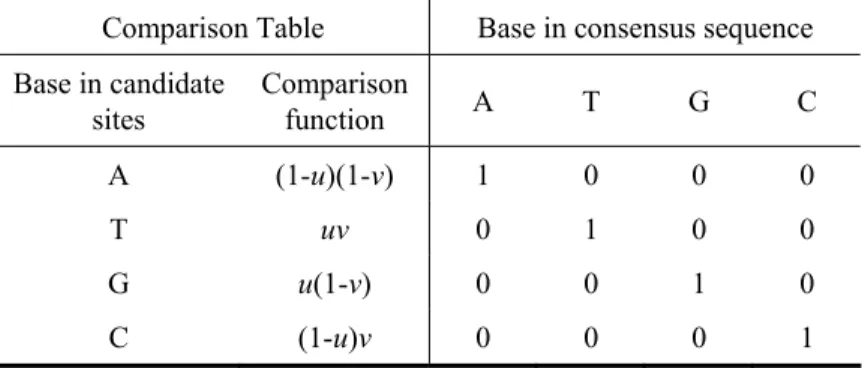
summing up base matches compared with the best fitting candidate site in every sequences. To formulate the comparison, every base appearing in a candidate site is represented by four kinds of comparison functions for different base types, as defined in follows. . ) 1 ( ), 1 ( , ), 1 )( 1 ( , C , G , T , A i i i i i i i i i i i i v u y v u y v u y v u y − = − = = − − = (3)
The illustrative table for the base comparison between the consensus sequence (a)
Sequence 1 (cole1): TAATGTTTGTGCTGGTT Sequence 2 (eco arabop): GACAAAAACGCGTAACA (b) (l, s) = (1,1) TAATG...CTGGT (l, s) = (1,2) AATGT...TGGTT (l, s) = (1,3) ATGTT...GGTTT (l, s) = (1,4) TGTTT...GTTTT (l, s) = (2,1) GACAA...GTAAC (l, s) = (2,2) ACAAA...TAACA (l, s) = (2,3) CAAAA...AACAA (l, s) = (2,4) AAAAA...ACAAA
Figure 6 Site extraction for fixed-pattern TFBS finding (The first two sequences in Appendix): (a) original sequence data; (b) schematic representation of the candidate sites.
and candidate sites is listed in Table 3.
Illustrative formulation for maximizing matches
The objective of a fixed-pattern TFBS finding is to find the consensus sequence best conserved among all the input DNA sequences. For every single DNA sequence, the scoring criterion is to compare its best fitting candidate sites with the consensus sequence and to count the base matches. This can be formulated as below:
⎭ ⎬ ⎫ ⎩ ⎨ ⎧ = + + + =
∑
∑
s s l s s l s l s l s l l z θ θ θ z Score max , ( ,,1 ,,2 ... ,,10) , 1 , (4)where zl,s is the binary indicator of whether the candidate site at (l, s) is chosen to
compare with the consensus. A candidate site is scored only when its corresponding z
equals 1. All other non-basic candidate sites will have its corresponding z valued 0.
For the assumption of one occurrence per sequence (OOPS), only the candidate site
Table 2 The binary coding for each four bases Base in consensus sequence u v A 0 0 T 1 1 G 1 0 C 0 1
Table 3 Illustrative table of base comparison
Comparison Table Base in consensus sequence Base in candidate sites Comparison function A T G C A (1-u)(1-v) 1 0 0 0 T uv 0 1 0 0 G u(1-v) 0 0 1 0 C (1-u)v 0 0 0 1
that best fits the consensus is to be scored in a sequence. That means for all l, . } 1 , 0 { , 1 , ,
∑
= ∈ s s l s l z z (5)In sequence scoring fiunction (4), θl,s,i is the comparison function defined by the ith base in the candidate site from (l, s). That is,
⎪ ⎪ ⎩ ⎪ ⎪ ⎨ ⎧ = = = = = G. if C if T if A if , , , G , , , C , , , T , , , A , , i s l i i s l i i s l i i s l i i s l d y d y d y d y θ (6)
Every candidate site is evaluated by summing up θl ,,si’s for a given (u, v) pair. For example of the first candidate site in Figure 6, “TAATG...CTGGT”, the site score (i.e. number of base matches) is obtained from
∑
= 10 1 ,, i θlsi = ) (yT,1+ yA,2 + yA,3 +yT,4 +yG,5 +yC,6 +yT,7 +yG,8 + yG,9 + yT,10 . When comparing with a consensus “TCATG******CATGA”, this score function will give 6 as the site score for six matched letters.
