R E S E A R C H
Open Access
Protein kinases modulate store-operated channels
in pulmonary artery smooth muscle cells
I-Shan Chen
1, Zen-Kong Dai
2, Donald G Welsh
3*, Ing-Jun Chen
1, Bin-Nan Wu
1*Abstract
Background: This study investigates whether protein kinase G (PKG), protein kinase A (PKA) and protein kinase C (PKC) are involved in the regulatory mechanisms of store-operated channel (SOC) in pulmonary arteries.
Methods: Pulmonary artery smooth muscle cells (PASMCs) were enzymatically dissociated from rat intralobar pulmonary arteries. Whole cell, cell-attached and inside-out patch-clamp electrophysiology were used to monitor SOCs in isolated PASMCs.
Results: Initially the Ca2+-ATPase inhibitor cyclopiazonic acid (CPA, 10μM) initiated a whole cell current that was reduced by the SOC blocker SKF-96365 (10μM). Subsequent work using both cell-attached and whole cell configurations revealed that the PKG and PKA inhibitors, KT5823 (3μM) and H-89 (10 μM), also stimulated SOC activity; this augmentation was attenuated by the SOC blockers SKF-96365 (10μM) and Ni2+(0.1 mM). Finally using the inside-out configuration, the PKC activator phorbol 12-myristate 13-acetate (PMA, 10μM) was confirmed to modestly stimulate SOC activity although this augmentation appeared to be more substantial following the application of 10μM inositol 1,4,5-triphosphate (Ins(1,4,5)P3).
Conclusions: SOC activity in PASMCs was stimulated by the inhibition of PKG and PKA and the activation of PKC. Our findings suggest that the SOC could be a substrate of these protein kinases, which therefore would regulate the intracellular concentration of calcium and pulmonary arteriopathy via SOC.
Background
Intracellular calcium ([Ca2+]i) is an important second
messenger responsible for many physiological functions including contraction, cell growth and gene expression. Many agonists increase [Ca2+]iby mobilizing
intracellu-lar Ca2+stores, such as the sarcoplasmic (SR) or endo-plasmic (ER) reticulum. To maintain Ca2+signaling, these intracellular Ca2+ stores must be refilled and as such many agonists are thought to activate specialized plasma membrane channels termed store-operated channels (SOCs) [1]. By definition, SOCs are Ca2
+
-permeable cation channels which are activated by depletion of intracellular Ca2+stores [2]. The activation of SOCs is often termed‘capacitative Ca2+entry (CCE)’, as the principal function of these Ca2+ channels is to
refill the internal stores, as if they were in essence capa-citors [3]. Inhibitors of the sarco-endoplasmic reticulum Ca2+-ATPase pump (SERCA) are the most common tools used to deplete the intracellular Ca2+ stores and consequently activate these unique channels.
It is generally accepted that SOCs play an important role in regulating smooth muscle contraction and cellu-lar proliferation in the resistance vasculature [4,5]. In a similar, although less documented manner, SOCs have also been coupled to the genesis of pulmonary vascular tone and pulmonary artery smooth muscle cell (PASMC) proliferation [6]. Given their functional importance and their role in severe pulmonary arterio-pathies, there is considerable interest in defining how SOCs are regulated in PASMCs [7]. To date, literature specific to PASMCs has prominently stressed a role for IP3, PIP2 and other lipid products in the activation of
these channels. While important, few pulmonary studies have ventured beyond these confines to electrically address other aspects of SOC regulation, including the role of protein kinase G (PKG), protein kinase A (PKA)
* Correspondence: [email protected]; [email protected] 1
Department of Pharmacology, School of Medicine, College of Medicine, Kaohsiung Medical University, Kaohsiung, Taiwan
3
Smooth Muscle Research Group and Department of Physiology and Pharmacology, University of Calgary, Calgary, Alberta, Canada Full list of author information is available at the end of the article
© 2011 Chen et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
and protein C (PKC). This is surprising given the rich nature of the research performed on smooth muscle cells derived from resistance arteries isolated from the coronary, mesenteric and hepatic circulation [4,8,9].
The main objectives of this study were to isolate and characterize a SOC current in pulmonary artery myo-cytes and determine whether protein kinases (i.e. PKG, PKA and PKC) are involved in the activation of SOCs in PASMCs. Whole cell, cell-attached and inside out patch-clamp electrophysiology were used to monitor SOCs and the effects of various agents known to modu-late protein kinases were recorded. Like smooth muscle cells isolated from the coronary, mesenteric and hepatic circulation, SOCs in PASMCs were stimulated by PKG and PKA inhibition and PKC activation. The functional significance of these findings is discussed.
Materials and methods
Animal procedures and tissue preparations
All procedures and protocols were approved by the Ani-mal Care and Use Committee at Kaohsiung Medical University. Briefly, female Sprague-Dawley rats (250-350 g) were sacrificed with an overdose of urethane (1.25 g/ kg) via intraperitoneal injection. Lungs were carefully removed and placed in cold phosphate-buffered saline containing (in mM): 122 NaCl, 1 MgCl2, 0.5 KH2PO4,
10 HEPES, 5 KCl, 0.5 NaH2PO4, 11 Glucose, 0.1 EGTA,
0.1 CaCl2, with pH adjusted to 7.4 with NaOH.
Intralo-bar resistance pulmonary arteries (internal diameter 300-400μm) were dissected free of the surrounding tis-sue and cut into 1 mm segments.
Preparation of isolated pulmonary artery myocytes Pulmonary artery smooth muscle cells (PASMCs) from rat intralobar pulmonary arteries were enzymatically isolated as follows. Arterial segments were placed in a warm (37°C) cell isolation medium containing (in mM) 122 NaCl, 1 MgCl2, 0.5 KH2PO4, 10 HEPES, 5 KCl, 0.5
NaH2PO4, 11 Glucose, 0.1 EGTA (pH 7.4, NaOH) for
20 min. After this equilibration step, arterial segments were initially incubated (37°C) in 1 mg ml-1 papain and 0.85 mg ml-1dithioerythritol for 20-25 min. After enzyme treatment, the tissue was washed three times in ice-cold isolation medium and triturated with a fire-polished pipette to release the myocytes. Cells were stored in ice-cold isolation medium for use on the same day.
