PLEASE SCROLL DOWN FOR ARTICLE
This article was downloaded by: [Chen, Calvin Yu-Chian]
On: 14 April 2011
Access details: Access Details: [subscription number 936394022]
Publisher Taylor & Francis
Informa Ltd Registered in England and Wales Registered Number: 1072954 Registered office: Mortimer House,
37-41 Mortimer Street, London W1T 3JH, UK
Molecular Simulation
Publication details, including instructions for authors and subscription information: http://www.informaworld.com/smpp/title~content=t713644482
Novel hemagglutinin inhibitors for H1N1 influenza virus screening from
TCM database
Tung-Ti Changab; Mao-Feng Sunac; Hsin-Yi Chend; Fuu-Jen Tsaide; Mark Fisherf; Jaung-Geng Lina;
Calvin Yu-Chian Chenadfg
a Laboratory of Computational and Systems Biology, School of Chinese Medicine, China Medical
University, Taichung, Taiwan, ROC b Department of Chinese Pediatrics, China Medical University
Hospital, Taichung, Taiwan, ROC c Department of Acupuncture, China Medical University Hospital,
Taichung, Taiwan, ROC d Department of Bioinformatics, Asia University, Taichung, Taiwan, ROC e
Department of Medical Genetics, China Medical University, Taichung, Taiwan, ROC f Harvard-MIT
Division of Health Sciences and Technology, Cambridge, MA, USA g Computational and Systems
Biology, Massachusetts Institute of Technology, Cambridge, MA, USA Online publication date: 13 April 2011
To cite this Article Chang, Tung-Ti , Sun, Mao-Feng , Chen, Hsin-Yi , Tsai, Fuu-Jen , Fisher, Mark , Lin, Jaung-Geng and Chen, Calvin Yu-Chian(2011) 'Novel hemagglutinin inhibitors for H1N1 influenza virus screening from TCM database', Molecular Simulation, 37: 5, 361 — 368
To link to this Article: DOI: 10.1080/08927022.2010.543973
URL: http://dx.doi.org/10.1080/08927022.2010.543973
Full terms and conditions of use: http://www.informaworld.com/terms-and-conditions-of-access.pdf This article may be used for research, teaching and private study purposes. Any substantial or systematic reproduction, re-distribution, re-selling, loan or sub-licensing, systematic supply or distribution in any form to anyone is expressly forbidden.
The publisher does not give any warranty express or implied or make any representation that the contents will be complete or accurate or up to date. The accuracy of any instructions, formulae and drug doses should be independently verified with primary sources. The publisher shall not be liable for any loss, actions, claims, proceedings, demand or costs or damages whatsoever or howsoever caused arising directly or indirectly in connection with or arising out of the use of this material.
Novel hemagglutinin inhibitors for H1N1 influenza virus screening from TCM database
Tung-Ti Changa,b†, Mao-Feng Suna,c†, Hsin-Yi Chend, Fuu-Jen Tsaid,e, Mark Fisherf, Jaung-Geng Linaand Calvin Yu-Chian Chena,d,f,g*a
Laboratory of Computational and Systems Biology, School of Chinese Medicine, China Medical University, Taichung 40402, Taiwan, ROC;bDepartment of Chinese Pediatrics, China Medical University Hospital, Taichung 40402, Taiwan, ROC;cDepartment of Acupuncture, China Medical University Hospital, Taichung 40402, Taiwan, ROC;dDepartment of Bioinformatics, Asia University, Taichung 41354, Taiwan, ROC;eDepartment of Medical Genetics, China Medical University, Taichung 40402, Taiwan, ROC;f Harvard-MIT Division of Health Sciences and Technology, 77 Massachusetts Avenue, Cambridge, MA 02139, USA;gComputational and Systems Biology, Massachusetts Institute of Technology, Cambridge, MA 02139, USA
(Received 5 September 2010; final version received 23 November 2010)
The emergence of Tamiflu (oseltamivir)-resistant viral strains in pandemic of H1N1/09 influenza virus has raised global awareness of anti-viral drug resistant issue. There is an urgent demand for developing new anti-influenza compound. The purpose of this research is to design novel haemagglutinin (HA) inhibitor for inhibiting viral entry into the host cell. We performed structure-based drug design to analyse interactions between the potent inhibitor and HA. A traditional Chinese medicine (TCM) database was used for in silico screening process. The docked TCM constituents were input into de novo evolution to generate derivatives. Selected derivatives were then docked back to HA binding site. We identify four key features from top 10 docked derivatives’ binding conformations and structure scaffolds. The addition of 2-aminopyridinium group has the greatest influence in the binding ability of TCM derivatives and is, therefore, suggested to be the key point in designing HA inhibitors.
