Arsenic induces pancreatic β-cell apoptosis via the oxidative stress-regulated mitochondria-dependent and endoplasmic reticulum stress-triggered signaling pathways
Tien-Hui Lua,1, Chin-Chuan Sub, Ya-Wen Chenc,1, Ching-Yao Yangd, Chin-Ching Wue, Dong-Zong Hunga,f, Chun-Hung Chena,g, Po-Wen Chengh, Shing-Hwa Liud,i,1, Chun-Fa Huangj,*
aGraduate Institute of Drug Safety, College of Pharmacy, China Medical University,
Taichung 404, Taiwan
bDepartment of Otorhinolaryngology, Head and Neck Surgery, Changhua Christian
Hospital, Changhua 500, Taiwan
cDepartment of Physiology, and Graduate Institute of Basic Medical Science, College
of Medicine,China Medical University, Taichung 404, Taiwan
dDepartment of Surgery, National Taiwan University Hospital, and Department of
Surgery, College of Medicine, National Taiwan University, Taipei 100, Taiwan
eDepartment of Public Health, China Medical University, Taichung 404, Taiwan fToxicology Center, China Medical University Hospital, Taichung 404, Taiwan gDepartment of Emergency, China Medical University Hospital, Taichung 404,
Taiwan
hDepartment of Otolaryngology, Far Eastern Memorial Hospital, Taipei 220, Taiwan iInstitute of Toxicology, College of Medicine, National Taiwan University, Taipei 100,
Taiwan
jSchool of Chinese Medicine, College of Chinese Medicine, China Medical
*To whom corresponding author should be addressed to Chun-Fa Huang, Ph.D.,
School of Chinese Medicine, College of Chinese Medicine, China Medical University, No.91 Hsueh-Shih Road, Taichung 404, Taiwan.
E-mail: [email protected] Tel.: +886 4 22053366 ext. 3323. Fax: + 886 4 22333641
Abbreviations:
ROS, reactive oxygen species; MDA, malondialdehyde; MAPKs, mitogen-activated protein kinases; PARP, poly (ADP-ribose) polymerase activation; ER stress, endoplasmic reticulum stress; GRP, glucose-regulated protein; CHOP, C/EBP homologue protein; XBP-1, X-box binding protein 1; JNK, c-Jun N-terminal kinases; ERK, extracellular signal-regulated kinases
Abstrct:
Arsenic (As), a ubiquitous toxic metal, is an important environmental and industrial pollutant throughout the world. Inorganic As (iAs) is usually more harmful than organic ones and with a high risk of diabetes incidence by exposure. However, the toxicological effects of iAs on growth and function of pancreatic β-cells still remain unclear. Here, we found that iAs significantly decreased insulin secretion and cell viability, and increased ROS and MDA formation in pancreatic β-cell-derived RIN-m5F cells. iAs also induced the increases in sub-G1 hypodiploids, annexin V-Cy3 binding, and caspase-3 activity in RIN-m5F cells, indicating that iAs could induce β-cell apoptosis. Moreover, iAs induced MAPKs activation, mitochondria dysfunction, p53 up-regulation, Bcl-2 and Mdm-2 down-regulation, PARP, and caspase cascades, which displayed features of mitochondria-dependent apoptotic signals. In addition, exposure of RIN-m5F cells to iAs, could trigger ER stress as indicated by the enhancement in ER stress-related molecules induction (such as GRP78, GRP94, CHOP, and XBP1), procaspase-12 cleavage, and calpain activation. The iAs-induced apoptosis and its-related signalings could be effectively reversed by antioxidant N-acetylcysteine. We next observed that exposure of mice to iAs in drinking water for 6 consecutive weeks significantly decreased the plasma insulin and elevated glucose intolerance, plasma lipid peroxidation, and induced islet cells apoptosis, which accompanied with arsenic accumulation in the whole blood and pancreas. N-acetylcysteine effectively antagonized the iAs-induced responses in mice. Taken together, these results suggest that iAs-induced oxidative stress causes pancreatic β-cells apoptosis via the mitochondria-dependent and ER stress-triggered signaling pathways.
Keyword: Arsenic; Pancreatic β-cells; Oxidative stress; Mitochondria dysfunction;
1. Introduction:
Arsenic (As), a naturally occurring toxic metalloid, is ubiquitous in the environment in both inorganic and organic forms. Inorganic As (iAs) is the predominant form of As in soil, groundwater reservoirs, and industrial pollutants-all of which are important sources of As in cases of human exposure (Ng et al., 2003; Smith et al., 2002). Moreover, significant exposure to As is also an occupational hazard for copper or lead smelters, glass workers, and agricultural workers who use As-containing agricultural pesticides or herbicides, etc (Otles and Cagindi, 2010; Rahman and Axelson, 1995). Many epidemiological studies have reported that in the United States, China, and other countries, chronic exposure to As in drinking water is associated with increased rates of serious health problems, including incidences of various cancers, neurotoxicity, liver injury, peripheral vascular disease (e.g., blackfoot disease (BFD)), and endocrine dysfunction (Chiou et al., 2005; Mazumder, 2005; Meliker et al., 2007). Recent studies have provided growing evidence of a significant dose-response relationship between As exposure and the prevalence of diabetes mellitus in high-iAs exposure areas in Taiwan and Bangladesh (Lia et al., 1994; Rahman et al., 1998). Notably, diabetes mellitus has been a substantially increasing world health problem, and As exposure is an environmental risk factors for diabetes mellitus development and/or pancreatic β-cell dysfunction (Tseng et al., 2002).
The endoplasmic reticulum (ER), a central intracellular organelle, is a quality control system for intracellular protein homeostasis: it controls protein synthesis, folding, and delivery of biologically active proteins (Schorder and Kaufman, 2005; Sitia and Braakman, 2003). The accumulation of unfolded or misfolded proteins in the lumen of the ER, which induces a coordinated adaptive program called the unfolded protein response (UPR), causes ER stress (Mori et al., 2000). Increasing evidence indicates that ER stress plays a critical role in the regulation of apoptosis caused by a
variety of toxic insults that damage mammalian cells, including hypoxia, chemicals, Ca2+ homeostasis imbalance, and heavy metals (Biagioli et al., 2008; Kubota et al., 2005). On the other hand, the production of ROS is induced by misfolded proteins in the ER, which leads to the activation of UPR and contributes to the induction of apoptosis (Hung et al., 2010; Malhotra et al., 2008). ROS induce a wide variety of undesirable biological reactions and functional cell damage, including pancreatic β-cell dysfunction and/or apoptosis caused by cytokines or autoimmune attack in type 1 diabetes. Pancreatic β-cells are reported to be vulnerable to ROS, which elicits oxidative stress damage (Hotta et al., 2000; Kaneto et al., 2005). iAs has been reported to induce toxic effects via oxidative stress causing the disruption of cellular functions and eventually resulting in cell apoptosis and pathophysiological injury (Mishra and Flora, 2008; Shi et al., 2010). iAs exposure is a critical risk factor for developing diabetes; however, only a few studies have shown that iAs can induce the dysfunction and apoptosis of pancreatic β-cells in vitro and in vivo (Diaz-Villasenor et al., 2006; Izquierdo-Vega et al., 2006; Yen et al., 2007). Moreover, the toxic effects of iAs in pancreatic β-cells and the possible mechanisms underlying these effects are mostly unclear.