Patch-clamp electrophysiology
SOC currents in PASMCs were recorded in voltage-clamp mode using whole cell, cell-attached and inside-out configurations [10]. When employing whole cell patch-clamp electrophysiology, PASMCs were placed in a recording dish and perfused with a bath solution
containing (in mM): 120 sodium methanesulfonate, 20 Ca(OH)2, 0.5 3,4-diaminopyridine, 10 HEPES and 10
glucose (pH 7.4, HCl). A recording electrode pulled from borosilicate glass (resistance, 4-7 MΩ for whole cell recordings; 8-12 MΩ for cell-attached and inside-out patches) was coated with sticky wax to reduce capa-citance [11,12] and backfilled with pipette solution con-taining (in mM): 138 CsOH, 2.5 EGTA, 1 Ca(OH)2(free
internal [Ca2+] ~100 nM as calculated using EQCAL software), 10 HEPES and 2 Na2ATP (pH 7.2, HCl). This
pipette was gently lowered onto a PASMC, negative pressure was briefly applied to rupture the membrane and a gigaohm seal was obtained. Cells were voltage clamped at 0 mV while resting membrane currents were recorded on an Axopatch 700A amplifier (Axon Instru-ments, Union City, CA, USA). Cells were subsequently equilibrated for 25 min and then exposed to a series of voltage ramps (-100 mV to +100 mV, 0.2 Vs-1) or step protocols (20 mV increments from -80 to +20 mV for 200 ms). These voltage protocols were performed under resting conditions and in the presence of cyclopiazonic acid (CPA, 10 μM) ± 1-[b-(3-(4-Methoxyphenyl)pro-poxy)-4-methoxyphenethyl]-1H-imidazole HCl (SKF-96365, 10μM) [13,14], (9S,10R,12R)-2,3,9,10,11,12-hexa- hydro-10-methoxy-2,9-dimethyl-1-oxo-9,12-epoxy-1H- diindolo-[1,2,3-fg:30,20,10-kl]pyrrolo[3,4-i][1,6]benzodia-zocine-10-carboxylic acid methylester (KT5823, 3 μM) or N-[2-((p-Bromocinnamyl)amino)ethyl]-5-isoquinoli-nesulfonamide (H-89, 10μM). Whole cell currents were then filtered at 1 kHz (low-pass Bessel filter), digitized at 5 kHz and stored on a computer for subsequent ana-lysis with Clampfit 9.0. A 1 M NaCl-agar salt bridge between the bath and the Ag-AgCl reference electrode was used to minimize offset potentials [12,15]. All elec-trical recordings were performed at room temperature.
When using the inside-out and the cell-attached patch clamp configurations to monitor single channel activity, recording pipettes were backfilled with a solution con-taining (in mM): 126 CsCl, 10 HEPES, 11 Glucose, 1.5 CaCl2, 10 TEA, 5 4-AP, 0.0002 Iberiotoxin, 0.1 DIDS,
0.1 Niflumic acid and 0.005 Nifedipine (pH 7.2, NaOH). The bath solution for the inside-out configuration con-tained (in mM): 18 CsCl, 108 C2H3CsO2, 1.2 MgCl2, 10
HEPES, 11 Glucose, 1 BAPTA, 0.48 CaCl2, 1 Na2ATP
and 0.2 NaGTP (pH 7.2, Tris). The bath solution for the cell-attached configuration contained (in mM): 126 KCl, 1.5 CaCl2, 10 HEPES, 11 Glucose and 0.01 Nifedipine
(pH 7.2, Tris). Following a 25 min equilibration period, single channel activity in excised patches was recorded at -80 mV, filtered at 100 Hz and digitized at 50 kHz. These recordings were collected under resting condi-tions and in the presence of phorbol 12-myristate 13-acetate (PMA, 10 μM), inositol-1,4,5-triphosphate (Ins (1,4,5)P3, 10μM), KT5823 (3 μM) and H-89 (10 μM).
Data analysis and statistics
Whole cell SOC currents were analyzed from the base-line-to-peak amplitude within 50 ms. Single SOC cur-rent amplitudes were calculated from idealized traces of at least 60 s in duration using the 50% threshold method and analyzed using Clampfit 9.0 as previously described [8]. For single channel analysis, SOC activity (NPo) was determined from continuous gap-free data
using Clampfit 9.0. The NPo was calculated from the
following equation: NPo= (Σtii)/T, where i is the
num-ber of channels open, tiis the open time for each level
i and T is the total time of analysis. Data are expressed as means ± SE, n indicating the number of cells. Repeated measures analysis of variance (ANOVA) compared values at a given voltage. When appropriate, a Tukey-Kramer pairwise comparison was used for post hoc analysis. ANOVA followed by Dunnett’s test was performed to statistically compare the open prob-ability of SOCs. P ≤ 0.05 was considered statistically significant.