The matching score of sequence 1 in Figure 6 is formulated as
1 Score = max
{
z1,1(yT,1 +yA,2 +yA,3+ yT,4 + yG,5 + yC,6 + yT,7 + yG,8 +yG,9 +yT,10) ) ( A,1 A,2 T,3 G,4 T,5 T,6 G,7 G,8 T,9 T,10 2 , 1 y y y y y y y y y y z + + + + + + + + + + ) ( A,1 T,2 G,3 T,4 T,5 G,6 G,7 T,8 T,9 T,10 3 , 1 y y y y y y y y y y z + + + + + + + + + + ) ( T,1 G,2 T,3 T,4 T,5 G,6 T,7 T,8 T,9 T,10 4 , 1 y y y y y y y y y y z + + + + + + + + + +}
1∑
sz1,s = . (7)For a fixed-pattern TFBS finding problem, the objective is to maximize the total matches among all the sequence, i.e.,
∑
lScorel
sites in E.Coli (see Appendix for complete data set), the mixed 0-1 nonlinear program is formulated as shown in Figure 7. There are 18 sequences each of which 105-bp long in this example. Because the length of the given pattern is 16 (i.e. “NNNNN******NNNNN”), we have 90 candidate sites in each sequence. The independent variables include 20 binary variables (i.e. u and v) for consensus sequence and 18*90 binary variables (i.e. z) for indicating proposed sites. The notation y in this program is used as comparison function for different cases and the number of these constraints for comparison is 40. This program has 18 conservation
{
}
{
}
{ }
{
}
{
}
{
1,...,18}
,{
1,...,90}
. , 10 ,..., 1 , C G, T, A, ], 1 , 0 [ , 1 , 0 , , 10 ,..., 1 ), 1 ( , ) 1 ( , ), 1 )( 1 ( , 18 ,..., 1 1 ..., Score ..., Score ], ... [ ... ] ... [ ] ... [ Score ], ... [ ... ] ... [ ] ... [ Score s.t. Score Max , , G , C , T , A 90 1 , 18 3 10 , G 5 , T 4 , T 3 , T 2 , A 1 , C 90 , 2 10 , A 5 , A 4 , A 3 , A 2 , C 1 , A 2 , 2 10 , C 5 , A 4 , A 3 , C 2 , A 1 , G 1 , 2 2 10 , G 5 , C 4 , A 3 , C 2 , C 1 , T 90 , 1 10 , T 5 , T 4 , G 3 , T 2 , A 1 , A 2 , 1 10 , T 5 , G 4 , T 3 , A 2 , A 1 , T 1 , 1 1 18 1 ∈ ∈ ∈ ∈ ∈ ∈ ∈ ∀ ⎭ ⎬ ⎫ − = − = = − − = ∈ ∀ = = = + + + + + + + + + + + + + + + + + + + + + = + + + + + + + + + + + + + + + + + + + + + =∑
∑
= = s l i b z v u i v u y v u y v u y v u y l z y y y y y y z y y y y y y z y y y y y y z y y y y y y z y y y y y y z y y y y y y z s l i i i i i i i i i i i i i i s s l l l Mconstraints (i.e.
∑
, =1 ss l
z ) for assumption of one occurrence per sequence (OOPS)
and 18 scoring constraints. And so the total number of constraints is 76.
General formulation of fixed-pattern TFBS finding
The objective of fixed-pattern TFBS finding is to maximize the total matches among all the sequence, i.e.,
∑
lScorel
max . With sequence score defined as
(
)
{
∑
∑
}
= s ls i lsi
l z θ
Score max , ,, , we have the objective function more precisely described as:
∑∑
∑
⎟⎟ ⎠ ⎞ ⎜⎜ ⎝ ⎛ l s i i s l s l θ z, , , max . (8)Therefore a mixed 0-1 nonlinear program for fixed-pattern TFBS finding problem can be generally formulated as program (P1).