Taken together, in the current study, we try to explore the role of ROS in iAs-induced pancreatic β-cell apoptosis. To this aim, we investigated the in vitro effects of iAs on ROS generation, insulin secretion, mitochondrial dysfunction, and ER stress-related molecules. In addition, we tested whether exposure to iAs, in a way that mimics the route of human exposure, alters the regulation of insulin secretion, the disturbance of blood glucose tolerance, generate lipid peroxidation, and islet cells apoptosis in mice. Moreover, we also investigated the potential protective effects of the antioxidant N-acetylcysteine on iAs-induced pancreatic β-cell damage in vitro and
2. Materials and Methods:
2.1. Materials
Arsenic trioxide (As2O3), 3-(4, 5-dimethyl thiazol-2-yl-)-2, 5-diphenyl
tetrazolium bromide (MTT), dimethyl sulfoxide (DMSO), N-methyl-2-phenylindole, 1, 1, 3, 3-tetramethoxypropane (MDA), Annexin V-Cy3TM apoptosis detection kit, propidium iodide (PI), Z-Val-Phe-CHO (MDL 28170), and D-glucose were purchased from Sigma-Aldrich (St. Louis, MO). 2’, 7’-dichlorofluorescin diacetate (DCFH-DA), and 3,3′-dihexyloxacarbocyanine iodide (DiOC6) were purchases from Molecular Probes 2’, 7’-dichlorofluorescin diacetate (DCFH-DA), and 3,3′-dihexyloxacarbocyanine iodide (DiOC6) were purchases from Molecular Probes (Eugene, OR). CaspACETM fluorometric activity assay kit, AMV RTase (reverse transcriptase enzyme), RNasin (RNAase inhibitor), and DeadEndTM Colorimetric terminal deoxynucleotidyl transferase mediated dUTP nick end labelling (TUNEL) system (TUNEL assay kit) were purchases from Promega Corporation (Promega Corporation, Pty. Ltd.). Mouse- or rabbit-polyclonal antibodies specific for cytochrome c, caspase-3, caspase-12, c-Jun N-terminal kinases (JNK)-1, extracellular signal-regulated kinases (ERK)1/2, p38, glucose-regulated protein (GRP)78, (GRP)94, XBP-1, α -tubulin, and secondary antibodies (goat anti-mouse or anti-rabbit IgG-conjugated horseradish peroxidase (HRP)) were purchased from Santa Cruz Biotechnology (Santa Cruz, Biotechnology, Inc.), and caspase-7, caspase-9, phosphor-JNK, phosphor-p38, phosphor-ERK1/2, C/EBP homologue protein (CHOP), were purchased from Cell Signaling Technology (Cell Signaling Technology, Inc.).
2.2. Cell culture
RIN-m5F rat insulioma pancreatic β-cell line is a clone derived from the RIN-m rat islet cells (Bhathena et al., 1984), which has property to produce and secrete
insulin (Cai and Lin, 2009; Matias et al., 2006). Cells were purchased from American Type Culture Collection (ATCC, CRL-11605; with Mycoplasma test: negative) and maintained in RPMI-1640 medium (Gibco BRL, Life Technologies) supplemented with 10 % fetal bovine serum (FBS) and antibiotics (100 U/ml of penicillin and 100 μg/ml of streptomycin) in a humidified chamber with a 5 % CO2-95 % air mixture at
37 ℃. The culture medium and supernatant from adherent cells were performing test for Mycoplasma contamination by Mycoplasma Detection Kit (Roche Applied Science, Indianapolis, USA) at every month.
2.3. Cell viability
Cells were washed with fresh media and cultured in 96-well plates (2×104/well) and then stimulated with As2O3 (1-10 μM) for 24 h. After incubation, the medium was
aspirated and fresh medium containing 30 μL of 2 mg/mL 3-(4, 5-dimethyl thiazol-2-yl-)-2, 5-diphenyl tetrazolium bromide (MTT) was added. After 4 h, the medium was removed and replaced with blue formazan crystal dissolved in dimethyl sulfoxide (100 μL). Absorbance at 570 nm was measured using an enzyme linked immunosorbent assay microplate reader (Bio-Rad, model 550, Hercules, CA).
2.4. Measurement of insulin secretion
To measure the amount of insulin secretion in RIN-m5F cells after exposure to As2O3, cells were performed in Krebs Ringer buffer (KRB), as previously described
(Chen et al., 2006b and 2010). In briefly, after iAs exposure, cells were switched to Krebs Ringer buffer (KRB) supplemented with 0.5 % BSA and low levels glucose (2.8 mM) for 1 h. Afterward, cells were challenged to secrete insulin in 16.7 mM glucose for 1 h. Supernatants were collected, centrifuged, and frozen at -70 ℃ until used. To measure the amount of insulin secretion, aliquots of samples were collected
from the plasma or experimental solutions at indicated time points, and subjected to insulin antiserum immunoassay according to the manufacturer's instructions (Mercodia AB, Sweden).
2.5. Flow cytometry analysis of ROS production, sub-G1 DNA content, and mitochondrial membrane potential
2.5.1. Determination of reactive oxygen species (ROS) production
Intracellular ROS generation was monitored by flow cytometry using the peroxide-sensitive fluorescent probe: 2’, 7’-dichlorofluorescin diacetate (DCFH-DA), as described Chen et al., 2010. In brief, cells were coincubated with 20 μM DCFH-DA at 37 ℃. After incubation with the dye, cells were resuspended in ice-cold phosphate buffered saline (PBS) and placed on ice in a dark environment. The intracellular peroxide levels were measured by flow cytometer (FACScalibur, Becton Dickinson, Sunnyvale, CA), that emitted a fluorescent signal at 525 nm. Each group was acquired more than 10000 individual cells.
2.5.2. Measurement of sub-G1 DNA content
RIN-m5F cells were detached and washed with PBS after treatment with for As2O3 24 h, then resuspended in 1 mL of cold 70% (v/v) ethanol and stored at 4 °C
for 24 h. After they were washed with PBS, the cells were stained with PI [50 μg/mL PI and 10 μg/mL ribonuclease (Rnase) in PBS] at 4 °C for 30 min in dark conditions. The cells were washed and subjected to flow cytometry analysis of DNA content (FACScalibur, Becton Dickinson). Nuclei displaying hypodiploid, sub-G1 DNA contents were identified as apoptotic. The sample of each group was collected more than 10,000 individual cells.
2.5.3. Determination of mitochondrial membrane potential
DiOC6, with a positive charge of a mitochondria-specific fluorophore. Briefly, RIN-m5F cells were plated in 12-well culture plate. After treatment, cells were harvested and loaded with 40 nM DiOC6 for 30 min and analyzed with FACScan flow cytometer (Becton Dickinson). The sample of each group was collected more than 10,000 individual cells.
2.6. Lipid peroxidation analysis
The formation of MDA, a substance produced during lipid peroxidation, was determined by using the commercial LPO assay kit (Calbiochem, San Diego, USA) as described by Huang et al. (2007) and Lu et al. (2010). Briefly, after exposure to As2O3
alone or in combination with NAC for 24 h, RIN-m5F cells were harvested and homogenized in 20 mM Tris-HCl buffer, pH 7.4, containing 0.5 mM butylated hydroxytoluene to prevent sample oxidation. The equal volumes samples (cell supernatant or plasma) were added 3.25 volumes of diluted R1 reagent (10.3 mM N-methyl-2-phenylindole in acetonitrile). After mixing, the mixture was added with 0.75 volumes of 37% HCl was added to the mixtures, which was then incubated at 45°C for 60 min. After cooling, the absorbance of the clear supernatant was read at 586 nm. The linearity of the standard curve was confirmed with 0, 1, 2.5, 5, 10, 20, 40 μM MDA standard (1, 1, 3, 3-tetramethoxypropane in Tris-HCl). The protein concentration was determined using the bicinchoninic acid protein assay kit with an absorption band of 570 nm. (Pierce, Rockford, IL, UAS). LPO level was expressed as nanomoles (nmol) MDA per milligram protein and estimated from the standard curve.
2.7. Detection of apoptotic cells
Apoptosis was assessed using Annexin V, a protein that binds to phosphatidylserine (PS) residues which are exposed on the cell surface of apoptotic
cells (Yen et al., 2007). Cells were treated with or without As2O3 for 24 h. After
treatment, cells were washed twice with PBS (ph 7.4), and incubated with Annexin V-Cy3 (Ann Cy3) and 6-carboxy fluorescein diacetate (6-CFDA) simultaneously (Annexin V-Cy3TM apoptosis detection kit). After labeled at room temperature, the cells were immediately observed by fluorescence microscopy (Axiovert 200, Zeiss, 200x). Ann Cy3 was available for binding to PS, which was observed as red fluorescence. In addition, cell viability could be measured by 6-CFDA, which was hydrolyzed to 6-CF and appears as green fluorescence. Cells in the early stage of apoptosis would be labeled with both Ann Cy3 (red) and 6-CF (green).
