國
立
交
通
大
學
電
子
物
理
研
究
所
碩 士 論 文
三氧化鎢/氧化銥水勵起固態元件電性研究
Water Activated Solid Device Based on WO
3/IrO
2研究生:蘇頤中
指導教授:趙書琦 博士
三氧化鎢/氧化銥水勵起固態元件電性研究
Water Activated Solid Device Based on WO
3/IrO
2研 究 生:蘇頤中 Student: Yi-Chung Su
指導教授:趙書琦 博士 Advisor: Dr. Shu-chi Chao
國 立 交 通 大 學
電 子 物 理 研 究 所
碩 士 論 文
A Thesis
Submitted to Institute of Electrophysics College of Science
National Chiao-Tung University in Partial Fulfillment of the Requirements
for the Degree of Master of Science
in
Electrophysics July 2008
Hsinchu, Taiwan, Republic of China
誌 謝
在研究所兩年的學習使我獲益匪淺,首先誠摯的感謝指導教授趙 書琦博士,老師悉心的教導使我得以瞭解半導體元件領域的深奧,不 時的討論並指點我正確的方向,使我能平安順利的完成論文。另外感 謝口試委員趙天生教授、莊振益教授及謝文峰教授對於論文的建議與 指導。本論文的完成另外亦得感謝玻璃工廠的洪師傅大力協助,因為 有您的幫忙,使本論文能夠更完整而嚴謹。 感謝淳政學長、夢嘉、昭廷、志遠、及學弟們的幫忙,因為有你 們的共同砥礪,讓兩年的研究生活變得絢麗多彩,恭喜我們都能順利 走過這兩年。 另外我還要感謝大學同學肆華、元彬、建良在實驗上給予的建議 與協助,讓我在實驗上有更多的靈感與創意,也感謝士豪同學在英文 上的幫忙。 最後,我要感謝我的父母親,因為有你們讓我能無憂無慮的完成 碩士學位,也因為有你們的支持,使我能期許自己更進步與成長。 最後,謹以此文獻給我摯愛的雙親、師長、朋友們,將此成果與 你們共享。三氧化鎢/氧化銥水勵起固態元件電性研究
研究生:蘇頤中
指導教授:趙書琦 博士
國立交通大學電子物理研究所
中 文 摘 要
本論文的內容主要介紹,以三氧化鎢/氧化銥材料為基礎的新元 件研究。我們在白金電極上先後鍍上三氧化鎢與氧化銥薄膜,並讓兩 薄膜中間部分重疊,使得薄膜重疊的接面在不同的位置上。這兩種薄 膜都以濺鍍的方式,最後以環氧樹脂封裝完成。本元件的工作機制, 是由於三氧化鎢與氧化銥薄膜都是雙載子注入,而我們濺鍍出來的氧 化銥薄膜內可能含有氫離子,當氫離子進入薄膜內與外加的偏壓下, 會使薄膜產生可逆式的氧化還原反應。我們將不同接面的元件分別做 加水與不加水的實驗,在本實驗中發現某種接面位置的元件加水可形 成二極體現象,稱為水勵起元件,另一種接面可在無電解質下也能形 成二極體現象,另外我們也對無電解質元件做溫度實驗,並寫出二極 體電流方程式。Water Activated Solid Device Based on WO
3/IrO
2Student: Yi-Chung Su Advisor: Dr. Shu-Chi Chao
Institute of Electrophysics
National Chiao-Tung University
Abstract
This article made a main research in the new elements based on WO3
and IrO2 materials. We plated platinum electrodes with WO3 and IrO2
films whose central parts were overlapped. The two kinds of the films, both fabricated by sputtering, were finally packaged with epoxy. The mechanism of the device showed that the films, both with double
injection, produced reversible redox reaction because H+, probably
included in IrO2 film, entered the films and was under the condition of the
additional bias. We experimented on the devices with different junction
positionsrespectively: one was with water; the other was not. The result
presented that adding water in the device with certain junction position produced junction behavior, called water activated device; another junction position without electrolyte also formed junction behavior. And, we also experimented on devices without electrolyte by controlling temperature, and derived current equation of the diode.
目錄
第一章 緒論
... 1
第二章 原理
... 3
2.1 三氧化鎢材料的特性...3 2.2 氧化銥材料的特性...4 2.3 三氧化鎢/氧化銥二極體工作原理...6 2.4 元件工作原理...8第三章 實驗裝置及方法
... 10
3.1 實驗器料...10 3.1.1 晶片之製作...10 3.1.2 靶材...11 3.1.3 光罩之製作...11 3.2 實驗裝置...11 3.2.1 晶片的清潔...12 3.2.2 濺鍍三氧化鎢...12 3.2.3 濺鍍氧化銥...13 3.2.4 元件封裝...14 3.3 三氧化鎢/氧化銥元件...15 3.3.1 二極體 I-V 特性...15 3.3.2 不同溫度下二極體 I-V 特性...15 3.3.3 在高真空度下二極體 I-V 特性...15 3.3.4 接面在不同位置的二極體 I-V 圖 ...16 3.3.5 加水的二極體 I-V 圖 ...16第四章 結果與討論
... 17
4.1 WO3/IrO2 不同接面位置 I-V 圖...17 4.1.1 第一種結構--接面在白金電極之間...17 4.1.2 第二種結構---在三氧化鎢上加上一個負電壓...18 4.1.3 第三種結構—氧化銥薄膜跨越兩端電極...21 4.2 WO3 /IrO2 二極體元件在溫度下的變化...21 4.2.1 使用 Origin 做 fitting ...21 4.2.2 飽和電流與溫度關係圖...22 4.2.3 二極體電流關係式...22第五章 總結
... 23
參考文獻... 24
圖目錄