Keywords: H1N1; Tamiflu; docking; structure-based drug design; traditional Chinese medicine
1. Introduction
With the advancement in computational technology, molecular modelling is becoming an integral part of material science, new polymer design and even pharmaceutics development [1– 6]. In particular, in silico screening, or structure-based drug design, that utilises computational chemistry and molecular mechanics, permits detailed investigation of potential drug– receptor interactions, while also offering speed enhancement in drug discovery.
The need for rapid assessment of compound libraries to search for potential medicinal compound is most recently reflected in the pandemic of H1N1 influenza virus, in which the viral strain resistant to the front-line antiviral drug Tamiflu was identified [7 – 9]. Tamiflu, a neuraminidase (NA) inhibitor, inhibits the spread of new influenza viral progeny by binding to viral surface glycoprotein, NA [10 – 12]. However, the segmented RNA genome of influenza viruses is prone to antigenic drift (sequence base mutations) and antigenic shift (genetic recombination), both of which are known causes for NA inhibitor resistance [13 – 16].
In addition to NA, influenza surface membrane also contains another glycoprotein, haemagglutinin (HA). HA
facilitates viral entry through binding to host surface sialic acid residues [17]. Hence, blocking of HA sialic acid binding site could inhibit viral entry into the host cell and then, indirectly, could prevent viral replication. When compared to NA inhibitors, HA inhibitor, theoretically, should be a more effective anti-influenza strategy, since NA inhibitor, in case of Tamiflu, are ineffective in the late stage of viral infection [18,19]. This lack of efficacy is most attributed to the build-up of virus number in the host body. Presently, there are no clinically available HA inhibitor despite numerous efforts. In the hope of finding new anti-viral compounds, we conducted structure-based drug design to analyse interactions between potential ligands and HA sialic acid binding site. There were many successful cases of applying structure-based drug design to investigate ligand – protein or protein – protein inter-actions [20,21], and in the past, our group has applied the computer-aided drug design technology to develop new therapeutic compounds and has also developed new scoring function for assessing binding affinity [22 – 38]. Of all the different HA subtypes (H1 – H16), we concentrated our efforts on H1, the HA subtype found on the recent pandemic H1N1/09 virus. We performed
ISSN 0892-7022 print/ISSN 1029-0435 online
q2011 Taylor & Francis
DOI: 10.1080/08927022.2010.543973 http://www.informaworld.com
†
Both authors contributed equally to this work.
*Corresponding author. Email: [email protected]; [email protected] Vol. 37, No. 5, April 2011, 361–368
in silico screening of a traditional Chinese medicine (TCM) database (Available at http://tcm.cmu.edu.tw/) to find constituents most fitted to the binding pocket of H1. TCM constituents have been studied in the past to investigate anti-cancer [39] and anti-inflammatory [40,41]; however, their anti-influenza potencies have not been closely examined. Natural compounds, such as those from TCM, cover diverse range of chemical structures and, therefore, could be extremely novel compounds for protein targets.
