Gallic acid ameliorated impaired glucose and lipid homeostasis in high fat diet-induced NAFLD mice.
Jung Chaoa, Teh-Ia Huoab, Hao-Yuan Chengc, Jen-Chieh Tsaide, Jiunn-Wang Liaof,
Meng-Shiou Leeg, Xue-Mei Qinh,Ming-Tsuen Hsiehg, Li-Heng Paoij*, Wen-Huang
Pengg*
a Institute of Pharmacology, College of Medicine, National Yang-Ming University, Taipei, Taiwan
b Department of Oncology and Internal Medicine, National Taiwan University Hospital, Taipei, Taiwan.
c Department of Nursing, Chung Jen College of Nursing, Health Sciences and Management, Chia-Yi, Taiwan
d Department of Health and Nutrition Biotechnology, College of Health Science, Asia University, Taichung, Taiwan
e Jen-Teh Junior College of Medicine, Nursing and Management, Miaoli, Taiwan f Graduate Institute of Veterinary Pathology, National Chung Hsing University,
Taichung, Taiwan
g Department of Chinese Pharmaceutical Sciences and Chinese Medicine Resources, College of Pharmacy, China Medical University, Taichung, Taiwan
h Modern Research Center for Traditional Chinese Medicine of Shanxi University, Taiyuan, China
i Research Center for Industry of Human Ecology, Chang Gung University of Science and Technology, Taoyuan, Taiwan
j School of Pharmacy, National Defense Medical Center, Taipei, Taiwan
* Corresponding authors: Dr. Wen-Huang Peng, [email protected] and Dr. Li-Heng Pao
Running title: Metabolomic profiles of NAFLD and GA intervention
Abbreviations: GA, Gallic acid; NAFLD, nonalcoholic fatty liver disease; HFD, high
fat diet; NASH, nonalcoholic steatohepatitis; NOAEL, no-observed-adverse-effect
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31
level; MS, mass spectrometry; NMR, nuclear magnetic resonance; OPLS-DA,
orthogonal partial least squares discriminant analysis; MCA, multi-criteria
assessment; VIP, variable importance in projection; TSP, trimethylsilane propionic
acid sodium salt; AST, aspartate aminotransferase; ALT, alanine aminotransferase;
HDL, high density lipoprotein-cholesterol; TG, triglycerides; TCHO, total
cholesterol; CPMG, Carr-Purcell-Meiboom-Gill; NOESY, nuclear overhauser
enhancement spectroscopy; BPP-LED, bipolar-pair longitudinal-eddy-current,
FWHM, full width at half maximum; FIDs, free induction decays; NS, number of
scans; DS, dummy scans; FT, Fourier transformation; PUFA, polyunsaturated fatty
acids; MUFA, monounsaturated fatty acids; UFA, unsaturated fatty acids; PCA,
principal components analysis; PLS-DA, partial least squares discriminant analysis;
IR, insulin resistance; BCAAs, branched-chain amino acids; TMAO, trimethylamine
N-oxide; TCA cycle, tricarboxylic acid-cycle; DMA, dimethylamine; TMA,
trimethylamine; SV coefficient plot, S-plot combined with the VIP plot and color
coefficient scale bar; PC1, contribution of first components; p(corr), Pearson's
correlation coefficient values; PPAR, peroxisome proliferator-activated receptor,
NAD+, nicotinamide adenine dinucleotide; NADP+, nicotinamide adenine
dinucleotide phosphate. 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50
Abstract
Gallic acid (GA), a naturally abundant plant phenolic compound in vegetables
and fruits, has been shown to have potent anti-oxidative and anti-obesity activity.
However, the effects of GA on nonalcoholic fatty liver disease (NAFLD) are poorly
understood. In this study, we investigated the beneficial effects of GA administration
on nutritional hepatosteatosis model by a more “holistic view” approach, namely 1H
NMR-based metabolomics, in order to prove efficacy and to obtain information that
might lead to a better understanding of the mode of action of GA. Male C57BL/6
mice were placed for 16 weeks on either a normal chow diet, a high fat diet (HFD,
60%), or a high fat diet supplemented with GA (50 and 100 mg/kg/day, orally). Liver
histopathology and serum biochemical examinations indicated that the daily
administration of GA protects against hepatic steatosis, obesity, hypercholesterolemia,
and insulin resistance among the HFD-induced NAFLD mice. In addition, partial least
squares discriminant analysis scores plots demonstrated that the cluster of HFD fed
mice is clearly separated from the normal group mice plots, indicating that the
metabolic characteristics of these two groups are distinctively different. Specifically,
the GA-treated mice are located closer to the normal group of mice, indicating that the
HFD-induced disturbances to the metabolic profile were partially reversed by GA
treatment. Our results show that the hepatoprotective effect of GA occurs in part
51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69
through a reversing of the HFD caused disturbances to a range of metabolic pathways,
including lipid metabolism, glucose metabolism (glycolysis and gluconeogenesis),
amino acids metabolism, choline metabolism and gut-microbiota-associated
metabolism. Taken together, this study suggested that a 1H NMR-based metabolomics
approach is a useful platform for natural product funtional evaluation. The selected
metabolites are potentially useful as preventive action biomarkers and could also be
used to help our further understanding of the effect of GA in hepatosteatosis mice.
Keywords: gallic acid (GA), nonalcoholic fatty liver disease (NAFLD),
metabolomics, 1H NMR, metabolic disease 70 71 72 73 74 75 76 77 78 79
Introduction
Nonalcoholic fatty liver disease (NAFLD) is a slowly progressive affliction
that includes a wide spectrum of liver diseases, ranging from simple fatty liver to
nonalcoholic steatohepatitis (NASH); these may eventually progress to liver cirrhosis,
and hepatocellular carcinoma [1]. As a primary cause of abnormal liver function tests
in Asia over the last few years, NAFLD has become an important clinical issue.
However, effective therapies for treating NAFLD have yet to be found [2] and this
has contributed to an increased use by sufferers of natural products.
Plant-derived polyphenol compounds possess a wide range of
pharmacological properties and their action has been the subject of considerable
interest in recent years. Gallic acid (GA), an endogenous plant phenol, is a naturally
abundant plant compound in vegetables, tea, grapes, berries, as well as wine [3-9].
GA have been reported to have potent free radical scavenging and anti-oxidative
activities [10,11] and therefore the study of the mechanism of action of GA has
received much attention recently. Many GA-rich plants exhibit protective effects
against liver injury [3-5]. In addition, GA seems to have a variety of different
pharmacological activities, including anti-inflammatory [9,12], anti-obesity [8,10,11],
and anti-cancer activities [13]. Furthermore, the protective effect of GA on hepatic
lipid peroxide metabolism, glycoprotein components and lipid peroxidation in the
80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98
STZ-induced diabetic rats has been reported [14]. The previous subchronic toxicology
study has suggested that GA is safe and seems to have a no-observed-adverse-effect
level (NOAEL) at doses of 119 and 128 mg/kg/day, respectively for male and female
rats [15]. Even though many reports have revealed that GA seems to play an
important role in the prevention of diabetes and metabolic disease development, direct
evidence of these effects and the mechanism underlying the action of GA on NAFLD
remain unclear.
Metabolomics is defined as the quantitative measurement of the time-related
multiparametric metabolic responses of multicellular systems to pathophysiological
stimuli or a genetic modification [16]. The metabolomics approach has demonstrated
potential in many fields, including disease diagnosis [17-19], investigations of
toxicological mechanisms [20,21], plant metabolomics [22,23], determination of the
mechanism of drug treatment and assessing the effect of nutritional intervention
[19,24-28]. The usually used analytical techniques of metabolomics can be classified
into mass spectrometry (MS)-based detection methods and nuclear magnetic
resonance (NMR)-based detection methods [29]. 1H NMR has been used as a major
analytical tool for many applications, because one of the major advantages of NMR is
that the biological fluid does not require any physical or chemical treatment before the
analysis [29]. In addition, NMR is a very useful technique for structure elucidation
99 100 101 102 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117
using various two-dimensional NMR measurements without the further fractionation
of the biological samples [22,30].
It is rational to propose that when trying to elucidate the preventive effects
and mechanisms of GA on NAFLD, the use of these results is likely to provide strong
evidence in support of, at least a part, the preventive effects on the metabolic diseases
of this functional food when used daily. The aim of this study is to investigate the
beneficial effects of GA on nutritional hepatosteatosis by 1H NMR-based
metabolomics using an animal model. The mechanisms by which GA affects the mice
were elucidated from a global perspective and used a metabolomics approach to
explore the complicated systematic changes that occur in HFD-induced nutritional
steatosis model mouse serum and urine samples. The experiment results and proposed
pathways will help to elucidate the multiple targets involved in the hepatoprotective
activities of GA.
