國 立 交 通 大 學
生物科技研究所
博士論文
利用突變策略針對氧化鯊烯環化酵素
進行其結構與功能及其產物專一性/
多樣性之研究
Mutagenesis Approach to Investigate the
Structure-Function Relationships and Product
Specificity/Diversity of Oxidosqualene Cyclase
研究生:劉媛婷
Student: Yuan-Ting Liu
指導教授:吳東昆
博士
Advisor: Prof. Tung-Kung Wu Ph.D
利用突變策略針對氧化鯊烯環化酵素進行其結
構與功能及其產物專一性/多樣性之研究
Mutagenesis Approach to Investigate the
Structure-Function Relationships and Product
Specificity/Diversity of Oxidosqualene Cyclase
研究生:劉媛婷
Student: Yuan-Ting Liu
指導教授:吳東昆
博士
Advisor: Prof. Tung-Kung Wu Ph.D
國 立 交 通 大 學
生物科技研究所
博士論文
A Dissertation
Submitted to Department of Biological Science and Technology College of Biological Science and Technology
National Chiao Tung University in partial Fulfillment of the Requirements
for the Degree of Doctor of Philosophy in Biological Science and Technology
Hsinchu, Taiwan, Republic of China
July, 2010
i
摘要
突變策略被視為是一種強而有力的工具,其廣泛地應用於針對蛋白質的結構、 功能、或反應機制的研究上。因此,在本論文中我們透過某些功能性已知的環化 酵素,對於其胺基酸序列和蛋白質結構間的關係進行比較,並利用丙胺酸掃描式 突 變 (alanine-scanning) 或 定 點 / 飽 和 突 變 的 策 略 (site-directed/saturated mutagenesis) 並配合基因互補和產物收集/鑑定的方法,針對啤酒酵母菌中的氧 化鯊烯環化酵素(Saccharomyces cerevisiae ERG7) 其活性區域內多個重要的胺基 酸位置,它們在環化酵素所負責的複雜環化與重組反應機制中所扮演的角色加以 確定。一開始的工作是針對活性區域內的組胺酸-234 號位置。當此位置被置換成 其它不同性質的胺基酸時,我們透過所獲得的多樣性產物,包括單環、三環、和 不同去質子化的四環產物進一步地闡明此胺基酸參予在環化酵素所催化之反應 中的角色。此外,利用相似的突變策略對於其它重要的胺基酸位置,我們可以透 過來自各突變株所收集到的不完全環化產物或是不同重組階段的替代產物加以 解釋各個胺基酸所賦予在各環化重組過程中的重要性。同時,為了要更進一步的 細看最後的去質子步驟,對於決定環化酵素其產物特異性的關係,我們建立多個 連續定點突變株。其中一個突變酵素上第384 號位置的蘇胺酸我們將之換成酪胺 酸,而谷氨醯胺450 位置將其換成組胺酸,最後我們將纈胺酸 454 位置換成異亮 胺酸 (也就是我們同時突變這三個胺基酸位置)。由實驗的結果發現,此突變株 劇烈地改變它的特異性,從原本的羊毛硬脂醇合成酵素變成一個極度精密的帕克 醇合成酵素,另外我們亦發現其產物中伴隨著兩個骨架已被下游連續酵素所修飾 的產物。這個現象不僅反映當有不尋常量的三萜類化合物累積在酵母菌體時,其 代謝流向的改變情況,而且也說明了下游酵素對於受質的選擇性效應。除了上述 的突變實驗外,我們也同步針對植物中的一個三萜類合成酵素,即 -香桂素合 成酵素進行系列的研究。包括從碗豆 (Pisum sativum) 種子中進行分子選殖以獲ii 得 -香桂素合成酵素的基因,並利用異質性的表現系統將其成功地在酵母菌中 表現,其後我們利用電腦模擬的方式建立其同源性結構,並透過上述的定點突變 實驗對於其重要的19 個胺基酸位置,進行研究並分析突變效應後所產生的環化 產物的樣式。雖然我們並沒有發現在各個突變株中有差異的非皂化脂質粗萃取物, 但這一系列的實驗說明當 -香桂素合成酵素中的重要胺基酸被置換時,會顯著 地影響此酵素之催化活性。因此,透過上述的實驗結果我們可以對於不同的氧化 鯊烯環化酵素其在環化/重組反應機制上提供更多的了解。
iii
謝誌 (Acknowledgement)
論文即將付梓之際,心中滿是感恩。感謝主的安排,匯集如此大的恩典,讓我 得以享受充實與精彩的過程,體會流淚灑種必歡笑收割的甘甜。 而論文能順利完成,最要感謝的是我的指導教授-吳東昆博士,八年前給了一 個不是出自名門學府、毫無化學背景、懵懂無知的我加入這個實驗室的機會。其 間老師對於整體研究方向、觀念的啟迪,實驗架構與邏輯的匡正,與對於結果的 辯證和思維,著實讓我受益良多。對於學術研究的熱情與堅持,以及正面積極、 不畏挫折的勇氣,更是學生學習與仿效的榜樣。另外對於學生求學態度的斧正, 以及給予生活細節的叮嚀與照顧,更是帶動學生持續往前走的動力,於此獻上最 深的敬意與謝意。口試委員李耀坤老師、鄭建中老師、林敬堯老師以及許鉦宗老 師陪我度過修業期間的幾個重要考試,謝謝您們不吝提供寶貴意見以及對於疏漏 處的指正,使得論文更臻完備。清大貴儀中心彭菊蘭小姐,因著有您對於NMR 光譜上的幫忙,才得以將那微乎其為的化合物驗明正身,謝謝您。師母賴美伶小 姐常以著開朗與樂觀的態度分享著實驗或生活上的建議,每每與您談話,總讓我 像是重新充電般,再次獲得能量,得以再出發。 漫長的旅途中,雖然總是挫折積累,幸運的是,有一群小天使,一路陪伴、鼓 勵著我,給予最溫暖的支持,使我得以堅強地走到現在,如今有個完美的結果, 您們功不可沒。程翔學長是我的良師益友,秉著熱情與耐心伴著我成長。每當徬 徨無措,總是理性的分析,像是盞明燈指引著我該走的方向。您那化腐朽為神奇 的魔力,著實令人讚嘆,從您的身上我真的獲得許多。晉豪是我的好夥伴。頭腦 聰穎的你,總是給予我不管是實驗還是生活上很大的幫忙。真的,還好有你Cover 我一切的不足。晉源是開心果。總是能讓煩悶的過程增添了許多的歡笑與樂趣, 有顆赤子之心的你,是很有魅力的喔!和小紅妳為室友的日子,夜裡不論是對於 實驗的討論,或是小女人間的話家常、說八卦,還是追星、瘋偶像劇,都是我難 忘的回憶。好麻吉怡親,總是給予我最真誠的關心。說心事、聊是非,一起逛街、 買拍賣,妳為我的研究生涯灑下了不一樣的精采。我的好鄰居文祥,超級行動派 的你總是實現每一個不可能。那段南征北討,通霄”學習”的歲月,現在想起來還 是覺得很酷!裕仁總是適時的給予我加油、打氣與肯定,重要時刻也沒缺席,你 就是一個這麼樣體貼的小男生。斯文的小宇學長在最初給了剛進實驗室懷著忐忑 不安心情的我那抹善意的微笑,至今我還記得。大景傻得可愛的氣息,讓我的生 活充滿著爆笑噴飯的事件。熱心的宏城和裕國是我的同窗伙伴,和你們之間的切 磋討論,讓我獲益匪淺。還有貼心的文鴻,不管是實驗還是生活的細節,總是幫 忙許多。告訴你喔,我真的覺得你很厲害,所以要繼續堅持喔!另外同屬Cyclase Project 的學弟妹美婷、文暄、皓宇、采婷、亦諄與宗璟,更是我學習生涯缺一不iv 可的好幫手。猶記得那段熬夜通銀染的日子,絞盡腦汁地玩著連連看,到最終產 物鑑定,並得以發表。這一切成就感的獲得,全因著你們的認真與執著,所收割 的豐碩果實,真的謝謝你們了。天昶是最挺我的學弟,總是努力認真的幫忙我完 成每一個實驗的假想。時常與奕齊你激辯、交鋒,也著實讓我成長。可愛的卡通 人靜婷,則是個認真有規劃的好學妹。怡臻的貼心,常為我枯竭的靈感注入新的 想法。新新人類欣怡,總是帶給我許多年輕人的新想法。和世穎一天一音樂的遊 戲,振奮夜裡萎靡的精神,也給了漫長的論文撰寫過程中一個期待。因著你們的 年輕有活力,使得實驗室充滿著生氣。也謝謝你們在庶務上面的幫忙,分擔了許 多的麻煩,真的謝謝了。而分屬其它計劃的衣鵑、Mili、妍希、佳宜、令宗、宏 明、庭翊、禕婷、書涵、宜芳與欣芳,謝謝你們豐富了我的生活。 另外還要感謝我的好朋友們- 建勝一路走來對我的關心與照顧,如果當初沒有 你,我想也不會有現在的我,謝謝。和智凱你那段慢跑的時光,以及之後書本小 說和音樂小品的分享,是我放鬆休憩的最佳良藥。千惇、佩陵與怡萱,常常給予 我加油、打氣,妳們給的溫暖,讓我明白這並不是個人單打獨鬥的旅程。晉嘉品 容雙寶,常常帶給我意外的捧腹大笑,使得原本抑鬱的心情展為笑顏。 最後,特將本論文獻給我最親愛的家人- 爸爸、媽媽以幾近縱容的態度,讓我 自由探索想走的方向。每每失去勇氣,您們的溫柔撫慰,總是再一次讓我獲得站 立的力量。因著您們的全然支持,是使得資質平庸的我,能夠獲得博士學位,最 重要的基石。妹妹和弟弟則是我最大的驕傲,更是促使我不斷向上的原動力。還 有已在天堂的爺爺和奶奶,您們溫良謙讓的處世態度,尤其令我銘記在心,也是 我日後為人處事的最大的後盾。還有要謝謝親朋好友給予的關心與代禱。 感謝所有關心我,陪伴著我的人。小小的版面,實在不足以聊表我最深忱的謝 意。 『參透為何,才能迎接任何』,願以此期勉自己的未來,能夠發揮所學,並開 創或將其應用至每一個可能性。加油! 媛婷 謹誌於 交通大學生物科技研究所 中華民國九十九年七月二十六日
v
Abstract
The mutagenesis is regarded as a powerful tool for investigating the structure, function, and reaction-mechanism relationships of proteins. Sequence alignment with other known cyclases and various forms of mutagenesis were used to identify and study catalytically important residues in Saccharomyces cerevisiae ERG7. Using mutagenic techniques coupled with genetic complementation and product characterization, it became possible to further characterize the cyclization and rearrangement mechanism of 2,3-oxidosqulene. Several mutations of the His-234 residue in ERG7 generated diverse product profiles with various monocyclic, tricyclic, and tetracyclic products while similar mutagenesis strategies on other catalytically important residues resulted in derailed cyclization and alternative deprotonation in other critical steps of the cyclization cascade. A series of site-directed mutations were also made to probe the crucial residues involved in the final deprotonation stage of the reaction for product specificity. For example, the ERG7T384Y/Q450H/V454I mutant changed its product specificity from the original lanosterol synthase into a parkeol synthase accompanied by two scaffold-modifying products that were generated speculatively by additional tailoring enzymes. The trend of metabolite flux changed when unusual levels of triterpene accumulated in the yeast cell and impacted the substrate selectivity of some downstream enzymes. A parallel experiment was conducted with a triterpene synthase, -amyrin synthase from Pisum sativum using functional expression by yeast and site-directed mutagenesis on 19 residues. Examination of its product profile revealed no divergence of the non-saponifiable lipid patterns among any of the mutants, suggesting that the exchange of the important functional residue of -amyrin synthase might influence its enzymatic
vi
activity dramatically. The aforementioned results, when combined, provides for a better understanding for the cyclization and rearrangement mechanism of various oxidisqualene cyclases.