Figure 3.1 白金微電極剖面圖………28 Figure 3.2 濺渡裝置圖………29 Figure 3.3 白金電極俯視圖………30 Figure 3.4 WO3/IrO2濺鍍圖………31 Figure 3.5 元件封裝圖………32 Figure 3.6 加熱裝置圖………33 Figure 3.7 不同接面位置剖面圖………34 Figure 4.1 結構一未加水I-V圖………35 Figure 4.2 結構一加水I-V圖………35 Figure 4.3 結構二I-V圖………36 Figure 4.4 結構三I-V圖……… 36 Figure 4.5 結構二溫度變化圖……… 37 Figure 4.6 溫度變化Fitting圖………38 Figure 4.7 IS溫度變化圖………47第一章 緒論
在可變色智慧型材料中,我們區分為電致色變(electrochromic, EC),光致色變(photochromism,PC),熱致色變(thermochromism,TC)。 三類: 1. 電致色變(electrochromic):當材料給予ㄧ外加偏壓時,顏色會有可 逆性的變化,可以利用電壓來控制顏色改變。 2. 光致色變(photochromism):當材料遇到特定波長的光時,會改變 起光學的特性,而產生顏色變化。 3. 熱致色變(thermochromism):當材料加熱到某特性溫度時,其光學 特性會產生變化,而有顏色上的改變,當溫度下降時,則會回到 原來的狀態。 光致色變的材料,雖然為無機材質,能長時間的工作,但是缺點是 在高溫下會有退色的現象。而熱致色變材料,要運用的溫度範圍與光 學的特性在生活實際上並不容易,因此這兩者都有所限制。而電致色 變的材料,則因為其反應速度快、有記憶性、穿透率可控制等優點, 所以具有比較大的發展空間。 電色元件(electrochromic device)是ㄧ種綜合光學、電學與材料科學 技術所製成的元件,在運用上非常地廣範,例如鏡面、光學鏡頭、顯 示器、建築玻璃、交通工具玻璃等等皆可用之。當元件加ㄧ偏壓上色 後,即使將外加偏壓除去,其顏色依然保持著色的狀態,故反應有記 憶性,還具有可逆性及持續性。而且電色反應具有連續調變的性質, 顏色的變化及顏色的深淺可以任意調整。在生活運用上有非常大的價 值。 電色(electrochromism)特性最早是在 1961 年,由 Platt 所發表的『電 致色變』著色的理論,說明當電子在激發狀態下,會顯示新的光吸收 帶,原因是由電子的轉移或氧化還原反應中,電子的捕抓與失去所造 成顏色的改變。到了1969 年,Deb 發現 WO3 薄膜顯示出具有電致色 變的性質,開始有很多人研究有關電色特性。在1975 年,Faughnan 和Crandall 運用 WO3 薄膜之技術,製作出WO3 薄膜與液態電解質 之元件,稱為電致色變元件。而由於液態電解質(H2SO4 )會使得 WO3 薄膜分解,因而此元件壽命不長,只能在短時間工作。顧名思義,電色是指在ㄧ定的條件下外加ㄧ偏壓或電流使得電色 物質氧化或還原反應,而讓電子侷限於新的空間中,形成顏色的變 化。電色的物質有很多,我們可以區分為有機物或無機物。有機物中 如anthraquinone,polymerized heterocycles 等等,而無機物又可分為 兩大類: 1.陰極著色(cathodic coloration): 其顏色的變化是由同時注入電子與離子,為還原的反應。此物質如 WO3,MoO3,V2O5等。 2.陽極著色(anodic coloration): 其顏色變化是同時移出電子及陽離子或移出電子和注入陰離子來 達成,此物質如IrO2 ,Rh2O3 ,Ni(OH)2 等。 從電致色變的特性被發現以來,在運用上還有需要突破的瓶頸, 原因是電色的特性需要藉由外界電解質交換IA 族離子來進行反應。 電色元件在需要液態電解質下,包裝上不容易,而且壽命也會比較 短。如果電色反應不需要電解質時,會更有其應用價值,例如:電色 顯示器(Electrochromic Display)、開關視窗(Switchable Window)、智慧 型視窗(Smart Window)、建築物玻璃等。 本實驗室在過去致力於三氧化鎢(WO3 )薄膜與氧化銥(IrO2 )薄膜 的研究,因為兩種金屬氧化物具有良好的特性且工作穩定,不容易受 外界環境而破壞其薄膜結構。而在老師與學長的努力下,將這兩薄膜 運用發展長二氧化碳感測器、葡萄糖感測器與PH 值感測器,其電性 及反應速度都有非常不錯的結果。 在偶然的情況下發現三氧化鎢與氧化銥兩種電色的材料製成元 件。在不加電解質,僅加水的情況下能夠量測到其I-V 二極體曲線, 雖然元件的特性並不好,需要1.5V 才能導通,在不斷得改變製程條 件及元件結構後,我們終於得到了一個不需加水且在0.5V 左右就能 導通的二極體元件。 本論文共分成五章,第二章將敘述三氧化鎢與氧化銥的反應機制 及工作原理,第三章則說明實驗儀器與元件的製作方法,以及量測裝 置,第四章對我們量測到的結果討論分析,第五章則是對整個實驗的 總結。
第二章 原理
三氧化鎢與氧化銥兩種材料都是具有可逆氧化還原反應(redox reaction) 及電色(electrochromic)的特性而廣受注目,其氧化還原反應 的機制主要是由氫離子和電子所共同達成,此ㄧ特性稱為雙重注入 (Double injection)兩者缺ㄧ不可。在以往實驗室所製成的感測元件都 是以外界溶劑電解質與固態電解質交換氫離子來工作,進而感測環境 中的氫離子濃度;但我們卻發現當我們不給予外界液態電解質時,三 氧化鎢與氧化銥兩個固態電解質之間會自行互相交換氫離子來達成 氧化還原反應。2.1 三氧化鎢材料的特性
三氧化鎢(tungsten oxide)是一種 n-type 半導體化合物及為陰極著色 的電色(Electrochromic,EC)材料。而且具有可逆氧化還原伴隨著電色 的反應。由於我們所給予的成長環境條件不同,就會使得形成的薄膜
成分不ㄧ樣,特性也不同。因此我們將其化學式表示為WOz,其中 z
稱之為 stoichiometric parameter , 接著 Sviridov 及 Kulak 等人以 Rutherford Backscattering Spectrum 分析發現,z 的值介於 2.72~3 之 間,且同ㄧ薄膜內的鎢與氧組成比例也不盡相同。ㄧ般在不需考慮此 特性的情況下,我們以WO3來表示三氧化鎢的化學式。 三氧化鎢的一個重要特性是:氧化還原反應會造成其導電度有大幅 度的變化,在酸性溶液中被還原後形成HxWO3,其中x 的大小由最 終的反應程度而定,實驗中發現x 值始終介於 0~0.5 之間,就ㄧ個 三氧化鎢分子能帶來看,其導電帶由空狀態而變成為有價電子之狀 態,而且x 的大小影響材料的導電性,當在酸性溶液中的反應是如下: (淺黃色絕緣體) WO3 + xH+ + xe- ⇔ HxWO3 (深藍色導體) 三氧化鎢的另ㄧ個重要特性是:電色,氧化態三氧化鎢的顏色為淡 黃色,當三氧化鎢處於酸性溶液中並給予外加負偏壓時,三氧化鎢分 子由WO3還原成 HxWO3,鎢的氧化數由六價變為五價(W+6 →W+5 ), 就整體來看還原態的三氧化鎢為HxWO3呈現深藍色,這種現象稱為 電色反應,此過程稱為上色(coloration)。而且電色反應具有記憶性,
即三氧化鎢變色後即使移去偏壓其顏色也不會消失,當外加ㄧ反向偏 壓時顏色才又變回淡黃色,此過程稱為消色(bleaching)。 三氧化鎢的可逆氧化還原反應受其所處環境的酸鹼值影響,目前已 建立的模型為離子和電子的雙重植入(double injection),當三氧化鎢所 處的環境有IA 族離子或 NH4+ 、Ag+ 時,藉由外加偏壓注入電子或 光學激發產生光電子,使得三氧化鎢可進行還原反應生成MxWO3, 在本實驗中的H+ 植入離子來是於三氧化鎢與氧化銥兩固態電解質之 間的交換,當氫原子進入三氧化鎢晶格內結合成HxWO3後,在三氧 化鎢分子的導電帶上提供一個價電子,於是三氧化鎢的導電帶能態密 度不為零,氫原子被注入越多,就有越多的三氧化鎢分子導電帶上具 有價電子,使得整體HxWO3的導電度上升。 解釋WO3電色機制的可能性有三種:
1.
電子移轉成色:電子由一個離子移轉到另一個離子時所引起的光 譜吸收。當電子吸收ㄧ光子時,W 會由 W+5 氧化至 W+6 。顏色變 化的深淺在ㄧ定範圍內是和每單位面積所注入的離子成正比。2.
Color center 成色:上色中心(color center)也會吸收可見光,引起電子能階躍升而使得晶體成色。ㄧ般認為MxWO3的藍色是由於注入 WO3的電子被區域陷入(locally trapped)後,在陰離子空缺處形成上 色中心,因為附近離子的晶體力場(crystal field)給予電子可佔用的 能階,這個電子若在這個可佔用能階躍升,便可產生顏色的變化。
3
. 能帶成色:當 WO3上色時,電子進佔未被佔領導電帶(conduction band)的底部,當電子在導電帶帶內(intraband)移轉從一個佔領態 到其他空態,便能吸收光子,這也可能為 WO3上色的原因。2.2 氧化銥材料的特性
電色物質為離子和電子的混合性導體,這兩種載子的傳輸速度率會 直接影響其氧化、還原反應的動力。氧化銥的可能氧化、還原反應式 有以下兩種: --在酸性環境中,以移出H+ 正離子的機制為主: (黑色導體) IrO2.H2O + H+ + e-⇔Ir(OH)3 (無色絕緣體)--在鹼性環境中,以注入 OH- 負離子機制為主:
(黑色導體) IrO2.H2O+ H2O + e-⇔OH- + Ir(OH)3 (無色絕緣體)
氧化銥電色機制的物理性解釋有下列幾種: (1)電子轉移成色:非晶態氧化銥薄膜的著色發生在氧化態而非還原 態。氧化銥顏色改變可能是由於三價氧化銥轉變成四價氧化銥氧化 態之間,價帶的電子轉移造成在可見光部分的吸收。 (2)上色中心成色:上色中心(color center)也會吸收可見光,造成薄膜 顏色改變。氧化銥的電色機制可能包含電子被氧空缺捕獲而引起 著色中心,使氧化銥產生顏色改變。因為附近離子的力場給予捕 獲電子可佔用的能階,這個電子若在這個可佔用能階躍升,便可 產生顏色變化。 (3)能帶理論模型解釋:氧化銥薄膜從導體著色態 Ir+4 轉移至絕緣消 色態Ir+3 可用理論模型加以解釋,這種模型假設費米能階從帶內至 帶隙間移位。 本實驗中採用濺鍍(銥靶材 + 氧/氬 電漿)的非晶態氧化銥薄膜製 備方式,形成為一般所稱的濺鍍氧化銥薄膜(sputtered iridium oxide film,SIROF)。非晶態和多晶態 SIROF 時,多晶態 SIROF 的電色反
應速度及電極催化特性較慢。但是,當以退火將SIROF 從非晶態轉 移為多晶態時,電阻的改變很小,因此可知,離子傳導速度有意義的 下降而非電子是其特性較慢的原因,非晶態的SIROF 結構提供了傳 輸速度及可逆注入、移出環境,可用以下幾點說明: (1)微隧道網路:非晶態SIROF 的結構中含有隨意的微隧道網路,其 中有些夠大,可允許各種容易接近離子進入內部。
(2)離子暫時的鍵結基位:晶態SIROF 為包含 IrO2、Ir(OH)3、H2O、
Ir 離子和 O 離子的混合體,其主要成份為 IrO2,而其它的殘留成 份,行為像雜質、晶格缺陷及顆粒邊界。而大量的雜質以及晶格 缺陷主導其導電機制。非晶態 SIROF 則提供了空的基位,讓某些 種類的離子暫時在基位上結鍵。陽離子 Ir3+ 則隨意的分佈於晶格 基位之間,並可能發展成顆粒鍊,形成網路,在平行路徑間存在 有許多的連接。因此當Ir3+陽離子的濃度時,被絕緣體Ir(OH)3所
包圍的金屬性IrO2所形成的島嶼便被發展出來。對於電色機制而 言,離子及電子的傳導必須同時進行,而非晶態的 SIROF 有很高 的離子及電子傳導速率,其電子傳導速率仍大到足以平衡所增加 的離子傳導速率。 (3)夾層水分子:SIROF 在經過了幾次的電色循環中,由於大量的水 分子併入薄膜的夾層內,使得薄膜的體積膨脹,而像海綿體的結 構。水分子合併進入SIROF 的多孔質微隧道網路時,反應速度就 會變快,因此,氧化銥的快速電色機制和薄膜夾層水分子合併可能 有關。 (4)金紅石結構本身的隧道:不含水的IrO2為金紅石結構,其[001]方 向有空的隧道被IrO4+ 離子所包圍,及在其上、下被六角最緊密堆 積之 O2- 離子面包圍。這些隧道可允許小型離子易於進入氧化銥 的晶格內部。所以在氧化銥的電色反應機制中,小型離子容易快 速進入和移出,可能和氧化銥本身是金紅石結構有相當密切的關 係。
2.3 三氧化鎢/氧化銥二極體工作原理
我們分別將三氧化鎢、氧化銥銥薄膜濺鍍於白金電極上其反應式如 下: (淺黃色絕緣體) WO3 + xH+ + xe- ⇔ HxWO3 (深藍色導體) (黑色導體) IrO2.H2O + H+ + e- ⇔ Ir(OH)3 (無色絕緣體) 三氧化鎢與氧化銥二極體施予偏壓下的結構如下圖:從以上的反應式與圖示可知道,當我們施予正向偏壓將負極連接至氧
化銥,正極連接至三氧化鎢時,WO3/IrO2接面附近的IrO2,釋放出氫
離子與電子到 WO3,則導電度就大幅的提升;氧化銥薄膜所釋放出的 氫離子擴散至三氧化鎢薄膜,同時加上負極所提供的電子,造成了三 氧化鎢薄膜由氧化態還原成HxWO3 ,薄膜則由黃色變成了深藍色, 導電度也相對的提升。氧化銥薄膜的標準電位比三氧化鎢薄膜的標準 電位還高,所以兩薄膜間形成能量位障(Barrier);當我們外加的偏壓 大於此位障時,元件則導通,電流增大,類似二極體順向偏壓下的電 性,因而我們將此狀態定義為順向偏壓。 反之,當我們施予負偏壓時,三氧化鎢薄膜的電子被正極所吸引,而
+
H
+Pt
Pt
IrO
2‧H
2O+H
+WO
3+
-
H
+Pt
Pt
IrO
2H
XWO
3-
由還原態變氧化成了WO3 ,同時釋放出氫離子與電子,導電度就下 降,而三氧化鎢薄膜所釋放出的氫離子擴散至氧化銥薄膜,電子只經 過三氧化鎢,這時候的負偏壓電流,只經過三氧化鎢,與氧化銥無關 。
2.4 元件工作原理
本元件的工作原理,主要機制是由三氧化鎢/氧化銥二極體中氫離子 擴散所造成的。我們以下圖來清楚說明: 其中Pt 為電極,WO3 /IrO2 為離子電子混和性導體。在實驗中,我們 所濺鍍出來的氧化銥薄膜為黑色導體,而三氧化鎢為淺黃色絕緣體, 氧化銥薄膜中可能含有H+ 離子,會擴散到 WO3薄膜,同時在外加 的正向偏壓下,Pt 電極提供了電子,則三氧化鎢薄膜就還原 HxWO3 , 我們由能士特方程式( Nernst equation )三氧化鎢與氧化銥薄膜反應 式來解釋,三氧化鎢薄膜電位的變化如下: 在固定順向偏壓下 WO3 + xH+ + xe- → HxWO3 EWO3 = E0WO3 - XF RT X X H WO WO H ] ][ [ ] [ ln 3 3 +( R = 8.3147 joule / K.mole;T = 298 K;F = 96493 coulomb )
+
-
H
+Pt
Pt
IrO
2‧H
2O + H
+WO