2.8. Measurement of Caspase-3 Activity
Caspase-3 activity was determined using the CaspACETM fluorometric activity assay kit as described by Chen et al., 2006. In brief, cell lysates were incubated at 37 °C with 10 μM Ac-DEVD-AMC, a caspase-3/CPP32 substrate for 1 h. The fluorescence of the cleaved substrate was measured by a spectrofluorometer (Spectramax, Molecular devices) with an excitation wavelength at 380 nm and an emission wavelength at 460 nm.
2.9. Quantitative Real-time PCR analysis
Expression of apoptosis-related genes was evaluated by quantitative real-time PCR (qPCR) analysis that carried out using Taqman® one-step PCR Master Mix (Applied Biosystems, Foster City CA). Briefly, total cDNA (100 ng) was added per 25-μl reaction with sequence-specific primers and Taqman® probes. Sequences for all target gene primers and probes were purchased commercially (β-actin, the housekeeping gene, was used as an internal control) (Applied Biosystems, CA). Quantitative RT-PCR assays were carried out in triplicate on StepOnePlus sequence
detection system. Cycling conditions were 10 min of polymerase activation at 95 °C followed by 40 cycles at 95 °C for 15 s and 60 °C for 60 s. After 40 cycles, samples were run on a 2 % agarose gel to confirm specificity. Data analysis was performed using StepOneTM software (Version 2.1, Applied Biosystems). All amplification
curves were analysed with a normalized reporter (Rn: the ratio of the fluorescence
emission intensity to the fluorescence signal of the passive reference dye) threshold of 0.2 to obtain the CT values (threshold cycle). The reference control genes were
measured with four replicates in each PCR run, and their average CT was used for
relative quantification analyses (the relative quantification method utilizing real-time PCR efficiencies (Pfaffl et al., 2002)). TF expression data were normalized by subtracting the mean of reference gene CT value from their CT value (ΔCT). The Fold
Change value was calculated using the expression 2-ΔΔCT, where ΔΔCT represents
ΔCT-condition of interest – ΔCT-control. Prior to conducting statistical analyses, the fold
change from the mean of the control group was calculated for each individual sample.
2.10. Calpain activity assays
Suc-Leu-Leu-Val-Tyr-AMC is a calpain protease substrate. Quantitation of 7-amino-4-methylcoumarin (AMC) fluorescence permits the monitoring of enzyme hydrolysis of the peptide-AMC conjugate and can be used to measure enzyme activity. Cells were prepared and treated on 24-well culture plates. Prior to addition of inhibitors cells were loaded with 40 μM Suc-Leu-Leu-Val-Tyr-AMC (Biomol) and treated with iAs for the indicated time at 37 °C in a humidified 5% CO2 incubator. Proteolysis of the fluorescent probe was monitored using a fluorescent plate reading system (FLx800, BioTek®
) with filter settings of 360±20 nm for excitation and 460±20 nm for emission.
2.11. Western blot analysis
The cellular lysates were prepared and western blotting was performed as previously described (Chen et al., 2006a). In brief, equal amounts of proteins (50 μg per lane) was subjected to electrophoresis on 10% (W/V) SDS-polyacrylamide gels and transferred to polyvinylidene difluoride (PVDF) membranes. The membranes were blocked for 1 h in PBST (PBS, 0.05% Tween-20) containing 5% nonfat dry milk. After blocking, the membranes were incubated with rabbit anti-rat antibodies against cytochrome c, PARP, caspase-3, caspase-7, caspase-9, caspase-12, PARP, phosphor-JNK, phosphor-p38, phosphor-ERK1/2, JNK-1, ERK1/2, p38, GRP-78, GRP-94, CHOP, XBP-1, calpain I, calpain II, or α-tubulin in 0.1% PBST (1:1000) for 1 h at room temperature. After they were washed in 0.1% PBST followed by two washes (15 min each), the blots were subsequently incubated with goat anti-mouse or anti-rabbit IgG-HRP secondary antibody (1:1000) for 1 h. The antibody-reactive bands were revealed by enhanced chemiluminescence reagents (Perkin-ElmerTM, Life Sciences) and were exposed on the Fuji radiographic film.
2.12. Animal preparation
We purchased normal male ICR mice (4weeks old, 20-25 g) from the Animal Center of College of Medical, National Taiwan University. The protocols used were approved by the Institutional Animal Care and Use Committee (IACUC) and the care and use of laboratory animals were conducted in accordance with the guidelines of the Animal Research Committee of China Medical University. Mice were housed in a room at a constant temperature (23 ± 2 ℃), 50 ± 20 % relative humidity, given a solid diet and tap water ad-libidum, and 12 hrs of light-ark cycles. Mice were acclimatized to the laboratory conditions prior to the experiments and all experiments were carried out between 8:00 AM and 05:00 PM. Mice were treated with 0, and 5
ppm arsenic trioxide (As2O3) in drinking water for 6 consecutive weeks in the
presence or absence of NAC (150 mg · kg-1 · day-1, oral application by gavage), respectively. Each group was contained 15 mice. All experimental mice were sacrificed by decapitation under pentobarbital anesthesia (80 mg/kg, i.p.), and the whole blood samples were collected from the peripheral vessels. Whole blood sample were centrifuged at 3000 x g for 10 min, and plasma was obtained, and insulin and lipid peroxidation (LPO) levels were assayed immediately. Pancreas were fixed in 4 % paraformaldehyde and embedded by paraffin for detecting islet apoptosis. At the same time, whole blood and pancreas were quickly removed and stored at liquid nitrogen until use, and then was analysis of As content.
2.13. Oral glucose tolerance test (OGTT)
Oral glucose tolerance testing was performed as previously described (Chen et al., 2006b). Mice were administrated with D-glucose by stomach tube after an overnight fast. Blood samples were collected (from an eyehole under anesthesia) before and 30, 60, 90, and 120 min after delivery of the glucose load. Blood glucose levels were determined using the Bayer blood glucose meter (
Ascensia ELITE,
Bayer).
2.14. Determination of arsenic content
300 mg of whole blood and pancreas were placed in a 15 ml polyethylene tube, and 0.5 ml of a 3:1 mixture of hydrocholic acid (35%) and nitric acid (70%) was added. The tube were capped and allowed to stand overnight at 50℃ oven. After cooling, suitable dilution buffer (0.3% nitric acid and 0.1% Triton X-100 in distilled water) was added to the digested material, and the total arsenic content was determined by inductively coupled plasma mass spectrometry (ICP-MS). The
detection limit for arsenic was ~0.1 ppb(ug/L).
2.15. Apoptosis analysis in mouse islet
Islet cells apoptosis was detected by TUNEL method utilizing the Dead End Colorimetic Apoptosis Detection system, as described previously (Kuwano et al., 1999). The deep brown of TUNEL-positive cells was imaged under the Nikon ECLIPSE 80i upright microscope equipped with a charge-coupled device camera (with ×200 magnification).
2.16. Statistical analysis
Data are presented as means ± S.E. The significance of difference was evaluated by the Student’s t-test. When more than one group was compared with one control, significance was evaluated according to one-way analysis of variance (ANOVA) was used for analysis, and the Duncans’s post hoc test was applied to identify group differences. The P value less than 0.05 was considered to be significant. The statistical package SPSS, version 11.0 for Windows (SPSS Inc., Chicago, IL, USA) was used for the statistical analysis.
