Unbinding of the streptavidin-biotin complex by atomic force microscopy:
A hybrid simulation study
Jian Zhou,a兲Luzheng Zhang, and Yongsheng Leng
Department of Chemical Engineering, University of Washington, Seattle, Washington 98195 Heng-Kwong Tsao
Department of Chemical Engineering, National Central University, Chung-li 320, Taiwan Yu-Jane Sheng
Department of Chemical Engineering, National Taiwan University, Taipei 106, Taiwan Shaoyi Jiangb兲
Department of Chemical Engineering, University of Washington, Seattle, Washington 98195 共Received 13 June 2006; accepted 25 July 2006; published online 13 September 2006兲
A hybrid molecular simulation technique, which combines molecular dynamics and continuum mechanics, was used to study the single-molecule unbinding force of a streptavidin-biotin complex. The hybrid method enables atomistic simulations of unbinding events at the millisecond time scale of atomic force microscopy 共AFM兲 experiments. The logarithmic relationship between the unbinding force of the streptavidin-biotin complex and the loading rate共the product of cantilever spring constant and pulling velocity兲 in AFM experiments was confirmed by hybrid simulations. The unbinding forces, cantilever and tip positions, locations of energy barriers, and unbinding pathway were analyzed. Hybrid simulation results from this work not only interpret unbinding AFM experiments but also provide detailed molecular information not available in AFM experiments. © 2006 American Institute of Physics.关DOI:10.1063/1.2337629兴
I. INTRODUCTION
In many physiological processes, cell adhesion plays an essential role in diverse biological phenomena including in-flammation and cancer metastasis, where the binding and unbinding of receptor-ligand pairs are extensively involved. The receptor-ligand interactions 共e.g., in the streptavidin-biotin complex兲 are also very important in applications such as biosensors and biomaterials. Thus, it is essential to study the receptor-ligand interactions.
In the past decade, there are increasing interests in de-termining the single-molecule forces of receptor-ligand complexes1–4 by dynamic force spectroscopy 共DFS兲. DFS experimental tools include atomic force microscope5–9,9–12 共AFM兲, biomembrane force probe13 共BFP兲, and optical tweezer14 共OT兲. Among these methods, AFM is the most widely used technique. To determine the unbinding force by AFM, a receptor is anchored to a surface while a ligand is covalently linked to an AFM tip. The force probe coated with the ligand is brought into contact with the substrate covered with the receptor共binding兲. On retraction of the tip from the surface 共unbinding兲, the spring force is recorded as a func-tion of cantilever posifunc-tion共or time兲. In such experiments, the unbinding共or rupture兲 force is defined as the largest spring force applied during the process.
The streptavidin共or avidin兲-biotin system is one of the
strongest noncovalent interactions in nature. It is also a model system for studying receptor-ligand interactions. Ex-perimentally, Lee et al.5demonstrated the capability of AFM to measure discrete and biologically specific rupture forces of this system at the single-molecule level. They found that surfaces functionalized with biotin and streptavidin exhibited adhesive forces three to eight times greater than nonspecific interactions observed between blocked streptavidin and bi-otinylated surfaces. The obtained unbinding forces for streptavidin-biotin are between 300 and 400 pN for cantile-vers with different spring constants. Gaub and co-workers6,7 showed the direct measurements of the rupture of individual biologically specific bonds; the unbinding forces are 160 and 257 pN for avidin-biotin and streptavidin-biotin, respec-tively. The unbinding forces of the streptavidin 共or avidin兲-biotin system were further investigated with statistical analysis8–10 and discrete methods.11,12 Patel et al.15 high-lighted the influence of molecular architecture on the kinetic stability of this system.