Mixed 0-1 Nonlinear Program for Fixed-pattern TFBS Finding
Maximize
∑∑
∑
⎟ ⎠ ⎞ ⎜ ⎝ ⎛ l s i i s l s l θ z, ,, (P1) Subject to{
1,...,}
, , ) 1 ( ), 1 ( , ), 1 )( 1 ( , C , G , T , A M i v u y v u y v u y v u y i i i i i i i i i i i i ∈ ∀ ⎪ ⎪ ⎭ ⎪ ⎪ ⎬ ⎫ − = − = = − − = ⎪ ⎪ ⎩ ⎪ ⎪ ⎨ ⎧ = = = = = G, if C, if T, if A, if , , , G , , , C , , , T , , , A , , i s l i i s l i i s l i i s l i i s l d y d y d y d y θ ∀l∈{
1,...,L}
∀s∈ℵ ∀i∈{
1,...,M}
,{
1,...,}
, , 1 , l L z s s l = ∀ ∈∑
{ }
{
}
{
}
{
1,...,}
, . , ,..., 1 , C G, T, A, ], 1 , 0 [ , 1 , 0 , , ℵ ∈ ∈ ∈ ∈ ∈ ∈ s L l M i b z v ui i lsChapter 2 Propositions
In the previous chapter we formulate a nonlinear program (P1) for fixed-pattern TFBS finding on DNA sequences. Unfortunately, (P1) is very hard to solve with current optimization tools because of numerous binary variables. On the other hand, with natures of nonlinear program containing product terms, (P1) can only obtain a local optimum. These defects quality make (P1) impractical. In this chapter we discuss techniques utilized to make (P1) solvable and even to conduct linearization which can obtain the global optimal solution.
2.1
Relaxation of Binary Indicator z
Program (P1) contains many binary variables which make it difficult to solve. The largest part of binary variables is from the indicators z. Every candidate site has a binary variable z indicating whether it best fits the consensus sequence. For example of finding CRP-binding sites, the formulation as program (P1) will have 1620 z’s. This large number of binary variables makes (P1) intractable. A linear relaxation on z is applicable to make (P1) solvable. For a TFBS finding problem, this relaxation provides a very tight bound to (P1). In fact, by the following proposition, it is proven having the same optimal value as (P1) has.
Proposition 1 A selection problem as
has a linear relaxation by loosing x to be continuous from 0 to 1 which shares i
Maximize (or minimize)
∑
icixiSubject to
∑
ixi =1,the same optimal value i
i c
max (or i
i c
min ). ■
Proof The proof is trivial. □
For Program (P1), the objective function (8) is separable by sequences, i.e.
(
)
∑
(
∑
(
∑
)
)
∑ ∑
∑
=l s ls i lsi l s zl,s iθl,s,i max z, θ,,
max . Because only one chosen (u, v)
pair is involved in each iteration,
∑
iθl,s,i can be regarded to as a constant. With Proposition 1 we can have the result that Program (P1) share the same optimal value,
(
)
∑
(
∑
(
∑
)
)
∑
(
∑
)
∑ ∑
l s zl,s∑
iθl,s,i = l max s zl,s iθl,s,i = l max iθl,s,imax , with a
relaxation where z’s are loosen to as continuous variables between 0 and 1. The enormous binary variables are therefore eliminated successfully and Program (P1) becomes manageable.
2.2
Disaggregated Nonlinear Formulation
To obtain the global optimal solution, program (P1) needs reformulated to a mixed 0-1 linear program. Before utilizing the linearization approach proposed in the following section, Program (P1) is firstly transformed to another formulation for effective elimination on all product terms. The formulation underlying the linearization process discussed in this chapter can be viewed as a disaggregated version of Program (P1).