3其中E0WO3 表示WO3 標準電位,XF 表示電量( 庫倫 ),在固定偏 壓下薄膜處於暫時的平衡態,我們不考慮電子增減,所導致三氧化鎢 薄膜產生的氧化還原反應,由上式我們可以知道,三氧化鎢薄膜電位 會隨著氫離子濃度升高而變大,隨氫離子濃度下降而變小,假設 H+ 濃 度 一 定 , 則 我 們 可 以 清 楚 的 知 道 三 氧 化 鎢 的 電 位 為 隨 著 HxWO3/WO3 濃度的比例而改變;同理氧化銥薄膜電位變化如下,假 設元件中氧化銥薄膜以氫離子的反應為主: 在固定順向偏壓下 Ir(OH)3 → IrO2.H2O + H+ + e- EIrO2 = E0IrO2 - F RT ] ) ( [ ] ][ [ ln 3 2 OH Ir H IrO + 其中 E0IrO3 表示 IrO2 標準電位,在固定偏壓下薄膜處於暫時的平衡 態,不考慮電子增減,所導致氧化銥薄膜產生的氧化還原反應, 由 上式我們可以知道,氧化銥薄膜電位會隨著氫離子濃度升高而變小, 隨氫離子濃度下降而變大。H+濃度固定,可以由 IrO2 /Ir(OH)3 濃度的 比例,知道IrO2 的電位。 由於兩者的H+ 濃度是互相擴散交換,所以當氧化銥失去多少H+ , 三氧化鎢就獲得多少的H+ ;而我們濺鍍出來的氧化銥為IrO2導體,
在正偏壓下Ir(OH)3 不斷得氧化成IrO2 ,因此我們可以知道IrO2 濃
度會很高。反之,我們濺鍍出來的三氧化鎢是WO3 是絕緣體,WO3 還 原成HxWO3。 氧化銥薄膜的標準電位比三氧化鎢薄膜還高,當外加固定正向偏壓 大於兩薄膜形成的能障時,就會有電流的導通,而[HxWO3 ] / [WO3 ] 會比[IrO2 ] / [Ir(OH)3 ]還要小,因此三氧化鎢的電位相對就上升,使 得兩薄膜間的電位差變小,能障變小,就有更多的電子跨越過能障, 電流就跟著變大。 反之,在負偏壓下,三氧化鎢薄膜的電位會下降,而氧化銥薄膜的 電位會上升,使得兩薄膜間的電位差變大,能障變大,因此可跨越能 障的電子變少了,所以電流隨之變小。
第三章 實驗裝置及方法
3.1 實驗器料
3.1.1 晶片之製作
(一)基材
本實驗室所使用晶片是由昇美達公司所代理的 4 吋矽晶片,直徑 為100±0.5 mm、晶向為(100)±0.5 deg、磷摻雜 N 型、電阻率約 1~20 ohm.cm、厚度 525±25 μm。(二) 光罩設計
光罩是由林志強學長利用 L-edit 軟體設計,單一微電極長 1400 μm、寬 600 μm、前端圓頭部分直徑 400 μm,兩相鄰電極間距 400 μm, 上下各四個互相對稱,組成一個我們切割晶片的最小單位即一個晶 方,如 Fig.3.1(a) 所示,大小約為 4.5 mm × 3 mm,此光罩橫向有 15 個晶方,縱向有 22 個晶方,總共有 330 個晶方,以此光罩設計交由 國家奈米中心( NDL )製作成一玻璃光罩。(三) 晶片製作
晶片的製程是由本實驗室設計,交由交通大學半導體中心代工製 成。晶片首先放在溫度75~85℃的 H2SO4 和H2O2 ( 3:1 )混合液中煮 20 分鐘,然後以去離子水( DI water )沖洗 5 分鐘,接著放入氧化/擴 散系統( 高溫爐 )中,以濕氧方式長氧化矽( SiO2 ) 2000 Å,再放入 低壓化學氧相沈積系統( LPCVD )中,長氮化矽( Si3N4 ) 4000 Å,藉 此使基材矽晶片和鍍在其上的白金微電極達到絕緣效果,以防止元件 漏電流發生。 接著作剝落製程( lift-off ),在晶片上塗布光阻,由罩幕對準系統 ( Aligner )作光罩對準,曝光顯影出我們所需的微電極陣列圖形,以 雙電子槍( Dual E-Gun )鍍鉻( Cr )50 Å 及白金( Pt )1200 Å,因為白金 薄膜和氮化矽無法緊密結合,因此在中間先鍍一層鉻增強附著力,最 後洗去光阻既完成陣列型白金微電極,如 Fig.3.1 所示,整個電極結 構由下到上是,矽/氧化矽/氮化矽/鉻/白金。3.1.2 靶材
本實驗鍍膜使用的靶材是由PURE TECH 公司生產。銥( Ir )純度 為99.95﹪,直徑 1 英吋厚度 0.125 英吋,密度 21.8 g/cc。三氧化鎢 ( WO3 )純度為 99.99﹪,直徑 2 英吋厚度 0.125 英吋,密度 4.77 g/cc, 與相同大小的銅背板壓製而成。3.1.3 光罩之製作
用鎢鋼筆將矽基板切成長條狀,接著將矽基板以去離子水、三氯 乙烷( TCA )、丙酮( ACE )、去離子水的順序,放入超音波震盪器 (ultra-sonic cleaner)中清洗,各震盪十分鐘後取出以氮氣槍將水分吹 掉,然後放入120℃的烤箱中烤 30 分鐘。3.2 實驗裝置
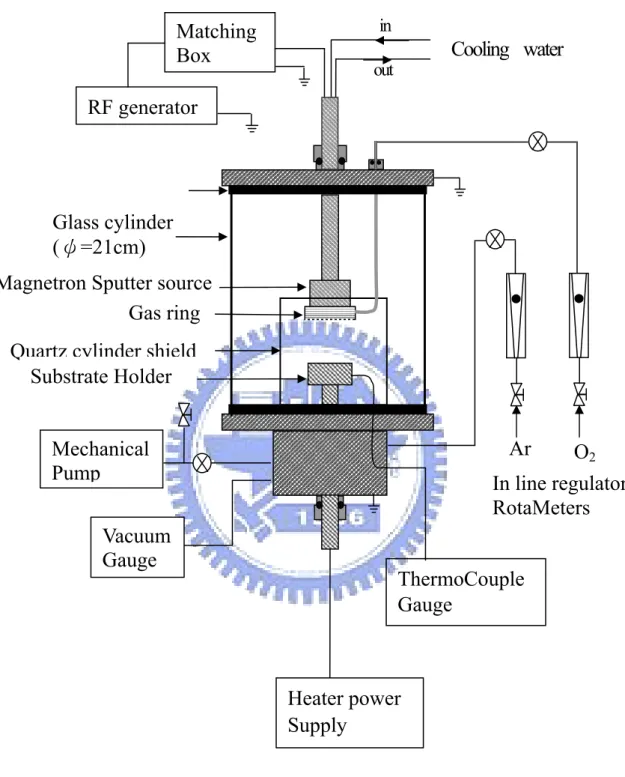
本實驗室所用到的薄膜,都是以反應式濺鍍法(Reactive sputtering) 採用由上往下 ( down hill )的方式所濺鍍。目前本實驗室擁有三套濺 鍍系統,裝置如 Fig.3.2 所示,分別用來濺鍍三氧化鎢薄膜、氧化銥 薄膜。 濺鍍系統主要由真空室、RF 產生器( RF generator )、匹配器 (Matching box)、磁控濺鍍槍( Magnetron sputtering gun )、氣體流量計 ( Gas flow meter ) 、機械式真空幫浦( Mechanical vacuum pump ) 、電 容式真空計( Capacitance vacuum gauge ) 、加熱器及溫度量測系統 等,所組成。真空室由一中空玻璃圓柱( Pyrex cylinder ),及上下各一片不銹鋼
體所構成,Pyrex 玻璃圓柱內部直徑 200 mm、壁厚 5 mm、高 300 mm﹔
在濺鍍槍和玻璃圓柱間另放置一石英罩,方便真空腔的清洗。磁控濺 鍍槍,可將電磁場所產生的電漿集中在基材和靶材之間減低功率的消
耗﹔RF 來源由一最大輸出為 600 W 之 RF 產生器,操作頻率為 13.56 MHz,經匹配器將電磁場送至濺鍍槍輸出,如 Fig.3.2。
3.2.1 晶片的清潔
(1)以鎢鋼筆將晶片切割成 4.5 mm × 3 mm 晶方,如 Fig.3.3(a)(b)所示。 (2)將切割好之晶方放入燒杯中,加入 D.I Water(De-ionized Water)以超
音波震盪20 分鐘,以除去晶片上的顆粒較大之灰塵。 (3)將晶方取出並放入裝有三氯乙烷溶液的燒杯中,再次震盪 10 分 鐘,除去晶方上的有機污垢。 (4)將晶方取出並用 D.I Water 沖洗,然後放入裝有丙酮溶液的燒杯中 震盪 10 分鐘,藉以除去晶方上殘留的三氯乙烷。 (5)再以 D.I Water 將晶方震盪 20 分鐘,除去晶方表面上的丙酮,再用 氮氣將晶片吹乾。 (6)接著將晶方放入體積比(95%H2SO4 ):(30%H2O2 ) = 3:1 的混合溶 液中蝕刻(etching)10 分鐘,除去白金表面上的氧化層,以增加白金 微電極的導電性。 (7)將晶方用 D.I Water 清洗 10 分鐘,然後用氮氣將晶方吹乾後放入烤 箱,以 120℃以上的高溫烘烤 3 小時,完全除去晶方所含的水分。
3.2.2 濺鍍三氧化鎢
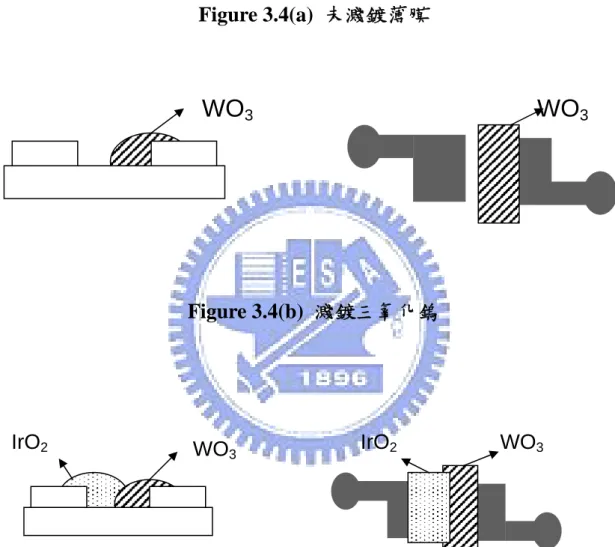
(1)將白金電極如圖 Fig.3.4(a)用矽基板光罩遮蓋放入三氧化鎢濺鍍 腔,靶材到基板距離 15 公分,加熱器加熱到約 420℃以加強三氧 化鎢在白金電極上的附著力,使得三氧化鎢薄膜形成我們要的多晶 型態(polycrystalline)。 (2)打開真空幫浦抽氣約至 2~3 mTorr,關閉幫浦的閥門停止抽氣,並充入氬氣到濺鍍腔內,直到壓力大於10 Torr 為止,接著關閉氬 氣,並打開幫浦閥門繼續抽氣,反覆抽放氣動作五次以降低濺鍍 腔內的水氣,純化氬氣,此過程稱純化(purge)。 (3)用微氣體流量計調整氣體流量,控制氬氣和氧氣的流量,使真空 腔內兩氣體之流量比維持在 30 mTorr : 15 mTorr。 (4)打開冷卻水(水溫設定為 18℃),調整 RF generator 和匹配器,使得 濺鍍功率為 130 W、反射功率為 0 W 開始濺鍍,濺鍍的時間為 20 小時。 (5)濺鍍完成後,將氣體以原比例 30 mTorr : 15 mTorr 放置約 3 小時。 (6)3 小時後,將氣體依原比例沖到 1000 mTorr,關掉加熱器放置約 20 個小時。 (7)降到室溫後,將晶片取出用丙酮、去離子水洗乾淨後,放到烤箱 以 120℃烘烤完,準備濺渡氧化銥,如 Fig.3.4(b)。