Chemicals
Buffer reagents, papain, dithioerythritol, H-89, KT5823, PMA, CPA, Ins(1,4,5)P3,NiCl2 and SKF-96365 were
obtained from Sigma-Aldrich Chemical Co. (St Louis, MO, USA). All drugs and reagents were dissolved in distilled water unless otherwise noted. CPA, PMA and KT5823 were dissolved in dimethylsulphoxide at 10 mM. Serial dilutions were made in phosphate-buffered solution to a final solvent concentration of≤0.01%. Results
CPA evoked whole cell currents in rat PASMCs
Our investigation of SOCs in PASMCs first began by monitoring the whole cell currents evoked by CPA. This assessment involved the application of voltage ramps (-100 mV to +100 mV, 0.2 Vs-1) every 30 s from a holding potential of 0 mV in order to inactivate vol-tage-dependent Na+ and Ca2+ channels. The recording solutions dictate that inward currents at negative poten-tials should be the result of Ca2+ and Na+ influx, while outward current at positive potentials should be puta-tively generated by Cs+ efflux [13]. Figure 1 illustrates that bath application of 10 μM CPA induces a whole cell SOC current that displays modest outward rectifica-tion and which reverses at -2 ± 1 mV (n = 6). In the presence of CPA, current density at -100 mV and +100 mV peaked at -7.3 ± 1.1 pA pF-1 and 13.9 ± 1.1 pA pF
-1
(n = 6), respectively. These values were significantly greater (P < 0.01) than control (-1.6 ± 0.1 pA pF-1and 3.5 ± 0.3 pA pF-1 at -100 mV and +100 mV, respec-tively). CPA evoked whole cell currents were subse-quently attenuated by the addition of SKF-96365 (10 μM, a SOC inhibitor) to the perfusate. In the presence
of CPA and SKF-96365, current density at -100 mV and +100 mV was 4.6 ± 0.8 pA pF-1and 6.5 ± 1.1 pA pF-1, respectively.
Activation of SOCs by a PKG inhibitor
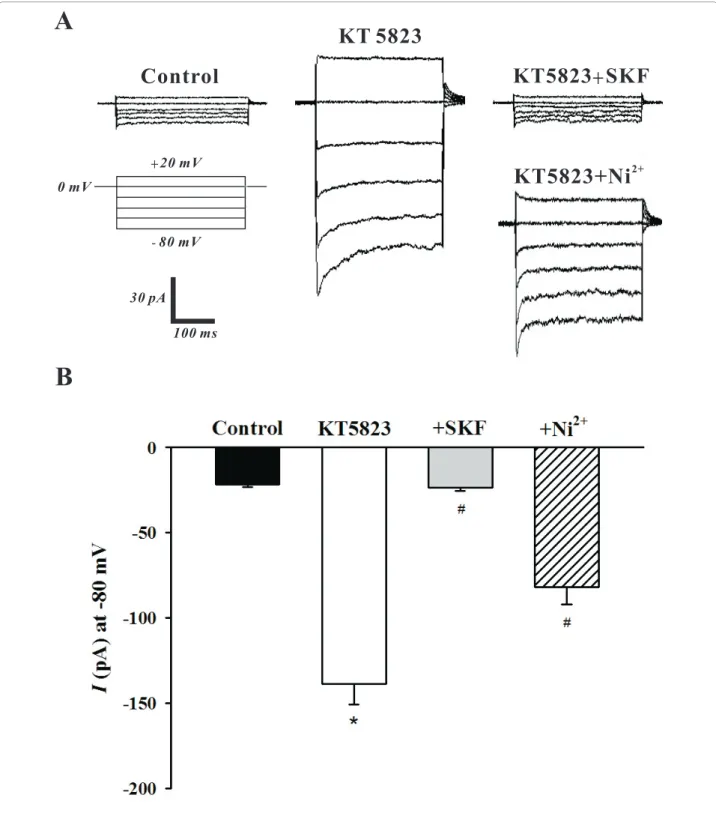
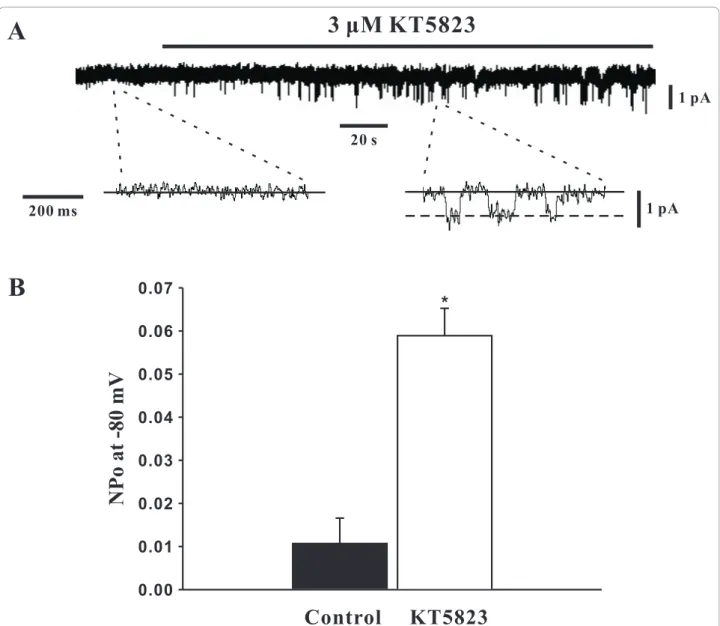
To determine whether PKG can modulate SOCs, this study first employed whole cell patch clamp electrophy-siology to monitor SOC currents in the absence of pre-sence of KT5823. The representative trace in Figure 2A illustrates that the bath application of KT5823 (3 μM) elevated SOC current and that SOC inhibitors SKF-96365 (10μM) and Ni2+(0.1 mM) [13,16] reversed and attenuated this effect respectively. As shown in Figure 2B, the mean inward current (at -80 mV) increased from -21.6 ± 1.4 pA to -138.8 ± 12.0 pA (n = 6, P < 0.01) in the presence of KT5823 and this effect was lar-gely eliminated by SKF-96365 (-23.5 ± 2.1 pA, n = 6, P < 0.01) or Ni2+(-82.0 ± 10.1 pA, n = 6, P < 0.01). With whole cell measurements suggesting that PKG likely inhibits SOCs in PASMCs, the cell-attached configura-tion was subsequently employed to assess single channel SOC activity prior to and following PKG inhibition. Representative traces in Figure 3A show on two differ-ent time scales that the bath application of KT5823 (3 μM) increases SOC activity. Summary data in Figure 3B highlights that KT5823 elevated the mean open prob-ability (NPo) from 0.0107 ± 0.0059 to 0.0589 ± 0.0063
(n = 6, P < 0.01).