Denote Sb,i as the index set of candidate sites having their ith base as
nucleotide type b, as defined as follows:
{
}
{
( , ) , , , A,T,G,C}
, = l s d =b b∈
Sbi lsi . (9)
) , ( all for , , , ,si bi bi l y l s S θ = ∈ . (10)
Then, with (9) and (10) the objective function (8)
∑
∑
⎟⎟ ⎠ ⎞ ⎜⎜ ⎝ ⎛ s l ls i lsi θ z , , ,, maxhas an equivalent disaggregated formulation as
∑
∑
⎟⎟ ⎠ ⎞ ⎜ ⎜ ⎝ ⎛ ∈ i b ls S s l i b i b z y , (,) , , , max . (11)Therefore program (P1) is reformulated as program (P1a) shown below:
Disaggregated version of (P1) Maximize
∑
∑
⎟⎟ ⎠ ⎞ ⎜ ⎜ ⎝ ⎛ ∈ i b ls S s l i b i b z y , (, ) , , , (P1a) Subject to{
1,...,10}
, , ) 1 ( ), 1 ( , ), 1 )( 1 ( , C , G , T , A ∈ ∀ ⎪ ⎪ ⎭ ⎪ ⎪ ⎬ ⎫ − = − = = − − = i v u y v u y v u y v u y i i i i i i i i i i i i{
(, ) ,,}
{
A,T,G,C}
{
1,...,10}
, , = l s d =b ∀b∈ ∀i∈ Sbi lsi{
1,...,}
, , 1 , l L z s s l = ∀ ∈∑
{ }
{
}
{
}
{
1,...,}
, . , 10 ,..., 1 , C G, T, A, ], 1 , 0 [ , 1 , 0 , , ℵ ∈ ∈ ∈ ∈ ∈ ∈ s L l i b z v ui i lsAn important progression from (P1) to (P1a) is elimination of an ambiguous term θl,s,i. This is very important for further linearization because it makes the
product term
∑
i lsi sl θ
z, ,, more explicit to eliminate.
2.3
Replacement of Mixed 0-1 Product Terms
Program (P1a) cannot find the global optimum because the product terms contained in the formulation. There are two kinds of product terms, uivi and
∑
ls ib z
y , , , conducting nonlinearity of (P1a). To make the program globally solvable, here we discuss how to eliminate product terms by a series of constraints.
The first kind of product term is u which exists in ivi yb,i. This product term consists only by binary variable and can be replaced by a continuous variable based on the following proposition.
Proposition 2 A general binary product
∏
=
n j 1uj
α where uj ∈
{ }
0,1 and α is nonzero constant can be replaced by a continuous variable w accompanied with the following bounding constraints:. 1 (iii) , 0 (ii) , (i) ⎟⎟ ⎠ ⎞ ⎜⎜ ⎝ ⎛ + − ≥ ≥ ∀ ≤
∑
u n w w j u w j j j α α ■ Proof Consider∏
= = n j uj u f 1 )( α . Because product of binary variables is also binary, there are only two possible values for f: 0 and α. Because α is
nonzero, the case of f = 0 implies 0
1 =
∏
=n
j uj and there must be at least one
0 =
j
u . The bounding constraints (i) and (ii) can make w = 0 = f when any of 0
=
j
u and meanwhile constraints (iii) and (iv) are inactive. The other case of
α
=
f implies
∏
nj=1uj =1 and uj =1 for all j. Consider α(
∑
juj −n+1)
. If f =α (i.e., all uj =1 ) then α(
∑
juj −n+1)
=α . If f =0 then(
∑
juj −n+1)
≤0α . That means with constraints (i) and (ii), constraint (iii) can make w=α when f =α but will become inactive when f =0. Therefore, with constraints (i), (ii) and (iii) the nonnegative variable w can
completely substitute f. □
Therefore, the first kind of product term u is a simplified case with ivi α =1 and can be replaced by a continuous variable w accompanied with the following i
constraints . 1 , 0 , , − + ≥ ≥ ≤ ≤ i i i i i i i i v u w w v w u w (12)
The second kind of product terms to eliminate is
∑
∈Sbi s l ls i b z y , ) , ( , , . In the
relaxation version of Program (P1a),
∑
∈Sbi s l,) , zls
( , is a continuous variable within [0,
L] (i.e., L is the number of sequences). This kind of mixed 0-1 product terms can be
eliminated with Corollary 1.
Corollary 1 A mixed 0-1 product term
∏
= n j uj x 1 α where uj ∈
{ }
0,1 ,(
0,ς]
∈x and α is nonzero constants can be replaced by a continuous variable w accompanied with the following bounding constraints:
(
)
(
)
. (iv) , 0 (iii) , (ii) , (i) n u x w w x w j u w j j j − + ≥ ≥ ≤ ∀ ≤∑
ς α α ς α ■ Proof Denote∏
= = nj uj up( ) 1 and obviously p(u) is also binary. From the proof of Proposition 2 we know that when any uj =0, p(u) becomes 0 and
so does
∏
=
n j uj
x 1
without violating (ii) and (iv). In the other case of uj =1 for all j, p(u) becomes 1 and therefore
∏
=
n j uj
x
1
α equals αx. For this case constraints (ii) and (iv) make w tightly bounded to αx without violating (i) and (iii). □
In Program (P1a), the second kind of product terms to eliminate is yb,i
∑
zl,s . The upper bound of∑
∈Sbi s l,) , zls
( , is L, the number of sequences, because every
sequence has only one candidate site to propose, i.e. one occurrence per sequence (OOPS). With Corollary 1, yb,i
∑
zl,s can be replaced by a continuous variable qb,i accompanied with following constraints:. 0 , ) 1 ( , , , , ) , ( , , , , ) , ( , , , , ≥ − + ≥ ≤ ≤
∑
∑
∈ ∈ i b i b S s l ls i b i b i b S s l ls i b q L y z q L y q z q i b i b (13)where L is the number of sequences.
