行政院國家科學委員會專題研究計畫 成果報告
自由基的環化反應在高度官能基化的碳環及雜環合成上之
運用
計畫類別: 個別型計畫 計畫編號: NSC91-2113-M-002-029- 執行期間: 91 年 08 月 01 日至 92 年 07 月 31 日 執行單位: 國立臺灣大學化學系暨研究所 計畫主持人: 蔡蘊明 計畫參與人員: 蔡蘊明,陳明仁,吳坤亮,翁紹華,潘己全,王農森,張哲健, 廖芳瑜 報告類型: 精簡報告 處理方式: 本計畫可公開查詢中 華 民 國 92 年 10 月 31 日
行政院國家科學委員會補助專題研究計畫
5 成 果 報 告 □期中進度報告自由基的環化反應在高度官能基化的碳環及雜環合成
上之運用
計畫類別:; 個別型計畫 □ 整合型計畫
計畫編號:NSC 91-2113-M -002 -029-
執行期間:91 年 08 月 01 日至 92 年 07 月 31 日
計畫主持人:蔡蘊明
成果報告類型(依經費核定清單規定繳交):
;精簡報告 □完整報告
本成果報告包括以下應繳交之附件:
□赴國外出差或研習心得報告一份
□赴大陸地區出差或研習心得報告一份
□出席國際學術會議心得報告及發表之論文各一份
□國際合作研究計畫國外研究報告書一份
處理方式:除產學合作研究計畫、提升產業技術及人才培育研究
計畫、列管計畫及下列情形者外,得立即公開查詢
執行單位:國立台灣大學化學系
中 華 民 國 九十二 年 十 月 三十 日
中文摘要 關鍵字:α-矽基自由基,α-矽基醛,1,5-氫轉移,天然植物鹼,自由基環化 Part I 我們企圖利用 1,3−矽基轉移的方式來促成羰基環合的可能性。首先合成了幾種 具有α-矽基的醛類化合物,我們發現這類化合物雖可環合,然而其環化的氧自 由基中間體又會進行開環而得到另一重排的產物。另一種系統是有關α-矽基自 由基與醛的環化,在此處雖然在羰機的α-位放置了一個強拉電子基,但此類的 環化仍因 1,5-氫轉移的發生造成只能得到直接還原的產物。 Part II 我們從一個單一的對掌的亞醯胺化合物合成了天然植物鹼(+)-heliotridine 與 (−)-retronecine。在此亞醯胺化合物中的不對稱中心最後成為此二天然物的 C-1,我們發展了方法能成功的利用此一不對稱中心架構出在其旁邊的 C-7a 的 立體化學,其選擇性極高。此二植物鹼的 B-環則是藉著自由基環化的策略來達 成。 英文摘要
關鍵字:α-silylaldehydes, α-silyl radical, 1,5-hydrogen transfer, (+)-heliotridine, (−)-retronecine, total synthesis, stereoselectively , radical cyclization.
Part I
We studied the possibility of using 1,3-silyl shift to promote carbonyl radical cyclization. Two types of cyclization systems were examined. The first type involved the cyclizations of α-silylaldehydes, and the other one dealt with α-silyl radical cyclizations with α-sulfonylaldehydes. The α-silyl radical cyclization system gave preferentially the straight reduction product. This is probably due 1,5-hydrogen transfer problem even when we designed a sulfonyl substituent at the α-position of the carbonyl group. The α-silylaldehyde cyclization system also met with failure. The cyclized intermediate alkoxy radical underwent β-scission to give a rearranged straight chain product.
Part II
Formal total synthesis of (+)-heliotridine (4) and total synthesis of (−)-retronecine (5) were accomplished by starting from the same enantio-pure imide 6. The
stereogenic center of 6 ended up as C-1 in both alkaloids. The chiral centers at C-7a of the alkaloids were stereoselectively constructed through the help of the adjacent functionality at C-1. The B-rings of the alkaloids were formed through α-sulfonyl radical cyclization strategy.