3.2.3 濺鍍氧化銥
(1)以濺鍍三氧化鎢的同樣方式濺鍍氧化銥,將白金電極放到濺鍍腔 裡,靶材到基板距離 15 公分,打開加熱器加熱到 100℃,可加強氧 化銥的附著力。 (2)打開真空幫浦抽氣約至 4~5 mTorr,關閉幫浦的閥門停止抽氣, 關 閉幫浦的閥門停止抽氣,並充入氬氣到濺鍍腔內,直到壓力大於 10 Torr 為止,接著關閉氬氣,並打開幫浦閥門繼續抽氣,反覆抽放 氣動作五次以降低濺鍍腔內的水氣,純化氬氣,此過程稱純化(purge)。 (3) 用微氣體流量計調整氣體流量,控制氬氣和氧氣的流量,使真空 腔內兩氣體之流量比維持在 35 mTorr : 20 mTorr。 (4)打開冷卻水(水溫設定為 18℃),調整 RF generator 和匹配器,使得 濺鍍功率為 50 W、反射功率為 0 W 開始濺鍍,濺鍍的時間為 2 小 時。(5)2 小時後關閉濺鍍系統,在 100℃的環境下氣體依照原比例放置 2 小時。 (6)接著依原比例通入氣體到 1000 mTorr,關閉加熱器冷卻到室溫, 再拿出濺鍍腔,開始封裝步驟,如 Fig.3.4(c)。
3.2.4 元件封裝
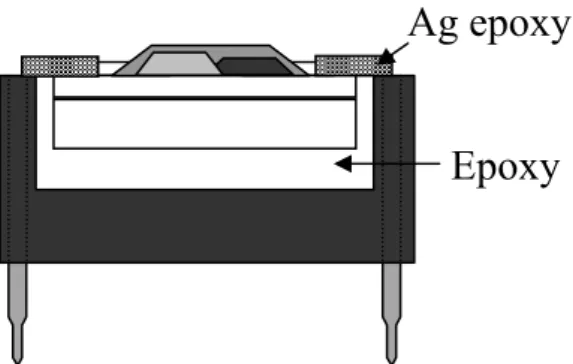
(1)將絕緣的環氧樹脂( epoxy ,南寶樹脂 no.980 )兩劑型,均勻混合, 填滿8-pin IC 腳座中間缺口部份,在 80℃烤箱中烤 1 小時使環氧 樹脂固化(cure),以便讓晶方放置其上。 (2)將濺鍍完成之晶方以環氧樹脂固定於 IC 座上,用針尖輕壓晶方使 其水平置於IC 座上,並用環氧樹脂將晶方四周完全覆蓋,包裝之 主要目的在於環氧樹脂的抗水性和絕緣性,以避免量測時電流經 由晶方周圍斷面上的表面能態( surface state )流通,而造成漏電 流,將覆蓋好環氧樹脂的晶方以80℃烤 2 小時,使其固化。 (3)本元件是以銀膠( 藤倉化成,FA-705 )做為接線,由於銀需低溫冷 藏,因此使用前半小時須先從冷凍庫中取出放在室溫下,待溫度 回升到和室溫相同時才可打開使用,避免水氣凝結而造成銀膠變 質。 (4)等晶方烤完環氧樹脂固化並冷卻至室溫後,才將元件取出,避免 環氧樹脂與晶方間和濺鍍完成的薄膜產生應力( stress ),以針尖挑 起銀膠略成線狀,將銀膠一端沾黏在晶方電極連接埠,另一端拉 至預定之IC 腳座接點上,為了配合量測基座的使用,我們將銀膠 的另一端,拉到腳座上半圓形缺口數過來的第二個腳座接點,然 後以120℃烤 2 小時,使銀膠固化並產生導電性,如 Fig3.5(a)。 (5)接下來把露在外面的銀膠及延伸電極以環氧樹脂覆蓋,使元件只 露出薄膜之工作區,否則元件可能因為外界微粒接觸到銀膠、電極, 而影響到量測的結果,甚至有漏電流產生使元件失效,在塗佈環氧 樹脂時需特別小心以避免蓋住過多的薄膜,然後放置室溫 24 小時, 其在常溫下固化,因為在高溫下固化會使得環氧樹脂黏滯力下降流 動性就增加,而可能會讓環氧樹脂覆蓋整個工作區,使得我們無法觀察,則完成整個元件的製作,Fig.3.5(b) 所示,元件製作流程如 Fig.3.5 所示,由切割好的晶方到元件封裝完成,各個步驟的剖面圖。
3.3 三氧化鎢/氧化銥元件
3.3.1 二極體 I-V 特性
將封裝好的三氧化鎢/氧化銥元件插於 8-pin IC 長腳座上,使用量 測電流對電壓曲線(I-V curve)的儀器為 Keithley Model 237 source measure units,將 Keithley 237 的 Output Hi 與 Sense Hi 的鱷魚夾接頭,
共同夾於一長腳上,連接到氧化銥薄膜端的IC 接腳上,將 Output Lo
與 Sense Lo 的鱷魚夾接頭,共同夾於另ㄧ長腳上,連接到氧化鎢薄
膜端的 IC 接腳上,將接地與接機殼的鱷魚夾接頭共同夾於一裸銅線
上;利用Gpib 介面卡將 Keithley 237 與電腦連線,使用 Labview 5.1
軟體寫控制程式,將它稱為Keithley 237 Sweep,此程式的主要功能 是控制Keithley 237 掃描電壓量測電流,並將結果作圖顯示。
3.3.2 不同溫度下二極體 I-V 特性
為了證明三氧化鎢/氧化銥元件在沒有液態電解質下,可以進行 氧化還原的反應,因此我們將元件加熱。將元件放進杜爾瓶(Dewars) 中,分別在300K、320K、340K、350K、380K、400K、420K、440K下用Keithley 237 量測元件 I-V 曲線,量測如 Fig3.6 所示。從 27℃~
170℃量測 I-V 曲線。
3.3.3 在高真空度下二極體 I-V 特性
為了避免空氣中所含的水氣,所以我們使用 ALCATEL 出產的分 子幫浦(Turbomolecular Pump,ATP 100)將真空腔抽半小時以去除可能
3.3.4 接面在不同位置的二極體 I-V 圖
在多次的實驗中發現三氧化鎢與氧化銥在白金上的接面位置在 量測I-V 圖時有不同的現象,在特定的位置下才能形成二極體 I-V 圖, 這對整個實驗有很重大的影響,在這個實驗也發現了在濺鍍氧化銥的 時候,並不是所有的氧化銥都只鍍在三氧化鎢上面,有些氧化銥會進 入三氧化鎢中,形成電流的通道,我們將接面位置分成三種形式:(1)
氧化銥濺鍍位置超過三氧化鎢端的白金電極,如Fig3.7(a)。(2)
氧化銥濺鍍位置到兩白金電極之間,如 Fig3.7(b)。(3)
氧化銥濺鍍位置在氧化銥端的白金電極上,如Fig3.7(c)。
3.3.5 加水的二極體 I-V 圖
在實驗的過程中,我們發現水裡面的氫離子對元件有很大的影響, 水可以提供氫離子,使WO3變成 HXWO3 。 加水的實驗分成兩個部分: (1) 在原本不能形成二極體 I-V 圖的元件上,加上少許的水,是否能 使元件中的三氧化鎢得到氫離子形成二極體I-V 圖。 (2) 在原本不加水就有二極體現象的元件上加上水在量測 I-V 圖,在 將元件烤乾,量測元件是否有重覆性。第四章 結果與討論
4.1 WO
3/IrO
2不同接面位置 I-V 圖
4.1.1 第一種結構--接面在白金電極之間
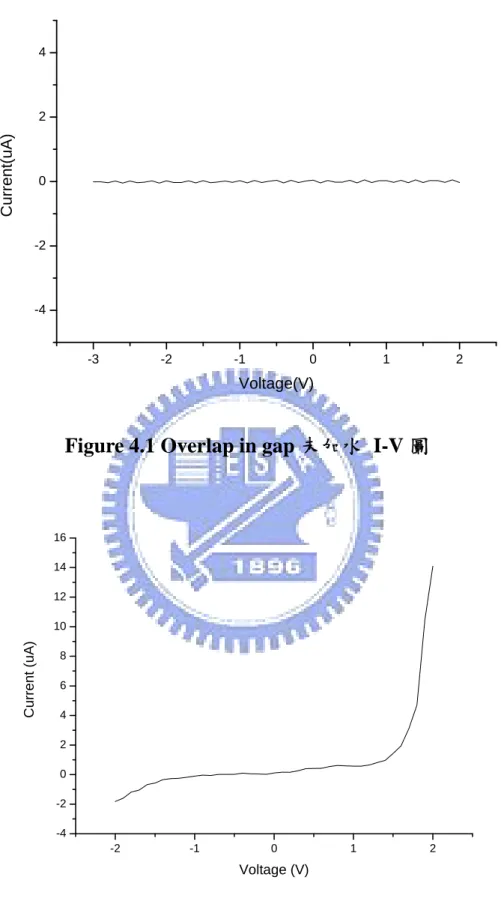
本元件是以三氧化鎢/氧化銥二極體為基本架構,一開始我們用 了下面的元件結構: 圖一 圖二 如圖一,在二極體兩端施予順偏,在三氧化鎢加上負電位,在氧 化銥加上正電位,氫離子受負電壓吸引進入三氧化鎢,WO3→HXWO3, 形成導通狀態;如圖二在二極體兩端施予逆偏壓,在三氧化鎢加上正 電位,在氧化銥上加上負電位,沒有電流通過元件,我們量測到的 IR~0 WO3 H+ - + Si3N4 Pt Pt Ir(OH)3 SMU Keithley 236 IF - + Si3N4 Pt Pt H+ IrO2 HxWO3 SMU Keithley 236I-V 圖(如Fig 4.1),並沒有出現我們預期的結果,電流無法經過這個 元件。 由Fig 4.1 得知 WO3沒有從氧化銥得到氫離子形成 HXWO3,我們 試著加上一些水看是否能藉由水提供氫離子使WO3 變成HXWO3,以 下是量測到的I-V 圖,如 Fig 4.2,我們發現元件形成了二極體,氫離 子來源是水,因此我們稱這個元件為WO3/IrO2水勵起二極體元件。
4.1.2 第二種結構---在三氧化鎢上加上一個負電壓