Therefore, all the product terms in Program (P1a) can be successfully replaced by other single continuous variables and a globally optimal solution is then available for fixed-pattern TFBS finding. In fact, based on the techniques discussed in this chapter, more flexible and complicated TFBS finding problems can also be formulated as mixed 0-1 linear programs. In the following chapters we have a more detailed discussion on these formulations.
Chapter 3 Model 1: Fixed-pattern TFBS
Finding
With linearization techniques discussed in Chapter 2, program (P1) can be transformed into a mixed 0-1 linear program which is solvable and promising on global optimum. In this chapter we firstly illustrate the mixed 0-1 linear program for fixed-pattern TFBS finding. Then, more details on TFBS finding is discussed and formulated to appropriated logical constraints which help accuracy. Finally, software designed using the proposed mixed 0-1 linear program is introduced and we discuss on the experimental results about searching for TFBS by this software.
3.1
Mixed 0-1 linear program for Fixed-pattern TFBS
Finding
After applying relaxation and linearization discussed in Chapter 2 on (P1), we have (P2), a mixed 0-1 linear program for finding fixed-pattern TFBS. From the nature of binary variable and mixed 0-1 linear program, (P2) has advantages over many current methods:
(i) A globally optimal solution is promised. Because the nonlinear formulation is successfully replaced by a linear relaxation proven exactly to match the original formulation at optimal points, this program can provide globally optimal solution.
(ii) Logical constraints are applicable for better searching quality. With binary variables utilized, structured information profiting accuracy can be formulated as logical constraints like structural constraints and exception rules. Some of
these constraints, especially structural constraints, can also notably reduce the searching space and computation.
(iii) The program can be extended for more complicated formulation with considerations of practical use. For situations of poor information of target TFBS, e.g. spacer number unknown, this program can still find the TFBS with some modification on the formulation.
(iv) Suboptimal solutions are available by excluding specific solutions. For case of searching for weak signals in DNA sequences, this program can find more than one solution to help explore the correct binding targets with further empirical examinations.
Mixed 0-1 Linear Program for Fixed-pattern TFBS Finding
Maximize
∑
i b i b q , , (P2) Subject to{
1,...,10}
, , 1 , 0 , , , , , , 1 , C , G , T , A ∈ ∀ ⎪ ⎪ ⎪ ⎪ ⎭ ⎪⎪ ⎪ ⎪ ⎬ ⎫ − + ≥ ≥ ≤ ≤ − = − = = + − − = i v u w w v w u w w v y w u y w y w v u y i i i i i i i i i i i i i i i i i i i i{
}
{
A,T,G,C}
{
1,...,10}
, ) , ( , 0 , ) 1 ( , , , , , , , ) , ( , , , , ) , ( , , , , ∈ ∀ ∈ ∀ ⎪ ⎪ ⎪ ⎪ ⎭ ⎪ ⎪ ⎪ ⎪ ⎬ ⎫ = = ≥ − + ≥ ≤ ≤∑
∑
∈ ∈ i b b d s l S q L y z q L y q z q i s l i b i b i b S s l ls i b i b i b S s l s l i b i b i b{
1,...,}
, , 1 , l L z s s l = ∀ ∈∑
{ }
{
}
{
}
{
1,...,}
, . , 10 ,..., 1 , C G, T, A, ], 1 , 0 [ , 1 , 0 , , ℵ ∈ ∈ ∈ ∈ ∈ ∈ s L l i b z v ui i ls(v) It is very straightforward to find the complete set of the second, third, etc. best consensus sequences.
For utilizing information which helps accuracy, we discuss the formulation of several types of logical constraints in the following sections.