本實驗室過去利用矽基酮作為自由基受體1
,進行自由基環合反應。自由基加成 到羰基,生成β-矽基烷氧自由基的中間產物,烷氧自由基可進一步的進行自由 基型態的 Brook 重排反應,矽基從碳上移到鄰位的氧上,得到一個新的α-矽 氧基碳自由基,因 O-Si 鍵結的鍵能(111 kcal/mol)遠大於一般 C-Si 鍵能(90 kcal/mol)2 ,成為極佳的反應趨動力,也使此一重排反應為不可逆反應,在這 方向上本實驗室已完成不少初步的研究。基於上述的矽基酮化學,藉由 1,2− 矽基轉移而成功的將環化反應推向環合的一邊,這讓我們想到是否可以利用 1,3−矽基轉移的方式來促成環合的可能性。 Scheme 1 我們首先合成了環合前驅物 1,接著進行自由基環化反應(流程 1),所使用的條 件為將前驅物 1 溶於苯中加熱至 80℃,再將另一溶於苯中的三丁基錫烷 (1.2 equiv.)與 AIBN (0.1 equiv.)經過定量加液馬達以兩小時的時間緩慢加入上述 之預熱溶液中,其最終濃度相對於矽基醛為 0.05 M,錫烷溶液加完之後再加熱 迴流二小時。在此反應中得到了兩個產物,一個是直接還原的α-矽基醛 2,另 一個是末端帶有矽基的醛 3。此反應並沒有得到我們所要的環合產物,從反應 可能的機制來看(流程 1),當α-矽基醛 1 與三丁基錫自由基作用形成自由基 4 後,可能進行的路徑為直接由錫烷上抓取氫原子即得到還原產物 2,或環合得 到自由基中間體 29 再進行β-消去反應,此時有兩種可能性,一為形成原先的 自由基 4,另一可能則為生成α-矽基取代的自由基 6,如果形成自由基 6,因 為它是一個α-矽基自由基,抓氫的速度非常快3 ,所以會直接抓取錫烷上的氫 原子而得到矽基轉移的還原產物 3,不過,3 的來源也有可能由 6 透過分子內 1,5-氫轉移後所形成的醯自由基 7 抓取氫原子而得到。至於,到底是直接還原, 還是經由 1,5-氫轉移才得到,並不易判定。由於反應的主要產物為化合物 3, H O I TBS AIBN (cat.), Bu3SnH PhH, 80 ℃, 4 h H O TBS + H O TBS 1 2 3 1 : 6.8 ( 28 % ) H O TBS 4 cyclization TBS O β -scission 5 1,5-H shift O TBS Bu3SnH Bu3SnH 6 8 Bu3SnH β -scission cyclization Bu3Sn O TBS H 1,5-H shift O TBS 7 26 Bu3SnH
這樣的反應結果表示在α-矽基醛 1,5-環化的系統中,自由基加成到羰基的效 率還不錯,可惜由於矽基轉移的速度太慢或是根本不轉移,導致開環得到α-矽基自由基 6,自由基 6 的環化顯然是很困難的。 Scheme 2 另外亦合成了溴化物 9,產率雖不高,但已可提供足夠的物質研究環化的可能 性。實驗的結果與之前五員環的系統類似,只得到直接還原產物 10 和重排後的 還原產物 11。產物 10 的產生可能是α−矽基醛 9 經由三丁基錫烷作用產生自由 基 12 後直接抓取錫烷上的氫而得到的(流程 2),但也可能透過 1,5-氫轉移得到 α位的自由基 15,然後抓取氫原子得到還原產物 10。至於 11,是自由基 12 加 成到羰基再開環後形成α−矽基自由基 14,之後再抓氫原子所得到的。另一個 11 的來源也有可能是自由基 14 經由分子間 1,5-氫轉移得到α位自由基 16 再 抓氫原子而得到的。從反應結果來看,矽基轉移的速度還是太慢,即使在六員 環開環速率比較慢的條件下,還是沒有發生 1,3-矽基轉移反應。與上一節 1,5-環化的系統做比較,1,6-環化的系統中直接還原的比例較高,這可能的原因有 二,一為環化的速率較慢,一則為自由基 12 進行 1,5-氫轉移的速率較快。從 Beckwith 等人所報導的醛基與自由基的環化反應速率來看4 ,1,5-與 1,6-環化 速率差異不大,因此就自由基 12 而言, 1,5-氫轉移可能是一個較嚴重的競爭 反應。 此外我們也合成了α−碸基醛 17,接著進行關鍵的自由基環化反應,如流程 3 所示,其反應條件與之前α−矽基醛的環化反應條件一樣,使用的 Bu3SnH 當量 數為 1.55 當量。反應的結果並沒有得到我們所要的環合產物,而是得到直接還 原的產物 18。還原產物的來源可能是α−碸基醛 17 經錫自由基作用後形成自由 基 19,之後再抓取三丁基錫烷上的氫而得到化合物 18,另一來源可能是形成自 H O TBS AIBN (cat.), Bu3SnH PhH, 80 ℃, 4 h H O TBS + H O Br TBS 9 10 11 2 : 1 H O TBS Bu3Sn Bu3SnH O TBS H O TBS O TBS cyclization cyclization β -scission β -scission 1,5-H shift Bu3SnH Bu3SnH 12 13 14 15 (67%) H O TBS 10 Bu3SnH 16 1,5-H shift H