vii
Table of Contents
Chapter 1. General introduction. ... 1
1.1 Overview of oxidosqualene cyclase ... 1
1.2 Historical hypothesis of the cyclization mechanism ... 5
1.2-1 Mechanistic and stereochemical insights relative to the 2,3-oxidosqualene cyclizationcascade ... 5
1.2-2 The theoretical models of cylase enzymes ... 11
1.3 Crystallization and structural characterization of cyclase ... 15
Chapter 2. Thesis organization ... 21
Chapter 3. Site-deirected mutagenesis of oxidosqualene cyclase to characterize the plasticity of the protein diversity of product ... 24
3.1 Research background and aim ... 24
3.2Results and Discussion. ... 27
3.2-1 Alignments of multiple amino acid sequences for sterol cyclases and triterpene cyclases. ... 27
3.2-2 Nine site-directed mutants of ERG7 gene from S. cerevisiae ... 28
3.2-3 Principle of the plasmid shuffle method ... 31
3.2-4 Screening inactive ERG7 mutants via plasmid shuffle method ... 33
3.2-5 Lipid extraction, column chromatography, and product characterization of yeast transformants ... 34
3.2-6 Characterizatio of the mutant products in the novel gene disruption strain, TKW14c2 strain ... 37
3.2-7 Site-saturated mutagenesis of His-234 to investigate its importance for ERG7 activity... 40
Chapter 4. Site-directed mutagenesis and product characterization to study the putative active-site residues from S. cerevisiae oxidosqualene cyclase ... 52
4.1 Research background and aim. ... 52
4.2 Results and Discussion. ... 53
4.2-1 Generation of a homology model for S. cerevisiae OSC and functional analysis of alanine-scanning mutants of putative active-site residues ... 53
4.2-2 Site-saturated mutagenesis and functional analysis of active-site residues from S. cerevisiae ERG7.. ... 55 4.2-3 Correlation between theoretical model results and experimential evidance to investigate cyclization/rearrangement mechanism of oxidosqualene cyclase. 59
viii
4.3 Conclusion. ... 65
Chapter 5. Site-directed mutagenesis study on the deprotonation course of the oxidosqualene cyclization ... 66
5.1 Research background and aim ...66
5.2 Results and Discussion. ...69
5.2-1 Generation of site-directed mutants on the S. cerevisiae erg7 gene and functional analysis of site-directed mutations via plasmid shuffle method and product characterization ... 69
5.2-2 Homology modeling illustration of critical residues on enzyme function . 73 5.2-3 Investigation of product modification by subsequent triterpene tailoring enzymes in S.cerevisiae by isolation and identifying unexpected downstream products. ... 74
Chapter 6. Homology modeling coupled with site-directed mutagenesis study on plant oxidosqualene -amyrin synthase to investigate the relationship between substrate folding geometry and the resulting diverse products ... 82
6.1 Research background and aim. ...82
6.2 Results and Discussion. ...85
6.2-1 Putative -amyrin synthase cDNA amplified from P. sativum. ... 85
6.2-2 Heterologous expression of -smyrin synthase in yeast ... 87
6.2-3 Study of enzyme activity using homology modeling coupled with site-directed mutagenesis approach. ... 89
6.2-4 Functional analysis of artificial enzymes via plasmid shuffle method and the product characterization ... 93
6.3 Conclusion. ... 96
Chapter 7. Future perspective ... 98
Chapter 8. Experimental section ... 101
8.1 Material. ...101
8.1-1Bacterial strains and molecular cloning/expression vectors. ... 101
8.1-2 Enzyme, chemicals, equipments, and reagents ... 101
8.2 general experimental procedure. ...104
8.2-1 Construction of site-directed/saturated mutahenic plasmids. ... 104
8.2-2 Preparation of competent yeast cell (CBY 57 and TKW14c2 strain) ... 107
8.2-3 Cyclase activity assay by using plasmid shuffle method in CBY57 strain ... 107
8.2-4 Cyclase activity assay by using ergosterol compementation in TKW14c2 strain. ... 108
ix
8.2-5 Lipid extraction, and column chromatography ... 109
8.2-6 Acetylationmodification and the alkine hydrolysis reaction. ... 109
8.2-7 GC and GC-MS column chromatography ... 110
8.2-8 Sequence alignment and molecular modeling ... 110
8.2-9 Molecular cloning of P. sativum -amyrin synthase. ... 111
8.2-10 Subcloning of full-length of P. sativum -amyrin synthase ... 112
8.2-11 Functional expression of P. sativum -amyrin synthase ... 113
References. ... 114
Appendix. ... 119
Table of Figures
Figure 1.1. The complexity of the cyclization cascade from 2,3-oxidosqualen to lanosterol and the end product,cholesterol, in the biosynthesis pathway for cholesterol in human ... 2Figure 1.2. Product diversity of oxidosqualene cyclae are acquired by the species- dependent cyclizations and stereochemical configuration of carbocationic intermediates ... 4
Figure 1.3. Isolation of a novel bicyclic triterpenoid from gum mastic of Pistacia lentiscus ... 7
Figure 1.4. Conversion of 18,19-dihydrooxidosqualene into a 6,6,5-ring fused products by rat liver OSC ... 7
Figure 1.5. Enzyme converted a substrate analogue, 20-oxa-2,3-oxidosqualene, into a product with a p-bromobenzoate derivative ... 7
Figure 1.6. Enzymatic transformation of an oxidosqualene analogues, 20-oxo-2,3- oxidosqualene, into a protosterol derivatives with 17 side-chain conformation... 9
Figure 1.7. Enzyme conversion of the 2,3-oxidosqualene analogue, which lacks the methyl substituent at C-10 and C-15, into an unrearrangement protosterol derivative ... 9
Figure 1.8. Hypothesized cyclization cascade of 2,3-oxidosqualene ... 10
Figure 1.9. Three cation-stabilizing auxiliaries that were proposed by Johnson in the cyclase enzyme ... 12
Figure 1.10. Summary of the highly-conserved QW motifs in OSC for four species . 12 Figure 1.11. Aromatic hypothetical model for the cyclization mechanism ... 13 Figure 1.12. Affinity labeling experiments for cyclase enzymes with mechanistic
x
suicide inhibitors ... 14
Figure 1.13. The overall structure of A. acidocaldarius SHC. ... 16
Figure 1.14. Crystal structure of human OSC ... 20
Figure 1.15. Local view of crystal structure of human OSC ... 20
Figure 3.1. The A. thaliana CAS mutants changed their product specificity from cycloartenol to lanosterol ... 25
Figure 3.2. Single amino acid substitution converted the product specificity between lupeol synthase and -amyrin synthase. ... 26
Figure 3.3. The mutagenesis approach might provide the opportunity to understand how the cyclase enzyme force the substrate into two different conformation ... 27
Figure 3.4. Alignment of multiple amino acid sequences for screening the putative residues involved in the product specificity/diversity ... 29
Figure 3.5. Comparison of the GC patterns of LA fraction from the NSL extrates among ERG7WT and nine mutatants ... 36
Figure 3.6. Structure characterization of protosta-12,24-dien-3-ol ... 39
Figure 3.7. Structure characterization of protosta-20,24-dien-3-ol ... 47
Figure3.8. Characterization of the structure of 13(H)-isomalabarica-14(26),17E,21- trien-3-ol ... 48
Figure 3.9. Product profile produces drom TKW14c2 expressing the ERG7H234X site-saturated mutations ... 51
Figure 4.1. Superimposition of the S. cerevisiae ERG7 homology modeling structure with crystal structure of human OSC. ... 54
Figure 4.2. Local view of homology model structure of S. cerevisiae ERG7 with reactant, lanosterol ... 56
Figure 4.3. Structures of various products isolated from S. cerevisiae ERG7WT and of various ERG7mutants ... 57
Figure 4.4. Placement and function of critical amino-acid residues inside the S. cerevisiae central cavity ...65
Figure 5.1. Alignment of multiple sequences of cycloartenol synthase and oxidosqualene cyclase ...68
Figure 5.2. Local view of the homology modeled S. cerevisiae ERG7 structure ...69
Figure 5.3. Oxidosqualene cyclization products formed by oxidosqualene cyclase mutants from S. cerevisiae ...72
Figure 5.4. Characterization of the structure and the result of NOE correlation of 4,14-dimethyl-24-methylene-5-cholest-9(11),24-dien-3-ol ...77