2. Methods 2.1 Software
Structure modelling, validation of homology model, virtual screening and generations of derivatives were performed by Discovery Studio Client v2.5.0.9164 (Dock Score 2.5). The 2D and 3D structures of TCM constituents within the TCM database were built by using Chem-BioOffice 2008 (CambridgeSoft, Inc., Cambridge, MA, USA). The TCM database can be accessed at http://tcm. cmu.edu.tw/
2.2 Sequence comparison and homology modelling of H1N1/09 HA
As shown in Figure 1, sequence alignment was performed prior to homology modelling to ensure the sequence identity and similarity shared between the template sequence and the target sequence. The gene sequence of HA, subtype H1, was obtained from GenBank: ADI99650. The H1 template was downloaded from the Protein Data Bank, PDB ID:
1RDB [42]. After homology modelling, Ramachandran plot and Profiles-3D were used to validate the reliability and the compatibility of the modelled H1 structure.
2.3 In silico screening (docking)
LigandFit module of DS 2.5 was used to dock TCM constituents into H1. All protein and small molecules were pre-applied with a forcefield of Chemistry at Harvard Macromolecular Mechanics. Ligand binding site of H1 was set to the N-Acetyl-D-Glucosamine (NAG) binding site found in the HA crystal. NAG molecule was also used as a control to filter TCM constituents.
2.4 Derivatives generation (de novo evolution) Candidate compounds obtained from previous docking screening were input into de novo evolution, in which the TCM compound scaffolds were used as the starting basis for further modification. Fragments that comp-lement the H1 binding sites were fused to the TCM scaffolds.
After de novo evolution, Lipinski’s Rule of Five [43] was used to screen out derivatives that are orally unstable or inapplicable compounds. Derivatives past the screen were then docked back to H1 ligand binding site to assess binding affinity and receptor – ligand interactions.
3. Results and discussion 3.1 Homology modelling
The sequence alignment of H1 protein and target sequence gives a sequence identity of 83.2% and a similarity of 92.1% (Figure 2). This high sequence identity and similarity between the target and the template sequence provide supports for using the template structure (H1: 1RD8) as modelling basis.
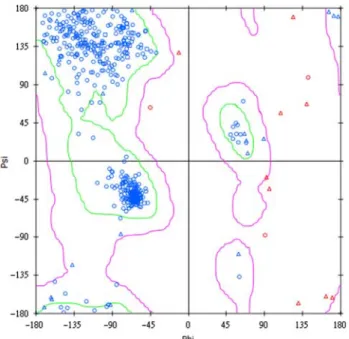
To validate the modelled H1 structure, Ramachandran plot was used to examine the compatibility of dihedral angles of protein backbone atoms. As the plot illustrates in Figure 3, 94.4% of residues are found in the acceptable regions, while only 2.5% of residues are in disallowed regions (Figure 3). As for Profiles-3D validation of protein structure, all the binding site residues (sequence 12 – 65) are found to have compatibility scores greater than zero (Figure 4). Based on the above results, we have confidence in the reliability of our H1 model (Figure 5).
3.2 Docking results
From docking, we selected the top 20 TCM constituents (Table 1) for further structural modification. The de novo evolution products of these TCM were screened by Lipinski’s Rule of Five and were docked back to the H1 Figure 1. Flowchart of the entire experiment.
T.-T. Chang et al. 362
protein. We used DS as the primary scoring function for ranking derivatives. The DS of the control, NAG, was used as the filtering basis. Derivatives with DS higher than control were further judged based on piecewise linear
potential (PLP), PLP2, and -potential of mean force. The top 10 derivatives are shown in Table 2. The top 10 de novo products are mostly derived from xylopine and rosmaricine, and they all share the addition of benzene ring and positive charged 2-aminopyridinium (Table 3).