因此,必須改變元件的結構,新的元件結構圖如下: 圖三+
-
H
+Pt
Pt
IrO
2‧H
2O + H
+WO
3圖四 如圖三,順偏情況下,在靠近接面的三氧化鎢上加上與氧化銥相 同的負電壓,在另一端的三氧化鎢施予正電壓,這與之前的元件結構 的 電 壓 極 性 恰 好 是 相 反 的 , 另 外 也 改 變 了 電 極 的 間 距 如 Figure.3.3(a)(b),從原本的 0.4mm 縮短到 0.2mm,大約縮短了一半的 距離。 在接面處三氧化鎢加上負電壓的目的是為了使氫離子更容易進入 三氧化鎢,縮短白金電極間距的目的是為了使電子更容易從一端的電 極到達另一端的電極,另一方面因為我們沒辦法確認氫離子擴散進入 三氧化鎢的長度有多大,所以縮短白金電極間距對我們的實驗是有利 的。 如圖四,在逆偏情況下氫離子會離開三氧化鎢回到氧化銥,這時 電子的流動只會經過三氧化鎢,氧化銥的作用只剩下接收離開三氧化 鎢的氫離子,這是與一般傳統二極體的最大差別。 這與一般二極體的差別如下圖: 圖五
+
-
H
+Pt
Pt
IrO
2H
XWO
3p
n
+
-
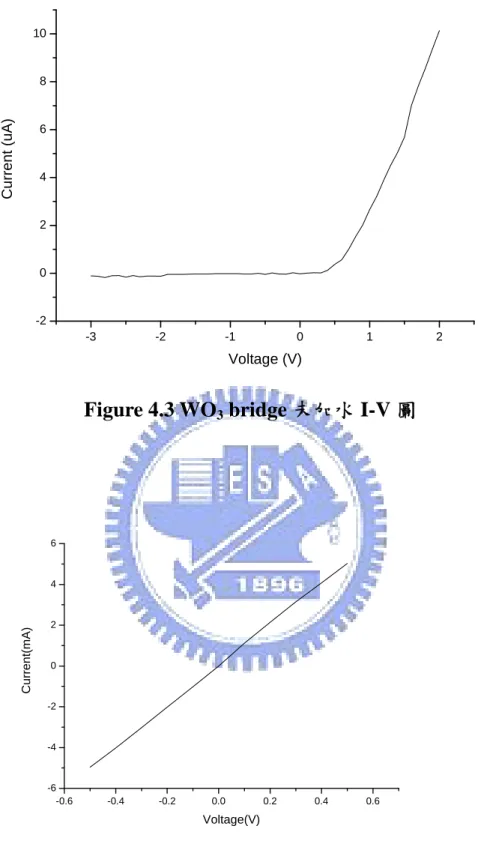
圖六 一般的 PN 二極體一邊只有一跟接腳,三氧化鎢/氧化銥二極體上 多了一跟接腳,這跟接腳的目的是為了使氫離子進入三氧化鎢,這種 二極體在順偏的情況下與一般二極體並無差別,只有在逆偏的時候量 到的 I-V 圖像是只有在白金電極上度上三氧化鎢一樣,在室溫的情況 下三氧化鎢的電阻是相當大的,所以逆偏電流是很小的,氧化銥在這 樣的元件結構上只是用來提供它所含的氫離子。 在經過不斷的實驗驗證下得到了 I-V 圖如Fig4.3,從圖上可以清 楚的看到類似二極體的趨勢圖,電流大約在 10-6A,導通電壓在 0.5V 以下,這樣的結果證明了這個特殊的元件結構是正確的,也達到了本 論文的目的,在不加任何電解質或水的情形下,三氧化鎢能夠從氧化 銥薄膜中得到氫離子,這與一般常見的三氧化鎢應用不同。 比較這兩種結構在第一種結構如圖一,雖然在三氧化鎢上加上了 負電壓,為何仍然無法使氫離子離開氧化銥進入三氧化鎢,我們猜測 因為三氧化鎢本身的電阻太大(1010Ω-cm),所以在接面處電壓並不如 在電極上的負電壓那麼大,而氧化銥是個電阻很小的材料,在接面處 造成的電位與白金電極上的正電壓幾乎是一樣的,所以在接面處的電 位是正的,因此氫離子無法進入在正電壓下的三氧化鎢,當我們改變 了結構,如圖三,雖然三氧化鎢上接的是正電壓,乍看之下氫離子理 應無法進入三氧化鎢,但在接面處三氧化鎢是負電壓,而這個負電壓 與第一種結構相比距離少了很多,所以受到電阻產生的壓降也少了很 多,使得氫離子能夠進入三氧化鎢。 前面兩種結構說明了三氧化鎢/氧化銥元件的接面位置與元件的
WO
3+
-
IrO
2電性有關,因此我們對下面這種接面位置也有了興趣,如果氧化銥薄 膜鍍到三氧化鎢端的白金電極。
4.1.3 第三種結構—氧化銥薄膜跨越兩端電極
在這個實驗中採用了Figure 3.7(a)的濺鍍方式,得到了 Figure 4.4
的I-V 圖,根據這個 I-V 圖元件看起來是一個導體,電阻大約是 100Ω,這樣的結果我們推測在濺鍍氧化銥的時候,部分的氧化銥會 穿過三氧化鎢薄膜形成電流的通道,如下圖七 圖七
4.2 WO
3/IrO
2二極體元件在溫度下的變化
這個實驗採用 4.1.1.2 的元件,為了證明三氧化鎢/氧化銥元 件不需要藉由外界的液態電解質下也能工作,因此我們杜爾瓶中將三 氧化鎢/氧化銥元件加熱且在量測的環境中,分別在300K、320K、 340K、350K、380K、400K、420K、440K 下量測我們發現在不同溫 度下所得到的I-V 曲線都有二極體的特性(如圖 Fig 4.5),溫度越高二 極體的電流就越大,因為當溫度升高時,電子與離子的傳輸速度會變 快,所以導通電壓會隨溫度上升越來越小,接下來對這些溫度變化 I-V 圖作數據分析。4.2.1 使用 Origin 做 fitting
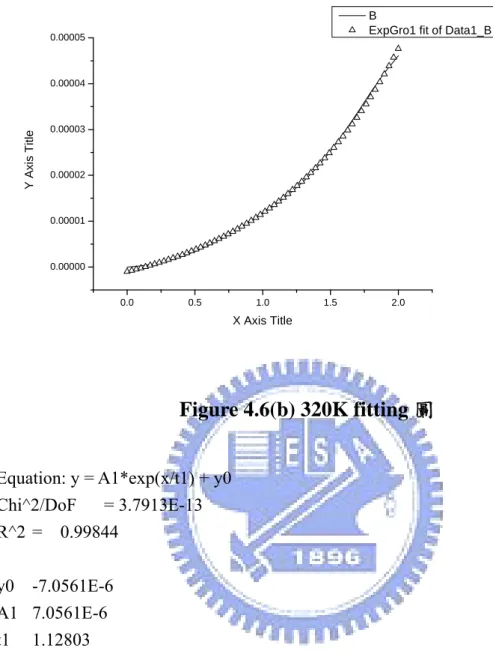
使用 Origin75 的 Fit Exponential Growth 功能作 fitting 如圖
Fig4.6(a)~(h),在 V>0 的部份將 I-V curve 寫成 Equation: y =
A1*exp(x/t1) + y0,根據Table 4.1,t1 在 300K 到 400K 呈現上升的趨
IrO
2 電流通道勢。
4.2.2 飽和電流與溫度關係圖
將y = A1*exp(x/t1) + y0 改寫成 I= IS [exp(V/nKT) -1] ,取出 IS
對溫度作圖如Fig 4.7。
4.2.3 二極體電流關係式
在V>0 的部份將 I-V curve 寫成 Equation:
I= IS [exp(V/nKT) -1],經過計算得到 n=40.1~44.6,IS 與溫度關係圖
如圖Fig4.7(T<400K),IS與溫度關係式IS= 0.0688T - 21.024 (10-5 A)
(T<400K),IS與溫度成直線上升的關係,WO3/IrO2固態二極體元件
電流關係式:
I=[0.0688T - 21.024 (10-5 A)] [exp(V/nKT) -1], n=40.1~44.6 (T<
第五章 總結
本實驗中製作的兩種新元件分別是WO3/IrO2水勵起二極體元件 及WO3/IrO2固態二極體元件,在水勵起二極體元件中用加水的方式 使元件從原本的絕緣體變成二極體,原因是水中的氫離子進入了 WO3,使WO3變成了HXWO3,白金電極到接面處的壓降大幅縮小, 使電子能夠通過薄膜形成二極體現象;而在固態二極體元件中使用在 WO3接面處加上負電壓的方式使氫離子進入WO3,使WO3變成 HXWO3,進而讓元件中的壓降變低,壓降變低電子就能通過元件上 的薄膜,這與水勵起元件不同的地方氫離子的來源,水勵起的來源是 水中的氫離子而固態元件不需要外加水及電解質就能形成二極體的 現象,兩者不同處在所需要的電壓不同及接面位置上的結構不同,水 勵起元件約在1.7V~1.8V,固態元件約在 0.3V~0.4V,水勵起元件 的接面上不需加上電壓,而固態元件上需在加上使氫離子轉移的負電 壓。 為了證明固態元件中的氫離子是氧化銥薄膜所提供的我們做了 抽真空的實驗在10-3 torr 下雨在空氣中並沒有什麼差別,因此可以用 來驗證氫離子的來源是薄膜內的氫離子而不是空氣中的水氣。 在溫度實驗中因為水勵起元件不適合在高溫下量測,過高的溫度 會使水蒸發掉,因此我們僅對固態元件做溫度實驗,在固態元件溫度 實驗中發現飽和電流會隨溫度上升而升高,這與氫離子的注入有關, 氫離子的注入效率會影響電子的的通過,當飽和電流在400K 以上 時,飽和電流便不在上升,這可能是質量傳輸達到上限,本實驗並沒 有對氫離子與電子的雙注入作更深入的分析。 目前所遇到的最大缺點是固態元件在常溫下只能承受 4V~5V 的電壓,顯示這個元件無法在常溫下通過大量的電子,在未來製作這 個元件的時候應設法改進這個缺點。 水勵起元件未來應可製作成水的感測元件,元件上有水的時候才 有電流通過,上面無水時電流便不會通過,固態元件中的 WO3和IrO2 在本實驗室中都曾分別做為氣體感測元件,可通入不同氣體量測固態 元件中的二級體現象與氣體種類、濃度的關係。參考文獻
[1] A.Hauch ,A.Georg , Electrochimica Acta,46,2001,p.2131. [2] Y.G.Mo ,R.O.Dillon , J.Vac.Sci.Technol,A17,5,p.2933.