Figure 5.5. Characterization of the structure and the result of NOE correlation of 4,14-dimethyl-5-cholest-9(11),24-dien-3-ol ...78
xi
Figure 5.7. Proposed biosynthetic pathway in the S. cerevisiae ERG7T384Y/Q450H/V454I80 Figure 6.1. Formation of diverse triterpene skeletons from oxidosqualene cyclization in plants ...83 Figure 6.2. Nucleotide sequence and the deduced amino acid sequence of P. sativum -amyrin synthase ...86 Figure 6.3. GC-MS analysis of the NSL isolated from the petroleum ether extraction
of pYES2-PSY in the yeast strain, TKW14c2 ...88 Figure 6.4. Superimposition of the S. cerevisiae ERG7 homology modeling structure
with P. sativum -amyrin synthase. ...91 Figure 6.5 After amino acid substitutions by computational simulation, the
superimposed orientation of the functional residues of “modified -amyrin synthase” is similar to the native S. cerevisiae ERG7. ...93
Table of Schemes
Scheme 3.1. The plasmid shuffle strategy. ... 33 Scheme 3.2. Preliminary analysis and separation of products on TLC plate... 35
Table of Tables
Table 3.1. Construction strategies and individual silent mutation site of the nine consenved residues ... 31 Table 3.2. 1H- and 13C-NMR data of protosta-12,24-dien-3-ol ... 39 Table 3.3. Complementary results of ERG7H234X mutants in an erg7 hem1 double-
knockout strain, TKW14c2. ... 42 Table 3.4. 1H- and 13C-NMR data of protosta-20,24-dien-3-ol ... 47 Table 3.5. 1H- and 13C-NMR data of (13H)-isomalabarica-14(26),17E,21-trien-3-ol ...48 Table 3.6. Product profile of S. cerevisiae TKW14c2 expressing the ERG7H234X
site-directed mutants ...49 Table 5.1. nProduct distribution and its ratio of S. cerevisiae ERG7 mutants ...71
Table 5.2. 1H- and 13C-NMR data of 4,14-dimethyl-24-methylene-5- cholest- 9(11),24-dien-3-ol ...77 Table 5.3. 1H- and 13C-NMR data of 4,14-dimethyl-5-cholest-9(11), 24-dien-
xii
Table 6.1. The relative distances between active-site residues with the docked
substrate,lanosterol in -amyrin synthaes ... 92
Table 6.2. Product profile of P. sativum AS mutants ... 95
Table 8.1. Primer sequence in the site-directed muatagenesis experiment ... 105
Table 8.2. Primer sequence in the site-saturated muatagenesis experiment ... 107
Table 8.3. All of the primers were used to clone of -amyrin synthase from P. sativum ... 113
xiii
List of Abbreviations
A, Ade Adenine
A. acidocaldarius Alicyclobacillus acidocaldarius A. thaliana Arabidopsis thaliana
Ala Alanine Arg Arginine Asn Asparagine
Asp Aspartic acid
bp Base Pair
cDNA Complementary DNA
Cys Cystine ddH2O Double Distilled Water
DEPC Diethylpyrocarbonate
dNTP Deoxynucleoside triphosphate
GC-MS Gas Chromatography-Mass Spectromerty Gln Glutamine
Glu Glutamic acid
Gly Glycine H, His Histidine
H. sapiens Homo sapiens
HPLC High Performance Liquid Chromatography Ile Isolucine kb(s) kilobase(s) L, Lys Lysine LA Lanosterol LB Luria-Bertani Leu Leucine M, Met Methonine
NMR Nuclear Magnetic Resonance NOE
NSL
Nuclear Overhauser Effect Non-Saponifiable Lipid
O. europa Olea europa
OS Oxidosqualene
P. ginseng Panax ginseng P. sativum Pisum sativum
xiv Phe Phenylanine Pro Prolne rpm revolutions per minute
RT Room Temperature
S. cerevisiae Saccharomyces cerevisiae
Ser Serine
ssDNA Single-stranded DNA
T, Trp Tryptophan
Thr Threnine TLC Thin Layer Chromatography Tyr Tyrosine U, Ura Uracil
Val Valnine WT Wild-type
X-gal 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside
YNB Yeast Nitrogen Base
List of Genes or proteins
AS -amyrin synthase CAS Cycloartenol synthase
ERG1 Squalene epoxidase
ERG6 Sterol C-24 methyltransferase
ERG7 Oxidosqualene Cyclase ; Lanosterol Synthase ERG11 Lanosterol 14-demethylase
ERG25 Sterol C-4 demethylase ERG26 Sterol C-3 dehydrogenase
LUS Lupeol synthase
OSC Oxidosqualene cyclase
SHC Squalene-hopene cyclase
SMT Steroll methyl-transferase
URA3 Orotidine-5’-Phosphate (OMP) Decarboxylase gene
1
CHAPTER 1
General introduction
1.1 Overview of oxidosqualene cyclase
The 2,3-oxidosqualene cyclization is the most remarkable example of a single enzyme-catalyzed reaction that triggers a common polyolefin for the production of a vast diversity of tetracyclic and pentacyclic products with exquisite control of stereoselective cyclization and skeletal rearrangements. This impressive enzymatic reaction catalyzes an acyclic squalene or 2,3-oxidosqualene with one or no stereogenic center into polycyclic sterols and triterpenes that contain multiple rings and several chiral centers, along with at least 10 formal covalent-bond cleavages and formations. The cyclization cascades include an initial epoxide protonation, four or five cation-olefin mediated ring annulations, multiple hydride/methyl group rearrangements, and the final termination either by deprotonation or water addition.1 The lanosterol is further converted to the end product, cholesterol, via additional 18 enzymatic reactions in the human steroid framework, which serves as the membranous components, precursors of steroid hormones, and other secondary metabolites. Certainly, the oxidosqualene cyclase (OSC) is responsible for the most amazing step among the natural chemical reactions (Figure 1.1).
Over a half century ago, this powerful and efficient chemical transformation catalyzed by OSC intrigued and fascinated scientists. The first rudimentary model was proposed in 1934, and, in addition, other different radioisotopic feeding experiments were used to demonstrate the final carbon and hydrogen source of cholesterol.2,3 In 1953, Woodward and Bloch suggested a hypothesis regarding lanosterol formation
2
derived from squalene.4 However, further experiments demonstrated that 2,3-oxidosqualene, rather than squalene, is the direct intermediate in the biosynthesis of lanosterol in which the 3S isomer of oxidosqualene was used as the exclusive substrate.5,6 Moreover, the more detailed information about lanosterol production, such as the [1,2]-hydride shifts and the migration of methyl groups, was examined from the trapping of truncated intermediates in the substrate-analogue mediated cyclization.5-8 These early pioneering chemical studies of OSC provided fundamental background information concerning its biological importance and the stereochemical course of the cyclization.
Figure 1.1. The complexity of the cyclization cascade from 2,3-oxidosqualene to lanosterol and the end product, cholesterol, in the biosynthesis pathway for cholesterol in human.
3
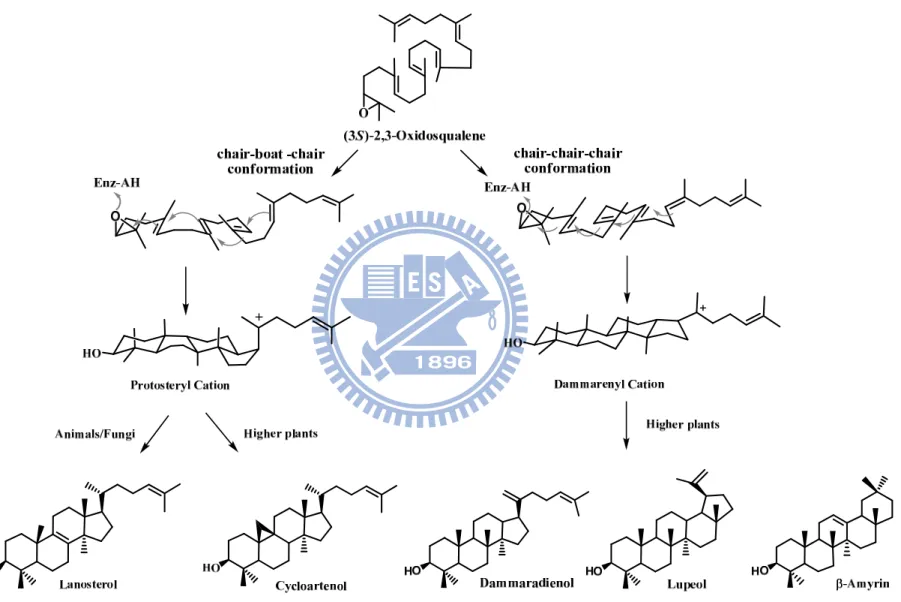
A wide array of tetracyclic or pentacyclic backbones is generated by the species-dependent cyclization manners. It is widely accepted that the distinct stereochemical configuration of the second cyclohexyl B-ring of 2,3-oxidosqualene in the active-site cavity of the enzyme resulted in two major cationic intermediates for its corresponding cyclization pathways. In the protosteryl cationic pathway, the six-membered B-ring is used as the “boat” form, whereas the “chair” form of the cyclohexyl B-ring takes place in the dammarenyl cationic pathway. Thus, either for lanosterol production in animals or for cycloartenol production in plants, the substrate, 2,3-oxidosqualene, proceeds via an energically-unfavorable pre-folded conformation, i.e., the “chair-boat-chair conformation” to produce a protosteryl C-20 cation, followed by a series of hydride and methyl group shifts and the elimination of an individual final proton. In parallel, after generation of the tetracyclic dammarenyl C-20 cation from the “all-chair” conformation, the substrate is triggered to form dammaradienol, lupeol, -amyrin, or -amyrin in the higher plants, algae through similar rearrangements, ring expansions, and final elimination steps (Figure 1.2).1
4
Figure 1.2. Product diversity of oxidosqualene cyclase is acquired by the species-dependent cyclizations and stereochemical configuration of carboncationic intermediates.