3. Results
3.1. Effects of iAs on cell viability, insulin secretion, and ROS production in RIN-m5F cells
iAs, the predominant form of As in soil, underground water reservoirs, and industrial pollutants, is the main type of As to which millions of people worldwide have been exposed. Therefore, iAs (arsenic trioxide, As2O3) was used in this study for
investigating the toxic effects of iAs in the pancreatic β-cells. To examine iAs-induced pancreatic β-cell cytotoxicity, we first investigated the effect of iAs on cell survival in rat pancreatic β-cell-derived cell line (RIN-m5F). Treatment of RIN-m5F cells with iAs for 24h reduced the number of viable cells in a concentration-dependent manner with a range from 1 to 10 μM using the MTT assay (Figure 1A). Moreover, to evaluate the effect of iAs on the insulin secretion, the short-term response of iAs on insulin secretion from β-cells was dete4cted. After 4 h of treatment, iAs (2 and 5 μM) effectively suppressed insulin secretion in RIN-m5F cells (2 μM iAs, 93.77 ± 1.46; 5 μM iAs, 75.93 ± 2.77; control, 119.81 ± 2.23 pM, n = 6, ∗p < 0.05) that was not induced the reduction of viable cells (2 μM iAs, 98.68 ± 1.07; 5 μM iAs, 98.14 ± 1.21% of control) (Figure 1B), indicating iAs could induced the dysfunction of pancreatic β-cells insulin secretion.
To investigate the effect of iAs on ROS formation, we treated RIN-m5F cells with iAs (2 and 5 μM) and determined ROS production and MDA formation. After exposure of RIN-m5F cells to iAs for 0.5-2 h, the intracellular ROS levels were significantly increased (using DCF fluorescence as an indicator of ROS formation) (Figure 2A). It also induced the production of high levels of MDA at the cell membrane (an index of oxidative damage to membrane lipids) by exposing of cells to iAs for 24 h (exposure to 2 and 5 μM As2O3 led to MDA levels of 2.95 ± 0.12 and
MDA levels of 1.91 ± 0.06 nmole-MDA/mg protein, n = 6, ∗p < 0.05) (Figure 2B).
The antioxidant N-acetylcysteine (NAC, 10 mM) could effectively reverse iAs-induced ROS formation and cell viability reduction (Figure 2).
3.2. iAs-induced apoptosis is mediated by a mitochondrial-dependent pathway in RIN-m5F cells
To determine whether iAs-induced cell death occurs through an apoptosis, Annexin V-Cy3/6-CFDA double-staining was used for detecting of phosphatidyl serine (PS) externalization, which is a hallmark of early events in apoptosis (Yen et al., 2007). As shown in Figure 3A, cells treated with iAs (2 and 5 μM) for 24 h were significantly labeled with both Ann-Cy3 (red) and 6-CF (green) fluorescence, which meant that these cells were in the early stage of apoptosis. Moreover, we examined the effect of iAs-induced apoptosis by analyzing the sub-G1 hypodiploid cell population. As compared to vehicle-treated RIN-m5F cells, the increase in the number of sub-G1 hypodiploid cells was markedly triggered in cells treated with iAs for 24 h (Figure 3B). To further evaluate apoptotic signaling by iAs, caspase-3 activity was measured. Caspase-3 activity is an integral step in the majority of apoptotic events. Here, treatment of cells with iAs (2 and 5 μM) induced remarkable caspase-3 activation (Figure 3C). Pretreatment with NAC (10 mM) could effectively prevent iAs-induced β-cell apoptosis (Figure 3). These results suggest that iAs can induce apoptosis in pancreatic β-cells.
Next, to determine whether iAs-induced apoptosis was mediated through mitochondrial dysfunction, we measured mitochondrial membrane potential (MMP) by using flow cytometry with a mitochondria-sensitive dye, DiOC6. As shown in
Figure 4A, exposure of RIN-m5F cells to iAs for 8 h induced a dose-dependent decrease in MMP, and the decrease in MMP levels was more significant after
exposure to iAs for 24h. We next investigated the release of cytochrome c from the mitochondria into the cytosol in iAs-treated RIN-m5F cells. Exposure of cells to iAs for 8 h effectively increased the cytochrome c level in the cytosolic fraction (cytosolic cytochrome c level: 2 μM As2O3, 1.87 ± 0.17; 5 μM As2O3, 2.58 ± 0.45 fold of control,
n = 6, ∗p < 0.05) (Figure 4B). Meanwhile, we also examined the change in the
expression of pro-apoptotic and anti-apoptotic mRNA. Treatment of RIN-m5F cells with iAs (2 μM) induced an increase in p53 (pro-apoptotic) and a decrease in Mdm2 (anti-apoptotic) mRNA levels. These changes were accompanied by significant down-regulation of Bcl-2 (anti-apoptotic) mRNA expression; however, Bax
(pro-apoptotic) expression was not altered (Figure 4C). Thus, iAs induced a marked shift in the Bcl-2/Bax expression ratio toward an apoptosis-associated state.
Moreover, PARP and caspase cascades activation were also detected. As shown in Figure 5A, the levels of 89-kDa cleaved PARP fragment (the active form) was significantly increased after exposure of RIN-m5F cells to iAs for 8-12 h. The activation of cysteine proteases is one of the critical biomarkers of apoptosis. We observed a marked increase in the activation of caspase-3, caspase-7, and upstream caspase-9 in iAs-treated RIN-m5F cells (Figure 5B). These iAs-induced responses could be reversed by NAC (10 mM) (Figures 4, 5A, and 5B).
3.3. iAs-activated phosphorylation of MAPKs in RIN-m5F cells
Mitogen-activated protein kinases (MAPKs) play important roles in many apoptosis signaling pathways, and ROS are known to induce MAPK activation in many kinds of cells. Therefore, the possible role of MAPKs in iAs-induced pancreatic β-cell death was investigated. As shown in Figure 5, the phosphorylation of JNK, p38 MAPK and ERK1/2 were significantly increased within 30 mins in iAs (2 and 5 μM)-treated RIN-m5F cells compared with control (Figure 5). In addition, the effect
of iAs on the MAPK pathway could be effectively abrogated by co-treatment with NAC (10 mM), which indicated that ROS played a key role in iAs-induced MAPKs activation (Figure 5C).
3.4. iAs also induced ER stress response and increased calpain activity in RIN-m5F cells
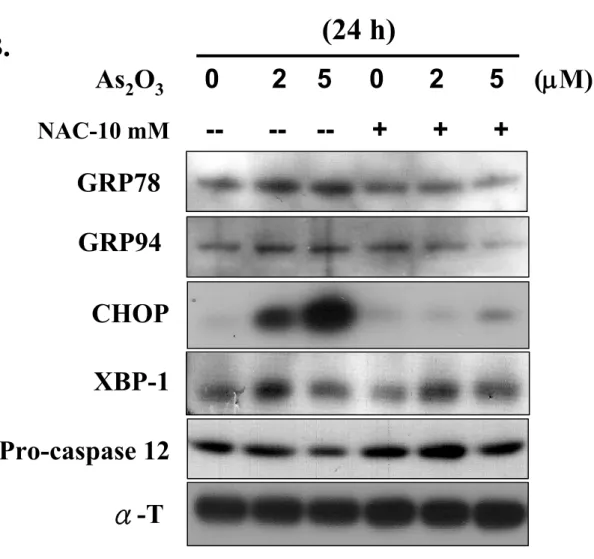
To further evaluate the involvement of ER stress signaling in responses triggered by iAs-induced oxidative stress damage, the expression of ER stress markers was examined after treatment of RIN-m5F cells with iAs. As shown in Figure 6A, iAs significantly increased the protein expression levels of GRP78, GPR94, CHOP, and XBP-1 within 8 h of exposure, and this increased expression was maintained for up to 24 h. It was also markedly triggered the degradation of the 55-kDa full-length caspase-12 in iAs-treated cells. Notably, the effect of iAs on the response of ER stress proteins, i.e., GRP78, GRP94, and CHOP, in RIN-m5F cells could be reversed by NAC (10 mM); however, the effect on the ER stress protein XBP-1 could not be reversed (Figure 6B). Next, we determined whether the activity of calpain (a calcium-dependent thiol proteases) could be induced in iAs (2 and 5 μM)-treated RIN-m5F cells. Calpain I and II protein expression and calpain activity were increased after iAs treatment in a time-dependent manner (Figures 7A and 7B). Furthermore, the calpain inhibitor, Z-Val-Phe-CHO (MDL 28170), and NAC, effectively inhibited the increase in calpain activity induced by iAs in RIN-m5F cells (Figure 7C). These results suggest that ER stress and calpain activity are involved in iAs-induced oxidative stress damage.