3.3 Characteristics of de novo product binding conformations
Our docking results show that all the top 10 derivative compounds have binding interaction with residues, Arg238 and Asp103, that were deemed important in the previous literature [42] (Figure 6). Further analyses of derivative binding conformations suggest that salt bridge Figure 2. Sequence alignment of H1 (sequence identity: 83.2% and similarity: 92.1%).
Figure 3. Ramachandran plot of H1. Residues of about 94.4% are in the allowed region, while only 5.6% of residues are in the semi-allowed region or disallowed region.
–0.5 0 0.5 1 1.5 0 100 200 300 400 500 Amino acid Score H1 1RD8
Figure 4. Result of 3D-profile for H1 model structure. A score greater than zero indicates high reliability in the modelling structure.
interaction, hydrophobic interaction, hydrogen-bond inter-action and p-stacking interaction are the most important non-bonded interactions. Based on these non-bonded interactions and derivative binding conformations, we summarised four key features (Figure 7) that are important for ligand – H1 binding.
3.3.1 NH2 þ
and NH3 þ
groups
Xylopine, rosmaricine and their derivatives all contain protonated amine group that can form interaction with hydrogen bond or salt bridge interaction with binding site residues. As shown in Figure 6, the protonated amine group could form salt bridge interaction with Glu83 or with Asp103.
3.3.2 2-Aminopyridinium and benzene groups
2-Aminopyridinium group is found on the top three derivatives, Xylopine_2, Rosmaricine_14 and Rosmar-icine_15, while the benzene ring is present in the rest. Addition of 2-aminopyridinium group gives significant increase in binding affinity and also influences the binding conformations. Xylopine_2, Rosmaricine_14 and Rosmar-icine_15 all have salt bridge interactions with Asp103 (Figure 6(a) and (b)). In contrast, the other derivatives lack the positive charge characteristic of 2-aminopyridinium and, therefore, can only form hydrophobic interactions with hydrophobic residues. These results demonstrate the importance of the 2-aminopyridinium in the H1 inhibitor design.
3.3.3 Isopropyl group
Presence of isopropyl group is important for 2-aminopyridinium-substituted rosmaricine derivatives to have hydrophobic interaction with hydrophobic residues of the H1 binding site (Figure 6(a) and (b)). This interaction is suggested to stabilise the previously discussed salt bridge interaction with Asp103 and to prevent flipping in the binding conformation.
3.3.4 Carbonyl group
Carbonyl group is a common characteristic found in rosmaricine derivatives. The electronegative oxygen of Figure 5. Modelling structures of H1. The binding sites are
indicated in green (colour online).
Table 1. The docking results of nature compounds. Only top 20 compounds of the database are shown here.
Name DS PLP1 PLP2 PMF Glycyrrhizic acid 94.86 55.27 57.91 107.59 Cynarin 93.52 54.20 62.49 68.70 Rosmarinic acid 92.10 71.65 76.59 42.55 Ergotamine 91.27 52.73 52.63 76.26 Tangshenoside I 87.77 64.95 78.17 64.28 Amentoflavone 86.28 56.85 61.44 95.28 Digallic acid 85.08 60.46 58.72 52.13 Glutinic acid 84.01 38.45 41.30 67.78 Eugeniin 83.74 62.25 70.18 68.99 Xylopine 79.59 30.64 32.89 59.54 Picrocrocinic acid 79.16 37.20 36.88 62.62 Rosmaricine 79.09 38.12 36.51 71.28 3-O-feruloylquinic acid 79.07 73.57 59.51 55.08 Procyanidin C1 79.06 46.63 51.98 74.49 Songbeisine 78.65 30.74 32.56 60.77 Galloyl-oxypaeoniflorin 78.32 81.94 88.67 82.60 Heliotrine 78.17 28.52 26.83 50.39 Schaftoside 77.57 62.71 63.10 77.72 Cholic acid 76.65 67.88 67.88 54.08 Rutin 74.47 80.21 87.42 76.89 NAG* 54.82 62.68 65.35 73.39
Notes: DS, dock score; PLP, piecewise linear potential; PMF, potential of mean force and NAG*, control set.