5
1.2 Historical hypothesis of the cyclization mechanism
1.2-1 Mechanistic and stereochemical insights relative to the 2,3-oxidosqualene cyclization cascade
Converting a mobile and linear polyolefinic substrate into tetracyclic and pentacyclic triterpene products is a highly exothermic chemical reaction with a low activation energy requirement.9 Thus, it is hard to control tendency for complex reactions in the non-enzymatic cyclization of oxidosqualene.9 In the biological system, OSC can trigger an efficient cyclization reaction and lead to a variety of product specificities by overcoming the energy barrier and diminishing the activation energy. Therefore, many experiments have been performed, mainly the synthesis of substrate analogues or inhibitors, to understand the relationship between the cyclization mechanism and the enzyme itself.
According to the “biogenic isoprene rule,”10 cyclizing 2,3-oxidosqualene to lanosterol should proceed via a direct, concerted, and “non-stop” processes. However, van Tamelen and his-coworker proposed that the cyclization proceeded through a series of discrete, conformationally-rigorous, and partially-cyclized carbocationic intermediates.11,12 These debates have been clarified by direct experimental evidence.
The reaction rate of the cyclization of various polyene monoepoxides with a Lewis acid suggested that the cleavage of the oxirane ring, accompanied by A-ring cyclization, occurs via a high degree of anchimeric assistance from the neighboring double bond (C-6/C-7).12 The enzymatic cyclization of two 2,3-oxidosqualene analogs with the C-6 methyl group replacement by either hydrogen or chloride, i.e, 6-desmethyl-2,3-oxidosqualene and 6-chloro-6-desmethyl-2,3-oxidosqualene, demonstrated that electrophilic oxirane-ring cleavage and cyclization of the A-ring are concerted and initiated by a particularly acidic and highly conserved aspartic acid
6
residue, Asp-456.13 Moreover, the Vmax/KM values of the enzymatic conversion of
2,3-oxidosqualene, 6-desmethyl-2,3-oxidosqualene, and 6-chloro-6-desmethyl-2,3- oxidosqualene are 138, 9.4, and 21.9, respectively. This implied that the -nucleophilicity of the proximate double bond might influence the rate of cleavage of the oxirane ring.13
The boat conformation of the B-ring might occur temporally once a positive charge has built at the C-5 of the A-ring. It is still unknown exactly how the cyclase enzyme mediates this process. One possibility is that cyclase may lower the activation energy of the boat form by delivering a negative point charge to the –face at C-8.14 In
addition, the isolation of “trapped” products from natural sources has provided direct experimental evidence that cyclization might proceed via different, stepwise, cyclized stages (Fig 1.3).15,16
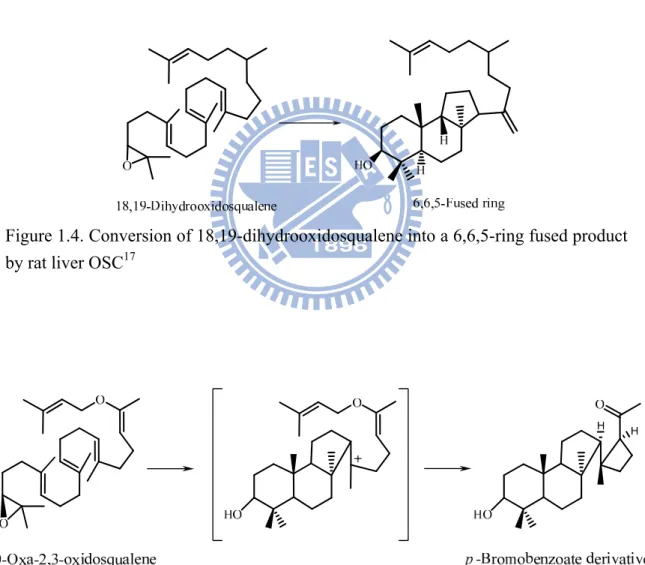
A secondary 6,6,6-tricyclic cation with a unfavorably thermodynamic tendency was proposed to exist via an anti-Markovnikov addition during the closure of the six-membered C-ring.14 However, a 6,6,5-tricyclic product was found via a thermodynamically-favorable, tertiary, cationic intermediate in the cyclization of 18,19-dihydro-2,3-oxidosqualene (Fig 1.4).17 In addition, the enzymatic cyclization of 20-oxa-2,3-oxidosqualene produced a 6,6,5,4-tetracyclic compound that further exhibited the precursor scaffold with a 6,6,5-ring (Fig 1.5). These observations strongly indicated that the cyclization of 2,3-oxidosqualene must proceed via a discrete tricyclic cation, from a Markovnikov closure of the cyclopentylcarbinyl cation, followed by a subsequent ring expansion to form a tricyclic anti-Markovnikov cyclohexyl C-ring intermediate cation.18,19
7
Figure 1.3. Isolation of a novel bicyclic triterpenoid from gum mastic of Pistacia
lentiscus15,16
Figure 1.4. Conversion of 18,19-dihydrooxidosqualene into a 6,6,5-ring fused product by rat liver OSC17
Figure 1.5. Enzyme converted a substrate analogue, 20-oxa-2,3-oxidosqualene, into a product with a p-bromobenzoate derivative.18
8
The stereochemistry at C-20 during the oxidosqualene cyclization has also been illustrated.1,20 Isolation of the trapped protosteryl derivatives from the enzyme-catalyzed cyclization of 20-oxa-2,3-oxidosqualene and (20E)-20,21-dehydro- 2,3-oxidosqualene, respectively, demonstrated that the stereochemical side chain conformation, but not a side chain conformation, is formed at C-17 of the protosteryl derivative (Figure 1.6). These findings further explained that only a smaller rotation angle (60o) of the C-17 side chain is required prior to the leading of the natural 20R stereo configuration during the D-ring closure of the C-20 protosteryl cation.21,22
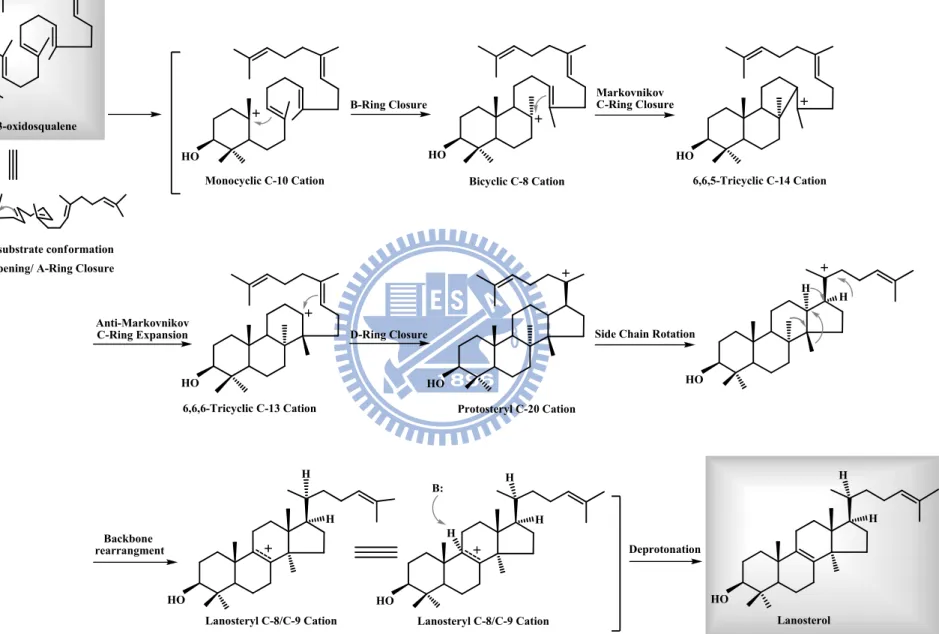
On the other hand, lanosterol-like products that contained an unnatural 6,6,5-tricyclic scaffold accompanied by various methyl group migrations, were also found in the cyclase-triggered catalysis.23 In parallel, the cyclase enzyme could convert the 2,3-oxidosqaulene analogues, which lack the methyl substituent at C-10 and C-15, into a cyclized product without rearrangement of carbon and hydrogen skeleton (Figure 1.7).24 This phenomenon indicated that the methyl group at C-10 of 2,3-oxidosqualene is very critical for the cyclization-rearrangement step.25 After the skeletal rearrangement goes through a series of [1,2]-hydride/methyl group shifts, the lanosteryl C-8 or C-9 cation is formed. The cyclization is terminated by a proton removement step. The deprotonation of the intermediate cation was performed by either water addition or proton abstraction from a well-positioned enzymatic base, which is elegantly controlled by the cyclase enzymes to avoid the disruption of the early product prior to the final reaction step. Therefore, efforts were made to understand the more-detailed, and diagrammatic elucidation of the oxidosqualene-catalyzed cyclization reaction. The extensive mechanistic hypothesis of the formation of lanosterol is summarized in Figure 1.8.
