腈正離子-烯烴環反應之研究:多取代喹啉與環亞胺的合成
94
0
0
全文
(2) 摘要 在有機合成方法學中,如何快速且高效率地合成含氮雜環分子一直都是相當 重要的研究課題,此類合成方法可被應用於有機材料的合成、藥物製程以及天然物 全合成中。在本論文中,我們透過不同種類之腈正離子與烯烴進行環化反應,成功 合成 3,4-二氫喹啉、3-羥基喹啉與環亞胺三種含氮雜環。 根據之前劉緒宗教授實驗室已發表的芳香基重氮氟硼酸鹽、腈以及炔類分子 的一鍋化合成喹啉反應進行修改,成功透過易製備且價格低廉的芳香基重氮氟硼 酸鹽與有機腈與烯類分子,進行不含金屬三成分一鍋化的合成反應以得到 3,4-氫化 喹啉,並觀察到 3,4-氫化喹啉與氧進行氣氧化後生成 3-羥基喹啉,進行最佳化條件 測試後,完成不含金屬三成分一鍋化的 3-羥基喹啉合成研究,並經實驗結果證實 其反應機制可能是涉及了自由基的氧化反應。 透過腈正離子的高活性,本研究成功設計出全新之腈正離子-烯烴環化反應, 使用高烯丙基醇或松油醇進行測試,成功分離出螺環與[3,3,1]壬烯產物並提出未來 可發展的腈正離子-多烯烴環化合成策略。. 關鍵字:腈正離子、環化反應、芳香基重氮鹽、3-羥基喹啉、環亞胺、里特反應、 有機合成方法學. i.
(3) Abstract In this study, the syntheses of 3,4-dihydroquinoline, 3-hydroxyquinoline and cyclic imine were developed based on different types of nitrilium-ene cyclization. This strategy utilized the in situ formation of reactive N-arylnitrilium intermediate, which then undergoes tandem cyclization with the consecutive formation of C-N and C-C bonds. Furthermore, unconventional oxidation of 3,4-dihydroquinolinium salts leading to 3hydroxyquinolines was revealed. Under basic conditions, 3,4-dihydroquinolinium salts could be transformed into the 3-hydroxyquinoline in the presence of oxygen gas. Such mild oxidation conditions and simple operation procedures provided this method a useful synthetic approach. Through mechanism studies, it was confirmed that this strange oxidation process may involve radicals. Lastly, a new type of nitrilium-ene cyclization reaction as a novel synthetic approach toward azacyclic structure was presented. Spirocyclic imine and 3-aza-bicyclo[3.3.1]nonene skeletons were prepared and isolated easily. Considering that the skeletons of quinoline and cyclic imine were widely found in compounds such as natural products and organic materials, this approach has the potential to benefit the society of synthetic chemistry.. Keywords: nitrilium ion, cyclization, aryldiazonium salt, 3-hydroxyquinoline, cyclic imine, Ritter reaction, organic methodology.. ii.
(4) 目錄 摘要………………………………………………………………....................................i Abstract…………...………………………………………………….............................ii 目錄………………………………………………………………..................................iii 圖目錄………………………………………………………………...............................v 表目錄………………………………………………………………..............................vi 流程目錄……………………………………………………………….........................vii 第一章. 緒論…………………………………………………………….……..... 1. 1.1. 研究動機……………...………………………………..………….….. 1. 1.2. 喹啉與環亞胺衍生物及其應用...………….…..……..………....…….3. 1.3. 3-羥基喹啉與環亞胺合成方法之文獻回顧………..…………………5. 1.4. 與腈正離子相關之研究回顧...…………………………………...…...8. 第二章. 3,4-氫化喹啉的合成與純化………………………….……………… 10. 2.1. 3,4-氫化喹啉之合成…………………..……………………...…....... 10. 2.2. 3,4-氫化喹啉的純化與氧化產物的鑑定………………..…….......... 11. 2.3. 多取代 3,4-氫化喹啉之合成………………………………………….12. 第三章. 3-羥基喹啉之合成…………………………………………………… 15. 3.1. 各種氧化劑之反應性測試……………..………….………….…….. 15. 3.2. 以氧氣作為氧化劑之進行最佳化…..…………..………………….. 17. 3.3. 3-羥基喹啉官能基耐受性測試…..…………………………………. 18. iii.