Table 2. The docking results of top 10 derivatives.
Name DS PLP1 PLP2 PMF Xylopine_2 144.18 37.33 40.03 45.62 Rosmaricine_14 138.92 31.03 31.41 82.25 Rosmaricine_15 135.67 37.86 37.50 99.24 Rosmaricine_5 97.92 79.98 84.81 93.56 Rosmaricine_16 96.53 75.60 78.85 88.38 Rosmaricine_23 95.85 69.71 67.90 91.22 Rosmaricine_12 95.52 38.66 37.16 81.00 Rosmaricine_6 95.43 41.85 39.70 81.50 Rosmaricine_21 95.23 42.88 41.21 79.77 Rosmaricine_11 94.73 74.06 76.23 91.65 NAG* 54.82 62.68 65.35 73.39
Notes: DS, dock score; PLP, piecewise linear potential; PMF, potential of mean force and NAG*, control set.
T.-T. Chang et al. 364
Table 3. The scaffold of original compounds and top 10 derivatives. Name Derivatives Xylopine Xylopine_2 Rosmaricine_14 Rosmaricine_15 Rosmaricine_5 Rosmaricine Rosmaricine_16 Rosmaricine_23 Rosmaricine_12 Rosmaricine_6 Rosmaricine_11 Rosmaricine_21 H2+ N O O O H2+ N O O O H + N NH 2 O O OH OH +H 3N NH+ NH2 O O OH OH +H 3N NH+ NH2 O O OH OH +H 3N O O OH OH OH OH +H 3N O O OH OH +H 3N OH O O OH OH +H 3N +H3N HO O O OH OH N H O O OH OH +H 3N N H O O OH OH +H 3N O O OH OH +H 3N OH
carbonyl could act as a hydrogen bond acceptor in forming hydrogen bond interaction. The presence of carbonyl group in Rosmaricine_14 and Rosmaricine_15 allows with hydrogen bond interaction the Lys71 of H1.
4. Conclusion
Our docking and de novo design give 10 candidate compounds that bind to key residues, as found in the
previous literature and as well as non-mutated residue in H1 binding site. Of the four key features obtained from structural analysis, the addition of 2-aminopyridinium has positive contribution in the binding affinity of xylopine and rosmaricine to H1. We hope that these results can have positive contribution to H1 inhibitor design and that the compounds suggested in this study can be further developed into anti-influenza compounds.
Figure 6. Docking pose of top 10 derivatives in H1 – (a) Xylopine_2, (b) Rosmaricine_14 (purple) and Rosmaricine_15 (green), (c) Rosmaricine_6 (purple) and Rosmaricine_11 (yellow), (d) Rosmaricine_16 (yellow) and Rosmaricine_21 (green), (e) Rosmaricine_5 (green), Rosmaricine_12 (yellow) and Rosmaricine_23 (purple) (colour online).
T.-T. Chang et al. 366
Acknowledgements
The research was supported by grants from the National Science Council of Taiwan (NSC 99-2221-E-039-013-), China Medical University (CMU98-TCM, CMU99-S-02) and Asia University (CMU98-ASIA-09). This study was also supported in part by the Taiwan Department of Health Clinical Trial and Research Center of Excellence (DOH99-TD-B-111-004) and the Taiwan Depart-ment of Health Cancer Research Center of Excellence (DOH99-TD-C-111-005). We are grateful to the National Center of High-performance Computing for computer time and facilities.
References
[1] A. Sharadadevi and R. Nagaraj, A molecular dynamics study of human defensins HBD-1 and HNP-3 in water, J. Biomol. Struct. Dyn. 27 (2010), pp. 541 – 550.