3.5. Effects of iAs on blood and pancreas arsenic levels, plasma insulin secretion,
production in mice
Mice exposed to iAs (5 ppm As2O3 in drinking water) during a 6-week period
had elevated whole blood As (age-matched control group: 9.43 ± 0.17 μg/L; iAs group: 35.28 ± 4.76 μg/L) and pancreatic As (age-matched control group: 8.85 ± 1.35 ng/g w.t.; iAs group: 68.75 ± 4.71 ng/g w.t.) levels (Table 1). Thus, these results provide evidence that even the exposure to iAs occurs over a long period, As could still absorbed by the gastrointestinal (G-I) tract and significantly accumulated in the whole blood and pancreas.
The next aim of our investigation was to ascertain whether iAs-induced oxidative stress might alter the function of pancreatic β-cells in vivo. As shown in Figure 8A, there was a significant decrease in the plasma insulin levels of mice after exposure to iAs for 6 consecutive weeks. iAs-exposed mice alsoappeared an elevation in glucose intolerance as compared with vehicle control (Figure 8B). Furthermore, the results of the plasma lipid peroxidation assay and islet cells apoptosis determination showed that the plasma malondialdehyde (MDA) levels were markedly increased, and the deep brown of TUNEL-positive cells was significantly induced after mice exposed to iAs for 6 consecutive weeks (Figures 8C and 8D). These iAs-triggered responses in mice were effectively reversed by the treatment with NAC (150 mg · kg-1).
4. Discussion:
Diabetes mellitus is a group of metabolic diseases characterized by hyperglycemia caused by defects in insulin secretion by pancreatic β-cells and/or insulin action on peripheral tissues. The crucial feature of type 1 (insulin-dependent) diabetes is severe insulin deficiency, which is caused by the autoimmune destruction of pancreatic β-cells. In contrast, type 2 (non-insulin-dependent) diabetes typically shows a partial decrease of β-cell mass, while insulin resistance may be exist, and the insufficiency of insulin production is relative (American Diabetes Association, 2005). iAs is an important environmental pollutant, and that exposure to iAs from drinking water is significantly associated with the prevalence and incidence of diabetes mellitus (Lia et al., 1994; Rahman et al., 1998). In addition, many studies have shown that iAs is a potent inducer of oxidative stress, which causes the important cascade activation during iAs-induced cellular damage (Chowdhury et al., 2009; Han et al., 2010). The deleterious effect of oxidative stress has been found to be induced during the progression of pancreatic β-cell dysfunction under diabetic conditions and apoptosis triggered by toxic insults (such as mercury exposure and high glucose levels) (Chen et al., 2006a; Zhang et al., 2010). Despite many studies indicated that iAs can induce oxidative stress damage, subsequently causing apoptosis in several kinds of cells and organs (Chowdhury et al., 2009; Han et al., 2010; Singh et al., 2010), the precise mechanisms underlying the pancreatic β-cell dysfunction and cell death caused by iAs-induced oxidative stress are mostly unclear. The main finding of this study was that iAs induced β-cells dysfunction, ROS generation, and apoptotic cascades activation in RIN-m5f cells. One of the mechanisms underlying iAs directed cell apoptosis was through mitochondrial dysfunction and ER stress. These iAs-induced cytotoxic responses could be reversed by treatment with the antioxidant NAC. Therefore, these findings indicate that oxidative stress mediated
mitochondria-dependent and ER stress-activated apoptotic signals are involved in iAs-induced pancreatic β-cell apoptosis.
It has been reported that the excess ROS caused the perturbations in mitochondrial function has been identified and affect the cause and complications of diabetes (Hotta et al., 2000; Kaneto et al., 2005). Mitochondria are very sensitive to oxidative stress that caused by various factors, and mitochondrial dysfunction caused by mitochondrial DNA mutation has been implicated in the pathogenesis of many diseases, including diabetes (Kang and Hamasaki, 2005). Mitochondrial dysfunction has been demonstrated to play a crucial role in mammalian cell apoptosis. Recent studies have indicated that iAs induces cell apoptosis by disrupting mitochondrial function (disruption of MMP and cytochrome c release from the intermembrane space of mitochondria to the cytosol) and activating caspase cascade signals in several types of cells (Singh et al., 2010; Tang et al., 2009; Yan et al., 2006). In this study, the results shown that iAs was capable of inducing RIN-m5f cell apoptosis by reducing MMP and increasing cytochrome c release. Meanwhile, we also found a significant increase in the levels of 89-kDa fragment (active form) of PARP, associated with the activation of caspase-3, -7, and -9 proteases. NAC could effectively prevent these iAs-induced responses. Furthermore, Bcl-2 family proteins have been reported to be the central regulators of the mitochondria-dependent (intrinsic) apoptotic pathway, with cell death being dependent on the balance of anti- and pro-apoptotic members (Leibowitz and Yu, 2010). p53, a tumor suppressor gene, is also a critical activator in intrinsic apoptosis. It has been reported that Bcl-2 and Bax gene expression is differentially regulated by p53 activation (Miyashita et al., 1994). Here, exposure RIN-m5f cells to iAs resulted in a marked decrease in Bcl-2 expression and an increase in p53 expression, but not Bax expression was not altered, which was reversed by NAC. Our results suggest that changes in the expression ratio of pro- and
anti-apoptotic Bcl-2 family members, concomitant with increased p53 expression, might contribute to the promotion of apoptotic activity by iAs. On the basis of these results, we implicate that iAs induces an oxidative stress-regulated pancreatic β-cell apoptosis through the mitochondria-dependent apoptosis pathway.
ROS can elicit oxidative stress, which serves as a trigger for cell death and affects many pathological and physiological processes, including pancreatic β-cell dysfunction and apoptosis (Chen et al., 2006a; Kaneto et al., 2005). Oxidative stress induces cellular responses by activating several protein phosphorylation pathways, including JNK, ERK1/2, and p38 MAPK pathways (McCubrey et al., 2006). It has been indicated that activation of the JNK pathway, which is mediated by oxidative stress, is involved in the progression of β-cell death (Hou et al., 2008). However, to the best of our knowledge, there is no literature to explore the role of MAPKs activation in iAs-induced oxidative stress with respect to cause pancreatic β-cell death. The results of the present study revealed that iAs significantly enhanced the phosphorylation of JNK, ERK, and p38 MAPK in RIN-m5f cells. The antioxidant NAC prevented iAs-induced MAPKs activation. These results suggest that iAs-triggered MAPKs activation may be involved in pancreatic β-cell death that is regulated by oxidative stress.