Activation of SOCs by a PKA inhibitor
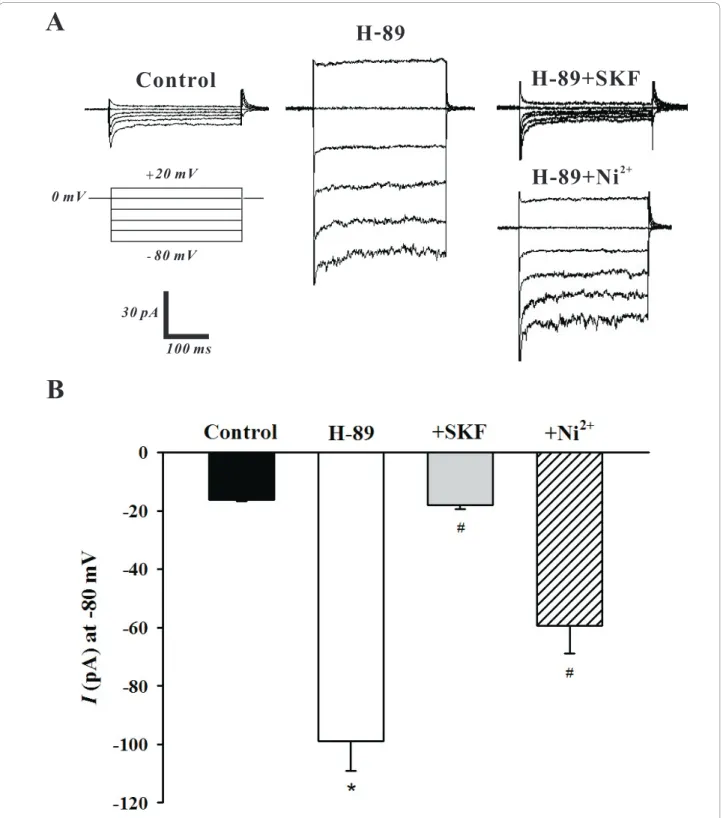
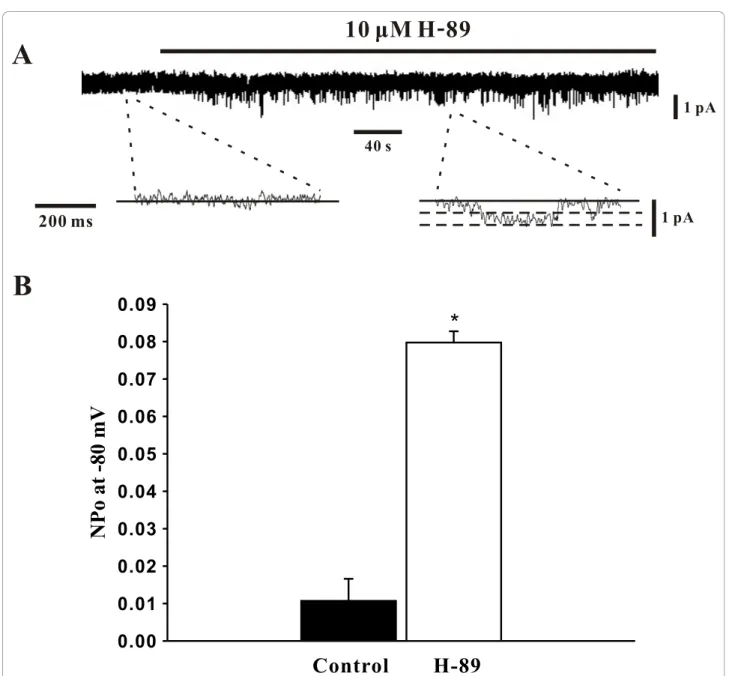
To ascertain the role of PKA in the modulation of SOCs, this study again employed whole cell patch clamp electrophysiology to monitor SOC currents in the absence and presence of H-89. The representative trace in Figure 4A shows that the addition of H-89 (10 μM) significantly increased the SOC current and that the bath application of SKF-96365 (10 μM) and Ni2+ (0.1 mM) abolished and attenuated this elevation respectively. Mean inward current (at -80 mV) plotted in Figure 4B further emphasized that the H-89 induced increase in SOC activity (-16.2 ± 0.5 pA to -98.8 ± 9.1 pA, n = 6, P < 0.01) was indeed effectively blocked by SKF-96365 (-17.5 ± 1.6 pA, n = 6, P < 0.01) or Ni2+ (-59.4 ± 9.5 pA, n = 6, P < 0.01). With whole cell mea-surements indicating that PKA likely inhibits SOCs in PASMCs, the cell-attached configuration was once again employed to monitor single channel SOC activ-ity. The representative trace in Figure 5A nicely illus-trates, at two different time scales, that the bath application of H-89 (10 μM) elevates SOC activity. Mean NPowas plotted in Figure 5B and reinforces that
single channel SOC activity rose from 0.0106 ± 0.0056 to 0.0798 ± 0.0028 (n = 6, P < 0.01) in the presence of H-89.
10 m
M SKF
10 m
M CPA
Figure 1 Cyclopiazonic acid (CPA) evokes an SOC conductance. Using whole cell patch clamp electrophysiology, PASMCs were voltage clamped and then periodically exposed to voltage ramps (-100 mV to +100 mV) in the absence and presence of 10μM CPA ± 10 μM SKF-96365 (SKF). A, original traces showing that CPA activates whole cell SOC currents in PASMCs. SKF-SKF-96365 was added to the perfusate 12 min after CPA. B, individual I-V relationships under resting conditions (a, Control), and in the presence of CPA (b) and SKF-96365 (c). C, mean I-V relationships of whole cell SOC currents. Data are means ± SE, n = 6. * denotes significant difference CPA alone.