[2] T.C. Ramalho, M.S. Caetano, E.F.F. da Cunha, T.C.S. Souza, and M.V.J. Rocha, Construction and assessment of reaction models of class I EPSP synthase: Molecular docking and density functional theoretical calculations, J. Biomol. Struct. Dyn. 27 (2009), pp. 195 – 207.
[3] C.D. Yoo, S.C. Kim, and S.H. Lee, Molecular dynamics simulation study of probe diffusion in liquid n-alkanes, Mol. Simul. 35 (2009), pp. 241 – 247.
[4] D.X. Li, G.L. Chen, B.L. Liu, and Y.S. Liu, Molecular simulation of -cyclodextrin inclusion complex with 2-phenylethyl alcohol, Mol. Simul. 35 (2009), pp. 199 – 204.
[5] F. Luan, H.T. Liu, Y. Gao, and X.Y. Zhang, QSPR model to predict the thermal stabilities of second-order nonlinear optical (NLO) chromophore molecules, Mol. Simul. 35 (2009), pp. 248 – 257. [6] J.H. Jing, G.Z. Liang, H. Mei, S.Y. Xiao, Z.N. Xia, and Z.L. Li,
Quantitative structure – mobility relationship studies of dipeptides in capillary zone electrophoresis using three-dimensional holographic vector of atomic interaction field, Mol. Simul. 35 (2009), pp. 263 – 269.
[7] P.K. Cheng, T.W. Leung, E.C. Ho, P.C. Leung, A.Y. Ng, M.Y. Lai, and W.W. Lim, Oseltamivir- and amantadine-resistant influenza viruses A (H1N1), Emerg. Infect. Dis. 15 (2009), pp. 966 – 968. [8] L. Guo, R.J. Garten, A.S. Foust, W.M. Sessions, M.
Okomo-Adhiambo, L.V. Gubareva, A.I. Klimov, and X. Xu, Rapid identification of oseltamivir-resistant influenza A(H1N1) viruses with H274Y mutation by RT-PCR/restriction fragment length polymorphism assay, Antiviral Res. 82 (2009), pp. 29 – 33. [9] A. Moscona, Global transmission of oseltamivir-resistant influenza,
New Engl. J. Med. 360 (2009), pp. 953 – 956.
[10] E. De Clercq, Antiviral agents active against influenza A viruses, Nat. Rev. Drug Discov. 5 (2006), pp. 1015 – 1025.
[11] C.U. Kim, W. Lew, M.A. Williams, H. Liu, L. Zhang, S. Swaminathan, N. Bischofberger, M.S. Chen, D.B. Mendel, C.Y. Tai, W.G. Laver, and R.C. Stevens, Influenza neuraminidase inhibitors possessing a novel hydrophobic interaction in the enzyme
active site: Design, synthesis, and structural analysis of carbocyclic sialic acid analogues with potent anti-influenza activity, J. Am. Chem. Soc. 119 (1997), pp. 681 – 690.
[12] M. von Itzstein, W.Y. Wu, G.B. Kok, M.S. Pegg, J.C. Dyason, B. Jin, T. Van Phan, M.L. Smythe, H.F. White, S.W. Oliver, P.M. Colman, J.N. Varghese, D.M. Ryan, J.M. Woods, R.C. Bethell, V.J. Hotham, J.M. Cameron, C.R. Penn, Rational design of potent sialidase-based inhibitors of influenza virus replication, Nature 363 (1993), pp. 418 – 423.
[13] J.S. Oxford, Influenza A pandemics of the 20th century with special reference to 1918: Virology, pathology and epidemiology, Rev. Med. Virol. 10 (2000), pp. 119 – 133.
[14] R. Salomon and R.G. Webster, The influenza virus enigma, Cell 136 (2009), pp. 402 – 410.
[15] N.J. McDonald, C.B. Smith, and N.J. Cox, Antigenic drift in the evolution of H1N1 influenza A viruses resulting from deletion of a single amino acid in the haemagglutinin gene, J. Gen. Virol. 88 (2007), pp. 3209 – 3213.