3.2
Structural Constraints
Most TFBS are not only conserved signals but having some specific features reflecting structures of the corresponding regulatory proteins. The proposed mixed 0-1 linear program is convenient for embedding logical constraints to elucidate specific TFBS precisely and efficiently. Structural features of various types of TFBS can be formulated as logical constraints to help facilitate the search process. There are three general types of TFBS: mono-type TFBS like binding sites for homeodomains, dyad-type TFBS like bHLH and bZIP binding sites, and serial-type TFBS like zinc-finger binding sites. To find TFBS with specific structures, program (P2) is further modified with several logical constraints incorporated.
The most often seen TFBS are dyad-type. This is because most gene regulators are dimers or tetramers. This kind of TFBS usually has a length less than or equal to 22 and has two symmetric half parts forming an inverted palindrome or direct repeats. For an inverted palindrome the homologous nucleotide bases are supposed complement, i.e. adenine (A) should be paired with thymine (T) and guanine (G) should be paired with cytosine (C). The logical constraint set describing inverted palindrome, for example of CRP-binding sites, can be formulated as:
. 1 , 1 11 11 = + = + − − i i i i v v u u (14)
Another type of TFBS for binding dimerized protein has direct repeats where the same sequence repeats tandem. The logical constraint set of direct repeat can be formulated as: . , 5 5 i i i i v v u u + + = = (15)
Obviously, this kind of logical constraints establishes tight relationships between two half sites and prunes a very large portion of searching space. Therefore, applying such a constraint can notably improve both accuracy and computational performance.
3.3
Suboptimal Consensus
The proposed program can find the globally optimal solution. But practically TFBS finding need more than one solution for further verification. This is because there may be more than one kind of regulatory binding sites and the target TFBS may relatively weaker than other signals. To find the suboptimal solutions, we need to embed exception constraints to banish previously obtained solutions and iteratively run the program with these exception constraints.
To exclude one or more solutions previously determined meaningless or not of interest (e.g. *ATGT******ACAT*), a constraint to be involved is as follows:
, 8 9 , T 8 , A 7 , C 6 , A 5 , T 4 , G 3 , T 2 , A +y + y +y + y +y + y +y ≤ −δ y (16)
where 0≤δ ≤8 is exclusiveness degree.
matches with “*ATGT******ACAT*” will be filtered. δ = 0 means no exclusion. In this example the right hand side of (16) is set as 8-δ because the number of reactive letters in the excluded solution is 8. Note that this number need not equal to the number of reactive bases.
3.4
General Exception Constraints
A reality among regulatory TFBS is that the background nucleotides are not independently and identically distributed. There are always other noises than the target TFBS in the data. When finding a weak signal, we will need exception constraints to help dig out the target. For instance, the often seen poly-A and poly-T tails should be excluded when searching for a direct repeat.
Solutions to be excluded may come from several sources: meaningless repeats, binding sites for a co-regulator, and regions to form stem-loops in mRNA when searching for binding sites of negative regulators, etc. Two kinds of exception constraints are formulated for different cases of noise source:
Repeats with uncertain length
Repeats of arbitrary length like poly-A tail or poly-T tail are meaningless and should be filtered out. For instance of poly-A tail, the constraint should be formulated as follows to banish all the possible solutions containing too many ‘A’:
δ − ≤ + + + + A,2 A,3 ... A,10 10 1 , A y y y y . (17)
Empirical exception rules
conveniently. For instance, consensus sequences consisting only of weak bases (A and T) or only of strong bases (C and G) are usually not a regulatory site of concern. If this kind of solutions is not expected, exclusive constraints can be attached as:
(
2)
1 for allweak consensusexclusion,10 1 -w v u i i i i + − ≥
∑
= (18)(
2)
9 for allstrongconsensusexclusion.10 1 -w v u i i i i + − ≤
∑
= (19)By utilizing binary variables, any if-then rules can also be formulated as logical constraints. These constraints vary by cases and they notably help discriminate weakly conserved TFBS.