Materials and Methods
Chemicals and Reagents
Gallic acid (98%), D2O (99.9%), and chloroform-d containing
tetramethylsilane (TMS) (99.9%) were purchased from Sigma-Aldrich (St. Louis,
MO). Trimethylsilane propionic acid sodium salt (TSP) was purchased from Merck
118 119 120 121 122 123 124 125 126 127 128 129 130 131 132 133 134 135 136
(Darmstadt, Germany).
Animals Treatment and Sample collection
Male 10-week old C57BL/6 mice were purchased from BioLASCO Taiwan
Co., Ltd. All mice were housed alone in standard cages for one week at least before
the experiments began The animals were kept at a constant temperature of 22 ± 1 °C,
relative humidity of 55 ± 5% and under a 12 h light–dark cycle (08:00 to 20:00). They
had free access to food and water. The animals were divided into three groups
(Figure S1A): (1) a normal chow diet (normal group), n=10, (2) a high fat diet (HFD
group) n=11, and (3) a high fat diet treated with GA (treatment group, high fat diet +
GA 50 and 100 mg/kg/day, orally), n=10. The high fat diet consisted of food with
60% of the calories coming from fat (5.24 kcal/g, 60% kcal from lard/soybean 9.8:1,
D12492; Research Diets, New Brunswick NJ) (Table S1). This diet has previously
been demonstrated to induce obesity in C57BL/6 mice [31]. The normal chow diet
consisted of food with 12.7 % of the calories coming from fat (4.14 kcal/g, LabDiet
5010 Rodent Diet, Richmond, IN, USA).
The normal and HFD groups of mice were gavaged with the same volume
water as the treatment group, while mice in treatment group were gavaged with water
containing GA. Mice were maintained on the treatment for 16 weeks and were then
137 138 139 140 141 142 143 144 145 146 147 148 149 150 151 152 153 154 155
sacrificed under isoflurane anesthesia after 16 hr fasting. Tissues were then rapidly
removed, immediately frozen in liquid nitrogen, and stored at ‒80°C until needed for
the metabolomics analysis. Other tissues were sampled and fixed in 10% neutral
buffered formaldehyde for histological analysis. Serum samples were collected before
the animals were sacrificed. Urine samples were collected before sacrifice and
between 18:00 p.m. and 00:00 a.m. These samples were then snap-frozen in liquid
nitrogen and stored at ‒80°C. Some urine samples were suspected to be contaminated
based on some spurious fecal sample signals observed in their 1H NMR spectra.
Therefore, these samples were excluded from the multivariate analysis.
The animals used in this study were housed and cared for in accordance with
the NIH Guide for the care and use of laboratory animals. The experimental protocol
was approved by the Animal Research Committee of National Defense Medical
Center (IACUC-11-051).
Serum biochemistry analysis
The activity levels of aspartate aminotransferase (AST) and alanine
aminotransferase (ALT), and the levels of high density lipoprotein-cholesterol (HDL),
triglycerides (TG), and total cholesterol (TCHO) were determined using an automatic
blood chemistry analyzer Dry-Chem 4000i (Fujifilm, Saitama, Japan). Hemolysis was
156 157 158 159 160 161 162 163 164 165 166 167 168 169 170 171 172 173 174
found to have occurred in some blood samples, which is known to interfere the AST and ALT measurement using blood samples. As a result the number of mice in the GA treatment group for AST and ALT analysis is nine. During insulin analysis, two serum samples were found to be too small to be analyzed. As a result number of
animals in GAH and GAL groups is eight. All procedures completely complied with
the manufacturer's guidelines. Blood glucose concentrations were determined by a
blood glucose meter (Accu-Check® Advantage, Roche). Serum insulin levels were
measured by ELISA kit (Linco Research, St. Charles, MO).
Histological analysis of liver
For histopathological examination, the liver tissue samples were fixed in 10%
neutral buffered formaldehyde, embedded in paraffin, and sectioned (4 μm). Some
sections were stained with hematoxylin and eosin, while others were processed for
immunohistochemistry staining.
Sample preparation and NMR analysis of serum, urine and tissue samples
Sample preparation of the serum, urine and tissue samples for the
metabolomics analysis were slightly modified from those previously described [32].
Serum and urine samples were thawed at room temperature, and then centrifuged at
13,000 rpm for 15 minutes to remove insoluble material. For serum preparation, 100
175 176 177 178 179 180 181 182 183 184 185 186 187 188 189 190 191 192 193 194
µl of serum was mixed with 500 µl of 0.9% NaCl (saline) in D2O. For urine
preparation, 100 µl of urine was mixed with 300 µl D2O and 200 µl phosphate buffer
(2.885 g Na2HPO4 and 0.525 g NaH2PO4 in 100 ml D2O, 1 mM TSP). Finally, 550 µl
of each sample supernatants was placed in a 5 mm NMR tube for NMR analysis.
Liver tissue samples (about 50 mg) were extracted with 0.4285 mL of precooled
methanol−water mixture (4/2.85, v/v) using a tissue lyser. After adding 0.4 ml
chloroform to the methanol−water mixture, the solutions were separated into an upper
methanol/water phase (with polar metabolites) and a lower chloroform phase (with
lipophilic compounds). The chloroform phase solution was collected after
centrifugation (1000 × g, 4°C, 10 min) and chloroform was then removed in vacuo.
The lipophilic extract was reconstituted using 600 µL of chloroform-d containing
TMS. Then 550 µl of each sample was transfered to a 5 mm NMR tube for NMR
analysis.
Analysis of the samples was performed as described previously [32] on an
AVANCE AV-600 MHz spectrometer with a cryogenic probe. The serum was
analyzed using the Carr-Purcell-Meiboom-Gill (CPMG) pulse sequence together with
the one-dimensional nuclear overhauser enhancement spectroscopy (NOESY)-presat
sequence in order to detect low molecular weight metabolites and using the
bipolar-pair longitudinal-eddy-current (BPP-LED) pulse sequence in order to detect high
195 196 197 198 199 200 201 202 203 204 205 206 207 208 209 210 211 212 213
molecular weight metabolites. The 1D J-resolved projection spectra were also used to
help identify metabolites. The urine and lipophilic tissue extract was analyzed using
the 1D NOESY-presat sequence and the 1D J-resolved projection spectrum.
All experiments were performed at 300 K. Manual shimming was performed
on each samples to reach full width at half maximum (FWHM) ≦ 10 Hz on water peak of serum sample (using normal one-pulse sequence (zg) and with a line
broadening of 0.3 hz) or ≦ 2.5 Hz on TSP peak of urine sample (using normal one-pulse sequence with water saturation (zgpr) and with a line broadening of 0.3 hz).
The 90˚ pulse length (~14.0 µs) was adjusted individually for each sample.
The free induction decays (FIDs) were acquired using 32 K data points with a spectral
width of 20 ppm, and were zero-filled to 65536 points. A relaxation delay of 2.0 s was
used. The other parameters were: number of scans (NS) = 128 and number of dummy
scans (DS) = 16 for the CPMG experiments; NS = 128 and DS = 4 for the NOESY
experiments; NS = 64 and DS = 4 for the LED-BPP experiments; and NS = 16 and
DS = 16 for the J-resolved experiments.
Data processing and analysis of NMR data
The NMR spectra were automatically phased and baseline corrected using
MestReNova software (8.0.2 Varian, Inc.). The FIDs were multiplied by an
exponential line-broadening factor of 0.3 Hz before Fourier transformation (FT). All
214 215 216 217 218 219 220 221 222 223 224 225 226 227 228 229 230 231 232 233
spectra were referenced to the CH3 resonance of lactate at δ 1.33 ppm for the spectra
obtained from plasma and to TSP at δ 0.00 ppm the spectra obtained from urine.
Selected metabolite peaks were identified by comparing the results with the published
literature (serum [33-36] and urine [33,37-39]) and using the Chenomx NMR
software suite (Version 7.5, Chenomx, Inc.)