9 HO H H O H + 20-Oxa-2,3-oxidosqualene O H H H HO H
Major product with 17 side chain conformation Protosterol analogue
O
O
H
Figure 1.6. Enzymatic transformation of an 2,3-oxidosqualene analogue, 20-oxa-2,3- oxidosqualene, into a protosterol derivative with a 17side chain conformation.26
Figure 1.7. Enzyme conversion of the 2,3-oxidosqualene analogue, which lack the methyl substituent at C-10 and C-15, into an unrearranged protosterol derivative.24
10 2,3-oxidosqualene
O H+
Epoxide Opening/ A-Ring Closure Prefolded substrate conformation
Monocyclic C-10 Cation HO B-Ring Closure Bicyclic C-8 Cation HO Anti-Markovnikov C-Ring Expansion HO D-Ring Closure HO
Side Chain Rotation
6,6,6-Tricyclic C-13 Cation Protosteryl C-20 Cation
Backbone rearrangment HO HO H H H H Lanosteryl C-8/C-9 Cation HO H H Lanosteryl C-8/C-9 Cation HO H H Lanosterol Deprotonation H B: 6,6,5-Tricyclic C-14 Cation HO Markovnikov C-Ring Closure H+ O
11
1.2-2 The theoretical models of cyclase enzymes
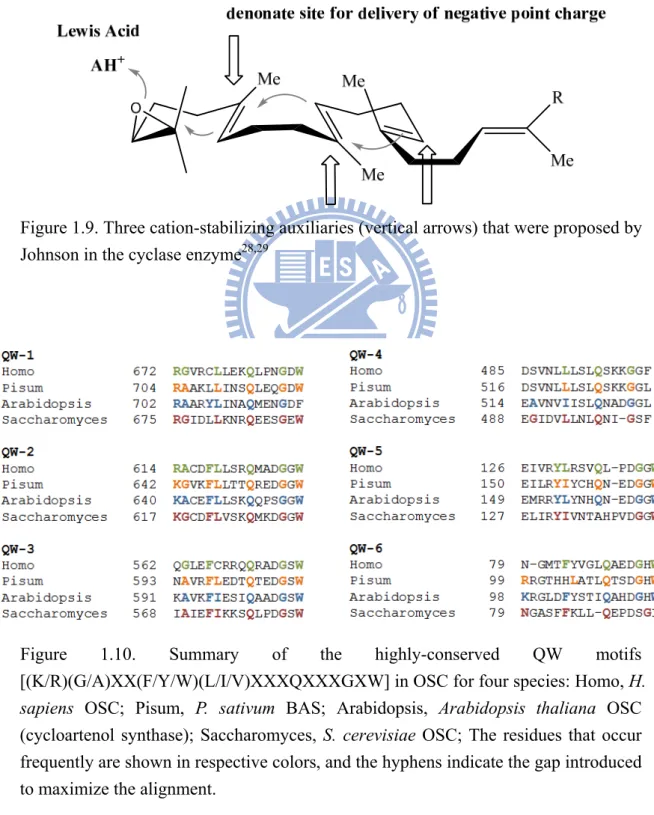
Due to the intrinsic difficulty of purifying active cyclase enzymes, the critical issue concerning the enzymes themselves is illusive and poorly understood. Many theoretical models of enzymatic processes have been developed. Ourisson established the phylogenetic relationship among all of the known cyclase families.27 It was suggested that, in the beginning, a primitive ancestral cyclase that contained several essential elements was responsible for the enzyme-catalyzed cyclization/rearrangement reaction. Through the divergent molecular evolution of these cyclase enzymes in different species, the shape of the cavities of active sites or some other critically important characteristics or functionalities were changed by mutations.27 Extensive research focused on substrate specificity and the stereochemistry of the cyclization reaction promoted Johnson to propose that cyclization is initiated by proton donation from a Lewis acid residue on an epoxide ring and that three anionic sites in the enzymatic active-site cavity are involved in axial delivery to form ion pairs, thereby stabilizing the highly-energetic and developing cationic center of the cyclized substrate (Figure 1.9).28,29
Comparison of the amino acid sequence among all of the known cyclases in bacterial sources or in the eukaryotic cell, one unique motif was observed for five or six repetitions, i.e., the “Q-W motif” with the highly-conserved sequence, [(K/R)(G/A)X2-3(F//Y/W)(L/I/V)3X3QX2-5GXW] (Figure 1.10).29,30 One of the “Q-W
motifs” was further labeled with a mechanistic inhibitor, suggesting that these “Q-W motifs” might play a role in stabilizing the enzyme structure through surface-connecting -helices or by mediating the carbocationic intermediate through cation- interactions.31,32,33 According to the “Aromatic hypothesis,” the essential
feature of the enzymatic active site is the presence of tryptophan and tyrosine, which possess the electron-rich indole and phenol groups. In addition, a positively-charged
12
transition state and highly-energetic intermediates occur during the cyclization/rearrangement processes.34 Thus, the cation- interaction from these highly aromatic functional groups is responsible for stabilizing the electron-deficient cationic intermediates (Figure 1.11).34
Figure 1.9. Three cation-stabilizing auxiliaries (vertical arrows) that were proposed by Johnson in the cyclase enzyme28,29
Figure 1.10. Summary of the highly-conserved QW motifs [(K/R)(G/A)XX(F/Y/W)(L/I/V)XXXQXXXGXW] in OSC for four species: Homo, H.
sapiens OSC; Pisum, P. sativum BAS; Arabidopsis, Arabidopsis thaliana OSC
(cycloartenol synthase); Saccharomyces, S. cerevisiae OSC; The residues that occur frequently are shown in respective colors, and the hyphens indicate the gap introduced to maximize the alignment.
13
Figure 1.11. Aromatic hypothetical model for the cyclization mechanism (Vertical arrows show the proposed stabilizing delivery force for the carbonium ions)
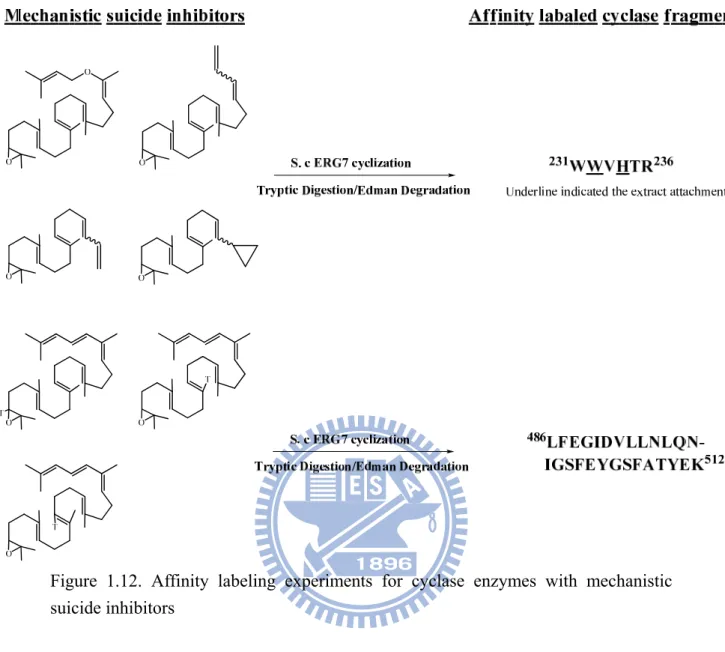
Also, the region of the sequence of amino acids from Val-454 to Glu-461, i.e., the VXDCATE motif in the eukaryotic cyclases, which correspond to the DXDDTAE motif in the bacterial cyclases, has also been characterized.31,35 According to the results obtained from affinity-labeling experiments, this peptide was specifically labeled with the first mechanism-based, irreversible inactivator, 29-methylidene-2,3-oxidosqualene (29-MOS), indicating the critical functional importance of this region in the oxidosqualene cyclization process.35 Successes in similar experiments have provided extensive and valuable insight into the mechanism of 2,3-oxidosqualene cyclization.36 The suicide substrates, which mimic monocyclic or bicyclic reactive intermediates, were specifically labeled on the fragments from Lys-489 to Lys-512 in Sacchomyces cerevisiae (S. cerevisiae) ERG7.21,36 In addition, the substrate analogues, which imitate the C-20 cation intermediates, revealed a capability for covalent modification at His-234 and Trp-232 residues in the cyclase enzyme (Figure 1.12).21,36
14
Figure 1.12. Affinity labeling experiments for cyclase enzymes with mechanistic suicide inhibitors
In addition, selective side-chain modifying agents have also been used to provide clues concerning the potential involvement of amino acid residues in the important aspects of the enzyme’s function. The compounds, p-(chloromercuri)-benzenesulfonic acid and N-ethylmaleimide, the SH-selective modifying reagents, presented strong inhibitory activity toward cyclases.37,38 It indicated that the presence of cysteinyl residue in the activity-site cavity of the enzyme is crucial for catalysis. Another chemical inactivation experiment was carried out to clarify the essentiality of histidine residue in cyclases.38 The discordant results from the chemical inactivation experiment on various source cyclases further imply that there are subtle differences
15
between the enzyme-active sites among these homologous cyclases.38,39
These early studies of cyclases focused mainly on the use of a variety of substrate analogues, which mimic various cyclic intermediates, to estimate the mechanism of structural requirement, the substrate’s specificity, and the stereochemistry of various stages in this chemical reaction. In addition, the conscientious analysis of the primary amino acid sequence among all known cyclases, in combination with the affinity labeling strategy, further elucidated the essential components of cyclase catalysis. However, the strict definition of which functional residues must be manipulated and how they are involved in the complex cyclization stages is still unknown at this time.