Evans and Ritchie16studied the strength of weak nonco-valent bonds in liquids. Although the streptavidin-biotin sys-tem is among the strongest noncovalent linkages in biology, these bonds can appear strong or weak in dynamic measure-ments, depending on how fast they are loaded. They16 used Kramers’ theory and derived a formula to predict the depen-dence of unbinding forces on loading rates. When their pre-dictions were applied to the range of loading rates attainable for AFM, a logarithmic dependence of unbinding force on loading rates was observed. They16further predicted that, for a range of loading rates covering 12 orders of magnitude, the
a兲Present address: School of Chemical and Energy Engineering, South China
University of Technology, GuangZhou, GuangDong Province, 510640, People’s Republic of China.
b兲Author to whom correspondence should be addressed. FAX:
共206兲-685-3451. Electronic mail: [email protected]
rupture force of an avidin-biotin complex could not be fitted with a single exponential. These predictions were confirmed by their BFP experiments13 later. The same dependence was also found by others from AFM experiments.9,12
For experimental studies, one difficulty is how to ensure that a particular force measurement actually deals with a single interacting pair between the tip and the substrate. An-other difficulty is to differentiate specific interactions from nonspecific interactions. When the tip approaches the sample, the adsorbed protein at the interface could experi-ence mild denaturation. It is hard to determine if the force curves obtained from AFM experiments truly correspond to the unbinding events of a native protein or the stretching of a partially denatured protein. The unbinding and mechanical properties of the receptor-ligand complex under an external force also largely depend on the orientation of the complex and its degrees of freedom. In addition to these difficulties, the atomic details of the unbinding process are not available from AFM experiments. Molecular simulations are well suited for studying truly single-molecule interactions, reveal-ing the molecular-level unbindreveal-ing mechanism and providreveal-ing detailed information during the unbinding process. Simula-tion results will complement those from experiments very well.
Grubmuller et al.17 pioneered the molecular simulation study of the unbinding of a streptavidin-biotin complex. A streptavidin monomer with a bound biotin was first equili-brated in a bath of water molecules. Force was then exerted on the biotin to unbind the complex. However, the unbinding process was simulated at very fast cantilever pulling veloci-ties for 1 ns in their molecular dynamics 共MD兲 study. Fur-thermore, a relatively stiff spring 共spring constant of 2.8 N / m兲 was used, which is much stiffer than that used in AFM experiments 共spring constant of ⬃0.05 N/m兲. Very fast pulling velocities and the stiff spring constant used lead to very high loading rates of 4.2– 112 N / s. A mathematical procedure was also used to process their raw data; they smoothed the force profile with a Gaussian distribution. The unbinding force obtained in this way was between 300– 650 pN. Izrailev et al.18 simulated the unbinding of an avidin-biotin complex in a continuum solvent with a slowly stiffening cantilever 共0–0.414 N/m兲 at very fast cantilever pulling velocities. The range of their loading rates is 1.3– 83 N / s. The obtained unbinding force for the avidin-biotin system was 450 pN. For these studies17,18 using con-ventional MD simulation methods, the loading rates共or can-tilever pulling velocities兲 are many orders of magnitude higher than those of AFM experiments. As suggested by Grubmuller et al.17 and discussed by Izrailev et al.,18 fric-tional effects are expected to contribute significantly to the unbinding force of receptor-ligand complexes at pulling ve-locities above 1.0 m / s 共friction region兲. In AFM experi-ments, activated processes dominate at slower velocities of ⬃1.0m / s 共activated region兲, for which frictional forces become negligible. Thus, it is hard to directly compare the unbinding forces obtained from MD simulations17,18 at very fast pulling velocities共⬃1.0 m/s兲 with those from AFM ex-periments at slow pulling velocities 共⬃1.0m / s兲. By con-sidering activated unbinding and frictional forces, Heymann
and Grubmuller19 used a simple model to extrapolate the simulated unbinding forces to the millisecond time regime of single-molecule AFM unbinding experiments for the AN02/ DNP-hapten system. However, their extrapolation could only be obtained for a simplified binding interaction potential and based on the assumption that there is only one single energy barrier along the unbinding trajectory. Galligan et al.20 adopted a combination of traditional reaction coordinate mapping and Brownian dynamics to simulate the dynamic strength of streptavidin-biotin interactions. They20 also ob-served the logarithmic dependence of rupture forces on load-ing rates.
In this work, a hybrid molecular simulation technique, which combines continuum mechanics and molecular dy-namics, was used to simulate the unbinding of a streptavidin-biotin complex21at the experimental AFM time scale. Previ-ous experiments9,12,13have shown that there is a logarithmic relationship between unbinding forces and loading rates. Thus, the objectives of this work are to simulate the single-molecule unbinding process at the AFM experimental time scale, to verify the loading-rate-dependent unbinding forces found in AFM experiments and to explore the unbinding pathway and locations of energy barriers of the streptavidin-biotin complex at the molecular level.