KT5823+SKF
Figure 2 PKG inhibition by KT5823 augments SOC whole cell currents. Using whole cell patch clamp electrophysiology, PASMCs were voltage clamped and then periodically exposed to a step protocol (-80 mV to +20 mV, 20 mV increments, 300 ms duration) in the absence and presence of 3μM KT5823 ± 10 μM SKF-96365 (SKF) or 0.1 mM Ni2+. A representative trace and summary data can be found in A &B,
Activation of SOCs by a PKC activator and IP3
Finally, to study the involvement of PKC and possibly Ins(1,4,5)P3 in SOC modulation, the inside-out
config-uration was utilized to monitor these channels in the absence and presence of PMA (a PKC activator) and Ins (1,4,5)P3. As the representative traces in Figure 6A
show, bath application of PMA (10μM) induced a mod-est increase in SOC activity that was further augmented by the addition of Ins(1,4,5)P3. The amplitude and NPo
histograms in Figure 6B and 6C statistical confirm this. Of particular note was the increase in NPofrom 0.0056
± 0.0023 to 0.2475 ± 0.0261 (P < 0.05) and to 0.8949 ± 0.1573 (n = 6, P < 0.05) as paired experiments advanced from rest, to PMA addition and finally to the dual
application of PMA and Ins(1,4,5)P3. Cumulatively,
these experiments support an important role for PKC and IP3 in the activation of SOCs in PASMCs.
Discussion
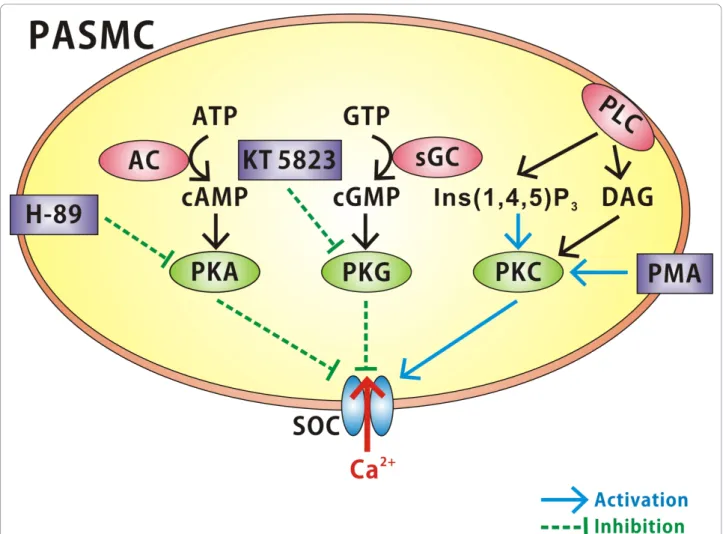
This study is the first to use patch clamp electrophysiol-ogy to investigate the role of protein kinases in the modulation of SOCs in rat PASMCs. To briefly sum-marize, this study observed that PKG and PKA elicited an inhibitory effect on SOC channels when measured at the whole cell and single channel level. Conversely, PKC appears to activate these channels and this augmenta-tion was enhanced by the addiaugmenta-tion of Ins(1,4,5)P3
(Figure 7). The findings show that SOCs in PASMCs are
NPo
at
-80
mV
0.00
0.01
0.02
0.03
0.04
0.05
0.06
0.07
Control
KT5823
*
Figure 3 PKG inhibition by KT5823 augments single channel SOC activity. Using cell attached patch clamp electrophysiology, single channel SOC activity in PASMCs was assessed (holding potential, -80 mV) in the absence and presence of 3μM KT5823. Representative traces and summary data can be found in A &B, respectively. Data are means ± SE, n = 6. * denotes significant difference from control.
H-89+SKF
Figure 4 PKA inhibition by H-89 augments SOC whole cell currents. Using whole cell patch clamp electrophysiology, PASMCs were voltage clamped and then periodically exposed to a step protocol (-80 mV to +20 mV, 20 mV increments, 300 ms duration) in the absence and presence of 10μM H-89 ± 10 μM SKF-96365 (SKF) or 0.1 mM Ni2+. A representative trace and summary data can be found in A &B, respectively. Data are means ± SE, n = 6. * and # denote significant difference from control and H-89, respectively.
diversely targeted by protein kinases. Such regulation likely plays an important role in setting pulmonary arterial tone under normal and pathophysiological conditions.
Type and character of SOCs in pulmonary artery myocytes
There are three types of SOCs described in vascular smooth muscles.ISOC1is the conductance (g = 2-3 pS)
described in rabbit portal vein myocytes. ISOC2 is the
conductance (g = 3 pS) described in aortic smooth mus-cle. ISOC3 is the conductance described in mouse
anococcygeus muscle due to its estimated conductance of less than 1 pS [4]. Single channel currents induced by CPA have also been recorded in cell-attached patches from cultured human PASMCs, which had a slope con-ductance of about 5 pS [17]. From the differences in the biophysical properties of SOCs recorded in smooth muscles, it is evident that there are different types of SOCs which probably reflect different molecular identi-ties and possibly physiological functions [4].
SOCs play an important role in controlling Ca2+ influx, arterial tone development and smooth muscle cell growth in the pulmonary vasculature [6,7]. While it
NPo
at
-80
mV
0.00
0.01
0.02
0.03
0.04
0.05
0.06
0.07
0.08
0.09
*
Control
H-89
Figure 5 PKA inhibition by H-89 augments single channel SOC activity. Using cell attached patch clamp electrophysiology, single channel SOC activity in PASMCs was assessed (holding potential, -80 mV) in the absence and presence of 10μM H-89. Representative traces and summary data can be found in A &B, respectively. Data are means ± SE, n = 6. * denotes significant difference from control.
is commonly agreed that these channels are activated by SR store depletion, their electrical properties do show some degree of variability. For example, theI-V relation-ship of SOC currents can be both linear in nature or outward rectifying [9]. Their relative permeability for monovalent and divalent cations has also been difficult to precisely define but appears to shift among published studies [17]. In this study, we report that CPA-evoked currents in rat PASMCs display, in essence, a linearI-V relationship at negative potentials and a limited degree of outward rectification at positive potentials. Although relative cation permeability was not directly ascertained, the composition of our recording solutions dictates that at negative potentials, Na+/Ca2+influx should dominate the whole cell current where as Cs+efflux will dominate at positive potentials. As such, the SOC channels noted in this investigation share some of the same biophysical characteristics as those previous isolated from rat and human pulmonary arteries [17,18].