1.3 Crystallization and structural characterization of cyclases
Cyclase enzymes are considered to be an integral membrane protein based on the biophysical characterization of OSC for either cellular localization or protein solubility.40 In order to access, steer, and release the lipidic substrate and the synthetic product, these enzymes permanently occupy the lipid bilayer. The membrane-binding region employs a large hydrophobic channel that connects the active-site cavity of the enzyme and the non-polar portion located on one leaflet of the bilayer. Thus, the protein structure is regarded as a path for interacting with the target substrate and delivering the reactants.9
In 1997, the X-ray structure of squalene-hopene cyclase (SHC) from
Alicyclobacillus acidocaldarius (A. acidocaldarius) identified the cyclase as a
homodimeric monotopic membrane protein.33 The polypeptide chain of each subunit is organized into two sets of - barrel domains, which further jointly form a dumbbell-shaped molecule (Figure 1.13). The unique sequence fingerprint, Q-W repeats, is distributed throughout the surface of the enzyme structure, where the
16
functional residues of these amino acid repeats form an intricate hydrogen-bonding network and connect all outer helices of the - barrel domains by hydrophobic interactions.33,41,42 It proposed that this arrangement might stabilize the whole protein structure against the reaction enthalpy that is released during the cyclization reaction.
Figure 1.13. (Left) The overall structure of A. acidocaldarius SHC33 (Right) The putative active-site cavity in the SHC41
Moreover, a large hydrophobic cavity located deep in the enzymatic active site of SHC is enclosed by long loops and several small sheets that link barrel domains.33
Several conserved aromatic amino-acid residues that are aligned in suitable positions in this cavity were assumed to access/constrict the mode of the substrate, thereby determining the manners in which the substrate is folded and stabilizing the individual carbocationic intermediates during the cyclization reaction.9 Two smaller polar patches formed by networks of hydrogen-bonding around Asp-376 (at the top of
17
cavity) and Glu-45 (at bottom of cavity) were believed to participate in the initiation of the cyclization cascade via olefin protonation and in the termination of the reaction by effectively abstracting the proton or hydroxylating the cationic position with water, respectively.9,33
The structure-based mutagenesis of conserved amino acid residues in the active site cavity was subsequently examined based on the structure information of A.
acidocaldarius SHC.43-48 Functional residues that participated in the SHC-catalyzed cyclization from squalene to hopene/hopanol were determined by isolating the resulting early-truncated products from different protein mutants. 43-48
The SHC in prokaryote is the counterpart of the OSC in eukaryotic cells, regardless of the variant amino acid sequences and the distinct enzyme-catalyzed mechanisms. Thus, the early three-dimensional structure and the mutagenesis studies of SHC provided a shallow insight for the first time that helped researchers to understand the relationship between functional residues in the active-site cavity of cyclase enzyme and the enzymatic reaction mechanism of the OSC in the eukaryotic cells (Figure 1.13).
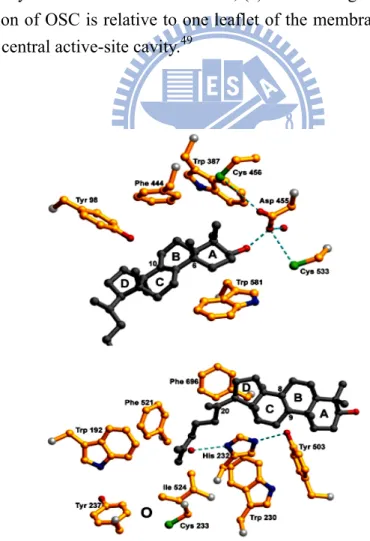
Recently, the long-awaited X-ray crystal structure of human OSC co-crystallized with the reaction product, lanosterol, was successfully determined (Figure 1.14).49 This was a significant milestone for understanding the molecular interactions between lanosterol and the enzymatic active site, and it also provided an important additional snapshot of the triterpene polycyclization cascades. Overall, the structure of human OSC has a topology that is almost identical to that of A. acidocaldarius SHC. OSC is a monotopic membrane protein, and its membrane-inserted plateau with a diameter of 25 Å serves as hydrophobic substrate entrance channel that connects the active-site cavity and non-polar interior of the membrane. On the basis of crystallographic analysis of human OSC structure, this channel is constricted by Tyr-237, Cys-233,
18
Ile-524, and the two stained loops from 516 to 524, and from 679 to 699. Once the immigrated substrate moves into the appropriate position in the enzymatic active-site cavity, the polycyclization reaction can be triggered. Asp-455, which is located at the top of the cavity and form a hydrogen-bonding network with the 3-hydroxyl group of the lanosterol, is thought to be responsible for the initiation of the reaction. The surrounding Cys-456 and Cys-533 residues also act as hydrogen-bonding partners with the Asp-455 to increase its acidity. Thus, Asp-455 performs the role of a general acid to donate a proton to the epoxide oxygen of the substrate and promotes the initiation of the cyclization cascade.33,41
As proposed in a previous theoretical model, the abundant, highly-electron-riched aromatic residues, which are located in the hydrophobic enzymatic cavity, are responsible for stabilizing the carbocation intermediate in different stages (Figure 1.15). Phe-444, Tyr-503, and Trp-581, which are oriented around the A/B ring of the lanosterol molecule, might stabilize the monocyclic C-10 and bicyclic C-8 cation (lanosterol numbering), respectively. Tyr-98 is located spatially at a better position to push the methyl group at C-8 below the molecular plane and enforces the formation of the energetically-unfavorable boat conformation of the B-ring. During formation of the C-ring, His-232 and Phe-696 are in suitable orientations to stabilize the unusual secondary anti-Markovnikov cyclohexyl carbocation at C-14. It has been reported that Trp-169 and Phe-605 in SHC stabilize the long-lived 6,6,6,5-fused tetracyclic secondary cation as required for the formation of the E-ring of hopene. The lack of aromatic residues in the corresponding position of the OSC may lead to the termination of the cyclization cascade at the tertiary D-ring protosteryl cation.49 Then the protosteryl C-20 cation undergoes a series of skeletal rearrangements to form the lanosteryl C-8/C-9 cation. Finally, His-232 and Tyr-503 are possible candidates for the role of a general base that extracts the proton from the lanosteryl C-8/C-9 cation to
19
terminate the cyclization/rearrangement reaction cascades.49-51
The insight on the biological and structural characteristics of the triterpene cyclase family provides a better understanding of the relationships between important catalytic residues in the active-site cavity and the mechanisms by which cyclization occurs. It also suggests that the strategies of rational, structure-based engineering of the cyclase enzymes and its experimental results might further illustrate the detailed interactions that occur between the enzyme itself and the reactants. Thus, in the following chapters, various mutagenesis approaches on OSC were carried out. The isolation/characterization of several novel truncated cyclizations, alternative deprotonations, as well as the distinct stereochemical conformation products from these mutations further demonstrated the importance and convenience of the mutagenesis strategy for developing a better understanding of the OSC-mediated reaction.
20
Figure 1.14. Crystal structure of human OSC; (a) Ribbon diagram of human OSC; (b) The orientation of OSC is relative to one leaflet of the membrane, and Ro48-8071 is bound in the central active-site cavity.49
Figure 1.15. Local view of crystal structure of human OSC49
.
21
CHAPTER 2
Thesis organization
Advances in crystallographic illustration, structure-based mutagenesis, chemical inhibition and modification of substrate analogues for SHC have provided the insight to understand the relationship between the structural and functional details of the catalytic mechanism of enzyme-templated consequent ring-formation reactions. SHC appear to be primitive ancestors of OSCs, and this link such allow for the collection of extensive, plausible information about comparable OSCs. Recently, a three-dimensional structure of a related OSC from Homo sapiens (H. sapiens) was elucidated, presenting structural clues for the molecular interactions between the substrate and crucial residues in the active-site. Despite this information, various catalytic mechanisms are still being proposed.
The identification of the important residues in OSC that participate in the course of the complex cyclization and rearrangement cascade is critical for understanding the relationships in the structure-function-reaction mechanism. This information can be used to identify the residues involved in specific rearrangement and deprotonation steps, as well as those that determine substrate folding and product diversity. Herein, computer-assisted homology modeling, site-directed mutagenesis and site-saturated mutagenesis, coupled with product characterization strategies, were used to understand and solve these fascinating puzzles.
Chapter 1 summarizes a historical review as well as the biomimetic approach toward OSC, whether in chemistry, molecular biology, or enzymology, to give a fundamental framework for exploring the squalene and OSC-mediated reaction.
Chapter 3 presents the alignments of multiple sequences among various cyclases, each with a distinct substrate pre-folding conformation, to determine which residues
22
are critical for product control. Out of nine selected residues, His-234 was found to play the greatest role in catalysis as evidenced by many truncated, incomplete products, including monocyclic, tricyclic, and alternatively rearranged products. Many of these products were identified for the first time, representing the strong catalytic role of His-234 in the intermediate steps of oxidosqualene cyclization.
Chapter 4 describes the use of a computer-based model of S. cerevisiae ERG7 to determine the potential performance of the active-site cavity. Also, site-saturated mutagenesis experiments coupled with product isolation and identification were used to illustrate the specific amino-acid residues of OSC that contribute to the stabilization of carbocationic intermediates, hydride and methyl group rearrangements, and the deprotonation reaction.
Chapter 5 investigates the catalytic motif specific to the rearrangement cascade and the final deprotonation reaction in sterol-producing cyclases. Three residues, Thr-384, Glu-450, and Val-454, whose conserved patterns are different between OSC and cycloartenol synthas (CAS), were chosen for the mutagenesis studies. Mutation of these residues in OSC into the corresponding substitutions in CAS produced the most accurate parkeol synthase in the study.
Chapter 6 details the efforts involved in the molecular cloning and sequencing of a plant 2,3-oxidosqualene -amyrin synthase from P. sativum (P. sativum). In addition, the structural comparison between P. sativum -amyrin synthase and S. cerevisiae OSC and their site-directed mutagenesis experiments were used to identify and confirm the possible role of the properties in specific amino acids in the mutations.
Chapter 7 is a discussion on the results from the previous chapters and describes concepts that could be used in future projects.
Chapter 8 presents the detailed experimental aspects of the study of 2,3-oxidosqualene cyclase enzymes. Fundamental molecular biology techniques,
23
general experimental materials, and the construction of specific recombinant plasmid are also described.