[3] 謝育忠,“電漿濺鍍電致色變氧化鎳薄膜特性研究與節能評估” , 中原大學 , 碩士論文 , 民國 91 年.
[4] C.G.Granqvist ,“Electrochromic tungsten oxide films:Review of progress 1993-1998",Solar Engery Materials&Solar Cells,60,2000,p.2001.
[5] G.Lenftheriotis, S.Papaefthimiou , “Effect of the tungsten oxidation states in the thermal coloration and bleaching of amorphous WO3 films " ,Thin Solid Films,384,2001,p.298.
[6] A.Georg, W.Graf, V.Wittwer , “Comparison of electrical conductivity and optical properties of subtoichiometrically and electrochemically colourced WO3 films of dufferent crystallinity" , Solar Energy Materials and Solar cells,51,1998,p.353. [7] L.M.Schiavone, W.C.Dautremont-Smith, G.Beni and J.L.Shay , Appl.phys.Lett. ,
35,1979,p.823.
[8] J.R.Platt, J.Chem , Phys ,34,1961,p.862. [9] S.K.Deb, Appl.Optics ,3,1969,p.192.
[10] B.W.Faughnan, R.S.Cradall and P.M. HeyMan , R.C.A. Rev , 36,1975,p.177. [11] 楊慧敏,“氧化鎢與氧化釩單層膜與多層膜之製備與特性分析及其電致色變
性質", 國立東華大學 , 碩士論文 , 民國 91 年. [12] A.Donnadien, Mater.Sci.Eng. , 133,1989,p.185.
[13] J.S.E.M.Svensson and C.G.Granqvisit , Appl .Phys.Lett. , 45,1984,p.828. [14] M.Tolgyesi and M.Novak , J.Appl.Phys. , 32,1993,p.93.
[15] J.N.Yao , B.H.Looand A.Fujishima , Ber. Bunsenges Phys.Chem.,94,1990,p.249. [16] Hackwood S., Dautremont-Smith W. C., Beni G., “Volume changes induced by
the electrochromic process in sputtered iridium oxide films”, J. Electrochem. Soc. ,128,1981,p.1212 .
[18] 陳百賢,“鎳氧化物電致色變性質研究", 逢甲大學 , 碩士論文 , 民國 91 年.
[19] S.Morisaki, K.Kawakami and N.Baba, Jpn. J.Appl.Phys.,27,1988,p.840. [20] 何國川,“電化學與無窗簾時代",化工,第 37 卷,第 3 期,1990,p.34
[21] 焦小浣,胡文玲,陳玲,“光窗透明材料的實研",太陽能學報,第 8 卷,第 4 期,1997,p.365.
[22] S.E.Selkowitz and C.M.Lampert , Large-Area,Chromogenics:Materials and Devics for Transmittance Control , C.M.Lampert and
C.G.Granqvist editors , PIE , 1988,p.22.
[23] 李淑端 , “有機固態電解質電致色變元件之製作",逢甲大學, 碩士論文 ,
民國92 年.
[24] Shuchi Chao ,“Electrical characteristics of WO3-based CO2-sensitive solid-state microsensor"Jpn.J.Appl.Phys,32,1993,p.1346
[25] Shuchi Chao, “Fabrication and characterization of IrO2-based microsensors for fast detection of carbon dioxide” , Jpn. J. Appl. Phys , 36 , 1997 , p.2292 .
[26] Shuchi Chao, “Electricl characteristics of CO2-sensitive diode based on WO3 and IrO2 for microsensor applications” , Jpn. J. Appl. Phys , 37 , 1998 , p.L245 . [27] 許閔壹 , “於液態環境中穩定工作之三氧化鎢 / 氧化銥二極體感測元件之 研究” , 國立交通大學 , 碩士論文 , 民國 86 年. [28] 鍾其龍 ,“三氧化鎢 / 氧化銥固態電化學二極體元件及其對二氧化碳氣體 感測之研究",國立交通大學,碩士論文,民國 85 年. [29] 盧兆晴,“氧化銥電位式固態微型二氧化碳感測元件",國立交通大學,碩士 論文,民國 84 年. [30] 謝學瀚, “以三氧化鎢 / 氧化銥二極體元件為基礎製成的葡萄糖陣列型感測 器” ,國立交通大學,碩士論文,民國 87 年. [31] 林志強 ,“液態環境中以三氧化鎢 / 氧化銥二極體元為基礎的陣列式葡萄 糖感測器之研究",國立交通大學,民國 88 年. [32] 莊益林 , “三氧化鎢 / 氧化銥二極體元件對 PH 值的感測研究" , 國立交 通大學 , 民國 92 年.
[33] C.Bechinger ,M.S.Burdis , “ Comparison between electrochromic and photochromic coloration efficiency of tungsten oxide thin film" , Vol.101
No.10 , 1997 , p.753.
[34] Irina V. Shiyanovskaya, “ Structure rearrangement and electrochromic properties of amorphous tungsten trioxide films", J.Non-Crystalline Solid , 187 , 1995 , p.420.
[35] S.Hashimoto , H.Matsuoka , “Mechanism of eletrochromism for amorphous WO3 thin films" , J.Appl.Phys. , 69 , 1991 , p.933.
[36] A.Donnadieu, Material Science and Engineering. , B3 , 1989 , p.185. [37] B. Reichman, A.J.Bard, J Electrochem.Soc. , 126 , 1979 , p.583. [38] S.K.Deb, Solar energy Materials and Solar Cells. , 39 , 1995 , p.191. [39] S.Hackwood, A.H.Dayem, and G. Beni , Phys.Rev.B. , 26 , 1982 , p.471.
[40]Mcintyre.J.D.E, Basu S., Peck W.F., “ Cation insertion reactions of electrochromic tungsten and iridium oxide films", Phy.Rev.B, Vol.25 No.25 , 1982 , p.7242.
[41] Mcintyre.J.D.E. , Basu S., Peck W.F., Brown W.L., Augustyniak W.M., “Cation insertion reactions of electrochromic iridium oxide films", Solid state
ionics , 5 , 1981 , p.359.
[42] Shay J.L., Beni G., Schiavone L.M., “Electrochromism of anodic iridium oxide films on transparent substrates", Appl.Phys.Lett. , 33 , 1978 , p.942.
[43] Gottesfeld S., “The anodic iridium oxide film,a two-color eletrochromic system" , J. Electrochem.Soc. , 127 , 1980 , p.1922.
[44] Mcintyre.J.D.E. , Basu S. , Peck W.F. , Brown W.L. , Augustyniak W.M. , Phs.Rev.B , 25 ,1982 , p.7242.
[45] T. Katsube. , I. Lauks, J.N. Zemel , Sensors & Actuators , 2 , 1982 , p.399. [46] I. lauks, M.F. Yuen, T. Dietz, Sensors & Actuator, 4 , 1983 , p.375.
[47] B. Scrosati, “Applications of Electroactive polymers”, Chapman&Hall , London , 1933 , p.256 .
[48] Gottesfeld S. , “Faradaic processes at the Ir / Ir oxide electrode” , J. Electrochem. Soc. , 126 , 1979 , p.742 .
[49] J.V.Grahn , P.E.Hellberg , “Effect of growth temperature on the properties of evaporated tatalum pentoxide thin films on silicon deposited using oxygen radicals" , Journal of Applied Ph. , Vol. 84 , No. 3 , 1998.