Activation of SOCs in pulmonary artery myocytes is regulated by PKG and PKA
The guanylate cyclase-cyclic GMP-protein kinase G sig-naling pathway plays a pivotal role in several physiologi-cal processes including vascular tone development and cell cycle progression. Broadly speaking, studies have shown that activation of this NO-dependent signaling pathway characteristically attenuates SOC activity in smooth muscle [19-21]. This is exemplified by the inhi-bition of SOCs not only in A7R5 cells [22] but in native smooth muscle cells derived from the mouse anococcy-geus and the rat systemic circulation [4,23,24]. In HEK-293 cells expressing recombinant canonical transient receptor potential isoform 3 (TRPC3), protein kinase G was reported to phosphorylate and inhibit Ca2+ influx through TRPC3 channels, the latter apparently function-ing in a store-operated mode [20,25]. On the other hand, cGMP did not affect the inactivation of Ca2+ release-activated Ca2+ current (ICRAC) in RBL-1 cells
Figure 6 Effects of PKC activation and Ins(1,4,5)P3on single channel SOC activity. Using inside out patch clamp electrophysiology, single channel SOC activity in PASMCs was assessed (holding potential, -80 mV) in the absence and presence of 10μM PMA ± 10 μM Ins(1,4,5)P3. Representative traces and amplitude histograms of single channel currents (a, control; b, PMA; c, PMA + Ins(1,4,5)P3can be found in A &B, respectively. Mean NPodata was plotted in C. Data are means ± SE, n = 6. * and # denote significant difference from control and PMA, respectively.
[26], nor did membrane-permeable analogs of cGMP alter store-operated Ca2+ entry (SOCE) in Xenopus oocytes [27], pancreatic acinar cells [28], or T lympho-cytes [29]. Regulation of SOCE by cGMP is likely there-fore to be cell type specific [25]. In a similar manner, it is thought that the adenylate cyclase-cyclic AMP-PKA signaling pathway relaxes vascular smooth muscle in part due to the inhibition of SOCs [30]. Indeed, in por-tal vein myocytes, b-adrenoceptor stimulation attenuates SOC activity whereas PKA inhibitors (H-89 and KT5720) elicit the reciprocal effect [31,32]. With regard to PKA-mediated inhibition of SOCs in portal vein myo-cytes there is some similarity to data obtained in Xeno-pus oocytes [27,31]. Petersen and Berridge [27] demonstrated that low concentrations of dibutyryl cAMP (1-10 μM) inhibited Ca2+influx but higher con-centrations (1-10 mM) potentiated SOCE. It should be noted that in corneal epithelial cells PKA has also been shown to inhibit an epidermal growth
factor-evoked Ca2+ influx pathway, attributed to opening of SOCs [33]. In this study, we extended the idea that both the NO-cGMP-protein kinase G and cAMP-protein kinase A signaling pathways effectively regulate SOCs by demonstrating that PKG and PKA inhibition (KT5823 and H-89, respectively) augment single channel activity in PASMCs. From a physiological perspective, this inhi-bition would likely raise intracellular calcium ([Ca2+]i),
an event intimately tied to both contraction and prolif-eration in PASMCs. Such altprolif-erations could in turn con-tribute to the constriction and medial hypertrophy that commonly underlies pulmonary arterial hypertension (PAH).
Activation of SOCs in pulmonary artery myocytes is regulated by PKC and IP3
Past studies have strongly linked PKC activation to the augmentation of SOC activity in vascular smooth mus-cle [32]. Particularly noteworthy has been the ability of
Figure 7 Diagram highlighting protein kinase modulation of SOCs in rat PASMCs. Note that protein kinase G (PKG) and protein kinase A (PKA) inhibit SOC currents whereas protein kinase C (PKC) appears to activate SOC currents. PKC activation of SOC currents is augmented by phospholipase C (PLC) activation and the production of Ins(1,4,5)P3. AC, adenylate cyclase; sGC, soluble guanylate cyclase.
phorbol esters and 1-oleoyl-sn-glycerol (diacylglycerol analogue) to activate and PKC inhibitors to attenuate SOCs [4,8,34]. Interestingly, this PKC-induced activation of vascular SOCs appears to be augmented by the pro-duction of IP3and/or the depletion of PIP2 [2,35]. Such
observations suggest some degree of cooperation among the various signaling events activated by vasoconstrictors via G-protein coupled receptors. In canine pulmonary vein smooth muscle cells, activation or inhibition of PKC was found to have no effect on SOCE [36]; thus, there appears to be considerable diversity in the role of PKC plays in regulating this Ca2+entry pathway in dif-ferent cells [34]. Perhaps SOCE pathways differ in differ-ent blood vessels. Indeed, smooth muscle cells from mesenteric and coronary arteries have store depletion activated cation channels with distinct properties, although both are activated by PKC [8]. A wide range of properties have been reported for SOCs in different smooth muscle preparations, suggesting that multiple cation channels can be opened by store depletion [37,38]. In this study of PASMCs, we observed a similar phenomenon whereby the PMA-induced increase in SOC activity was further enhanced by the application of IP3. Generally speaking, in physiological states this
acti-vation would be more important than PKG and PKA inhibition to induce both the contraction and prolifera-tion in PASMCs, which promotes the development of severe PAH.