The Appendix provides information on the 1H-NMR, DEPT, HSQC, and HMQC NMR spectra that characterize the protosta-12,24-dien-3-ol, protosta-20,24-dien-3- ol, (13H)-isomalabrica-14(26),17E,32-trien-3-ol, 4,14-dimethyl-5-cholest- 9(11)-dien-3-ol, and 4,14-dimethyl-24-methylene-5-cholest-9(11)-en-3-ol.
24
CHAPTER 3
Site-directed mutagenesis of oxidosqualene cyclase to characterize the plasticity of the protein and the diversity of the product
3.1 Research background and aim
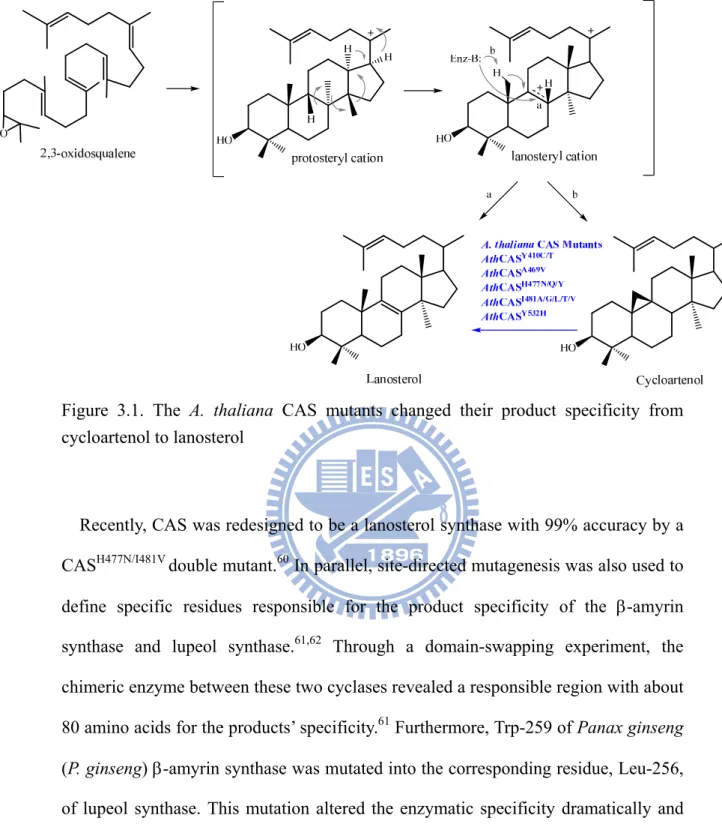
Nearly 200 distinct triterpenoid skeletons have been isolated from several sources.52-54 The diverse product profiles with regio- and stereo-selectivity of oxidosqualene-catalyzed cyclization reaction might be controlled precisely by the pre-organized substrate conformation and the plastic enzyme active site. Recently, mutagenesis studies have shown that a slight modification of the enzymatic active sites is sufficient to lead to different cyclization/rearrangement reaction products.55,56 For example, our group conducted random mutagenesis and in vivo selection experiments on Arabidopsis thaliana (A. thaliana) CAS to identify several critical residues that are responsible for the specific deprotonation at the C-8/C-9 position or for the formation of a strained cyclopropyl ring.57 Five product specificity-determining point mutations of A. thaliana CAS, including Tyr410Cys, Ala469Val, His477Tyr, Ile481Thr, and Tyr532His, produced lanosterol to complement the erg7-deficient strain (Figure 3.1).51 Subsequently, various other functional residues have been isolated independently by using similar strategies coupled with computer modeling illustration.58,59
25
Figure 3.1. The A. thaliana CAS mutants changed their product specificity from cycloartenol to lanosterol
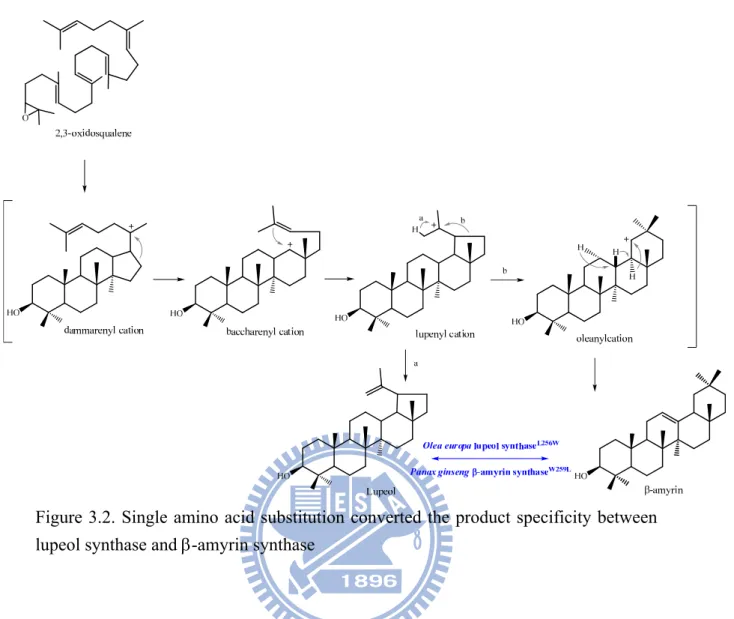
Recently, CAS was redesigned to be a lanosterol synthase with 99% accuracy by a CASH477N/I481V double mutant.60 In parallel, site-directed mutagenesis was also used to define specific residues responsible for the product specificity of the -amyrin synthase and lupeol synthase.61,62 Through a domain-swapping experiment, the chimeric enzyme between these two cyclases revealed a responsible region with about 80 amino acids for the products’ specificity.61 Furthermore, Trp-259 of Panax ginseng (P. ginseng) -amyrin synthase was mutated into the corresponding residue, Leu-256, of lupeol synthase. This mutation altered the enzymatic specificity dramatically and produced lupeol as its dominant product along with other minor compounds (less than 10%). Interestingly, the corresponding mutation of Leu256Trp in Olea europa (O.
europa) lupeol synthase became a functional amyrin synthase to further elucidate the important role of this residue in determining product specificity (Figure 3.2).62
26
Figure 3.2. Single amino acid substitution converted the product specificity between lupeol synthase and -amyrin synthase
The geometry of the enzymatic active site might adapt the substrate into a product-like, pre-folded conformation prior to the initiation of the reaction.However, there has been no study that focused on the relationship between substrate conformation and product specificity. How some ingenious performance of the enzyme forces the substrate into an energetically-unfavored boat form or the preferable chair form is very critical for cyclase to determine the following product formation in either the steroid or the triterpenoid biosynthesis pathway. Therefore, the alignment of the amino acid sequences among these cyclases with an opposite substrate conformation might provide valuable information for explaining the catalytic discrepancy among oxidosqualene-catalyzed reactions.51 Site-directed mutagenesis, genetic selection, and the product-characterization approach, were
27
conducted to examine those artificial cyclases with novel catalytic activity or novel product specificity/diversity (Figure 3.3).
Figure 3.3. The mutagenesis approach might provide the opportunity to understand how the cyclase enzyme forces the substrate into two different conformations, which is critical for determining the following product formation in either the sterol or the triterpene biosynthesis pathway.
3.2 Results and Discussion
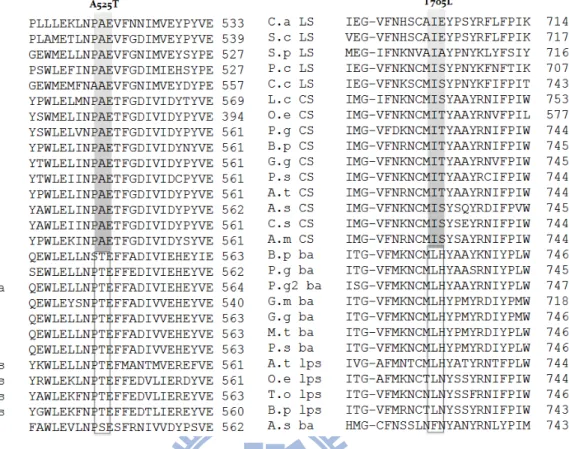
3.2-1 Alignments of multiple amino acid sequences for sterol cyclases and triterpene cyclases
Despite the low sequence identity (from 16% to 21%) in these two types of cyclases, which have the opposite conformations of the substrate, the triterpene cyclase families in prokaryotic cell and sterol cyclase in eukaryotic cell show similar structural topology.49,63It is considered that the conserved amino acid residues may be
28
responsible for the catalytic correlation and also for maintaining the secondary structure. The exclusively conserved amino-acid residues in either sterol OSCs or in triterpene OSCs might be required for the formation of the “boat” or “chair” substrate conformation, which further leads to a protosteryl cation or a dammarenyl cation, respectively. Therefore, the comparison of multiple amino acid sequences among all known cyclases was first utilized to elucidate the putative functional amino-acid residues that are critically involved in influencing the B-ring conformation (Figure 3.4).
3.1-2 Nine site-directed mutants of ERG7 gene from S. cerevisiae
The site-directed mutagenesis approach was first used to substitute the selected residues on ERG7 with various suitable amino acids. Nine conserved residues, i.e., Met-103, Gly-108, His-234, Ile-240, Thr-333, Pro-340, Gly-444, Ala-525, and Ile-705, from the ERG7 gene of S. cerevisiae were mutated into the corresponding residues that are conserved in the triterpene OSCs with a substrate that has an opposite, pre-folded chair-chair-chair conformation. These mutants, including Met103Leu, Gly108Pro, His234Tyr, Ile240Met, Thr333Ser, Pro340Cys, Gly444Thr, Ala525Thr, and Ile705Leu, were constructed by using the QuikChangeTM Site-Directed Mutagenesis System or the overlapping extension PCR methods with the respective mutagenic primer pairs. A silent mutation was introduced simultaneously for screening the desired mutants according to a restrictive endonucleases analysis. The presence of the mutations was verified further by using the sequencing determination (Table 3.1).