II. SIMULATION METHODOLOGY
Previously, a hybrid simulation method22was developed to study the adhesion and friction of self-assembled mono-layers on Au共111兲.22–24
Since the mass of an atom is many orders of magnitude different from that of an AFM tip, their characteristic vibration frequencies are very different. There-fore, the atomic motion and the tip motion can be decoupled. For hybrid simulations, the integration of the dynamic equa-tions of motion for the AFM tip and the complex system can be done separately, i.e., the equations of motion for the tip is integrated with a comparably larger time step共s兲 and then a fast relaxation of the atomic complex by MD simulations with a smaller time step共fs兲 is followed. Quasistatic assump-tion is adopted, during the tip moassump-tion, the molecular system should be well preserved in thermal fluctuation. In this way, we can simulate the streptavidin-biotin unbinding process by AFM at pulling velocities or loading rates close to AFM unbinding experiments.
In simulations of an unbinding process, the AFM tip is dragged by a support through a spring共kx, ky, kz兲 at a velocity ofv. The differential equations of motion of the AFM tip are
governed by
Mx¨t= kx共xM− xt兲 + Wx共xt,yt,zt;X共t兲兲, 共1兲
My¨t= ky共yM− yt兲 + Wy共xt,yt,zt;X共t兲兲, 共2兲
Mz¨t= kz共zM− zt兲 + Wz共xt,yt,zt;X共t兲兲, 共3兲 where the forces contributed by the receptor and streptavidin
are denoted by Wx, Wy, and Wz in the x, y, and z directions. These forces depend not only on the tip position 共xt, yt, zt兲 but also on the configuration of the receptor-ligand complex studied X共t兲 at time t. M is the effective mass25
of the tip,
M = 10−11kg. The position of the support is 共x
M, yM, zM兲 where zM=vt. The spring force is given by Fspring= kz共zM − zi兲 where kzis the spring constant of the cantilever. In this work, kxand kyare set to zero, i.e., the sideward共directions perpendicular to the pulling direction兲 motions of the biotin group in the binding pocket are not restrained, since such motions are essentially unconstrained in AFM unbinding ex-periments.
A tetramer structure21of the streptavidin-biotin complex 共Protein Data Bank code: 1swe兲 was used. The CHARMM
package26was used to add hydrogen atoms. The whole com-plex was then reoriented so that the possible unbinding path-way of biotin is parallel to the direction of the external force 共z direction designated in this work兲. The potential param-eters and charges of streptavidin are from theCHARMMforce field for protein26whereas the parameters and partial charges of biotin are the same as used by Izrailev et al.18 In AFM unbinding experiments, the biotin and the AFM tip are usu-ally connected by a reactive bifunctional covalent cross-linker and should move together. To simplify, we let the po-sition of the oxygen atom O2 of the biotin be exactly the same as the position of the AFM tip in our hybrid simula-tions as Grubmuller et al.17 did in their MD simulations. It has been argued27 that the binding pocket of avidin is inac-cessible to water. Izrailev et al.18 attempted to place water molecules in the binding pocket and they found that the tight contact between biotin and its binding pocket makes it un-likely for more than one water molecule to fit inside the binding pocket. Hence we do not include explicit water mol-ecules in this work. For the tetramer structure of the streptavidin-biotin complex,21it has been found that the 3-4 loop共residues 45–52兲 of streptavidin adopts an open confor-mation for the unbound state and a closed conforconfor-mation for the bound state. In AFM unbinding experiments,5–12 the bi-otin and the AFM tip are usually connected by a reactive bifunctional covalent cross-linker. Because of the existence of the cross-linker, the 3-4 loop of streptavidin may not close even after binding. Furthermore, during the experimental un-binding process, the 3-4 loop will adopt an open conforma-tion. To facilitate the exit of biotin from the binding pocket, in MD simulations by Izrailev et al.,18 the loop was pre-opened. Similarly, the open conformation of the 3-4 loop was adopted in this work.