由基 19 後先進行分子內 1,5-氫轉移得到自由基 20,再抓取氫原子而得到還原 產物 18。對於只得到直接還原產物的結果,這表示α−矽基自由基的抓氫速度 還是決定整個環化反應能不能成功的主要因素。 Scheme 3 由上面研究結果顯示在α−矽基醛的系統中,無法得到環合的產物,其可能原 因有二,一為矽基轉移的速率太慢或是根本不會轉移,才會使得當自由基加成 到羰基後形成烷氧自由基時,在矽基還沒發生轉移前就發生β−斷裂而開環, 所以得到的產物除了直接還原的產物外,還有自由基加成到羰基再開環後的產 物。二為第三丁基二甲基矽基的立體障礙太大,所以自由基加成到羰基時,烷 氧自由基與矽基大部分都為反式,所以無法得到環合產物。 在α−碸基醛的系統中,利用碸基去抑制 1,5-氫轉移的結果並不理想,仍然有 1,5-氫轉移的情形發生,在α−矽基自由基的系統中,沒有得到我們要的環合 產物,顯示α−矽基自由基抓氫的速度遠快於加成到羰基的速度,而且我們也 無法確定反應過程中有沒有發生環化。而在未含矽基的碳自由基系統中,其環 化反應的結果可得到些許的環合產物,顯示α−矽基自由基抓氫的速度是決定 環化反應能否發生的主要因素,而且自由基的立體障礙較小似乎有利於環化反 應的進行。 Part II
Pyrrolizidine alkaloids are important class of compounds that continued to attract the attention of synthetic chemists.1,2 Several years ago (Scheme 1), we developed an α-sulfonyl free radical cyclization strategy in the synthesis of supinidine (3), the most simple unsaturated necine base of the pyrrolizidine alkaloids.3 In this letter, we wish to report the successful stereoselective synthesis of (+)-heliotridine (4) and (−)-retronecine (5) using similar methodology.
Our synthetic strategy started from enantio-pure imide 6 (Scheme 2) which can TMS Br H O SO2Ph AIBN(cat.), Bu3SnH PhH, 80 ℃, 4 h TMS H O SO2Ph Bu3Sn TMS H O SO2Ph TMS H O SO2Ph 1,5-H shift Bu3SnH Bu3SnH 17 18 41 % 19 20
be synthesized easily from (−)-malic acid.4
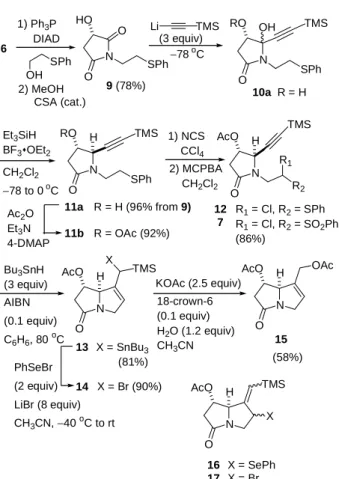
The pre-existing stereogenic center in imide 6 can be transformed into C-1 of (+)-heliotridine (4) and (−)-retronecine (5). Relying on the oxygen functionality of this stereogenic center, we planned to construct the adjacent chiral center in a controlled fashion. As shown in Scheme 3, imide 6 was coupled with 2-phenylthioethanol using triphenylphosphine and diisopropyl azodicarboxylate (DIAD).5 The resulting imide acetate product was then stirred in methanol with the presence of catalytic amount of camphor sulfonic acid (CSA) to afford imide alcohol 9 in 78% yield. Treatment of imide alcohol 9 with excess lithium trimethylsilylacetylide (3 equiv) gave a mixture of diastereomeric lactam diol 10a. This diol mixture was directly reduced with triethylsilane and borontrifluoride etherate to give lactam 11a as a single isomer (96% yield from 9).6 The stereocontrol of the reduction step was excellent.