Limitations
It is generally agreed that SOCs are relatively difficult to isolate from other conductance channels in the plasma membrane because of a deficiency of highly selective pharmacological agents and the lack of selectively char-acteristic electrophysiological properties. A previous report [4] demonstrated that cell-attached recording may be an improved method for studying SOCs in smooth muscle, and benefits from the advantage of not disturbing the intracellular milieu. In this study, we initially showed that PKG and PKA inhibition and PKC activation enhance the SOC currents in freshly dispersed rat PASMCs. Nevertheless, over the time course of typi-cal experiments, we cannot exclude the possibility that an induced current of such amplitude is due to a change in‘leak’ which might occur, for example, due to myocyte contraction. Accordingly, to further confirm our find-ings, cell-attached and inside-out configurations were used to measure single channel SOC activity with and without PKG and PKA inhibitors (KT5823 and H-89) and PKC activator (PMA) and IP3. The data obtained
from single channel SOC activity was consistent with those of whole cell SOC currents. However, we still need to interpret our findings cautiously because the supposed opposite effects on SOC channels with
activators of PKG and PKA and/or inhibitors of PKC in rat PASMCs remain unresolved. Also, it is not yet clear what upstream and/or downstream signaling molecules are involved in these protein kinase pathways.
Conclusions
In summary, this study presents evidence that protein kinases play an important role in regulating SOCs in smooth muscles derived from the pulmonary artery. While McElroy et al. [34] previously used Ca2+-imaging technique to show that protein kinases can regulate SOCs in rat PASMCs, this investigation appears to be the first to use patch-clamp electrophysiology to mea-sure similar SOC regulation at the whole cell and single channel level. Given that enhanced SOC activity has been linked to the development of pulmonary arteriopa-thies, we proposed that protein kinase modulation may provide a means of attenuating the progression of these debilitating disorders.
Abbreviations [Ca2+]
i: intracellular calcium; CCE: capacitative Ca2+entry; CIF: calcium influx factor; CPA: cyclopiazonic acid; ICRAC: Ca2+release-activated Ca2+current; iPLA2: Ca2+-independent phospholipase A2; Ins(1,4,5)P3: inositol 1,4,5-triphosphate; PASMC: pulmonary artery smooth muscle cell; PKA: protein kinase A; PKC: protein kinase C; PKG: protein kinase G; PMA: phorbol 12-myristate 13-acetate; SERCA: sarco-endoplasmic reticulum Ca2+-ATPase pump; SOC: store-operated channel; SOCE: store-operated Ca2+entry; TRPC3: canonical transient receptor potential isoform 3
Acknowledgements
The authors would like to thank Suzanne E. Brett Welsh for her help reading and discussing this manuscript. This study was supported by a grant NSC97-2320-B-037-006-MY3 to Dr Bin-Nan Wu from the National Science Council, Taiwan. Dr Donald Welsh is supported by the Natural Science and Engineering Research Council of Canada.
Author details
1Department of Pharmacology, School of Medicine, College of Medicine, Kaohsiung Medical University, Kaohsiung, Taiwan.2Department of Pediatrics, School of Medicine, College of Medicine, Kaohsiung Medical University, Division of Pediatric Pulmonology and Cardiology, Kaohsiung Medical University Hospital, Kaohsiung, Taiwan.3Smooth Muscle Research Group and Department of Physiology and Pharmacology, University of Calgary, Calgary, Alberta, Canada.
Authors’ contributions
ISC performed the experiments and drafted the manuscript. ZKD and IJC provided the ideas and participated in the design and coordination of this study, and helped to draft the manuscript. DGW and BNW designed and directed the experiments, interpreted the data and polished the paper to meet the scientific content. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests. Received: 17 May 2010 Accepted: 6 January 2011 Published: 6 January 2011
References
1. Salido GM, Sago SO, Rosado JA: TRPC channels and store-operated Ca2+ entry. Biochim Biophys Acta 2008, 1793:223-230.
2. Liu M, Albert AP, Large WA: Facilitatory effect of Ins(1,4,5)P3on store-operated Ca2+-permeable cation channels in rabbit portal vein myocytes. J Physiol 2005, 566:161-171.
3. Putney JW Jr: A model for receptor-regulated calcium entry. Cell Calcium 1986, 7:1-12.
4. Albert AP, Large WA: Store-operated Ca2+-permeable non-selective cation channels in smooth muscle cells. Cell Calcium 2003, 33:345-356. 5. Smani T, Zakharov SI, Csutora P, Leno E, Trepakova ES, Bolotina VM: A novel
mechanism for the store-operated calcium influx pathway. Nat Cell Biol 2004, 2:113-120.
6. Landsberg JW, Yuan JX: Calcium and TRP channels in pulmonary vascular smooth muscle cell proliferation. News Physiol Sci 2004, 19:44-50. 7. Lu W, Ran P, Zhang D, Peng G, Li B, Zhong N, Wang J: Sildenafil inhibits
chronically hypoxic upregulation of canonical transient receptor potential expression in rat pulmonary arterial smooth muscle. Am J Physiol Cell Physiol 2010, 298:C114-C123.
8. Saleh SN, Albert AP, Peppiatt CM, Large WA: Diverse properties of store-operated TRPC channels activated by protein kinase C in vascular myocytes. J Physiol 2008, 586:2463-2476.
9. Ng LC, Gurney AM: Store-operated channels mediate Ca2+influx and contraction in rat pulmonary artery. Circ Res 2001, 89:923-929. 10. Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ: Improved
patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch 1981, 391:85-100.
11. Luykenaar KD, El-Rahman RA, Walsh MP, Welsh DG: Rho-kinase-mediated suppression of KDRcurrent in cerebral arteries requires an intact actin cytoskeleton. Am J Physiol Heart Circ Physiol 2009, 296:H917-H926. 12. Wu BN, Luykenaar KD, Brayden JE, Giles WR, Corteling RL, Wiehler WB,
Welsh DG: Hyposmotic challenge inhibits inward rectifying K+channels in cerebral arterial smooth muscle cells. Am J Physiol Heart Circ Physiol 2007, 292:H1085-H1094.
13. Yu Y, Sweeney M, Zhang S, Platoshyn O, Landsberg J, Rothman A, Yuan JX: PDGF stimulates pulmonary vascular smooth muscle cell proliferation by upregulating TRPC6 expression. Am J Physiol Cell Physiol 2003, 284: C316-C330.