[16] M.F. Boni, Vaccination and antigenic drift in influenza, Vaccine 26 (2008), pp. C8 – C14.
[17] K.L. Hartshorn, L.S. Liou, M.R. White, M.M. Kazhdan, J.L. Tauber, and A.I. Tauber, Neutrophil deactivation by influenza A virus. Role of hemagglutinin binding to specific sialic acid-bearing cellular proteins, J. Immunol. 154 (1995), pp. 3952 – 3960.
[18] K.G. Nicholson, F.Y. Aoki, A.D. Osterhaus, S. Trottier, O. Carewicz, C.H. Mercier, A. Rode, N. Kinnersley, and P. Ward, Efficacy and safety of oseltamivir in treatment of acute influenza: A randomised controlled trial. Neuraminidase Inhibitor Flu Treat-ment Investigator Group, Lancet 355 (2000), pp. 1845 – 1850. [19] H. Chen, C.L. Cheung, H. Tai, P. Zhao, J.F. Chan, V.C. Cheng,
K.H. Chan, and K.Y. Yuen, Oseltamivir-resistant influenza A pandemic (H1N1) 2009 virus, Hong Kong, China, Emerg. Infect. Dis. 15 (2009), pp. 1970 – 1972.
[20] A.M. Al-Mekhnaqi, M.S. Mayeed, and G.M. Newaz, Prediction of protein conformation in water and on surfaces by Monte Carlo simulations using united-atom method, Mol. Simul. 35 (2009), pp. 292 – 300.
[21] M.L. Mihajlovic and P.M. Mitrasinovic, Applications of the ArgusLab4/AScore protocol in the structure-based binding affinity prediction of various inhibitors of group-1 and group-2 influenza virus neuraminidases (NAs), Mol. Simul. 35 (2009), pp. 311 – 324. [22] C.Y. Chen, Virtual screening and drug design for PDE-5 receptor from traditional Chinese medicine database, J. Biomol. Struct. Dyn. 27 (2010), pp. 627 – 640.
[23] C.Y. Chen, Y.H. Chang, D.T. Bau, H.J. Huang, F.J. Tsai, C.H. Tsai, and C.Y.C. Chen, Ligand-based dual target drug design for H1N1: Swine flu – A preliminary first study, J. Biomol. Struct. Dyn. 27 (2009), pp. 171 – 178.
[24] H.J. Huang, K.J. Lee, H.W. Yu, C.Y. Chen, C.H. Hsu, H.Y. Chen, F.J. Tsai, and C.Y.C. Chen, Structure-based and ligand-based drug design for HER 2 receptor, J. Biomol. Struct. Dyn. 28 (2010), pp. 23 – 37.
[25] H.J. Huang, K.J. Lee, H.W. Yu, H.Y. Chen, F.J. Tsai, and C.Y. Chen, A novel strategy for designing the selective PPAR agonist by the ‘sum of activity’ model, J. Biomol. Struct. Dyn. 28 (2010), pp. 187 – 200.
[26] C.Y.C. Chen, Weighted equation and rules – a novel concept for evaluating protein – ligand interaction, J. Biomol. Struct. Dyn. 27 (2009), pp. 271 – 282.
[27] C.Y.C. Chen, Discovery of novel inhibitors for c-Met by virtual screening and pharmacophore analysis, J. Chin. Inst. Chem. Eng. 39 (2008), pp. 617 – 624.
[28] C.Y.C. Chen, A novel perspective on designing the inhibitor of HER2 receptor, J. Chin. Inst. Chem. Eng. 39 (2008), pp. 291 – 299. [29] C.Y.C. Chen, Insights into the suanzaoren mechanism – From constructing the 3D structure of GABA-A receptor to its binding interaction analysis, J. Chin. Inst. Chem. Eng. 39 (2008), pp. 663 – 671.