Scheme 1 N O SiMe3 SO2Ph Cl 1 Bu3SnH (2 equiv) N O Bu3Sn SiMe3 N OH 2 3 supinidine N OH 4 (+)-heliotridine N OH 5 (−)-retronecine HO H HO H 2 5 3 7 1 6 7a 8 4 Scheme 2 N O SiMe3 SO2Ph Cl NH AcO H O O AcO N O SiMe3 SO2Ph Cl AcO H 8 6 7 5 4
In fact we have put different blocking group on the C-4 hydroxyl group of lactam diol 10a to examine the stereoselectivity of the reduction (Table 1). In the case with triethylsilane and borontrifluoride etherate (entries 1−4), the reduction of the unprotected lactam diol 10a (entry 1) gave the highest yield with excellent
trans-stereoselectivity. Even when we used a bulky t-butyldiphenylsilyl group to
major product (11d/cis isomer = 75/25). These stereochemical outcomes can be rationalized by the chelation effect of the C-4 oxygen substituent that directs the hydride attack from the same side.6a,7 The stereoselectivity was converted into slight preference of the cis-isomer (entries 5−7) when using sodium cyanoborohydride in acetic acid. However, surprisingly the reduction of 10d (entry 8) under this condition still gave 11d as the major product.
Scheme 3 N O X TMS N O TMS N RO OH O O HO N O TMS R2 R1 AcO H SPh 9 (78%) 6 SPh 10a R = H N O TMS RO H SPh 11a 11b R = H (96% from 9) R = OAc (92%) 12 7 R1 = Cl, R2 = SPh R1 = Cl, R2 = SO2Ph (86%) AcO H 13 14 X = SnBu3 (81%) N O OAc AcO H 15 N O TMS AcO H X 16 17 X = SePh X = Br 1) Ph3P DIAD SPh OH 2) MeOH CSA (cat.) Li TMS −78 oC (3 equiv) Et3SiH BF3OEt2 CH2Cl2 −78 to 0 oC Ac2O Et3N 4-DMAP 1) NCS CCl4 2) MCPBA CH2Cl2 Bu3SnH (3 equiv) AIBN (0.1 equiv) C6H6, 80 oC X = Br (90%) PhSeBr (2 equiv) LiBr (8 equiv) CH3CN, −40 oC to rt KOAc (2.5 equiv) 18-crown-6 (0.1 equiv) H2O (1.2 equiv) CH3CN (58%)
Table 1 The stereochemical outcome of the reduction of lactam carbinols 10a-d.
Entry Substrate Methoda Products (ratio of trans/cis)b Yields (%)
1 10a A 11ac 96
2 10b R = Ac A 11bc 17 3 10c R = Bn A 11cc 51 4 10d R = TBDPS A 11d + cis isomer (75/25) 68d 5 10a B 11a + cis isomer (40/60) 70d 6 10b B 11b + cis isomer (43/57) 72d 7 10c B 11c + cis isomer (40/60) 73d 8 10d B 11d + cis isomer (60/40) 40d
a
Method A: Et3SiH, BF3·OEt2, CH2Cl2, −72 to 0 o
C; Method B: NaBH3CN, HOAc, rt. b
The stereochemistry was determined by nOe experiments. cOnly observed the trans isomer.
d
The two isomers can be separated easily by silica gel column chromatography. The yields are combined isolation yields.