For hybrid simulations, the equations of motion for the tip were integrated using a backward differential algorithm either over 200⌬ttipor when the tip displacement during each
loop of tip motion exceeds 0.2 Å. If the tip displacement reaches 0.2 Å during the integration of tip motion equations, the hybrid simulation will return to MD relaxation. When stick-slip occurs, the tip will quickly slip from the binding pocket and the biotin moves a large distance even for a single step of cantilever motion. Thus, the criterion for a tip
displacement of less than 0.2 Å is critical; otherwise, the quasistatic assumption is violated. We have tried different maximum allowed tip displacements: 0.1, 0.2, 0.3, and 0.4 Å. It has been found that for the case of 0.4 Å, occasion-ally, there are spikes for potential energy-time curves, which indicate the violation of quasistatic assumption. The value of 0.2 Å for the maximum allowed tip displacement during the tip motion was used in the hybrid simulations, no spike was found from the potential energy evolution curves. The time step ⌬ttip used in this work is 0.25s 共equivalent to the
displacement of the support by 0.0025 Å at a pulling veloc-ity of 1.0m / s兲. Following the tip motion, the complex was relaxed by MD simulations with a time step⌬tMDof 1.0 fs.
We found that MD relaxation over 300⌬ttip, 600⌬ttip, and
1200⌬tMDyielded the same force-distance curve, indicating
that the relaxation of atomic system is quite fast. In this work, 400⌬tMDwas used for MD relaxation. For MD
simu-lations, initial velocities were assigned from a Maxwell-Boltzmann distribution at 300 K. The Berendsen method28 was used to keep the temperature of the system constant. The simulated system was coupled with a heat bath, using a cou-pling time of 0.1 ps. Bonds containing hydrogen are con-strained by using theRATTLEmethod29with a geometric tol-erance of 10−6. Cell-linked list30
was employed to accelerate the simulations. The short-range nonbonded interactions were calculated by a switched potential with a switching function starting at 10 Å and reaching zero at a distance of 11 Å. Electrostatic interactions were calculated by the shifted potential with a cutoff distance of 11 Å. For the streptavidin tetramer, only one pair of streptavidin-biotin was selected for unbinding. The center of mass of the streptavidin monomer to be unbound with the biotin group was fixed, which allowed the free rotational and internal motions of the system by external forces while the centers of mass of the other three streptavidin monomers were not fixed. With the alternative integration of the equations of motions for the tip and the atomic complex, the biotin was pulled out gradually. The hybrid simulations were performed up to a tip displace-ment of 15.0 Å. A graphical illustration of the hybrid simu-lation method is shown in Fig.1. For clarity, only one pair of streptavidin-biotin was shown.
III. RESULTS AND DISCUSSION
In this work, the single-molecule interactions between streptavidin and biotin were investigated with the hybrid simulation method. The problems encountered in experi-ments, such as multiple-pair interactions, nonspecific inter-actions, and protein denaturization, can be avoided. To ex-amine the loading-rate dependence of unbinding force found in AFM experiments, the unbinding of the streptavidin-biotin complex21 was simulated with an AFM cantilever 共spring constant kz= 0.039 N / m兲 under different cantilever pulling velocities of 1.0, 10, and 50m / s. This spring constant is in the range of spring constants for AFM cantilevers commonly used and the same as that used in AFM unbinding experi-ments by Lo et al.9
A. Force-distance relationship
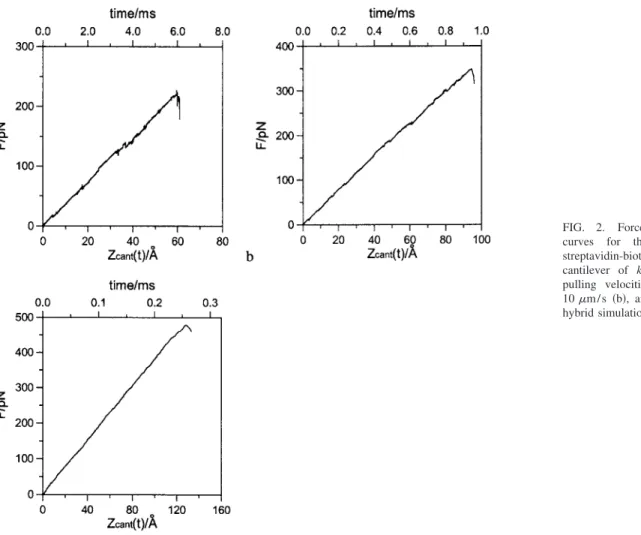
The simulated force-distance共or time兲 curves at pulling velocities of 1.0, 10, and 50m / s are shown in Figs. 2共a兲–2共c兲, respectively. As seen from these figures, the exter-nal spring forces increase almost linearly before reaching the maximum and then drop sharply. This typical stick-slip be-havior of force-distance curves observed in AFM experi-ments is well reproduced by the hybrid simulation method. The simulated unbinding共or rupture兲 force is defined as the largest spring force experienced during the unbinding simu-lation process. At pulling velocities of 1.0, 10, and 50m / s, the unbinding forces of the streptavidin-biotin complex are 227, 349, and 478 pN, respectively, whereas the rupture
times of the complex upon AFM pulling are 5.95, 0.94, and 0.257 ms, respectively. Thus, the time scale gap between AFM experiments and conventional MD simulations is bridged by the hybrid simulation method.