For serum samples, each spectrum range of δ 0.04–10.0 was divided into
integrated regions of equal width (0.005 ppm), whereas the range of δ 0.05–10.0 for
urine samples was bucketed into 9500 bins (0.005 ppm). The regions containing
resonance from residual water (δ 4.500–5.000) were excluded. When examining the
urine samples, the urea peak is influenced by water presaturation and therefore the δ
5.000–6.000 region that contains the urea resonance was also excluded. The integral
values of each spectrum of serum and urine samples were normalized to a total sum of
all integrals in the spectrum in order to reduce any significant concentration
differences between samples. For the tissue samples, the integral values of each
spectrum were normalized against the weight of the wet tissue. The relative integrals
of the liver cholesterol, liver triglyceride and liver fatty acids were calculated from the
spectral regions at δ 0.670−0.695 for liver cholesterol (C18-H3), at δ 4.120−4.170 for
liver triglyceride (Glycerol (C1-Hu) and (C3-Hu)) and at δ 0.81−0.93 for the methyl
groups of all fatty acids (-CH3). The polyunsaturated fatty acids (PUFA)-to-234 235 236 237 238 239 240 241 242 243 244 245 246 247 248 249 250 251 252
monounsaturated fatty acids (MUFA) ratio was calculated from the spectral regions at
δ 5.29−5.44 for unsaturated fatty acids (UFA) (-CH=CH-), at δ 2.73−2.88 for PUFA
(-C=C-CH2-C=C-) and at δ 0.81−0.93 for the methyl groups of all fatty acids (-CH3)
[33]. The resulting datasets were then imported into SIMCA-P version 13.0
(Umetrics, Umea, Sweden), and all variables were scaled to Pareto (par) for the
multivariate statistical analysis (principal components analysis (PCA), partial least
squares discriminant analysis (PLS-DA), and orthogonal partial least squares
discriminant analysis (OPLS-DA)). The quality of the fitting model can be explained
by the appropriate R2 and Q2 values. R2 is defined as the total amount variation
explained by the model and Q2 is the indicated predictability of the model under cross
validation [33,40]. As a result of the fact that ten mice were used to form the three
different groups in this study, a cutoff value of |r| > 0.576 (r > 0.576 and r < - 0.576)
was chosen for the correlation coefficient to be significant based on a discrimination
significance of p < 0.05. MCA were performed based on the followed criteria: 1.
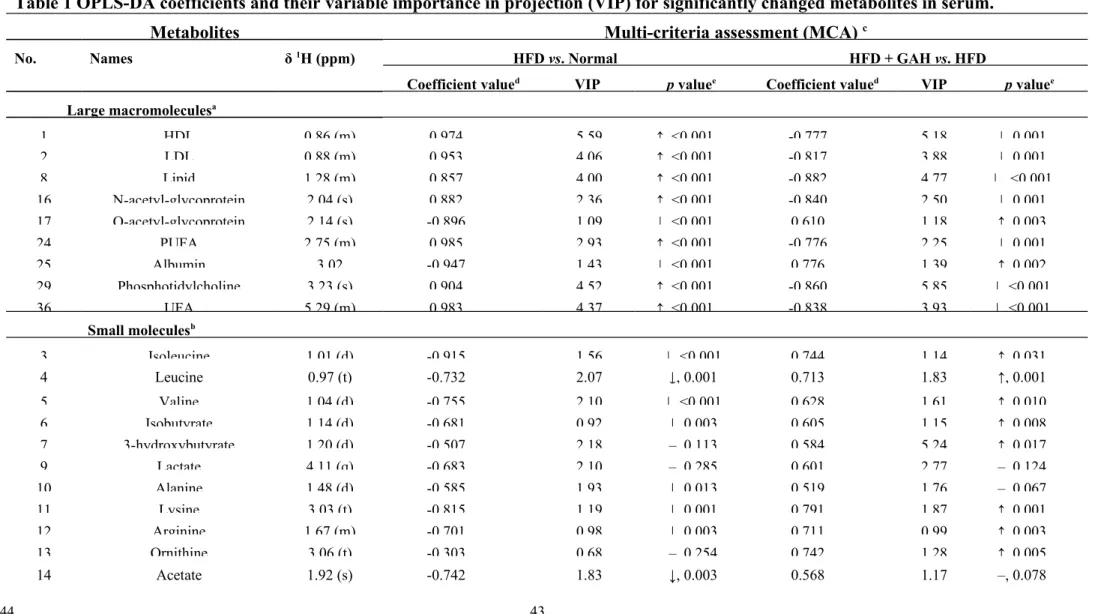
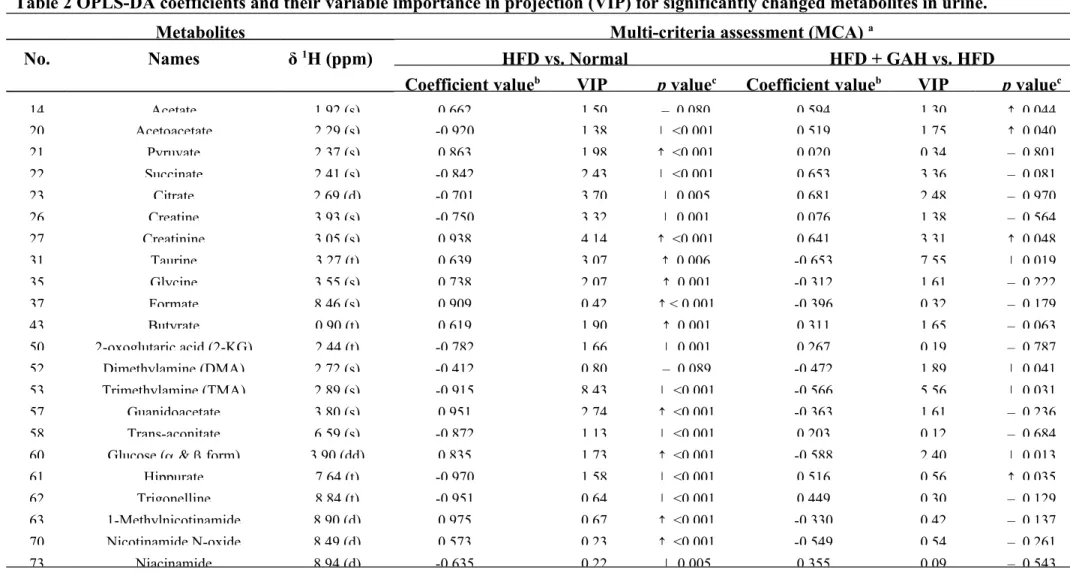
coefficient value |r| > 0.576, 2. VIP > 1, 3. p value < 0.05.
Statistical analysis
All the results are shown as mean ± SE. Statistical analysis was carried out
using one-way ANOVA followed by Bonferroni post hoc test. The criterion used for
statistical significance was p < 0.05.
253 254 255 256 257 258 259 260 261 262 263 264 265 266 267 268 269 270 271
Results and Discussion
As a common natural product, GA is a naturally abundant plant phenolic
compound in vegetables and fruits [3-6]. However, the effects and exact mechanisms
by which GA affects NAFLD have not been totally described. Many mechanistic
studies of GA have been performed using pharmacological methods [8,10,11], but
these may not have reflected the effects of GA on metabolite profiles of the test
organisms. In this study, we are the first to investigate the beneficial effects of GA
administration on nutritional hepatosteatosis via a more holistic approach that uses
NMR-based metabolomics.
GA ameliorates hepatic steatosis in HFD-induced NAFLD mice GA decreased body weight in HFD-induced NAFLD mice.
In order to evaluate the preventive effects of GA on NAFLD, male C57BL/6
mice were subjected to HFD for 16 weeks. Previous studies have been indicated that
hepatic steatosis is commonly associated with obesity [41]. Therefore, we measured
body weight changes twice per week from the start point of the experiments to the end
point of experiments. In this study we found that long-term HFD feeding resulted in a
progressive increase in the body weight (Figure S2A) of the HFD-fed mice.
Consistent with a previous study [10], we found that, compared with the HFD group
mice, the mice treated with GA showed a reduced HFD-induced body weight gain
272 273 274 275 276 277 278 279 280 281 282 283 284 285 286 287 288 289 290 291
(Figure S2A). In addition, food intake was not affected by GA treatment (Figure
S2B), which indicates that the decrease of body weight found to occur with the GA
treated mice were not due to changes in food consumption.
GA altered lipid homeostasis in HFD-induced NAFLD mice.
To evaluate whole-body glucose and lipid homeostasis, we next examined
various systemic parameters in the mice. As expected, the HFD group mice have
higher serum levels of HDL, TCHO, insulin and glucose than the normal diet group
(Figure 1A, 1B, Figure 2B, and 2C). Interestingly, the serum TG level was not
significantly affected by HFD feeding (Figure 2A). These clinical biochemistry
results indicated that long-term HFD feeding caused severe insulin resistance (IR) and
hypercholesterolemia, but did not induce hyperlipidemia. Compared with the HFD
group mice, the GA treated mice showed a significant decrease in these serum
metabolic parameters (Figure 1A, and Figure 2B, 2C and 2E). Although there are
no statistically significant differences in blood glucose between the GA treatment
group and the HFD group mice, the data showed that GA-treated mice have a
recovering trend compared to those in the HFD group mice. The GA-treated mice
developed only modest hypercholesterolemia, which demonstrates that GA treatment
produced an improved lipid homeostasis found in the NAFLD mice.
292 293 294 295 296 297 298 299 300 301 302 303 304 305 306 307 308 309 310
GA reduced hepatic steatosis in HFD-induced NAFLD mice.