14. Ng LC, McCormack MD, Airey JA, Singer CA, Keller PS, Shen XM, Hume JR: TRPC1 and STIM1 mediate capacitative Ca2+entry in mouse pulmonary arterial smooth muscle cells. J Physiol 2009, 587:2429-2442.
15. Smith PD, Brett SE, Luykenaar KD, Sandow SL, Marrelli SP, Vigmond EJ, Welsh DG: KIRchannels function as electrical amplifiers in rat vascular smooth muscle. J Physiol 2008, 586:1147-1160.
16. Wilson SM, Mason HS, Smith GD, Nicholson N, Johnston L, Janiak R, Hume JR: Comparative capacitative calcium entry mechanisms in canine pulmonary and renal arterial smooth muscle cells. J Physiol 2002, 543:917-931.
17. Golovina VA, Platoshyn O, Bailey CL, Wang J, Limsuwan A, Sweeney M, Rubin LJ, Yuan JX: Upregulated TRP and enhanced capacitative Ca2+ entry in human pulmonary artery myocytes during proliferation. Am J Physiol Heart Circ Physiol 2001, 280:H746-H755.
18. Guibert C, Marthan R, Savineau JP: 5-HT induces an arachidonic acid-sensitive calcium influx in rat small intrapulmonary artery. Am J Physiol Lung Cell Mol Physiol 2004, 286:L1228-L1236.
19. Moneer Z, Dyer JL, Taylor CW: Nitric oxide co-ordinates the activities of the capacitative and non-capacitative Ca2+-entry pathways regulated by vasopressin. Biochem J 2003, 370:439-448.
20. Kwan HY, Huang Y, Yao X: Regulation of canonical transient receptor potential isoform 3 (TRPC3) channel by protein kinase G. Proc Natl Acad Sci USA 2004, 101:2625-2630.
21. Yao X: TRPC, cGMP-dependent protein kinases and cytosolic Ca2+. Handbook Exp Pharmacol 2007, 179:527-540.
22. Moneer Z, Taylor CW: Reciprocal regulation of capacitative and non-capacitative Ca2+entry in A7r5 vascular smooth muscle cells: only the letter operates receptor activation. Biochem J 2002, 362:13-21. 23. Wayman CP, McFadzean I, Gibson A, Tucker JF: Two distinct membrane
currents activated by cyclopiazonic acid-induced calcium store depletion in single smooth muscle cells of the mouse anococcygeus. Br J Pharmacol 1996, 117:566-572.
24. Inoue R, Jian Z, Kawarabayashi Y: Mechanosensitive TRP channels in cardiovascular pathophysiology. Pharmacol Ther 2009, 123:371-385. 25. Parekh AB, Putney JW Jr: Store-operated calcium channels. Physiol Rev
2005, 85:757-810.
26. Parekh AB, Penner R: Depletion-activated calcium current is inhibited by protein kinase in RBL-2H3 cells. Proc Natl Acad Sci USA 1995,
92:7907-7911.
27. Petersen CC, Berridge MJ: G-protein regulation of capacitative calcium entry may be mediated by protein kinases A and C in Xenopus oocytes. Biochem J 1995, 307:663-668.
28. Gilon P, Obie JF, Bian X, Bird GS, Putney JW Jr: Role of cyclic GMP in the control of capacitative Ca2+entry in rat pancreatic acinar cells. Biochem J 1995, 311:649-656.
29. Bian X, Bird GS, Putney JW Jr: cGMP is not required for capacitative Ca2+ entry in Jurkat T-lymphocytes. Cell Calcium 1996, 19:351-354.
30. Smani T, Domínguez-Rodríguez A, Hmadcha A, Calderón-Sánchez E, Horrillo-Ledesma A, Ordóñez A: Role of Ca2+-independent phospholipase A2and store-operated pathway in urocortin-induced vasodilatation of rat coronary artery. Circ Res 2007, 101:1194-1203.
31. Liu M, Large WA, Albert AP: Stimulation ofβ-adrenoceptors inhibits store-operated channel currents via a cAMP-dependent protein kinase mechanism in rabbit portal vein myocytes. J Physiol 2005, 562:395-406. 32. Albert AP, Saleh SN, Peppiatt CM, Large WA: Multiple activation
mechanisms of store-operated TRPC channels in smooth muscle cells. J Physiol 2007, 583:25-36.
33. Yang H, Sun X, Wang Z, Ning G, Zhang F, Kong J, Lu L, Reinach PS: EGF stimulates growth by enhancing capacitative calcium entry in corneal epithelial cell. J Memb Biol 2003, 194:47-58.
34. McElroy SP, Drummond RM, Gurney AM: Regulation of store-operated Ca2 +entry in pulmonary artery smooth muscle cells. Cell Calcium 2009, 46:99-106.
35. Saleh SN, Albert AP, Large WA: Activation of native TRPC1/C5/C6 channels by endothelin-1 is mediated by both PIP(3) and PIP(2) in rabbit coronary artery myocytes. J Physiol 2009, 587:5361-5375.
36. Shimizu S, Ding X, Murray PA: Intravenous anesthetics inhibit capacitative calcium entry in pulmonary venous smooth muscle cells. Anesthesiology 2006, 104:791-797.
37. Beech DJ, Muraki K, Flemming R: Non-selective cationic channels of smooth muscle and the mammalian homologues of Drosophila TRP. J Physiol 2004, 559:685-706.
38. Leung FP, Yung LM, Yao X, Laher I, Huang Y: Store-operated calcium entry in vascular smooth muscle. Br J Pharmacol 1996, 153:846-857.
doi:10.1186/1423-0127-18-2
Cite this article as: Chen et al.: Protein kinases modulate store-operated channels in pulmonary artery smooth muscle cells. Journal of Biomedical Science 2011 18:2.
Submit your next manuscript to BioMed Central and take full advantage of:
• Convenient online submission • Thorough peer review
• No space constraints or color figure charges • Immediate publication on acceptance
• Inclusion in PubMed, CAS, Scopus and Google Scholar • Research which is freely available for redistribution
Submit your manuscript at www.biomedcentral.com/submit