With lactam 11a in hands, the hydoxyl group was then protected as acetate by the reaction with acetic anhydride to afford 11b (92%). Treatment of 11b with NCS in carbon tetrachloride gave the α-chlorosulfide8
12 that was directly oxidized with MCPBA without purification to give α-chloro sulfone 7 (86%). The reaction of 7 with excess tributyltin hydride (3 equiv) afforded bicyclic lactam 13 (81%) as a mixture of two isomers epimeric at the exo-cyclic chiral center. This process3 involved the generation of α-sulfonyl radical9,10
18 first (Scheme 4). Intramolecular cyclization of 18 gave allyl sulfone 19. Further addition of tributyltin radical to the allyl sulfone moiety of 19 yielded the SR2’ product 13.11
Scheme 4 N O H SiMe3 N O SiMe3 AcO H SO2Ph AcO H 7 Bu3Sn 18 Bu3SnH Bu3Sn −PhSO2 SO2Ph 13 19 Scheme 5 N O X Y N O TMS N HO H O OMe HO N O TMS SO2Ph Cl AcO H 20 (95%) 9 SPh AcO H 22 23 24 X = SnBu3, Y = SiMe3 (58%) X = Br, Y = SiMe3 (97%) X = OAc, Y = H (84%) 21 (80%) 8 (85%) 5 (63%) 1) NaBH4 0 oC THF/EtOH 2) p-TsOH MeOH 1) ClEt2Si(CCTMS) (1.5 equiv) Et3N, 4-DMAP CH2Cl2, rt SPh 2 2) TiCl4 (3.7 equiv) −78 oC 1) Ac2O 2) NCS 3) MCPBA 1) Bu3SnH (2.5 equiv) AIBN (cat.), C6H6, 80 oC 2) PhSeBr (2 equiv) LiBr (8 equiv) CH3CN, −40 oC to rt 3) KOAc, 18-crown-6 H2O, CH3CN, rt LAH, THF, ∆
The final stage of the synthesis requires the conversion of the allyl moiety to an allylic alcohol. This was accomplished by the reaction of bicyclic lactam 13 with two equivalents of phenylslenenyl bromide in acetonitrile to obtain bromide 14 in 90% yield.3 This process involved the formation of selenide 16 which reacted further with phenylslenenyl bromide to give bromide 14. Note that large excess of lithium bromide (8 equiv) was added to facilitate the reaction. Without the addition of lithium bromide the initial formation of selenide 16 was always containminated with allyl bromide 17. Bromide 14 was displaced with potassium acetate in the presence of 18-crown-6. The silyl group was also removed in the same step when small amount of water was present. This led to the formation of diacetate 1512,13 in 58% yield. The conversion of diacetate 15 to (+)-heliotridine (4) has been reported by Hart et. al.13a,b
Scheme 6 N O OMe O SPh 25 Si SiMe3 N O O SPh 26 Si SiMe3 TiCl4 N O SPh Si O SiMe3 27 19
As mentioned above, sodium cyanoborohydride reduction of lactam diol 10a in acetic acid (Table 1, entry 5) afforded the cis-isomer of lactam 11a as the major product. However, the stereoselectivity of this reduction is not satisfactory for the synthesis of (−)-retronecine (5). We decided to employ the methodology reported by Vaseela and Bürli14 to intramolecularly deliver the trimethylsilylacetylene group with the help of the adjacent hydroxyl group. In this direction (Scheme 5), we started from imide 9 and regioselectively reduced the carbonyl group adjacent to the hydroxyl group using sodium borohydride under the condition reported by Speckamp.15 The resulting crude α-acylamino alcohol was converted directly to the methyl ether 20 (95%) in methanol with catalytic amount of p-toluenesulfonic acid. The methyl ether 20 was first silylated with diethyl trimethylsilylethynyl chloro silane14 in the presence of triethylamine and catalytic amount of 4-DMAP in dichloromethane. The reaction mixture was then cooled to −78 o
C followed by the addition of excess titanium tetrachloride (3.7 equiv). This one pot process successfully gave the lactam alcohol 21 with a cis-relationship of the hydroxyl and acetylenic groups. This reaction involved the formation of silyl ether 25 (Scheme 6) first. The addition of titanium tetrachloride to the solution of 25 generated the acyliminium ion 26 that further cyclized to form the vinyl cation intermediate 27.
Desilylation of 27 regenerated the acetylenic group at the same side of the transporting oxygen atom. The rest of the synthesis was carried out in a similar fashion as in the case of (+)-heliotridine (4). Thus, as shown in Scheme 5, acetylation of lactam 21 followed by chlorination and oxidation afforded α-chloro sulfone 8 in an 85% overall yield. Radical cyclization of 8 provided bicyclic lactam 22 (58%). The bicyclic lactam 22 was then converted to allyl bromide 23 (97%). Substitution of bromide 23 with potassium acetate gave the desilylated diacetate 24 (84%). Finally, lithium aluminum hydride reduction of 24 produced (−)-retronecine (5)16
in 63% yield.
In summary, starting from (−)-malic acid derived imide 6 we were able to synthesize either (+)-heliotridine (4) or (−)-retronecine (5) with high stereoselectivity. Relying on the pre-existing stereogenic center in imide 6, we could construct the newly formed adjacent chiral center at will. The B-ring of the target alkaloids was formed through a key radical cyclization strategy involving α-sulfonyl radical. The methodology developed here has the potential to be extended to the synthesis of the more functionalized alkaloids of the same type.