B. Cantilever position versus tip position
The cantilever position versus tip position curves of the unbinding process are shown in Figs.3共a兲–3共c兲for different pulling velocities. These figures indicate the unbinding path-way of the streptavidin-biotin complex. This information cannot be readily extracted from experiments, since only the position of the support is known while one cannot locate the position of the AFM tip from unbinding experiments. At the pulling velocity of 1.0m / s, as shown in Fig.3共a兲, the bi-otin moves away from its initial position and fluctuates around the position of 0.3 Å 共the first local barrier兲. Later, the biotin jumps to a new position of 1.0 Å, which means that the biotin overcomes the first energy barrier, and moves to a new barrier at the position of 1.0 Å. With the further increase of the external force, the biotin overcomes this new barrier and reaches another barrier at 2.6 Å, then the biotin slips out of the streptavidin binding pocket quickly, which is reflected by the steep increase of tip distance. This steep increase of tip distance in Fig.3共a兲corresponds to the sudden
FIG. 1. 共Color online兲 Illustration of the hybrid simulation method. The streptavidin is in ribbon representation while the biotin is in ball-stick rep-resentation. For clarity, only one pair of streptavidin-biotin was shown.
FIG. 2. Force-distance 共or time兲
curves for the unbinding of a
streptavidin-biotin complex with a cantilever of kz= 0.039 N / m at the
pulling velocities of 1.0m / s 共a兲, 10m / s 共b兲, and 50m / s 共c兲 from hybrid simulations.
decrease of force in Fig. 2共a兲. At the pulling velocities of 10 and 50m / s, the simulated cantilever position versus tip position curves exhibit similar behaviors as those at the pull-ing velocity of 1.0m / s.
C. Unbinding pathway
From the ribbon representation structure of streptavidin in Fig. 1, it can be seen that streptavidin is organized as eight-stranded, sequentially connected, antiparallel sheets. The sheets are formed of coiled polypeptide chains with a staggered pattern of adjacent strand hydrogen-bond registra-tion. The biotin binds in pockets at the end of streptavidin barrels. The residues lining the pockets are primarily polar amino acids.21
Figures 4共a兲–4共e兲 show a few typical configurations found during the unbinding process at the pulling velocity of 1.0m / s. For the initial configuration in Fig.4共a兲, the hy-drogen bonds are distributed in three areas: the inner, the middle, and the outer of the binding pocket. In the inner part, an extensive pattern of five hydrogen bonds with the ureido group of biotin are found, i.e., the Ser45 OH with biotin N1, Asp128 OD2 with biotin N2, and Tyr43 OH, Asn23 NH, Ser27 OH with the biotin ureido oxygen. In the middle part, the biotin sulfur interacts with the OH of Thr90. In the outer part, the two valeryl oxygen atoms of biotin are hydrogen bonded to the backbone NH of Asn49 and the OH of Ser88. Under the force exerted by the support, the biotin moves from its original binding position gradually. When the tip moves 0.3 Å from its original position, two hydrogen bonds break first, i.e., the sulfur atom of biotin with the OH of Thr90 and one valeryl oxygen with the backbone NH of
Asn49. This is shown in Fig. 4共b兲. Figure 4共c兲 displays the configuration corresponding to the location of energy barrier at 1.0 Å. It can be seen that another two hydrogen bonds break, i.e., the Ser45 OH with biotin N1 and the Tyr43 OH with the biotin ureido oxygen. In Fig. 4共d兲, for the location of energy barrier at 2.6 Å, the biotin sulfur interacts with the OH of Thr90 and Ser45 OH interacts with the biotin ureido oxygen while the hydrogen bond interaction between Asn23 NH and the biotin ureido oxygen breaks. After this major
FIG. 3. Cantilever position vs tip po-sition curves for the unbinding of a streptavidin-biotin complex with a cantilever of kz= 0.039 N / m at the
pulling velocities of 1.0m / s 共a兲, 10m / s 共b兲, and 50m / s 共c兲 from hybrid simulations. The horizontal bars indicate the locations of energy barriers.