3.5
Experimental Results
CRP-binding sites
For the example of finding CRP-binding sites on DNA of E. Coli, after solving program (P2) we can obtain the globally optimum solution “TGTGA******TCACA” with objective value 147. The related nonzero zl,s values indicate the starting positions of the binding sites in the 18 sequences, as listed below:
. 1 81 , 18 87 , 17 56 , 16 20 , 15 74 , 14 51 , 13 44 , 12 64 , 11 17 , 10 12 , 9 42 , 8 27 , 7 63 , 6 53 , 5 66 , 4 79 , 3 58 , 2 64 , 1 = = = = = = = = = = = = = = = = = = z z z z z z z z z z z z z z z z z z (20)
Based on the solution, we can also apply an exception constraint to find suboptimal solutions. Program (P2) can find the exact global optimum solution. But in some cases this globally optimal solution may be only an overrepresented but
meaningless repeat more conserved than target TFBS. For further discovery of target TFBS, we can apply exclusive constraints to find the suboptimal solutions. For example of CRP-binding sites, the second best solution of (P2) can be obtained by adding a new constraint as
9 10 , A 9 , C 8 , A 7 , C 6 , T 5 , A 4 , G 3 , T 2 , G 1 , T +y + y + y +y + y +y + y +y +y ≤ y . (21)
The new constraint is used to force the program to find a new solution different from the solution of (P2). The found second best consensus sequence is “AAATT******AATTT” with score 129. This is a solution consisted by only weak bases (i.e. A and T), so we can regard it as a meaningless solution. Similarly we can find another solution by adding following constraint.
9 10 , T 9 , T 8 , T 7 , A 6 , A 5 , T 4 , T 3 , A 2 , A 1 , A + y +y + y +y + y + y +y +y + y ≤ y . (22)
The third best solution obtained is “TTTGA******TCAAA” with score 129.
Computational experiments
To analyze the effect of sequence length and number of sequences on the computational time, several experiments are tested using the example of CRP-binding sites. The solving engine for optimization is LINGO (Schrage, 1999), a widely used optimization software, on a personal computer with a Pentium 4 2.0G CPU.
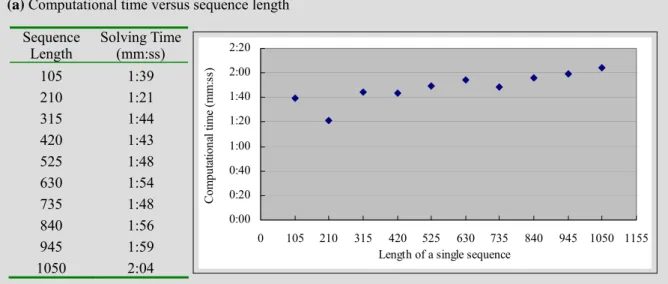
Figure 8 illustrates the experimental results for analyzing the time complexity. Figure 8(a) is the computational time given various sequence lengths, where the number of sequences is fixed at 18. The results show that the computational time changes slightly even if the sequence length is increased from 105 to 1050. Figure 8(b)
is the computational time with various numbers of sequences. It shows that the (a) Computational time versus sequence length
Sequence
Length Solving Time (mm:ss) 105 1:39 210 1:21 315 1:44 420 1:43 525 1:48 630 1:54 735 1:48 840 1:56 945 1:59 1050 2:04 0:00 0:20 0:40 1:00 1:20 1:40 2:00 2:20 0 105 210 315 420 525 630 735 840 945 1050 1155
Length of a single sequence
C omp ut atio na l t ime ( mm:s s)
(b) Computational time versus number of sequences Number of
Sequences Solving Time (mm:ss) 9 0:30 18 1:39 27 3:21 36 4:32 45 6:15 54 6:01 63 8:16 72 10:29 81 10:01 90 9:37 00:00 02:00 04:00 06:00 08:00 10:00 12:00 0 9 18 27 36 45 54 63 72 81 90 99 Number of sequences C omp ut at io na l t ime (mm: ss )
(c) Computational time versus number of independent positions Number of
Indep Pos Solving Time (h:mm:ss) 2 0:00:01 3 0:00:03 4 0:00:21 5 0:01:23 6 0:03:38 7 0:05:18 8 0:08:25 9 0:15:52 10 0:53:27 11 2:33:20 0.1 1.0 10.0 100.0 1000.0 10000.0 100000.0 1 2 3 4 5 6 7 8 9 10 11 12 13
Number of independent positions
C om pu ta tio na l ti m e ( seco nd s)
Figure 8 Computational experiments for fixed-pattern TFBS finding. The relationship between computational time and various factors involved in a motif finding problem. This figure illustrates the computational time of solving Program 2 with (a) various sequences sizes; (b) various number of sequences and (c) various independent positions.
solving time is roughly proportional to the number of sequences. The proposed model is quite promising for treating the TFBS finding problems with long sequences and a large number of sequences. Figure 8(c) shows that the computational time rises exponentially as the number of independent positions increases.