References of Part I:
1. Chang, S.-Y.; Jiaang, W.-T.; Cherng, C.-D.; Tang, K.-H.; Huang, C.-H.; Tsai, Y.-M. J. Org. Chem. 1997, 62, 9089. (b) Jiaang, W.-T,; Tang, K.-H.; Chang, L.-B.; Tsai, Y.-M. J. Org. Chem. 1999, 64, 618. (c) Huang, C.-H.; Chang, S.-Y.; Wang, N.-S.; Tsai, Y.-M.; J. Org. Chem. 2001, 66, 8983.
2. Jackon, R. A. J. Organomet. Chem. 1979, 166, 17.
3. (a)Wilt, J. W. J. Am. Chem. Soc. 1981, 103, 5251. (b) Wilt. J. W.; Lusztyk, J.;
Ingold, K. U. J. Am. Chem. Soc. 1988, 110, 281.
4. (a)Beackwith, A. L. J.; Hay, B. P. J. Am. Chem. Soc. 1989, 111, 230. (b) Beackwith, A. L. J.; Hay, B. P. J. Am. Chem. Soc. 1989, 111, 2674. (c) Beackwith, A. L. J., Raner, K. D. J. Org. Chem. 1992, 57, 4954.
References of Part II:
1. Mattocks, A. R. in Chemistry and Toxicology of Pyrrolizidine Alkaloids; Academic Press: New York, 1986.
2. Liddell, J. R. Nat. Prod. Rep. 2002, 19, 773−781.
3. Tsai, Y.-M.; Ke, B.-W.; Yang, C.-T.; Lin, C.-H. Tetrahedron Lett. 1992, 33, 7895−7898.
4. Chamberlin, A. R.; Chung, J. Y. J. Am. Chem. Soc. 1983, 105, 3653−3656. (b) Choi, J.-K.; Hart, D. J. Tetrahedron 1985, 41, 3959−3972.
5. Mitsunobu, O. Synthesis 1981, 1. (b) Hughes, D. L. Org. React. 1992, 42,
335−656.
J. X. Tetrahedron 1998, 54, 12547−12560. (b) Yoda, H.; Kitayama, H.; Yamada, W.; Katagini, T.; Takabe, K. Tetrahedron Asym. 1993, 4, 1451−1454. 7. For alternative explanations, see: (a) Cieplak, A. S. Chem. Rev. 1999, 99,
1265−1336. (b) Ohwada, T. Chem. Rev. 1999, 99, 1337−1375. 8. Dilworth, B. M.; Mckervey, M. A. Tetrahedron 1986, 42, 3731−3752. 9. Ke, B.-W.; Liu, C.-H.; Tsai, Y.-M. Tetrahedron 1997, 53, 7805−7826. 10. Paquette, L. A. Synlett. 2001, 1−12.
11. Ueno, Y.; Aoki, S.; Okawara, M. J. Am. Chem. Soc. 1979, 101, 5414−5415. 12. The spectroscopic data of this material is identical to that reported in the
literature (ref. 4, 13). [α]D22.8 = +34.7o (c, 0.92 in CHCl3) [lit.13b [α]D25 =
+34.4o (c, 2.2 in CHCl3)].
13. Dener, J. M.; Hart, D. J. Tetrahedron 1988, 44, 7037−7046. (b) Kametani, T.; Yukawa, H.; Honda, T. J. Chem. Soc. Perkin Trans. 1 1990, 571−577.
14. Vasella, A.; Bürli, R. Helv. Chim. Acta 1996, 79, 1159−1168.
15. Hubert, J. C.; Wijnberg, J. B. P. A.; Speckamp, W. N. Tetrahedron 1975, 31, 1437.
16. This material is identical to the reported product in all respect (ref. 17). [α]D23.1 = −53.2o (c, 1.15 in EtOH) [lit.18 [α]D20 = −52.9o (EtOH)].
17. Niwa, H.; Miyachi, Y.; Okamoto, O.; Uosaki, Y.; Kuroda, A.; Ishiwata, H.; Yamada, K. Tetrahedron 1992, 48, 393−412. (b) Drewes, S. E.; Antonowitz, I.; Kaye, P. T.; Coleman, P. C. J. Chem. Soc. Perkin Trans. 1 1981, 287−289.