HFD group mice exhibited increased liver weight (Figure 1C) and severe
hepatosteatosis by both gross morphological examination and histological
examination with the latter showing the liver as having hepatic vacuoles, lipid
droplets and hepatocyte swelling (Figure 1D, 1E). Liver injury was also confirmed by
significant increase of serum AST and ALT (Figure 2D, Figure 2E). In addition,
HFD feeding caused a significantly increased level of liver TG, cholesterol and fatty
acids and a significantly decreased ratio of PUFA to MUFA (Figure 3).These findings
indicated that HFD feeding resulted in significant hepatic steatosis and liver injury in
mice. Based on the results of the NAFLD diagnostic gold standard, namely the
histological analysis, these findings showed that the GA treatment had a significant
hepatoprotective effect on HFD-induced steatosis (Figure 1D). Administration of GA
reversed the excess fat accumulation in hepatic intracellular vacuole (Figure 1D), and
reversed the increased level of liver TG, cholesterol and fatty acids. The significant
decreased PUFA-to-MUFA ratio in HFD group was also reversed by GA treatment.
GA treatment also protected liver function and lowered the increase in ALT level
found in the HFD-fed mice (Figure 2E). Taken together, above results indicate that
GA ameliorates hepatic steatosis in HFD-induced NAFLD mice.
In the present study, the treatment doses of GA are 100 mg/kg and 50 mg/kg,
311 312 313 314 315 316 317 318 319 320 321 322 323 324 325 326 327 328 329
which are under the NOAEL of GA [42]. Based on formula from the FDA guidelines
[43] that is used to convert an animal dose of 100 mg/kg and 50 mg/kg of GA to a
human equivalent dose (HED), we calculated that the HED of GA are 487.8 mg/60 kg
and 243.9 mg/60 kg, respectively.
Metabolomics profiling in serum and urine by 1H NMR spectroscopy
To investigate the biochemical effects of HFD-induced hepatosteatosis and of
GA intervention in NAFLD mice, we performed an 1H NMR-based metabolomics
analysis combined with pattern recognition techniques to detect the endogenous
metabolites present in the serum and urine of the control, HFD and treatment group
mice (Figure S1B). Typical 1D 1H NMR spectra of the serum and urine taken from
normal group mice are presented in Figure 4. A total of 73 endogenous metabolites
were unambiguously assigned based on the published literature [33-39] and these
were confirmed by Chenomx 7.6.
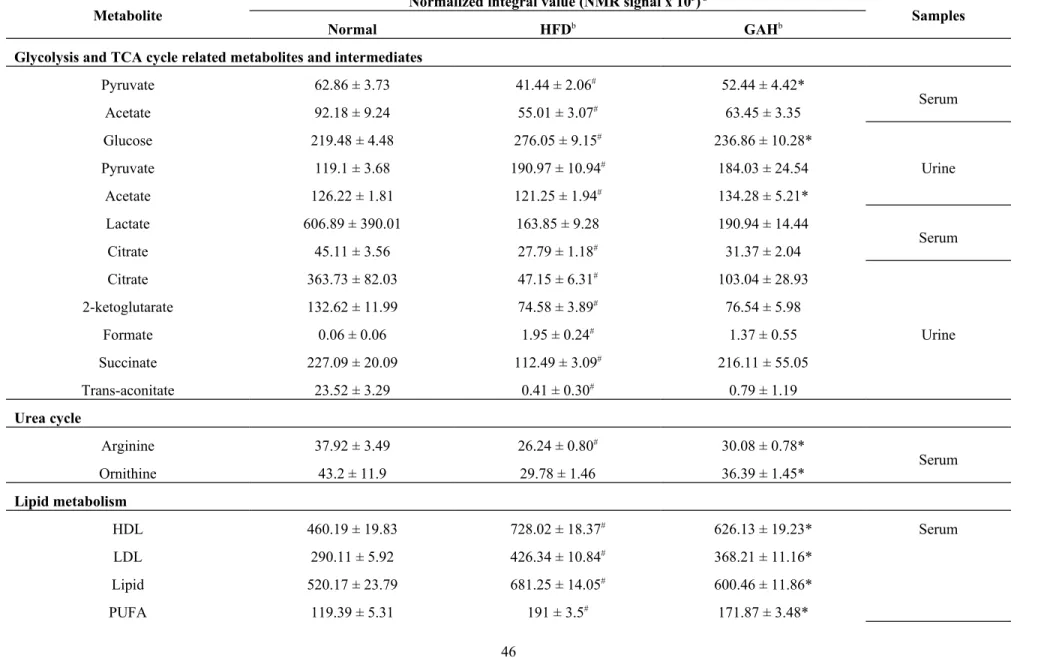
In this study, serum metabolic profiling provides information on lipid and
energy metabolism (Table S2). When a typical 600-MHS 1H NMR BPP-LED
spectrum is analyzed, serum signals characterizing common markers of CH3
resonance come from components of lipoprotein, such as cholesterol, HDL, LDL, and
phospholipids. In addition, the serum CPMG and NOESY spectra contained
330 331 332 333 334 335 336 337 338 339 340 341 342 343 344 345 346 347 348 349
resonances signals from low-molecular-mass metabolites, such as branched-chain
amino acids (BCAAs: valine, isoleucine, leucine), acidic amino acids (glutamate,
glycine), basic amino acids (lysine, arginine), aromatic amino acids (tyrosine,
phenylalanine), other aliphatic amino acids (alanine, proline), 1-methylhistidine,
ketone bodies (3-hydroxybutyrate and acetoacetate), several carboxylic acids (acetate,
formate), various choline-associated metabolites (choline, trimethylamine N-oxide
(TMAO), betaine) and taurine. A number of glycolysis and tricarboxylic acid-cycle
(TCA cycle) related metabolites and intermediates (glucose, pyruvate, lactate,
succinate, fumarate, citrate) were also detected in the serum.
The urine metabolic profiling provides information on intermediary
metabolism (Table S3). A typical urine 1H NMR spectrum was found to show a range
of different metabolites including amino acid (isoleucine, leucine, valine, lysine, and
arginine, glycine), organic acids (formate, acetate, butyrate), TCA cycle metabolites
(succinate, citrate, fumarate), gut microbiota-derived metabolites (methylamine,
dimethylamine (DMA), trimethylamine (TMA), TMAO, hippurate, formate,
benzoate), nicotinate and nicotinamide metabolism derived metabolites (trigonelline,
1-methylnicotinamide, nicotinamide N-oxide, niacinamide), choline, creatine,
creatinine, and urea.
350 351 352 353 354 355 356 357 358 359 360 361 362 363 364 365 366 367 368
Evaluation of the HFD-induced hepatosteatosis model using NMR-base metabolomics approach.
In order to identify the various different metabolic changes affecting the
NAFLD mice, the NMR spectrum were preprocessed in order to be able to carry out
multivariate statistical analysis (PCA, PLS-DA, and OPLS-DA). First, an
unsupervised pattern recognition method, PCA, was performed. Exploratory PCA was
employed to detect intrinsic clustering and possible outliers [44]. The different PCA
score plots of the serum illustrates that the HFD group is clearly separated from the
normal group (Figure S3A-C, (A) CPMG: R2X=0.589, Q2=0.448 ; (B) NOESY:
R2X=0.667, Q2= 0.539 ; (C)BPP-LED: R2X=0.742, Q2=0.475) or urine (Figure
S3D, NOESY: R2X=0.71, Q2=0.583). The major effect of component T1 is to
differentiate the HFD induction on the PCA score plot. Hotelling's T2 statistical
results indicated that only one outlier observation was found within the PCA score
plot of the BPP-LED spectrum. These findings demonstrated that the preprocessed
dataset from the NMR spectrum has good stability and low variation.
For the regression analysis, a supervised pattern recognition method,
PLS-369 370 371 372 373 374 375 376 377 378 379 380 381 382 383 384
DA, was used. The PLS-DA model was validated using a 7-fold cross validation
model and then was further evaluated using a permutation test (200 permutations).
The quality of the model was assessed by the cross-validation parameter (Q2Y), which
indicates the predictability of the model [45]. By applying PLS-DA, a reasonably
good separation was obtained for the scatter plots obtained from the (Figure S4A,
CPMG: R2X=0.41, R2Y=0.932, Q2=0.63; Figure S4C, NOESY: R2X=0.657,
R2Y=0.841, Q2=0.753; Figure S4E, BPP-LED: R2X=0.711, R2Y=0.929, Q2=0.873)
and urine (Figure S4G, NOESY: R2X=0.705, R2Y=0.988, Q2=0.973) samples. In a
similar manner to that of the PCA score plot, the major effect of principal component
T1 discriminates diet induction for the serum and urine samples. To further validate
the PLS-DA model, permutation tests were performed (Figure S4, right part). A
higher Q2 was obtained from the real model than was obtained as a Q2
max by the
permutation test when the normal group was compared with the HFD group.
Similarly, the R2 of the real model was higher than the R2
max obtained from the
permutation test. These findings imply that the PLS-DA model possessed great
predictability between the normal group and the HFD group and are not overfitted.