[30] C.Y.C. Chen, Chemoinformatics and pharmacoinformatics approach for exploring the GABA-A agonist from Chinese herb suanzaoren, J. Taiwan Inst. Chem. Eng. 40 (2009), pp. 36 – 47.
Figure 7. The four points in Xylopine_2 and Rosmaricine_14 for designing inhibitors of H1.
[31] C.Y.C. Chen, Pharmacoinformatics approach for mPGES-1 in anti-inflammation by 3D-QSAR pharmacophore mapping, J. Taiwan Inst. Chem. Eng. 40 (2009), pp. 155 – 161.
[32] Y.C. Chen, The molecular dynamic simulation of zolpidem interaction with gamma aminobutyric acid type A receptor, J. Chin. Chem. Soc. 54 (2007), pp. 653 – 658.
[33] C.Y.C. Chen, De novo design of novel selective COX-2 inhibitors: From virtual screening to pharmacophore analysis, J. Taiwan Inst. Chem. Eng. 40 (2009), pp. 55 – 69.
[34] C.Y.C. Chen, G.W. Chen, and W.Y.C. Chen, Molecular simulation of HER2/neu degradation by inhibiting HSP90, J. Chin. Chem. Soc. 55 (2008), pp. 297 – 302.
[35] C.Y.C. Chen, Inhibiting the vascular smooth muscle cells proliferation by EPC and DPPC liposomes encapsulated magnolol, J. Chin. Inst. Chem. Eng. 39 (2008), pp. 407 – 411.
[36] C.Y. Chen, Y.H. Chang, D.T. Bau, H.J. Huang, F.J. Tsai, C.H. Tsai, and C.Y.C. Chen, Discovery of potent inhibitors for phosphodi-esterase 5 by virtual screening and pharmacophore analysis, Acta Pharmacol. Sin. 30 (2009), pp. 1186 – 1194.
[37] C.Y.C. Chen, Weighted equation and rules – A novel concept for evaluating protein – ligand interaction, J. Biomol. Struct. Dyn. 27 (2009), pp. 271 – 282.
[38] C.Y.C. Chen, Computational screening and design of traditional Chinese medicine (TCM) to block phosphodiesterase-5, J. Mol. Graphics Model. 28 (2009), pp. 261 – 269.
[39] Y.J. Tang, J.S. Yang, C.F. Lin, W.C. Shyu, M. Tsuzuki, C.C. Lu, Y.F. Chen, and K.C. Lai, Houttuynia cordata Thunb extract induces apoptosis through mitochondrial-dependent pathway in HT-29 human colon adenocarcinoma cells, Oncol. Rep. 22 (2009), pp. 1051– 1056. [40] C.R. Su, Y.F. Chen, M.J. Liou, H.Y. Tsai, W.S. Chang, and T.S. Wu, Anti-inflammatory activities of furanoditerpenoids and other constituents from Fibraurea tinctoria, Biorg. Med. Chem. 16 (2008), pp. 9603 – 9609.
[41] Y.H. Hsieh, F.H. Chu, Y.S. Wang, S.C. Chien, S.T. Chang, J.F. Shaw, C.Y. Chen, W.W. Hsiao, Y.H. Kuo, and S.Y. Wang, Antrocamphin A, an anti-inflammatory principal from the fruiting body of Taiwanofungus camphoratus, and its mechanisms, J. Agric. Food Chem. 58 (2010), pp. 3153 – 3158.
[42] J. Stevens, A.L. Corper, C.F. Basler, J.K. Taubenberger, P. Palese, and I.A. Wilson, Structure of the uncleaved human H1 hemagglutinin from the extinct 1918 influenza virus, Science 303 (2004), pp. 1866 – 1870.
[43] C.A. Lipinski, F. Lombardo, B.W. Dominy, and P.J. Feeney, Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings, Adv. Drug Del. Rev. 46 (2001), pp. 3 – 26.
T.-T. Chang et al. 368