30
Figure 3.4. Alignment of multiple amino acid sequences for screening the putative residues involved in the product specificity/diversity. The sequences shown are
Candida albicans OSC (C.a LS), S. cerevisiae OSC (S.c LS), Schizosaccharomyces pombe OSC (S.p LS), Pneumocystis carinii OSC (P.c LS), Cephalosporium caerulens
OSC (C.c LS), Luffa cylindrica CAS (L.c CS), Olea europaea CAS (O.e CS), P.
ginseng CAS (P.g CS), Betula platyphylla CAS (B.p CS), Glycyrrhiza glabra CAS
(G.g CS), P. sativum CAS (P.s CS), A. thaliana CAS (A.t CS), Avena strigosa CAS (A.s CS), Costus speciosus (C.s CS), Abies magnifica CAS (A.m CS), Betula
platyphylla AS (B.p ba), Panax ginseng AS (P.g ba), Panax ginseng-2 AS (P.g2 ba), Glycine max AS (G.m ba), Glycyrrhiza glabraAS (G.g ba), Medicago
truncatula AS (M.t ba), P. sativum AS(P.s ba), A. thaliana LUS (A.t lps), Olea
europaea LUS (O.e lps), Taraxacum officinale LUS (T.o lps), Betula platyphylla LUS
31
Table 3.1. Construction strategies and individual silent mutation sites of the nine conserved residues
Constructs Amino acid
location Mutation Translation
Mutation strategies
Mapping site For silent mutation
pRSYTLOSCM103L : T1 103 ATG → CTG M → L QuikChange PCR Pvu l
pRSYTLOSCG108P : T2 108 GGT → CCT G → P QuikChange PCR Apa l
pRSYTLOSCH234Y : T3 234 CAT → TAT H → Y Overlapping extension PCR Xho l
pRSYTLOSCI240M : T4 240 ATT → ATG I → M QuikChange PCR Xho l
pRSYTLOSCT333S : T5 333 ACT → AGT T → S QuikChange PCR Pst l
pRSYTLOSCP340C : T6 340 CCT → TGT P → C QuikChange PCR Hind lll
pRSYTLOSCG444T : T7 444 GGC → ACC G → T QuikChange PCR Ban l
pRSYTLOSCA525T : T9 525 GCT → ACT A → T QuikChange PCR Nco l
pRSYTLOSCI705L : T11 705 ATT → CTT I → L QuikChange PCR Pvu ll
3-2.3 Principle of the plasmid shuffle method
The plasmid shuffle method, which involves the exchange of a plasmid bearing the wild-type gene sequence with a plasmid bearing the mutation of interest, is an effective method for analyzing the effect of mutants by their complementation ability to the erg7-deficient strain.64
Biosynthetic ergosterol, which is required for yeast viability, is generated through conversion of 2,3-oxidosqualene into lanosterol by OSC and another 18 additional enzymatic steps. Thus, erg7 is regarded as an essential gene. In order to demonstrate the functional role of ERG7 mutants, the plasmid shuffle method is used. First, the
erg7 gene of a haploid yeast cell was disrupted by means of one-step gene disruption
with a selectable genetic marker. Viability was maintained by transforming a plasmid bearing the wild-type gene, pZS11. Any random loss of the wild-type gene plasmid from the yeast cell would be lethal to the viability of the cell. The wild-type gene
32
plasmid also carries a second selectable/counter-selectable marker, URA3. This commonly used marker, the ura3 gene, encodes orotidine-5’-phosphate decarboxylase, which converts the 5’-fluoroorotic acid (5’-FOA) into a toxic compound, 5’-fluorouracil. Thus, yeast that lacks URA3 activity is resistant to 5’-FOA. Another pRS314 plasmid bearing a gene with a mutated sequence of interest and a third selectable genetic marker is transformed into a yeast cell containing a disrupted erg7 gene (erg7-) and the wild-type gene plasmid (pZS11). Viability is the result of the presence of the first wild-type gene plasmid. Placing a supplement of the exogenous uracil into the medium, which is the end product of the URA3-involved biosynthetic pathway, could facilitate the loss of the first wild-type gene plasmid with frequencies up to 1% per generation.
Counter-selection against the wild-type gene plasmid with 5’-FOA allows the yeast cell to retain only the second transformed mutated gene plasmid. Therefore, the survival or the death of the yeast transformant depends on the functional activity of the mutated enzyme. If the mutant abolishes the enzymatic activity, the yeast transformant is expected to die upon loss of the first plasmids. Conversely, if the functional activity is not dramatically influenced by the mutation, the yeast transformants will still remain viable. The selectable/counter-selectable plasmid shuffle strategy provides a powerful tool for functional analysis of the mutated gene of interest (Scheme 3.1).64
33 pZS11 URA3 OSCWT Yeast CBY57 pZS11 URA3 OSCWT YEAST CBY57 pRS314 Trp3 OSCmutant YEAST CBY57 pRS314 Trp3 OSCmutant pRS314 Trp3 OSCmutant
Transform into the ERG7 knockout strain
Yeast CBY57
(MAT a/ ERG7 ::LEU2 ade2-101 his3- 200 leu2- 1 lys2-801 trp1- 63 ura3-52 [pZS11]
5'-FOA N O N O F OH O 5-fluoroorotic acid ( Non-toxic ) Orotidine-5-phosphate decarboxylase N O N O F 5-fluorouracil acid ( toxic ) URA3
Scheme 3.1. The plasmid shuffle strategy. Two step methods are used to exchange the wild type plasmid with the mutant plasmid by 5’-FOA, which is converted into 5’-flurouracil by the URA3 enzyme.
3.2-4 Screening inactive ERG7 mutants via the plasmid shuffle method
The genome type of a haploid yeast strain, CBY57, is MATa/ ERG7∆::LEU2
ade2-101 his3-∆200 leu2-∆1 lys2-801 trp1-∆63 ura3-52. A CBY57[pZS11] with an erg7-disrupted genome and a pZS11 plasmid, which is a URA3 centromeric plasmid
with a wild-type erg7 gene, was used as the assay strain. The pRS314-derived ERG7-mutated plasmids with a selectable genetic marker of TRP1 were transformed into the yeast strain CBY57[pZS11] by using electroporation. The plasmid shuffle strategy, illustrated above, was used to analyze the mutation effect. Positive (pRS314ERG7WT) and negative (pRS314) controls were also carried out to confirm the accuracy of the plasmid shuffle. First, yeast transformants were selected on SD+Ade+Lys+His plates, and then, re-selected on SD+Ade+Lys+His+Ura+5’-fluoro- orotic acid media to elucidate the complementation effects. Among the results, all of the mutants grew colonies upon counter-selection for pZS11 with 5’-FOA, indicating
34
that Met103Leu, Gly108Pro, His234Tyr, Ile240Met, Thr333Ser, Pro340Cys, Gly444Thr, Ala525Thr, and Ile705Leu mutants are active and don’t dramatically influence the enzymatic activity. This finding suggested that the nine selected residues in the S. cerevisiae ERG7 do not impair the activity below the basal level or affect the growth of yeast cells. Mutation might slightly influence the active site or the enzyme structure, butlanosterol was still produced to maintain the yeast viability. This finding implied that these residues might not be responsible for the determination of product specificity/diversity.
3.2-5 Lipid extraction, column chromatography, and product characterization of the yeast transformants
To deduce the possible catalytic or structural role of the selected residues, the non-saponifiable lipid (NSL) extract was prepared from 2.5 liters of cultured yeast transformants and applied to analysis of the product profiles. NSL extracts were dissolved in CH2Cl2, spotted on thin-layer chromatography (TLC), developed with
20% Ethyl Acetate (EA)/Hexane, and observed by using anisaldehyde staining. Furthermore, to examine the chemical structure of these triterpene products, the NSL extracts were separated roughly into several fractions by silica gel chromatography.
Different product patterns were collected independently (Scheme 3.2) by comparing to the control strain, pRS314ERG7WT. In addition, gas chromatography-mass spectrometry (GC-MS) was utilized to screen all fractions of the product pattern in the wild-type ERG7 and mutants.
35
Scheme 3.2. Preliminary analysis and separation of products on TLC plate
Based on the results of TLC and GC-MS, the product patterns of all mutants were similar to that of the wild-type ERG7 except for the His234Tyr mutant, demonstrating that Met-103, Gly-108, Ile-240, Thr-333, Pro-340, Gly-444, Ala-525, and Ile-705 of ERG7 are not the critical determinative amino acids that affect or determine the specificity/diversity of the enzyme.
A novel product, with a smaller polarity than lanosterol, was detected in the His234Tyr mutant by analyzing the TLC and GC-MS spectra. In addition, the GC-MS spectra revealed two additional novel compounds that had an identical molecular mass of m/z = 426; these compounds were indistinguishable from TLC chromatography in the Lanosterol fraction (LA fraction) (Figure 3.5). By characterizing the product structure, the possible role of His-234 of ERG7 involved in the cascades of cyclization/rearrangement will be elucidated in the next sections.
36
Figure 3.5. Comparison of the GC patterns of LA fraction from the NSL extracts among ERG7WT and nine mutants
The product of His234Tyr mutant with higher Rf value on the TLC plates was
further isolated via silica gel chromatographic purification. After analyzing the GC-MS spectra and the 1H-NMR spectroscopic data for this compound and
ERG7Met103Leu - LA fraction ERG7Thr333Ser -LA fraction ERG7Gly108Pro - LA fraction ERG7Pro340Cys -LA fraction ERG7His234Tyr- -LA fraction ERG7Gly444Thr -LA fraction ERG7Ile240Met - LA fraction ERG7Ala525Thr -LA fraction ERG7Ile705Leu -LA fraction RS314WT- LA fraction LA ? ?