As a result of the above, the OPLS-DA method was employed in order to
maximize the covariance between the measured data (peak intensities in NMR
spectra) and the response variable (predictive classifications). The scores plot of the
385 386 387 388 389 390 391 392 393 394 395 396 397 398 399 400 401 402 403
OPLS-DA show a much clearer separation between the normal group and the HFD
group and have higher R2 and Q2 values than the other pattern recognition methods
(Figure 5A, serum CPMG spectra: R2X=0.481, R2Y=0.916, Q2=0.762; Figure 5D,
serum BPP-LED spectra: R2X=0.711, R2Y=0.929, Q2=0.879; Figure 5G, urine
NOESY spectra: R2X=0.705, R2Y=0.988, Q2=0.972). These findings indicated that
OPLS-DA should be able to help us to identify the important and latent variables
associated with liver steatosis and as well as those related to the drug-intervention
mechanism.
We further used OPLS-DA with coefficient plots to directly visualize the
results of the loadings and correlation coefficients (Figure 5B, 5E and 5H). The
color-coded correlation coefficients indicate the significance of the metabolites in
terms of their contribution to the separation between the different groups [46,47]. The
metabolites that are colored red and blue are more significant than those that are
colored green. Additionally, to exhibit how each of these variables is responsible for
the separation more intuitively, we created an S-plot that is combined with a VIP plot
and a color coefficient scale bar (we called this the “SV coefficient plot”) (Figure 5C,
404 405 406 407 408 409 410 411 412 413 414 415 416 417 418 419
5F, 5I). This is the first time that a “SV coefficient plot” has been used in 'omics
related papers. The SV coefficient plot is able to provide rich information, including
the metabolite contribution of first components (PC1), the VIP values of the
metabolites, and Pearson's correlation coefficient values (p(corr)), all in one figure.
Hence, the SV coefficient plot should be able to be applied generally to potential
biomarker selection in MCA.
The two different plots, the coefficient-coded loadings plots (Figure 5B, 5E,
5H), and the SV coefficient plot (Figure 5C, 5F, 5I), show that various latent
metabolites in the serum and urine have significant values. A series of key metabolites
contribute to the separation of the HFD group from the normal group; these, along
with their significance values (coefficient value, VIP value, and p value), are
summarized in Table 1 and Table 2. When the HFD group is compared with the
normal group, the levels of the metabolites with a positive coefficient value were
found to have been increased by HFD feeding, whereas those with negative values
were found to have been decreased (Table 1 and Table 2). These findings
demonstrate that the selected metabolites that have higher or lower coefficient and
VIP values are highly relevant biomarkers when explaining the discrimination
between the different groups. Moreover, in order to verify the results of the
OPLS-DA, the NMR spectra integrals of the altered metabolites were compared using
420 421 422 423 424 425 426 427 428 429 430 431 432 433 434 435 436 437 438
independent Student’s t-testa (Table 1, Table 2 and Table 3).
The observed latent metabolites identified as being associated with lipid
metabolism (HDL, LDL, TG, fatty acids, polyunsaturated fatty acids, unsaturated
fatty acids), ketogenesis (acetoacetate, 3-hydroxybutyrate), protein metabolism
marker (albumin), liver injury biomarker (albumin, taurine), glycolysis (lactate,
pyruvate) and TCA cycle intermediates (citrate, succinate, and 2-ketoglutarate),
amino acids metabolism (BCAAs, aromatic, acidic, basic, and other aliphatic amino
acid ), choline metabolism (Phosphotidylcholine, betaine) and gut-microbiota
metabolism (TMA, DMA, hippurate, butyrate, isobutyrate), nicotinate and
nicotinamide metabolism (trigonelline, 1-methylnicotinamide, nicotinamide N-oxide,
niacinamide), and creatine metabolism (creatine, creatinine, guanidoacetate) (Figure
7 and Table 3).
Evaluation of the GA therapeutic effect on the HFD-induced hepatosteatosis mice using a supervised pattern recognition method, PLS-DA
The serum PLS-DA model is able to discriminate the effect of HFD induction
and GA intervention on the score plot (Figure 6A-C). Using the CPMG score plot
(Figure 6A), the HFD group is clearly separated from the normal group in the
direction of component T1, which implies that the metabolic characteristics of the
439 440 441 442 443 444 445 446 447 448 449 450 451 452 453 454 455 456 457
various small molecules are distinctively different. However, a few GA-treated mice
are close to HFD-fed mice, while other GA-treated mice are close to the normal group
mice. These differences suggest that GA treatment is not able to restore the
homeostasis of all the disturbed metabolic pathways of the HFD-fed mice to the state
found in the normal group mice. Using the NOESY and LED-BPP score plots (Figure
6B, 6C), it can be seen that the GA-treated group is located in a distinct cluster that is
different to those of the HFD group and normal group; furthermore, the GA-treated
group is closer to the normal group than the HFD group. This supports the hypothesis
that GA treatment affects the NAFLD mice by improving the homeostasis of the
mice's serum at a macromolecular level.
The urine PLS-DA model also clearly differentiates the three different groups
(normal group, HFD group, and treatment group) based on the score plot result
(Figure 6D). The main effect of component T1 is to discriminate HFD induction,
whereas T2 seems to describe the response of GA intervention to HFD induction.
Taken together, the results of the metabolomic analysis of the metabolic effects of GA
support those obtained by biochemistry and histopathology and confirm the
hypothesis that GA has a significant therapeutic effect on NAFLD mice.
458 459 460 461 462 463 464 465 466 467 468 469 470 471 472 473 474 475 476
Metabolic effects of GA in high fat diet-induced NAFLD mice: traditional biochemical aspect
Lipid metabolism and ketogenesis
HFD feeding resulted in significant dyslipidemia, including elevated levels of
lipoprotein and fatty acids (Table 1, Figure 3). Compared with the normal group,
there were significantly increased levels of phosphotidylcholine and
O-acetyl-glycoprotein in the serum of the HFD-fed mice, which is consistent with the swelling
of hepatocytes (Figure 1D). Phophocholine is an abundant structural component of
the cell membrane [47,48]. Serum O-acetyl-glycoprotein is an “acute phase”
glycoprotein that is associated with inflammation of injury tissue in inflammatory
animal models [47,49]. Previous studies have suggested that elevated O-acetyl
glycoprotein fragment signals in the blood are associated with inflammatory
associated diseases, including cancer, certain liver diseases, and also surgical trauma
[49,50]. GA was able to reduce these increased levels of metabolites, which indicates
that GA ameliorates hepatosteatosis and protects the liver against injury during
HFD-feeding (Figure 1D and Figure 2E).
Ketone bodies, which contain acetone, acetoacetate and 3-hydroxybutyrate,
are important by-products of β-oxidation of fatty acids in the human body [51]. They
are produced from acetyl-CoA by ketogenesis and this mainly occurs in the
477 478 479 480 481 482 483 484 485 486 487 488 489 490 491 492 493 494 495
mitochondrial matrix of hepatocytes. Previous studies have been revealed that long
term HFD feeding causes hepatocyte mitochondrial DNA damage and dysfunction,
and that, as a result, there is increased oxidative stress in the liver [52]. In the present
study, the levels of acetoacetate and 3-hydroxybutyrate were lower in the HFD-fed
mice than in the normal diet-fed mice (Table 1, Table 2 and Table 3), which
suggests that HFD feeding caused a certain degree of mitochondria dysfunction in the
mouse hepatocytes, thereby decreasing the β-oxidation of fatty acids in the liver. It is
worth noting that GA treatment increased the levels of ketone bodies in the serum and
urine (Table 1, Table 2 and Table 3). In addition, an elevated concentration of
acetate, which is the end product of fatty acid oxidation in peroxisomes [53], was also
found in the GA-treated mice (Table 1 and Table 3). These results demonstrate that
the liver protective effect of GA is partially due to an increase β-oxidation of fatty
acids in the liver. Previous studies have suggested that a decreased PUFA/MUFA
ratio is indicative of excessive lipid peroxidation and oxidative stress in HFD-fed mice [54]. The PUFA/MUFA ratios in the liver were calculated (Figure 3D). HFD feeding caused a significant decreased the ratio of PUFA to MUFA (Figure 3D) and GA reversed this phenomenon. Our findings are consistent with previous studies [54].
496 497 498 499 500 501 502 503 504 505 506 507 508 509 510 511 512 513 514
Albumin
Long-term HFD feeding caused a significant decrease in the level of serum
albumin in HFD-fed mice (Table 1 and Table 3). Previous studies have demonstrated
that insulin dysfunction is caused by HFD feeding and that this inevitably results in
increased protein catabolism. This increase produces precursors for gluconeogenesis
and energy generation via the TCA cycle, and decreased protein production [55]. On
the other hand, albumin is a protein produced specifically by the liver. Therefore, the
serum level of albumin ought to reflect liver function [56]. In general, serum albumin
levels are decreased when chronic liver disease, such as hepatitis or liver cirrhosis, is
present. However, the relationship between serum albumin level and hepatic steatosis
is unclear. We suggested that the decreased level of serum albumin is probably a
symptom of reduced liver function in the hepatosteatosis mice (Table 1 and Table 3).