ER stress has been demonstrated to be involved in apoptosis induction that occurs during the pathophysiological progresses, including diabetes (Fonseca et al., 2009). GRP78 and GRP94, the major ER-resident chaperones and the most abundant glycoproteins in the ER, play critical roles in protein folding/assembly and ER calcium binding and are widely used as biomarkers of ER stress (Lee, 2001). Moreover, other ER stress-related chaperones, such as CHOP and XBP1, are also components of the apoptosis pathway mediated by ER stress. CHOP, known as growth arrest- and DNA damage-inducible gene 153 (GADD153), is a member of the
C/EBP transcription factor family. It has been reported that CHOP induction, which occurs during several responses to cellular stress, is involved in the ER stress-induced apoptosis pathway (Zinszner et al., 1998). In mammalian cells, upon ER stress, XBP1 mRNA is initiated the splice processing by the ER transmembrane kinase inositol-requiring enzyme 1 (IRE1), and the results in activation of XBP1, which can then bind ER stress response elements and activate the transcriptional set of ER chaperones, such as GRP and CHOP (Lee et al., 2003). Several studies have shown that the upregulation of these ER stress-related chaperones is believed to induce ER stress, leading to cell apoptosis by toxic insults (such as heavy metals and anit-tumor agents)(Shinkai et al., 2010; Yokouchi et al., 2007). Moreover, calpains are proteins that belong to the family of calcium-dependent intracellular cysteine proteases, which are ubiquitously expressed in mammalian cells and organisms. Activation of calpain-I (μ-calpain) and calpain-II (m-calpain) proteases has been implicated in the development of apoptosis (Huang and Wang, 2001). Caspase-12 is specifically localized to the ER and cleaved during the disruption of ER function. Calpain-mediated cleavage of caspase-12 is reported to be involved in the processes of ER stress-induced apoptosis (Sheu et al., 2007). However, it is still unclear whether iAs-induced oxidative stress causing the expression of ER chaperones and the activation of calpain in pancreatic β-cell apoptosis. In the current study, our results showed that the expression of the ER-related chaperones, including GPR78, GPR94, CHOP, and XBP1, and the degradation of caspase-12 were significantly increased after exposure of RIN-m5F cells to iAs for 8-24 h. These effects could be reversed by treatment of cells with NAC, with the exception of XBP1. Furthermore, iAs also increased calpain-I and calpain-II expression and calpain activity in a time-dependent manner. The pharmacological inhibitor MDL-28170 (calpain inhibitor) and NAC (antioxidant) could prevent iAs-induced calpain activity. These results implicate that
an increase in ER stress is involved, at least partially, in iAs-triggered pancreatic β-cell apoptosis.
Many epidemiological studies have shown that exposure to iAs via drinking water increases the rates of diabetes mellitus and other chronic diseases (Chiou et al., 2005; Lia et al., 1994; Meliker et al., 2007; Rahman et al., 1998). In 2001, the United States Environmental Protection Agency (US EPA) lowered the maximum contaminant level (MCL) of As permissible in drinking water from 50 μg/L to 10 μg/L. However, very high levels of iAs are still found in underground water reservoirs in the United States, Taiwan, and Bangladesh (Lai et al., 1994; Meliker et al., 2007; Rahman et al., 1998). In these areas, the As concentrations range from 0.35 to 2.1 mg/L (ppm), even well above 3 ppm in drinking water, and 5.3 to 11.2 mg/kg in soil (Alam et al., 2002; Lo, 1978; Rahman et al., 1998). Therefore, in this study, to investigate the effects of iAs on pancreatic β-cell dysfunction in vivo, we chose a dosage of 5 ppm iAs via drinking water exposure to mice, which mimics human exposure route and conditions Here, our results found a significant decrease in plasma insulin levels (age-matched control, 84.28 ± 3.38 pmol; 5 ppm iAs, 43.88 ± 5.28 pmol , respectively) and an elevation in glucose intolerance, which was accompanied by significant arsenic accumulation in the whole blood (age-matched control group: 9.43 ± 0.17 μg/L; iAs group: 35.28 ± 4.76 μg/L) and pancreas (age-matched control group: 8.85 ± 1.35 ng/g w.t.; iAs group: 68.75 ± 4.71 ng/g w.t.) after exposure of mice to iAs for 6 consecutive weeks. Moreover, the results of the plasma lipid peroxidation assay and islet cells apoptosis determination also revealed that the plasma MDA levels and the TUNEL-positive cells in islet were markedly increased in iAs-exposed mice. These iAs-induced responses could be effectively reversed by NAC. Thus, these results indicate that iAs-induced oxidative stress plays a key role in pancreatic β-cell dysfunction and injury.
5. Conclusion:
In conclusion, the results from the in vitro and in vivo experiments performed in this study indicate that iAs is capable of inducing the oxidative stress-related insulin secretion suppression in pancreatic β-cells. More importantly, further evidence demonstrates that iAs triggers oxidative stress to induce pancreatic β-cell apoptosis through MAPK activation and mitochondrial dysfunction leads to the activation of PARP and caspase cascades-mediated signaling pathway (Figure 9). iAs also induces the expression of ER stress-related markers (GRP78, GRP94, and CHOP), which subsequently triggers calpain activation resulting in apoptosis (Figure 9). Based on the aforementioned observations, there is beneficial evidence to suggest that iAs may be an importantly environmental risk factor for diabetes.
Conflict of interest statement:
Acknowledgments:
This work was supported by research grants form the National Science Council of Taiwan (NSC 98-2314-B-039-015 and NSC 98-2320-B-039-014-MY3), the National Health Research Institutes of Taiwan (NHRI-EX98-974SI and NHRI-EX99-9744SI), and the China Medical University, Taichung, Taiwan (CMU97-211).
References:
Alam, M.G., Allinson, G., Stagnitti, F., Tanaka, A., Westbrooke, M., 2002. Arsenic contamination in Bangladesh groundwater: a major environmental and social disaster. Int. J. Environ. Health Res. 12, 235-253.
American Diabetes Association., 2005. American Diabetes Association, Diagnosis and classification of diabetes mellitus. Diabetes Care 28 (Suppl. 1), pp. S37–S42.
Biagioli, M., Pifferi, S., Ragghianti, M., Bucci, S., Rizzuto, R., Pinton, P., 2008. Endoplasmic reticulum stress and alteration in calcium homeostasis are involved in cadmium-induced apoptosis. Cell Calcium 43, 184-195.
Bhathena, S. J., Awoke, S., Voyles, N. R., Wilkins, S. D., Recant, L., Oie, H. K., Gazdar, A. F., 1984. Insulin, glucagon, and somatostatin secretion by cultured rat islet cell tumor and its clones. Proc Soc Exp Biol Med 175, 35-38.
Cai, E. P., Lin, J. K., 2009. Epigallocatechin gallate (EGCG) and rutin suppress the glucotoxicity through activating IRS2 and AMPK signaling in rat pancreatic beta cells. J Agric Food Chem 57, 9817-9827.
Chen, Y.W., Huang, C.F., Tsai, K.S., Yang, R.S., Yen, C.C., Yang, C.Y., Lin-Shiau, S.Y., Liu, S.H., 2006a. Methylmercury induces pancreatic beta-cell apoptosis and dysfunction. Chem. Res. Toxicol. 19, 1080-1085.
Chen, Y.W., Huang, C.F., Tsai, K.S., Yang, R.S., Yen, C.C., Yang, C.Y., Lin-Shiau, S.Y., Liu, S. H., 2006b. The role of phosphoinositide 3-kinase/Akt signaling in low-dose mercury-induced mouse pancreatic beta-cell dysfunction in vitro and in vivo. Diabetes 55, 1614-1624.
Chen, Y.W., Huang, C.F., Yang, C.Y., Yen, C.C., Tsai, K.S., Liu, S.H., 2010. Inorganic mercury causes pancreatic beta-cell death via the oxidative stress-induced apoptotic and necrotic pathways. Toxicol. Appl. Pharmacol. 243, 323-331.
ingestion and increased microvascular disease risk: observations from the south-western arseniasis-endemic area in Taiwan. Int. J. Epidemiol. 34, 936-943. Chowdhury, R., Chowdhury, S., Roychoudhury, P., Mandal, C., Chaudhuri, K., 2009.
Arsenic induced apoptosis in malignant melanoma cells is enhanced by menadione through ROS generation, p38 signaling and p53 activation. Apoptosis 14, 108-123.
Diaz-Villasenor, A., Sanchez-Soto, M.C., Cebrian, M.E., Ostrosky-Wegman, P., Hiriart, M., 2006. Sodium arsenite impairs insulin secretion and transcription in pancreatic beta-cells. Toxicol. Appl. Pharmacol. 214, 30-34.