3.6
Software Package: “Global Site Seer”
A software package named “Global Site Seer” is developed based on program (P2) for solving fixed-pattern TFBS finding problems. This software is available from
Chapter 4 Model 2: Ambiguous-spacer TFBS
Finding
A more complicated TFBS finding problem is to find the consensus sequence in an uncertain pattern format where the number of ignored letters between the two half sites is unknown. In this chapter we introduce a modification of program (P2) to solve this kind of TFBS finding problems.
4.1
Problem of Ambiguous-spacer TFBS Finding
A TFBS finding problem with Ambiguous spacers is defined as follows:
Generally the TFBS for binding dimerized regulators have their length not more
than 22 bases. This is because of the space restriction on bindingα-helices of both protein units to the major groove of DNA double strand helix structure. That means, a reasonable range of spacer numbers is limited from 0 to 12 in the problem defined above. Therefore, the concept of mixed 0-1 linear programming approach for this kind of TFBS finding problem is to enumerate all possible spacer numbers k and reformulate program (P2) to cover these enumerations.
Given
(i) A sequence set containing L sequences co-regulated by a dimerized activator, (ii) An inverted palindrome shared pattern which has 5 adjacent reactive bases in each half sites but in-between spacers unknown, i.e. “NNNNN*…*NNNNN”. To find the best conserved consensus sequence
10 9 8 7 6 5 4 3 2 1x x x x (k)x x x x x x ,
where xi∈
{
A,T,G,C}
, i is the index of reactive base, k is the spacer number to find and comp(·) means a complement base.The data preparation step is similar to the preparation procedure of fixed-pattern TFBS finding. To enumerate all possible k, a candidate set D=
{
D0,D1,...,Dk,...,D10}
, where D is constructed as a fixed-pattern candidate set, is prepared for different k kfrom 0 to 10. The candidate sites are thus indexed by (l,s,k). A simple illustration of constructing D is shown in Figure 9. And therefore we can redefine the index set Sb,i as
{
( , , ) , ,}
, , l s k d b S k i s l i b = = (23) where k i s ld, , is the ith base of a candidate site contained by
k D . (a) Sequence #1: AAGACTGTTTTTTTGATC Sequence #2: … (b)
{
( , , ) 0}
0 = l s k k = D , (l, s, k) = (1,1,0) AAGACTGTTTTTTTGATC (l, s, k) = (1,2,0) AAGACTGTTTTTTTGATC … …{
( , , ) 1}
1 = l s k k = D , (l, s, k) = (1,1,1) AAGACTGTTTTTTTGATC (l, s, k) = (1,2,1) AAGACTGTTTTTTTGATC … …{
( , , ) 2}
2 = l s k k = D , (l, s, k) = (1,1,2) AAGACTGTTTTTTTGATC (l, s, k) = (1,2,2) AAGACTGTTTTTTTGATC … …Figure 9 Site extraction for ambiguous-spacer TFBS finding: (a) original sequence data; (b) schematic representation of the candidate sites.
4.2
Mixed 0-1 linear program for Ambiguous-spacer
TFBS Finding
To solve a TFBS finding problem with an ambiguous spacer number, we need to apply some modification on program (P2) to enumerate different k. Because the target consensus sequence is fixed on its spacer number, we need to find k with the maximum matching score. With the assumption of OOPS where each sequence has only the best fitting candidate site proposed, the conservation constraints of site indicator zl,s,k is reformulated as follows:
{
L}
l z k s k s l 1 1,..., , , , = ∀ ∈∑
, (24)∑
∑
∑
= = = s k s L s k s s k s z z z1,, 2,, ... ,, ∀k∈{
0,...,10}
. (25)Constraint set (24) is a modification of (5) which is based on the assumption of OOPS: only one zl,s,k is supposed to be nonnegative in a sequence. Constraint (25) is used to make sure that all the nonzero zl,s,k have their corresponding candidate sites
from the same set D . By applying these two constraints we can then obtain a k solution with a fixed spacer number.
Because this kind of TFBS finding problem is only for analyzing dimerized activator, structural constraint of inverted palindrome must be incorporated as
. 1 , 1 11 11 = + = + − − i i i i v v u u