It is worth noting that GA treatment is able to restore to some degree the reduced level
of serum albumin in HFD fed mice. The relationship between albumin and NAFLD
diagnosis remains unclear and should be investigated in the future.
Glycolysis and TCA cycle (energy metabolism)
HFD feeding induced a significant decrease in the levels of both anaerobic
(lactate) and aerobic glycolysis metabolites (pyruvate) (Table 1 and Table 3) as well
515 516 517 518 519 520 521 522 523 524 525 526 527 528 529 530 531 532 533
as increased levels of serum glucose and insulin (Figure 1B and 1C). These disorders
of glucose metabolism indicated that the occurrence of enhanced gluconeogenesis and decreased glycolysis in the HFD group mice. This is consistent with the fact that lipid
accumulation in the liver impairs insulin signaling and the ability of insulin to
regulate gluconeogenesis [57]. In addition to abnormal glucose metabolism, a
disordered energy metabolism is the other main biological phenotype associated with
long term HFD feeding. Lipid accumulation-induced mitochondria DNA damage
correlates with mitochondrial dysfunction and increased oxidative stress in skeletal
muscle and liver, which are associated with the induction of endoplasmic reticulum
stress markers ER stress, protein degradation and apoptosis [52]. In present study,
compared with the normal group mice, various TCA cycle intermediates, such as
citrate, succinate, and 2-ketoglutarate, were found to be decreased in HFD group mice
(Table 1, Table 2 and Table 3). These findings indicated that TCA cycle activity and
the homeostasis of energy metabolism were both affected by HFD feeding. In the
GA-treated group, the levels of metabolites related to anaerobic (lactate) and aerobic
glycolysis, such as pyruvate and lactate, show a recovering trend compared to those in
the HFD group mice, while other metabolites, such as citrate, succinate and
2-ketoglutarate, exhibit no manifest change (Table 1, Table 2 and Table 3). These
results demonstrated that GA treatment does seems to have an effect in NAFLD mice
534 535 536 537 538 539 540 541 542 543 544 545 546 547 548 549 550 551 552
and that this occurs via an improvement in glycolysis rather than via changes in the
metabolism associated with the TCA cycle.
Taurine and bile acid metabolism
Taurine is a most abundant amino acid-like compound that is involved in
many important physiological processes, including stabilization of the cellular plasma
membrane, osmorregulation, anti-oxidative effects, and hepatic detoxification [58]. In
the liver, either taurine or glycine can be conjugated with hepatic bile acids in order to
allow excretion into bile [58]. Previous studies have been suggested that urinary
taurine is a non-invasive biomarker for various types of liver damage and reflect
changes in protein metabolism [59-61]. This increase in urine taurine is a result of
leakage of taurine from damage hepatocytes, and an inhibition of protein synthesis by
hepatotoxicants, which has been shown to increase urinary taurine excretion in rats
[59-61]. Furthermore, a recent study has also proposed the preventive and therapeutic
effects of dietary taurine supplementation as a treatment for alcoholic steatohepatitis
and NAFLD [58]. In the current study, increased amounts of urinary taurine and
glycine were detected in the HFD-fed mice (Table 2 and Table 3), which indicates
that HFD feeding not only disturbs bile acid metabolism in the liver, but also leads to
hepatocyte destruction. The urine levels of taurine and glycine found in the samples
553 554 555 556 557 558 559 560 561 562 563 564 565 566 567 568 569 570 571
from GA-treated mice were significantly reduced compared with those from the urine
of animals fed the HFD (Table 2 and Table 3). These findings indicate that GA
treatment reversed the changes in urine taurine and glycine and that this probably
occurs through the hepatoprotective effect of GA, whereby there is an amelioration of
disordered bile acid metabolism.
Amino acids metabolism
The levels of the glucogenic amino acids (alanine, valine, glutamine, arginine,
glycine) as well as those of the ketogenic and glucogenic amino acids (isoleucine,
tyrosine, phenylalanine) were decreased in the HFD group mice compared with the
levels in the normal group mice (Table 1 and Table 3). Our results are consistent
with previous observation whereby a HFD cause an impairment of insulin signaling
and the ability of insulin to regulate gluconeogenesis [57]. The reduction in
glucogenic amino acids may reflect the promotion of gluconeogenesis, which is
observed when there is an increased level of glucose (Figure 1 and Table 3).
Additionally, it is now well established that skeletal muscle is the principle storage
target site for insulin-stimulated glucose uptake. In the IR state, skeletal muscle cells
shows impaired insulin activity with respect to both glucose transport and intracellular
glucose metabolism [62]. As a result of these changes, the aromatic amino acids
572 573 574 575 576 577 578 579 580 581 582 583 584 585 586 587 588 589 590
(tyrosine, phenylalanine), the BCAAs (valine, isoleucine, leucine), as well as
glutamate and glutamine are fed into TCA cycle in order to produce ATP and energy
for the skeletal muscle. Interestingly, the levels of amino acids in GA treatment mice
was found to show a tendency towards recovery compared with similar levels in the
HFD-fed mice (Table 1 and Table 3). These findings demonstrated that GA
treatment is able to ameliorate the IR state in the peripheral tissues, and that this then
affect the pathways associated with amino acids metabolism.
Glutamine and glutamate are both precursors of glutathione, the first line of
defense against free radicals in the liver [63]. A clinical investigation has indicated
reduced plasma glutamate is able to act as a biomarker for septic shock patients with
acute liver dysfunction [64]. In the present study, we noted that there was a
significantly decreased level of serum glutamate and glutamine in the HFD-fed mice
(Table 1 and Table 3), which probably reflects the presence of the HFD-induced
promotion of oxidative stress [65]. GA treatment reversed this significant decrease in
the level of glutathione-associated amino acids (Table 1 and Table 3). In agreement
with our findings, a previous study has also shown that GA enhances the level of
glutathione in the liver and reduces oxidative stress in HFD-fed rats [10]. These
results suggest that the hepatic protective effect of GA in this area of metabolism is
probable due to GA's anti-oxidative activity.
591 592 593 594 595 596 597 598 599 600 601 602 603 604 605 606 607 608 609
Choline metabolism
There are three different metabolic pathways involved in choline metabolism
[37] (Figure 7): In the first, the oxidized choline is excreted as betaine in the urine,
which ultimately leads to the production of creatine and creatinine. In the second,
choline is converted to methylamine (TMA, TMAO and DMA) by the gut microbiota.
While in the third, choline is phosphorylated by choline kinase to generate PC.
Betaine is an essential osmoregulatory compound and an important cofactor
in methylation during the methionine-homocysteine cycle [22,66]. A previous study
has shown that betaine insufficiency is associated with metabolic syndrome, lipid
disorders and, diabetes as well as playing a crucial role in vascular and other diseases
[66]. Moreover, betaine administration was found to significantly improve IR in a
NAFLD animal model [67], whereas betaine treatment of NASH patient was found to
decrease their steatosis indices [68]. However, the mechanisms by which betaine
ameliorates hepatic steatosis have not been fully understood. In this study, compared
with the normal diet group mice, a decreased level of betaine was observed in the
HFD-fed mice (Table 1 and Table 3), which seems to reflect the HFD-induced
promotion of oxidative stress; this then inhibits methylamine metabolism, which
might be implicated in the pathogenesis of fatty liver. Our analysis shows that the
610 611 612 613 614 615 616 617 618 619 620 621 622 623 624 625 626 627 628
levels of betaine in the GA-treated mice were found to recover and return towards
those found in the normal group mice (Table 1 and Table 3). Therefore, we
suggested that the methylamine metabolism pathway might be another treatment
target of GA.
Methylamine-associated metabolites, such as TMA, TMAO and DMA, are
the products of the metabolism of choline by gut microbiota [37]. Consistent with a
recent study [69], lower levels of TMA and DMA were found in both the HFD and
treatment groups (Table 2 and Table 3); these changes are most probably due to a
dietary effect. On other hand, when the HFD and GA treatment groups were
compared, the HFD group mice were found to have higher levels of
methylamine-associated metabolites (Table 2 and Table 3), which suggests that GA is able to
reduce the elevation in gut microflora choline metabolism present in HFD fed mice to
a similar level to that of the low-choline diet condition and thereby reduce the
induction of severe hepatic steatosis [37]. In addition, it is likely that other changes in
gut microbiota-related metabolites in the HFD-fed mice, including changes in
hippurate and various short chain organic acids (acetate, butyrate, and isobutyrate),
are also associated with the changes to gut microbiota (Table 1, Table 2 and Table
3). Our findings are consistent with previous studies [70,71] showing that short chain
organic acids are produced by gut bacterial fermentation of carbohydrates such as
629 630 631 632 633 634 635 636 637 638 639 640 641 642 643 644 645 646 647
cellulose and resistant starches. In present study, GA treatment not only reversed
elevated choline metabolism, but also seemed to improve disorders in gut
microbiota-related metabolites (Table 1, Table 2 and Table 3); this supports the hypothesis that
the gut microbiota are a probable target for GA treatment [72]. These findings
confirm those reported by Bialonska et al. [73] wherein GA-rich fruits seem to cause
an enhancement in the growth of probiotic bacteria. In future studies, how GA
affected gut microbiota should be further investigated using a metagenomic approach.