FIG. 4. 共Color online兲 Snapshots of the streptavidin-biotin binding pocket during an unbinding process at the initial stage共a兲, at 0.3 Å 共b兲, at 1.0 Å 共c兲, at 2.6 Å共d兲, and at the final unbinding stage 共e兲 from hybrid simulations with a cantilever of kz= 0.039 N / m at the pulling velocity of 1.0m / s. The
barrier, the biotin leaves out of the binding pocket quickly. The final unbinding configuration is shown in Fig. 4共e兲. In general, the unbinding of the streptavidbiotin complex in-volves the breakage and formation of multiple hydrogen bonds between the ureido group and valeryl oxygen of biotin and the binding site residues of streptavidin.
D. Loading-rate dependence of unbinding forces
To get good statistics of the unbinding forces, we per-formed eight hybrid simulations under each condition from different initial configurations 共results not shown兲. Under each simulation condition 共same cantilever spring constant and same pulling velocity兲, the shapes of the obtained force-distance curves and the tip position versus cantilever position curves are very similar to those in Figs.2共a兲–2共c兲and Figs. 3共a兲–3共c兲, respectively. Major locations of energy barriers around 0.3, 1.0, and 2.6 Å are found in all cases. The aver-age unbinding forces are 263± 36, 378± 42, and 494± 39 pN at pulling velocities of 1.0, 10, and 50m / s, respectively. Experimentally, it has been observed5–12 that there is a dis-tribution of determined unbinding forces at each condition.
One advantage of simulation studies is that each param-eter can be studied separately under well-controlled condi-tions. In AFM pulling experiments, loading rate is often used, which is the product of cantilever spring constant and pulling velocity. Previous AFM experiments show the loga-rithmic relationship between unbinding force and loading rate. The average unbinding forces by hybrid simulations of a cantilever 共spring constant of 0.039 N/m兲 at different cantilever pulling velocities are shown in Fig. 5 together with experimental results by AFM 共Ref. 9兲 and BFP.13 The hybrid simulation results show a similar logarithmic trend to the experimental data. Thus, the loading-rate dependence of unbinding forces predicted by theory16 and found by experiments9,12,13is also observed from the hybrid simulations.
IV. CONCLUSIONS
In this work, a hybrid molecular simulation method, which combines continuum mechanics and molecular dy-namics, was used to study the single-molecule unbinding force of a streptavidin-biotin complex at the time scale of AFM unbinding experiments. For the cantilever with a spring constant similar to that used in AFM experiments, the logarithmic relationship between unbinding forces of the streptavidin-biotin complex and loading rates was observed. The simulated force-distance curves display a typical stick-slip behavior as in AFM experiments. The three major loca-tions of energy barriers of the streptavidin-biotin complex around 0.3, 1.0, and 2.6 Å along the unbinding pathway were found. The streptavidin-biotin unbinding involves the breakage and formation of multiple hydrogen bond interac-tions between the ureido group and valeryl oxygen of biotin and the binding site residues of streptavidin. Our hybrid simulations cannot only interpret unbinding AFM experi-ments but also provide detailed molecular information not available in AFM experiments.
ACKNOWLEDGMENT
This work was supported by the National Science Foun-dation 共CMS-0302139兲.