Fonseca, S.G., Burcin, M., Gromada, J., Urano, F., 2009. Endoplasmic reticulum stress in beta-cells and development of diabetes. Curr. Opin. Pharmacol. 9, 763-770.
Han, Y.H., Moon, H.J., You, B.R., Kim, S.Z., Kim, S.H., Park, W.H., 2010. Effects of arsenic trioxide on cell death, reactive oxygen species and glutathione levels in different cell types. Int. J. Mol. Med. 25, 121-128.
Hotta, M., Yamato, E., Miyazaki, J.I., 2000. Oxidative stress and pancreatic β-cell destruction in insulin-dependent diabetes mellitus. In: Packer, L., Rosen, P., Tritschler, H., King, G.L., Azzi, A. (Eds.), Antioxidants and Diabetes Management. Marcel Dekker, New York, pp. 265–274.
Hou, N., Torii, S., Saito, N., Hosaka, M., Takeuchi, T., 2008. Reactive oxygen species-mediated pancreatic beta-cell death is regulated by interactions between stress-activated protein kinases, p38 and c-Jun N-terminal kinase, and mitogen-activated protein kinase phosphatases. Endocrinology 149, 1654-1665. Huang, C.F., Liu, S.H., Lin-Shiau, S.Y., 2007. Neurotoxicological effects of cinnabar
(a Chinese mineral medicine, HgS) in mice. Toxicol. Appl. Pharmacol. 224, 192-201.
Huang, Y., Wang, K.K., 2001. The calpain family and human disease. Trends. Mol. Med. 7, 355-362.
Hung, J.Y., Hsu, Y.L., Ni, W.C., Tsai, Y.M., Yang, C.J., Kuo, P.L., Huang, M.S., 2010. Oxidative and endoplasmic reticulum stress signaling are involved in dehydrocostuslactone-mediated apoptosis in human non-small cell lung cancer cells. Lung Cancer 68, 355-365.
Izquierdo-Vega, J.A., Soto, C.A., Sanchez-Pena, L.C., De Vizcaya-Ruiz, A., Del Razo, L.M., 2006. Diabetogenic effects and pancreatic oxidative damage in rats subchronically exposed to arsenite. Toxicol. Lett. 160, 135-142.
Kaneto, H., Kawamori, D., Matsuoka, T.A., Kajimoto, Y., Yamasaki, Y., 2005. Oxidative stress and pancreatic beta-cell dysfunction. Am. J. Ther. 12, 529-533. Kang, D., Hamasaki, N., 2005. Alterations of mitochondrial DNA in common diseases
and disease states: aging, neurodegeneration, heart failure, diabetes, and cancer. Curr. Med. Chem. 12, 429-441.
Kubota, K., Lee, D.H., Tsuchiya, M., Young, C.S., Everett, E.T., Martinez-Mier, E.A., Snead, M.L., Nguyen, L., Urano, F., Bartlett, J.D., 2005. Fluoride induces endoplasmic reticulum stress in ameloblasts responsible for dental enamel formation. J. Biol. Chem. 280, 23194-23202.
Kuwano, K., Hagimoto, N., Kawasaki, M., Yatomi, T., Nakamura, N., Nagata, S., Suda, T., Kunitake, R., Maeyama, T., Miyazaki, H., Hara, N., 1999. Essential roles of the Fas-Fas ligand pathway in the development of pulmonary fibrosis. J Clin Invest 104, 13-19.
Lai, M.S., Hsueh, Y.M., Chen, C.J., Shyu, M.P., Chen, S.Y., Kuo, T.L., Wu, M.M., Tai, T.Y., 1994. Ingested inorganic arsenic and prevalence of diabetes mellitus. Am. J. Epidemiol. 139, 484-492.
endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell Biol. 23, 7448-7459.
Lee, A.S., 2001. The glucose-regulated proteins: stress induction and clinical applications. Trends Biochem. Sci. 26, 504-510.
Leibowitz, B., Yu, J., 2010. Mitochondrial signaling in cell death via the Bcl-2 family. Cancer Biol. Ther. 9, 417-422.
Lu, T.H., Chen, C.H., Lee, M.J., Ho, T.J., Leung, Y.M., Hung, D.Z., Yen, C.C., He, T.Y., Chen, Y.W., 2010. Methylmercury chloride induces alveolar type II epithelial cell damage through an oxidative stress-related mitochondrial cell death pathway. Toxicol. Lett. 194, 70-78.
Malhotra, J.D., Miao, H., Zhang, K., Wolfson, A., Pennathur, S., Pipe, S.W., Kaufman, R.J., 2008. Antioxidants reduce endoplasmic reticulum stress and improve protein secretion. Proc. Natl. Acad. Sci. U S A 105, 18525-18530.
Matias, I., Gonthier, M. P., Orlando, P., Martiadis, V., De Petrocellis, L., Cervino, C., Petrosino, S., Hoareau, L., Festy, F., Pasquali, R., Roche, R., Maj, M., Pagotto, U., Monteleone, P., Di Marzo, V., 2006. Regulation, function, and dysregulation of endocannabinoids in models of adipose and beta-pancreatic cells and in obesity and hyperglycemia. J Clin Endocrinol Metab 91, 3171-3180.
Mazumder, D.N., 2005. Effect of chronic intake of arsenic-contaminated water on liver. Toxicol. Appl. Pharmacol. 206, 169-175.
McCubrey, J.A., Lahair, M.M., Franklin, R.A., 2006. Reactive oxygen species-induced activation of the MAP kinase signaling pathways. Antioxid. Redox. Signal. 8, 1775-1789.
Meliker, J.R., Wahl, R.L., Cameron, L.L., Nriagu, J.O., 2007. Arsenic in drinking water and cerebrovascular disease, diabetes mellitus, and kidney disease in Michigan: a standardized mortality ratio analysis. Environ. Health 6, 4.
Mishra, D., Flora, S.J., 2008. Differential oxidative stress and DNA damage in rat brain regions and blood following chronic arsenic exposure. Toxicol. Ind. Health 24, 247-256.
Mori, K., 2000. Tripartite management of unfolded proteins in the endoplasmic reticulum. Cell 101, 451-454.
Ng, J.C., Wang, J., Shraim, A., 2003. A global health problem caused by arsenic from natural sources. Chemosphere 52, 1353-1359.
Otles, S., Cagindi, O., 2010. Health importance of arsenic in drinking water and food. Environ Geochem. Health 32, 367-371.
Pfaffl, M. W., Horgan, G. W., Dempfle, L., 2002. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res 30, e36.
Rahman, M., Axelson, O., 1995. Diabetes mellitus and arsenic exposure: a second look at case-control data from a Swedish copper smelter. Occup. Environ. Med. 52, 773-774.
Rahman, M., Tondel, M., Ahmad, S.A., Axelson, O., 1998. Diabetes mellitus associated with arsenic exposure in Bangladesh. Am. J. Epidemiol. 148, 198-203. Schroder, M., Kaufman, R.J., 2005. ER stress and the unfolded protein response.
Mutat. Res. 569, 29-63.
Sheu, M.L., Liu, S.H., Lan, K.H., 2007. Honokiol induces calpain-mediated glucose-regulated protein-94 cleavage and apoptosis in human gastric cancer cells and reduces tumor growth. PLoS One 2, e1096.
Shi, Y., Wei, Y., Qu, S., Wang, Y., Li, Y., Li, R., 2010. Arsenic induces apoptosis of human umbilical vein endothelial cells through mitochondrial pathways. Cardiovasc. Toxicol. 10, 153-160.
endoplasmic reticulum chaperones GRP78 and GRP94 in vascular endothelial cells via the JNK-AP-1 pathway. Toxicol. Sci. 114, 378-386.
Singh, S., Greene, R.M., Pisano, M.M., 2010. Arsenate-induced apoptosis in murine embryonic maxillary mesenchymal cells via mitochondrial-mediated oxidative injury. Birth Defects Res. A Clin. Mol. Teratol. 88, 25-34.