Nicotinate and nicotinamide metabolism
Nicotinamide, also known as niacinamide and nicotinic acid amide, is the
amide derivative of nicotinic acid (vitamin B3 / niacin) [74]. Nicotinamide is the
precursor for two cofactors, NAD+ (nicotinamide adenine dinucleotide) and NADP+
(nicotinamide adenine dinucleotide phosphate), which both play essential roles in
redox reactions [75]. Through the nicotinamide metabolic pathway, nicotinamide is
able to be oxidized to nicotinamide N-oxide, methy1ated to 1-methy1nicotinamide, or
methy1ated to trigonelline, all of which can be excreted into urine [74].
1-Methy1nicotinamide has been suggested as a urine biomarker of peroxisome
proliferation in rats [76]. Compared with the normal group, there were relatively
decreased levels of nicotinamide and trigonelline observed in HFD-induced NAFLD
648 649 650 651 652 653 654 655 656 657 658 659 660 661 662 663 664 665 666
mice compared to control mice, together with increased levels of
1-methylnicotinamide and nicotinamide-N-oxide (Table 2, Table 3). These results
indicate that HFD feeding seems to alter nicotinate and nicotinamide metabolic
pathway. However, the levels of nicotinamide related metabolites in the GA-treated
mice did not show a significant recovery towards the levels from in the control mice.
Nonetheless, there was a trend towards recovery compared with the levels found in
the HFD group mice (Table 2, Table 3). This implies that GA treatment does not
have a primary effect on the metabolic pathways involved in nicotinate and
nicotinamide metabolism, but it is possible that there is a secondary effect.
Conclusions
On the basis of the changes in metabolites identified in this study, a series of
metabolic pathway that seem to be associated with HFD-induced hepatosteatosis are
proposed in Figure 7. These results are based on a 16 weeks HFD feeding regimen
that caused metabolome changes in the overall metabolic pathways of a NAFLD mice
model . Interestingly, it is important to note that the disturbed metabolic pathways are
able to be partially reversed by GA treatment. Our results indicate that the targets of
GA treatment are lipid metabolism and ketogenesis, glycolysis, amino acids
667 668 669 670 671 672 673 674 675 676 677 678 679 680 681 682 683 684 685
metabolism, choline metabolism, and gut-microbiota metabolism. These changes are
probably useful as novel preventive action biomarkers and also can be used to explore
the mechanism by which GA treatment restore normal metabolism. Finally, the
current investigation provides further evidence in support of GA as natural dietary
compound that is able to ameliorate NAFLD and other metabolic disorders.
Acknowledgments
The authors would like to thank Dr. Ralph Kirby for his editorial assistance. The
NMR spectra were obtained at the core facility for metabolomics analysis supported
by National Core facility Program for Biotechnology. This study is supported in part
by the National Science Council, Taiwan (NSC100-2320-B-039-013,
NSC101-2320-B-039-032-MY2) and Committee on Chinese Medicine and Pharmacy, Department of
Health, Executive Yuan (CCMP102-RD-104, CCMP102-RD-019).
686 687 688 689 690 691 692 693 694 695 696 697 698 699
References
1. Browning JD, Horton JD (2004) Molecular mediators of hepatic steatosis and liver injury. J Clin Invest 114: 147-152.
2. Cusi K (2009) Nonalcoholic fatty liver disease in type 2 diabetes mellitus. Curr Opin Endocrinol Diabetes Obes 16: 141-149.
3. Maheshwari DT, Yogendra Kumar MS, Verma SK, Singh VK, Singh SN (2011) Antioxidant and hepatoprotective activities of phenolic rich fraction of Seabuckthorn (Hippophae rhamnoides L.) leaves. Food Chem Toxicol 49: 2422-2428.
4. Peng CH, Liu LK, Chuang CM, Chyau CC, Huang CN, et al. (2011) Mulberry water extracts possess an anti-obesity effect and ability to inhibit hepatic lipogenesis and promote lipolysis. J Agric Food Chem 59: 2663-2671.
5. Wang SH, Kao MY, Wu SC, Lo DY, Wu JY, et al. (2011) Oral administration of Trapa taiwanensis Nakai fruit skin extracts conferring hepatoprotection from CCl4-caused injury. J Agric Food Chem 59: 3686-3692.
6. Lee JE, Lee BJ, Hwang JA, Ko KS, Chung JO, et al. (2011) Metabolic dependence of green tea on plucking positions revisited: a metabolomic study. J Agric Food Chem 59: 10579-10585.
7. Ma J, Luo XD, Protiva P, Yang H, Ma C, et al. (2003) Bioactive novel polyphenols from the fruit of Manilkara zapota (Sapodilla). J Nat Prod 66: 983-986.
8. Oi Y, Hou IC, Fujita H, Yazawa K (2012) Antiobesity effects of Chinese black tea (Pu-erh tea) extract and gallic acid. Phytother Res 26: 475-481.
9. Hsiang CY, Hseu YC, Chang YC, Kumar KJ, Ho TY, et al. (2013) Toona sinensis and its major bioactive compound gallic acid inhibit LPS-induced inflammation in nuclear factor-kappaB transgenic mice as evaluated by in vivo bioluminescence imaging. Food Chem 136: 426-434. 10. Hsu CL, Yen GC (2007) Effect of gallic acid on high fat diet-induced dyslipidaemia,
hepatosteatosis and oxidative stress in rats. Br J Nutr 98: 727-735.
11. Jang A, Srinivasan P, Lee NY, Song HP, Lee JW, et al. (2008) Comparison of hypolipidemic activity of synthetic gallic acid-linoleic acid ester with mixture of gallic acid and linoleic acid, gallic acid, and linoleic acid on high-fat diet induced obesity in C57BL/6 Cr Slc mice. Chem Biol Interact 174: 109-117.
12. Kroes BH, van den Berg AJ, Quarles van Ufford HC, van Dijk H, Labadie RP (1992) Anti-inflammatory activity of gallic acid. Planta Med 58: 499-504.
13. Inoue M, Suzuki R, Sakaguchi N, Li Z, Takeda T, et al. (1995) Selective induction of cell death in cancer cells by gallic acid. Biol Pharm Bull 18: 1526-1530.
14. Punithavathi VR, Stanely Mainzen Prince P, Kumar MR, Selvakumari CJ (2011) Protective effects of gallic acid on hepatic lipid peroxide metabolism, glycoprotein components and lipids in
700 701 702 703 704 705 706 707 708 709 710 711 712 713 714 715 716 717 718 719 720 721 722 723 724 725 726 727 728 729 730 731 732 733 734
streptozotocin-induced type II diabetic Wistar rats. J Biochem Mol Toxicol 25: 68-76.
15. Niho N, Shibutani M, Tamura T, Toyoda K, Uneyama C, et al. (2001) Subchronic toxicity study of gallic acid by oral administration in F344 rats. Food Chem Toxicol 39: 1063-1070.
16. Nicholson JK, Lindon JC, Holmes E (1999) 'Metabonomics': understanding the metabolic responses of living systems to pathophysiological stimuli via multivariate statistical analysis of biological NMR spectroscopic data. Xenobiotica 29: 1181-1189.
17. Wang X, Zhang A, Han Y, Wang P, Sun H, et al. (2012) Urine metabolomics analysis for biomarker discovery and detection of jaundice syndrome in patients with liver disease. Mol Cell Proteomics.
18. Holmes E, Loo RL, Stamler J, Bictash M, Yap IK, et al. (2008) Human metabolic phenotype diversity and its association with diet and blood pressure. Nature 453: 396-400.
19. Xuan J, Pan G, Qiu Y, Yang L, Su M, et al. (2011) Metabolomic profiling to identify potential serum biomarkers for schizophrenia and risperidone action. J Proteome Res 10: 5433-5443. 20. Wang X, Wang H, Zhang A, Lu X, Sun H, et al. (2012) Metabolomics study on the toxicity of
aconite root and its processed products using ultraperformance liquid-chromatography/electrospray-ionization synapt high-definition mass spectrometry coupled with pattern recognition approach and ingenuity pathways analysis. J Proteome Res 11: 1284-1301. 21. Yang HJ, Choi MJ, Wen H, Kwon HN, Jung KH, et al. (2011) An effective assessment of
simvastatin-induced toxicity with NMR-based metabonomics approach. PLoS One 6: e16641. 22. Kim HJ, Kim JH, Noh S, Hur HJ, Sung MJ, et al. (2011) Metabolomic analysis of livers and
serum from high-fat diet induced obese mice. J Proteome Res 10: 722-731.