1E. Evans, Annu. Rev. Biophys. Biomol. Struct. 30, 105共2001兲. 2M. Horton, G. Charras, and P. Lehenkari, J. Recept. Signal Transduct.
Res. 22, 169共2002兲.
3R. Lavery, A. Lebrun, J. F. Allemand, D. Bensimon, and V. Croquette, J.
Phys.: Condens. Matter 14, R383共2002兲.
4J. W. Weisel, H. Shuman, and R. I. Litvinov, Curr. Opin. Struct. Biol. 13,
227共2003兲.
5G. U. Lee, D. A. Kidwell, and R. J. Colton, Langmuir 10, 354共1994兲. 6E. L. Florin, V. T. Moy, and H. E. Gaub, Science 264, 415共1994兲. 7V. T. Moy, E. L. Florin, and H. E. Gaub, Science 266, 257共1994兲. 8Y. S. Lo, N. D. Huefner, W. S. Chan, F. Stevens, J. M. Harris, and T. P.
Beebe, Langmuir 15, 1373共1999兲.
9Y. S. Lo, Y. J. Zhu, and T. P. Beebe, Langmuir 17, 3741共2001兲. 10Y. S. Lo, J. Simons, and T. P. Beebe, J. Phys. Chem. B 106, 9847共2002兲. 11J. Wong, A. Chilkoti, and V. T. Moy, Biomol. Eng. 16, 45共1999兲. 12C. Yuan, A. Chen, P. Kolb, and V. T. Moy, Biochemistry 39, 10219
共2000兲.
13R. Merkel, P. Nassoy, A. Leung, K. Ritchie, and E. Evans, Nature
共London兲 397, 50 共1999兲.
14K. Svoboda and S. M. Block, Annu. Rev. Biophys. Biomol. Struct. 23,
247共1994兲.
15A. B. Patel, S. Allen, M. C. Davies, C. J. Roberts, S. J. B. Tendler, and P.
S. Williams, J. Am. Chem. Soc. 126, 1318共2004兲.
16E. Evans and K. Ritchie, Biophys. J. 72, 1541共1997兲.
17H. Grubmuller, B. Heymann, and P. Tavan, Science 271, 997共1996兲. 18S. Izrailev, S. Stepaniants, M. Balsera, Y. Oono, and K. Schulten,
Bio-phys. J. 72, 1568共1997兲.
19B. Heymann and H. Grubmuller, Chem. Phys. Lett. 303, 1共1999兲. 20E. Galligan, C. J. Roberts, M. C. Davies, S. J. B. Tendler, and P. M.
Williams, J. Chem. Phys. 114, 3208共2001兲.
21S. Freitag, I. LeTrong, L. Klumb, P. S. Stayton, and R. E. Stenkamp,
Protein Sci. 6, 1157共1997兲.
22Y. S. Leng and S. Y. Jiang, J. Chem. Phys. 113, 8800共2000兲. 23Y. S. Leng and S. Y. Jiang, J. Am. Chem. Soc. 124, 11764共2002兲. 24L. Z. Zhang, Y. S. Leng, and S. Y. Jiang, Langmuir 19, 9742共2003兲. 25L. Y. Li, Q. M. Yu, and S. Y. Jiang, J. Phys. Chem. B 103, 8290共1999兲. 26A. D. MacKerell, Jr., D. Bashford, R. L. Bellott et al., J. Phys. Chem. B
FIG. 5. Loading rate dependence of unbinding forces for a cantilever of kz= 0.039 N / m. Open circles are for the hybrid simulation results. Solid
circles are for the AFM data共Ref.9兲. Triangles are for the BFP data 共Ref. 13兲. The lines are drawn to guide eyes.
102, 3586共1998兲.
27L. Pugliese, A. Coda, M. Malcovati, and M. Bolognesi, J. Mol. Biol.
231, 698共1993兲.
28H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. Dinola, and
J. R. Haak, J. Chem. Phys. 81, 3684共1984兲.
29H. C. Andersen, J. Comput. Phys. 52, 24共1983兲.
30D. J. Auerbach, W. Paul, C. Lutz, A. F. Bakker, W. E. Rudge, and F. F.