Sitia, R., Braakman, I., 2003. Quality control in the endoplasmic reticulum protein factory. Nature 426, 891-894.
Smith, A.H., Lopipero, P.A., Bates, M.N., Steinmaus, C.M., 2002. Public health. Arsenic epidemiology and drinking water standards. Science 296, 2145-2146. Tang, C.H., Chiu, Y.C., Huang, C.F., Chen, Y.W., Chen, P.C., 2009. Arsenic induces
cell apoptosis in cultured osteoblasts through endoplasmic reticulum stress. Toxicol. Appl. Pharmacol. 241, 173-181.
Tseng, C.H., Tseng, C.P., Chiou, H.Y., Hsueh, Y.M., Chong, C.K., Chen, C.J., 2002. Epidemiologic evidence of diabetogenic effect of arsenic. Toxicol. Lett. 133, 69-76.
Yan, H., Wang, Y.C., Li, D., Wang, Y., Liu, W., Wu, Y.L., Chen, G.Q., 2007. Arsenic trioxide and proteasome inhibitor bortezomib synergistically induce apoptosis in leukemic cells: the role of protein kinase Cdelta. Leukemia 21, 1488-1495.
Yen, C.C., Lu, F.J., Huang, C.F., Chen, W.K., Liu, S.H., Lin-Shiau, S.Y., 2007. The diabetogenic effects of the combination of humic acid and arsenic: in vitro and in vivo studies. Toxicol. Lett. 172, 91-105.
Yokouchi, M., Hiramatsu, N., Hayakawa, K., Kasai, A., Takano, Y., Yao, J., Kitamura, M., 2007. Atypical, bidirectional regulation of cadmium-induced apoptosis via distinct signaling of unfolded protein response. Cell Death Differ. 14, 1467-1474. Zhang, Z., Liew, C.W., Handy, D.E., Zhang, Y., Leopold, J.A., Hu, J., Guo, L.,
glucose-6-phosphate dehydrogenase, leading to increased oxidative stress and beta-cell apoptosis. Faseb. J 24, 1497-1505.
Zinszner, H., Kuroda, M., Wang, X., Batchvarova, N., Lightfoot, R.T., Remotti, H., Stevens, J.L., Ron, D., 1998. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 12, 982-995.
Figure Legends:
Figure 1. Effects of arsenic on cell viability and insulin secretion in RIN-m5f cells. (A) Cells were treated with As2O3 (1-10 μM) for 24 h, and cell viability was determined
by MTT assay. (B) Cells were treated with or without As2O3 (2 and 5 μM) for 4 h.
The insulin secretion was detected under 16.7 mM glucose-stimulated condition. All data are expressed as mean ± S.E. for four independent experiments with triplicate determination. *P<0.05 as compared with vehicle control.
Figure 2. Effects of arsenic on oxidative stress damage generation in RIN-m5f cells. Cells were treated with As2O3 (2 and 5 μM) for various time courses in the absence or
presence of NAC (10 mM). (A) ROS was determined by flow cytometry, (B) oxidative damage to membrane lipid (lipid peroxidation) was measured the levels of MDA, and (C) cell viability was determined by MTT assay as described in the Materials and Methods section. Data are presented as mean ± S.E. for four independent experiments with triplicate determination. *P<0.05 as compared with vehicle control. #P<0.05 as compared with arsenic group alone.
Figure 3. Arsenic induced apoptosis production in RIN-m5f cells. Cells were treated with or without As2O3 (2 and 5 μM) for 24 h in the absence or presence of NAC (10
mM). (A) Apoptosis cells were be observed by fluorescence microscopy (100×) staining with fluorescent probe: Ann Cy3 (red fluorescence) and 6-CFDA (green fluorescence)(a and d, control; b and e, As2O3-2 μM; c and f, As2O3-5 μM; g and j,
NAC-10 mM; h and k, As2O3-2 μM + NAC; i and l, As2O3-5 μM + NAC).(B) Cells
with genomic DNA fragmentation (sub-G1 DNA content) were analyzed by flow cytometry. (C). Caspase-3 activity was examined by CaspACETM fluorometric activity assay kit as described in the Materials and Methods section. Data in B and C
are presented as mean ± S.E. for four independent experiments with triplicate determination. *P<0.05 as compared with vehicle control. #P<0.05 as compared with
arsenic group alone.
Figure 4. Arsenic induced mitochondrial dysfunction, cleavages of poly (ADP-ribose) polymerase (PARP), and caspase cascades activation in RIN-m5f cells. Cells were treated with or without As2O3 (2 and 5 μM) for different time intervals in the absence
or presence of NAC (10 mM). Mitochondrial membrane potential depolarization was determined by flow cytometry (A, for 8 and 24 h). Cytosolic cytochrome c release was examined by Western blot analysis (B, for 8 h). The expression of anti-apoptotic (Bcl2 and Mdm2) and pro-apoptotic (Bax and p53) genes was analyzed by real-time PCR (C). PARP cleavage (D, for 8 and 12 h) and the caspase-3, -7, and -9 expressions (E, for 16 and 24 h) were examined by Western blot analysis as described in the Materials and Methods section. Data in A and C are presented as mean ± S.E. for four independent experiments with triplicate determination. *P < 0.05 as compared with vehicle control. #P<0.05 as compared with arsenic alone. Results shown in B, D, and
E are representative of at last three independent experiments.
Figure 5. Analysis of mitogen-activated protein kinase (MAPK) activation in arsenic-treated RIN-m5f cells. Cells were treated with or without As2O3 (2 and 5 μM)
for 30 min in the absence or presence of NAC (10 mM), and analyzed the phosphorylation of JNK-, p38- and ERK1/2-MAPK by Western blotting analysis. Results are representative of at last three independent experiments.
Figure 6. Effects of arsenic on ER stress marker expressions in RIN-m5f cells. Cells were treated with or without As2O3 (2 and 5 μM) for 8, 16, and 24 h, the expressions
of GRP78, GRP94, CHOP, XBP1, and procaspase-12 were analyzed by Western blotting analysis (A). The effect of NAC (10 mM) on As2O3-induced ER stress
marker expressions was examined (B). Results shown are representative of at last three independent experiments.
Figure 7. Arsenic activated calpain activity. RIN-m5f cells were treated with or without As2O3 (2 and 5 μM) for different time intervals, (A) calpain I and II
expressions were examined by Western blotting analysis, and (B) calpain activity was measured with the fluorescent calpain substrate. (C) Calpain inhibitor (MDL-28170, 20 μM) and NAC (10 mM) significantly inhibited arsenic-increased calpain activity. Results shown in A are representative of at last three independent experiments. Data in B and C are expressed in term of fold of control conditions and presented as mean ± S.E. for four independent experiments with triplicate determination. *P < 0.05 as compared with vehicle control. #P<0.05 as compared with arsenic alone.
Figure 8. Plasma insulin levels, glucose tolerance test, plasma lipid peroxidation, and islet cells apoptosis production in arsenic-exposed mice. Mice were treated with 5 ppm arsenic trioxide (As2O3) in drinking water for 6 consecutive weeks in the
presence or absence of NAC (150 mg/kg/day, oral application by gavage). (A) Plasma insulin levels were determined by insulin assay ELISA kit. (B). Oral glucose tolerance tests were carried out in mice given vehicle or arsenic for 6 weeks and determined as described in the Materials and Methods section. (C) Plasma malondialdehyde (MDA) levels in vehicle or arsenic-exposed mice were examined by using the lipid peroxidation say kit. (D) Apoptosis of islet cells was detected by TUNEL assay as described in the Materials and Methods section. All data are presented as mean ± S.E.;
arsenic alone.
Figure 9. Schematic diagram of the signal pathways involved in iAs-induced apoptosis in pancreatic β-cells. Proposed models represent that iAs induces pancreatic β-cell apoptosis through ROS-regulated mitochondria-dependent and ER stress-triggered signaling cascades.