23. Lisec J, Schauer N, Kopka J, Willmitzer L, Fernie AR (2006) Gas chromatography mass spectrometry-based metabolite profiling in plants. Nat Protoc 1: 387-396.
24. McLoughlin GA, Ma D, Tsang TM, Jones DN, Cilia J, et al. (2009) Analyzing the effects of psychotropic drugs on metabolite profiles in rat brain using 1H NMR spectroscopy. J Proteome Res 8: 1943-1952.
25. Bao Y, Zhao T, Wang X, Qiu Y, Su M, et al. (2009) Metabonomic variations in the drug-treated type 2 diabetes mellitus patients and healthy volunteers. J Proteome Res 8: 1623-1630.
26. Wang Y, Holmes E, Tang H, Lindon JC, Sprenger N, et al. (2006) Experimental metabonomic model of dietary variation and stress interactions. J Proteome Res 5: 1535-1542.
27. Rezzi S, Ramadan Z, Fay LB, Kochhar S (2007) Nutritional metabonomics: applications and perspectives. J Proteome Res 6: 513-525.
28. Llorach R, Garcia-Aloy M, Tulipani S, Vazquez-Fresno R, Andres-Lacueva C (2012) Nutrimetabolomic strategies to develop new biomarkers of intake and health effects. J Agric Food Chem 60: 8797-8808.
29. Nicholson JK, Lindon JC (2008) Systems biology: Metabonomics. Nature 455: 1054-1056. 30. Kim HK, Choi YH, Verpoorte R (2011) NMR-based plant metabolomics: where do we stand,
where do we go? Trends Biotechnol 29: 267-275.
735 736 737 738 739 740 741 742 743 744 745 746 747 748 749 750 751 752 753 754 755 756 757 758 759 760 761 762 763 764 765 766 767 768 769 770 771 772
31. Sheng X, Wang M, Lu M, Xi B, Sheng H, et al. (2011) Rhein ameliorates fatty liver disease through negative energy balance, hepatic lipogenic regulation, and immunomodulation in diet-induced obese mice. Am J Physiol Endocrinol Metab 300: E886-893.
32. Beckonert O, Keun HC, Ebbels TM, Bundy J, Holmes E, et al. (2007) Metabolic profiling, metabolomic and metabonomic procedures for NMR spectroscopy of urine, plasma, serum and tissue extracts. Nat Protoc 2: 2692-2703.
33. Xu W, Wu J, An Y, Xiao C, Hao F, et al. (2012) Streptozotocin-Induced Dynamic Metabonomic Changes in Rat Biofluids. J Proteome Res.
34. He Q, Tang H, Ren P, Kong X, Wu G, et al. (2011) Dietary supplementation with l-arginine partially counteracts serum metabonome induced by weaning stress in piglets. J Proteome Res 10: 5214-5221.
35. He Q, Kong X, Wu G, Ren P, Tang H, et al. (2009) Metabolomic analysis of the response of growing pigs to dietary L-arginine supplementation. Amino Acids 37: 199-208.
36. Nicholson JK, Foxall PJ, Spraul M, Farrant RD, Lindon JC (1995) 750 MHz 1H and 1H-13C NMR spectroscopy of human blood plasma. Anal Chem 67: 793-811.
37. Dumas ME, Barton RH, Toye A, Cloarec O, Blancher C, et al. (2006) Metabolic profiling reveals a contribution of gut microbiota to fatty liver phenotype in insulin-resistant mice. Proc Natl Acad Sci U S A 103: 12511-12516.
38. Salek RM, Maguire ML, Bentley E, Rubtsov DV, Hough T, et al. (2007) A metabolomic comparison of urinary changes in type 2 diabetes in mouse, rat, and human. Physiol Genomics 29: 99-108.
39. Zhao XJ, Hao F, Huang C, Rantalainen M, Lei H, et al. (2012) Systems responses of rats to mequindox revealed by metabolic and transcriptomic profiling. J Proteome Res 11: 4712-4721. 40. Trygg J, Holmes E, Lundstedt T (2007) Chemometrics in metabonomics. J Proteome Res 6:
469-479.
41. Rolo AP, Teodoro JS, Palmeira CM (2012) Role of oxidative stress in the pathogenesis of nonalcoholic steatohepatitis. Free Radic Biol Med 52: 59-69.
42. Rajalakshmi K, Devaraj H, Niranjali Devaraj S (2001) Assessment of the no-observed-adverse-effect level (NOAEL) of gallic acid in mice. Food Chem Toxicol 39: 919-922.
43. http://www.fda.gov/cder/Guidance/5541fnl.pdf (2005) Guidance for Industry: Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers. US Food and Drug Administration. Center for Drug Evaluation and Research.
44. Carrola J, Rocha CM, Barros AS, Gil AM, Goodfellow BJ, et al. (2011) Metabolic signatures of lung cancer in biofluids: NMR-based metabonomics of urine. J Proteome Res 10: 221-230.
45. Bjerrum JT, Nielsen OH, Hao F, Tang H, Nicholson JK, et al. (2010) Metabonomics in ulcerative colitis: diagnostics, biomarker identification, and insight into the pathophysiology. J Proteome Res 9: 954-962.
46. Cloarec O, Dumas ME, Trygg J, Craig A, Barton RH, et al. (2005) Evaluation of the orthogonal
773 774 775 776 777 778 779 780 781 782 783 784 785 786 787 788 789 790 791 792 793 794 795 796 797 798 799 800 801 802 803 804 805 806 807 808 809 810
projection on latent structure model limitations caused by chemical shift variability and improved visualization of biomarker changes in 1H NMR spectroscopic metabonomic studies. Anal Chem 77: 517-526.
47. Zhang L, Ye Y, An Y, Tian Y, Wang Y, et al. (2011) Systems responses of rats to aflatoxin B1 exposure revealed with metabonomic changes in multiple biological matrices. J Proteome Res 10: 614-623.
48. Klein J (2000) Membrane breakdown in acute and chronic neurodegeneration: focus on choline-containing phospholipids. J Neural Transm 107: 1027-1063.
49. Grootveld M, Claxson AW, Chander CL, Haycock P, Blake DR, et al. (1993) High resolution proton NMR investigations of rat blood plasma. Assignment of resonances for the molecularly mobile carbohydrate side-chains of 'acute-phase' glycoproteins. FEBS Lett 322: 266-276.
50. Wang Y, Utzinger J, Saric J, Li JV, Burckhardt J, et al. (2008) Global metabolic responses of mice to Trypanosoma brucei brucei infection. Proc Natl Acad Sci U S A 105: 6127-6132.
51. Laffel L (1999) Ketone bodies: a review of physiology, pathophysiology and application of monitoring to diabetes. Diabetes Metab Res Rev 15: 412-426.
52. Yuzefovych LV, Musiyenko SI, Wilson GL, Rachek LI (2013) Mitochondrial DNA damage and dysfunction, and oxidative stress are associated with endoplasmic reticulum stress, protein degradation and apoptosis in high fat diet-induced insulin resistance mice. PLoS One 8: e54059. 53. Leighton F, Bergseth S, Rortveit T, Christiansen EN, Bremer J (1989) Free acetate production by
rat hepatocytes during peroxisomal fatty acid and dicarboxylic acid oxidation. J Biol Chem 264: 10347-10350.
54. Vinaixa M, Rodriguez MA, Rull A, Beltran R, Blade C, et al. (2010) Metabolomic assessment of the effect of dietary cholesterol in the progressive development of fatty liver disease. J Proteome Res 9: 2527-2538.
55. Gupte AA, Bomhoff GL, Swerdlow RH, Geiger PC (2009) Heat treatment improves glucose tolerance and prevents skeletal muscle insulin resistance in rats fed a high-fat diet. Diabetes 58: 567-578.
56. Lee WM, McLeod L, Martin K, Emerson DL, Galbraith RM (1987) Antibodies to polymerized human serum albumin in acute and chronic liver disease. Hepatology 7: 906-912.
57. Samuel VT, Shulman GI (2012) Mechanisms for insulin resistance: common threads and missing links. Cell 148: 852-871.
58. Miyazaki T, Matsuzaki Y (2012) Taurine and liver diseases: a focus on the heterogeneous protective properties of taurine. Amino Acids.
59. Waterfield CJ, Turton JA, Scales MD, Timbrell JA (1991) Taurine, a possible urinary marker of liver damage: a study of taurine excretion in carbon tetrachloride-treated rats. Arch Toxicol 65: 548-555.
60. Timbrell JA, Waterfield CJ (1996) Changes in taurine as an indicator of hepatic dysfunction and biochemical perturbations. Studies in vivo and in vitro. Adv Exp Med Biol 403: 125-134.
811 812 813 814 815 816 817 818 819 820 821 822 823 824 825 826 827 828 829 830 831 832 833 834 835 836 837 838 839 840 841 842 843 844 845 846 847 848