(5) 第四章. 里特反應衍生之腈正離子-烯烴環化反應…………...…………....... 22. 4.1. 里特反應衍生環化反應之反應設計………..……............…............ 22. 4.2. 高烯丙基醇與多取代松油醇之合成…………..…………………… 23. 4.3. 高烯丙基醇與有機腈類環化反應條件之最佳化………………….. 24. 4.4. 環亞胺產物之結構鑑定………………………..…………………… 28. 4.5. 環亞胺之生成機制…………………..……………………………… 33. 第五章. 腈正離子-烯烴環化的合成應用………..…………............................ 34. 5.1. Aristoteline 的簡潔合成策略探討…………………….……………. 34. 5.2. 以苯乙腈為模板的 Aristoteline 合成測試…………………..…….…. 37. 5.3. 松油醇之腈正離子-烯烴環化反應條件最佳化…………..………… 38. 5.4. 松油醇之腈正離子-烯烴環化的機制討論…………………..……… 39. 第六章. 結論…………………………………………………………………… 41. 第七章. 實驗部分……………………………………………………………… 43. 7.1. 實驗儀器……………………………………………………………. 43. 7.2. 試劑來源與前處理…………………………………………………. 43. 7.3. 實驗步驟與數據……………………………………………………. 44. 參考文獻……………………………………………………………………………… 56 附錄 化合物之光譜資料…………………………………………………………….. 59. iv.
(6) 圖目錄 Figure 1-1 以喹啉為骨架的衍生物..…………………………………………………. 3 Figure 1-2 以 3-羥基喹啉為骨架的衍生物................................................................... 3 Figure 1-3 具有生物活性之環亞胺結構…………………………………………....... 4 Figure 2-1 2-甲基-4-苯基-3-羥基喹啉質譜訊號(ESI-TOF) ……...…….………...… 11 Figure 4-1 環亞胺鹽 12’之 1H NMR 解析……………………………………....… 28 Figure 4-2 環亞胺鹽 12’之 13C NMR 解析……………………………………......… 29 Figure 4-3 環亞胺鹽 12’之 13C + DEPT NMR 解析…………………………............ 29 Figure 4-4 環亞胺鹽 12’之 HMQC 光譜解析……………………………….........… 30 Figure 4-5 環亞胺鹽 12’之 HMBC 光譜解析……………………………….........… 31 Figure 4-4 12’質譜訊號(ESI-TOF) ……………….………………………….........… 32 Figure 5-1 Aritoteline 與其他結構相近之 aristotelia-type 生物鹼………….........… 34. v.
(7) 表目錄 Table 2-1 欲研究之 3,4-氫化喹啉的合成……………………………….…………... 13 Table 2-4 烯烴合成氫化喹啉的失敗例子…………………………………...…….... 14 Table 3-1 各種氧化劑之反應性測試……………………………….………….......... 16 Table 3-2 以氧氣作為氧化劑之條件最佳化……………………….………….......... 17 Table 3-3 3,4-氫化喹啉氧化至 3-羥基喹啉的官能基耐受性…….…………............. 19 Table 3-4 一鍋化合成 3-羥基喹啉的官能基耐受性測試……...…………................ 20 Table 4-1 質子酸反應活性之測試……………………...…………..………….......... 24 Table 4-2 各種路易士酸反應活性之測試……………...…………..………….......... 25 Table 4-3 各種路易士酸在甲苯與乙腈混合溶液下反應活性之測試………........... 26 Table 4-4 三氟化硼-乙醚絡合物當量數對反應活性之測試……..…………........... 27 Table 5-1 不同溶劑、比例與溫度下環化反應的最佳化..…………..…………......... 38. vi.