[50] J.Y. Zhang, Boom Lin, I.W. Boyed, “Characteristics of high quality tantalum oxide films deposited by photoinduced chemical vapor deposition " , Appl.Phys.Lett. , Vol. 73 , No.16 , 1998.
[51] K.A. Mckinley, N.P. Sandler, “ Tatalum pentoxide for advanced DRAM applications", Thin Solid Films, 290-291, 1996, p.440.
[52] Paul M.S. Monk, Roger J. Mortimer, David R. Rosseinsky, Electrochromism: Fundamentals and Applications, VCH , 1995.
[53] 田福助 , “電化學基本原理應用” , 五洲出版社 , 中華民國八十三年八月 , p.55 .
[54] A. Georg, D. Schweiger, W. Graf, “The dependence of the chemical potential of WO3 films on hydrogen insertion”, Solar Energy Materials & Solar Cells , 70 , 2002 , p.437 .
[55] C. Bock, V. I. Birss, “Anion and water involvement in hydrous Ir oxide redox reactions in acidic solutions” , J. Electroanalytical Chemistry , 475 , 1999 , p.20 . [56] Yasushi Murakami , Shigeki Tsuchika and Yoshio Takasu , “Preparation of
ultrafine IrO2-Ta2O5 binary oxide particles by a sol-gel process " , Electrochimica , Vol. 39 , No.5 , 1994 , p.651.
[57] J. Krysa , L. Kule , R. Mraz , I. Rousar ,“Effect coating thickness and surface trearment of titanium on the properties of IrO2-Ta2O5 anodes" , Jolrnal of Applied electrochemistry , 26 , 1996 , p.999.
[58] J.L. He , M.C. Chiu ,“Effect of oxygen on the electrochromism of RF reactive magnetron sputter deposited tugsten oxide " , Surface and Coatings Technology , 127 , 2000 , p.43.
[59] A. Monterio, M.F. Costa, Vacuum, 64, 2002 , p.287. [60] L. Meda, Richars C., Thin Solid Films, 402 , 2002 , p.126.
Fig 3.1 白金電極剖面圖
SiO
2Si
3N
4N
-type
Silicon
Pt/Cr
Figure 3.2
The sputtering equipment setup.
Quartz cylinder shieldMagnetron Sputter source
In line regulator RotaMeters Glass cylinder (ψ=21cm) Substrate Holder Cooling water in out Matching Box RF generator Vacuum Gauge Mechanical Pump Ar Gas ring ThermoCouple Gauge Heater power Supply O2
Figure.3.3(a) 白金電極俯視圖 Figure.3.3(b) 白金電極俯視圖 0.6 mm 0.4 mm 0.6 mm 0.4 mm 0.3 mm 0.6 mm 0.4 mm 0.4 mm 0.4 mm 0.2mm
Figure 3.4(a) 未濺鍍薄膜 Figure 3.4(b) 濺鍍三氧化鎢 Figure 3.4(c) 濺鍍氧化銥
WO
3WO
3 WO3 WO3 IrO2 IrO2Figure 3.5(a) 晶片裝上 8 pin 腳座
Figure 3.5(b) 晶片用 AB 膠封裝
Epoxy Ag epoxy
Figure 3.6 加熱裝置圖
溫度計
Keithley237
元件 杜爾瓶 加熱平台WO3
IrO2 WO3
IrO2
Figure 3.7(a) IrO2 bridge
WO3
IrO2 WO3
IrO2
Figure 3.7(b) Overlap in gap
WO3
IrO2 WO3
IrO2
Figure 4.1 Overlap in gap 未加水 I-V 圖
Figure 4.2 Overlap in gap 加水 I-V 圖
-3 -2 -1 0 1 2 -4 -2 0 2 4 C u rrent (uA) Voltage(V) -2 -1 0 1 2 -4 -2 0 2 4 6 8 10 12 14 16 Cur ren t (uA ) Voltage (V)
Figure 4.3 WO3 bridge 未加水 I-V 圖
Figure 4.4 IrO2 bridge I-V 圖
-3 -2 -1 0 1 2 -2 0 2 4 6 8 10 Cu rr ent (uA) Voltage (V) -0.6 -0.4 -0.2 0.0 0.2 0.4 0.6 -6 -4 -2 0 2 4 6 Current(mA) Voltage(V)
-3 -2 -1 0 1 2 -0.6 -0.4 -0.2 0.0 0.2 0.4 0.6 0.8 1.0 1.2 1.4 1.6 Current (1 0E-4A) 300K 320K 340K 350K 370K 400K 420K 440K Voltage (V) Figure 4.5 二極體溫度變化圖
0.0 0.5 1.0 1.5 2.0 0.000000 0.000002 0.000004 0.000006 0.000008 0.000010 Y Axi s T itl e X Axis Title B
ExpGro1 fit of Data1_B
Figure 4.6(a) 300K fitting 圖
Equation: y = A1*exp(x/t1) + y0 Chi^2/DoF = 6.7001E-14 R^2 = 0.99471 y0 1.9249E-6 A1 1.9249E-6 t1 1.04834
0.0 0.5 1.0 1.5 2.0 0.00000 0.00001 0.00002 0.00003 0.00004 0.00005 Y Axi s Ti tle X Axis Title B
ExpGro1 fit of Data1_B
Figure 4.6(b) 320K fitting 圖 Equation: y = A1*exp(x/t1) + y0 Chi^2/DoF = 3.7913E-13 R^2 = 0.99844 y0 -7.0561E-6 A1 7.0561E-6 t1 1.12803
0.0 0.5 1.0 1.5 2.0 0.00000 0.00002 0.00004 0.00006 0.00008 Y Ax is T itle X Axis Title B
ExpGro1 fit of Data1_B
Figure 4.6(c) 340K fitting 圖 Equation: y = A1*exp(x/t1) + y0 Chi^2/DoF = 9.6083E-13 R^2 = 0.99877 y0 -0.00002 A1 0.00002 t1 1.20328
0.0 0.5 1.0 1.5 2.0 0.00000 0.00002 0.00004 0.00006 0.00008 0.00010 Y Axi s T it le X Axis Title B
ExpGro1 fit of Data1_B
Figure 4.6(d) 350K fitting 圖 Equation: y = A1*exp(x/t1) + y0 Chi^2/DoF = 2.0777E-12 R^2 = 0.99816 y0 -0.00003 A1 0.00003 t1 1.223
380K 0.0 0.5 1.0 1.5 2.0 0.00000 0.00002 0.00004 0.00006 0.00008 0.00010 0.00012 Y Axis T it le X Axis Title B
ExpGro1 fit of Data1_B
Figure 4.6(e) 380K fitting 圖
Equation: y = A1*exp(x/t1) + y0 Chi^2/DoF = 3.7555E-12 R^2 = 0.99737 y0 -0.00004 A1 0.00004 t1 1.377
0.0 0.5 1.0 1.5 2.0 -0.00002 0.00000 0.00002 0.00004 0.00006 0.00008 0.00010 0.00012 0.00014 Y Axis Ti tle X Axis Title B
ExpGro1 fit of Data1_B
Figure 4.6(f) 400K fitting 圖 Equation: y = A1*exp(x/t1) + y0 Chi^2/DoF = 7.3643E-12 R^2 = 0.99628 y0 -0.00007 A1 0.00007 t1 1.624
0.0 0.5 1.0 1.5 2.0 -0.00002 0.00000 0.00002 0.00004 0.00006 0.00008 0.00010 0.00012 0.00014 0.00016 Y Ax is Tit le X Axis Title B
ExpGro1 fit of Data1_B
Figure 4.6(g) 420K fitting 圖 Equation: y = A1*exp(x/t1) + y0 Chi^2/DoF = 1.2064E-11 R^2 = 0.9953 y0 -0.00007 A1 0.00007 t1 1.564
0.0 0.5 1.0 1.5 2.0 -0.00002 0.00000 0.00002 0.00004 0.00006 0.00008 0.00010 0.00012 0.00014 0.00016 Y A x is Ti tl e X Axis Title B
ExpGro1 fit of Data1_B
Figure 4.6(h) 440K fitting 圖 Equation: y = A1*exp(x/t1) + y0 Chi^2/DoF = 1.7961E-11 R^2 = 0.99386 y0 -0.00007 A1 0.00007 t1 1.537
Table 4.1 n IS nKT 300K 41.67 1.92E-06 1.04834 320K 40.54 7.06E-06 0.96803 340K 40.8 0.00002 1.20328 350K 40.1 0.00003 1.28937 380K 41.7 0.00004 1.48085 400K 44.6 0.00007 1.80345 420K 42.9 0.00007 1.75463 440K 40.23 0.00007 1.7171 I= IS [exp(V/nKT) -1]
Figure 4.7 IS溫度變化圖 300 320 340 360 380 400 0 10 20 30 40 50 60 70 80 C u rren t 0 μ A ) Temperature (K)



![HPSH [ 氧化數平衡反應式係數 ]](data:image/gif;base64,R0lGODlhAQABAIAAAP///wAAACH5BAEAAAAALAAAAAABAAEAAAICRAEAOw==)