(8) 流程目錄 Scheme 1-1 基於芳香基重氮氟硼酸鹽與腈的合成方法學……................................. 1 Scheme 1-2 本篇研究之 3,4-氫化喹啉與 3-羥基喹啉合成步驟……......................... 2 Scheme 1-3 氫化喹啉與環亞胺的合成策略……......................................................... 2 Scheme 1-4 弗里德蘭德喹啉合成反應…..................................................................... 5 Scheme 1-5 普菲青格反應............................................................................................. 5 Scheme 1-6 達參反應..................................................................................................... 6 Scheme 1-7 經由 3,4-氫化喹啉之喹啉合成方法.......................................................... 6 Scheme 1-8 喹啉官能基轉換......................................................................................... 7 Scheme 1-9 常見之環亞胺合成方法............................................................................. 7 Scheme 1-10 具有腈正離子中間體的 Name Reactions................................................ 8 Scheme 1-11 芳香基重氮化物與有機腈類合成菲啶................................................... 9 Scheme 2-1 席曼反應................................................................................................... 10 Scheme 2-2 本篇研究之 5aaa 合成方法..................................................................... 10 Scheme 2-3 芳香親電加成反應機制........................................................................... 12 Scheme 2-4 α-溴代苯乙烯生成喹啉............................................................................. 12 Scheme 3-1 生成 3,4-氫化喹啉與 3-羥基喹啉的反應機制........................................ 21 Scheme 4-1 里特反應與本研究之合成策略比較....................................................... 22 Scheme 4-2 高烯丙基醇 11 之合成方法...................................................................... 23 Scheme 4-3 松油醇衍生物 16a 之合成方法................................................................ 23 Scheme 4-4 環亞胺酸鹽 12’之合成方法..................................................................... 28. vii.
(9) Scheme 4-5 高烯丙基醇與腈生成環亞胺之反應機制................................................. 33 Scheme 5-1 Stevens 和 Kenney 的 Aritoteline 全合成.................................................. 34 Scheme 5-2 Darbre 等人的 Aritoteline 全合成............................................................. 35 Scheme 5-3 腈正離子-烯烴環化之 Aristoteline 合成策略......................................... 36 Scheme 5-4 苯乙腈之腈正離子-烯烴環化構想.......................................................... 37 Scheme 5-5 3-氮雜雙環[3.3.1]的建構與 21 的生成.................................................... 37 Scheme 5-6 化合物 21 的生成機制…………………………………………………. 39 Scheme 5-7 閉環機制的比較………………………………..………………………. 40. viii.
(10) 第一章 緒 論 1.1 研究動機 在過去五年來,劉緒宗教授實驗室陸續開發了以芳香基重氮氟硼酸鹽 (aryldiazonium tetrafluoroborate)與腈類(nitrile)為起始物合成喹啉(quinoline)1、喹唑 啉(quinazoline)2 與酮亞胺(ketimine)3 之合成方法(Scheme 1-1),本研究第一部分是 根據芳香基重氮氟硼酸鹽、烯烴與腈類合成 3,4-氫化喹啉的合成策略進行衍生化。 在合成 3,4-氫化喹啉的過程中,意外發現 3,4-氫化喹啉經氧化後可以得到少量的 3羥基喹啉產物,而目前文獻中對於 3-羥基喹啉的合成方法大多需要透過官能基的 轉換才能得到。若能尋找出從 3,4-氫化喹啉轉換到 3-羥基喹啉的最佳氧化條件,將 可以透過易於製備的芳香基重氮鹽、腈與烯烴,合成相對較為難合成的 3-羥基喹 啉,反應步驟如 Scheme 1-2 所示。此一方法可望提高 3-羥基喹啉之合成效率,可 應用於組合式化學合成(combinational synthesis),透過三成分一鍋化的合成策略大 量合成各種官能基排列組合的 3-羥基喹啉分子,擴大化學空間(chemical space),更 可能被應用於藥物與有機材料之合成。. Scheme 1-1 基於芳香基重氮氟硼酸鹽與腈的合成方法學. 1.
(11) Scheme 1-2 本篇研究之 3,4-氫化喹啉與 3-羥基喹啉合成步驟. 雖然芳香基重氮氟硼酸鹽與腈類提供了一個研究腈正離子的平台,但是芳香 基重氮氟硼酸鹽的使用也同時限制了其生成化合物之結構,即須帶有一個芳香環, 若能使接有飽和烷鏈的腈正離子與烯烴進行環化反應,便能合成環亞胺(cyclic imine)(Scheme 1-3),此一結構不僅大量出現於天然物與藥物之中,更可透過官能 基轉換合成醯胺、胺等含氮官能基,具有極高的應用性。於是本研究後半部分將根 據此一策略實現對環亞胺的合成。. Scheme 1-3 氫化喹啉與環亞胺合成策略. 2.
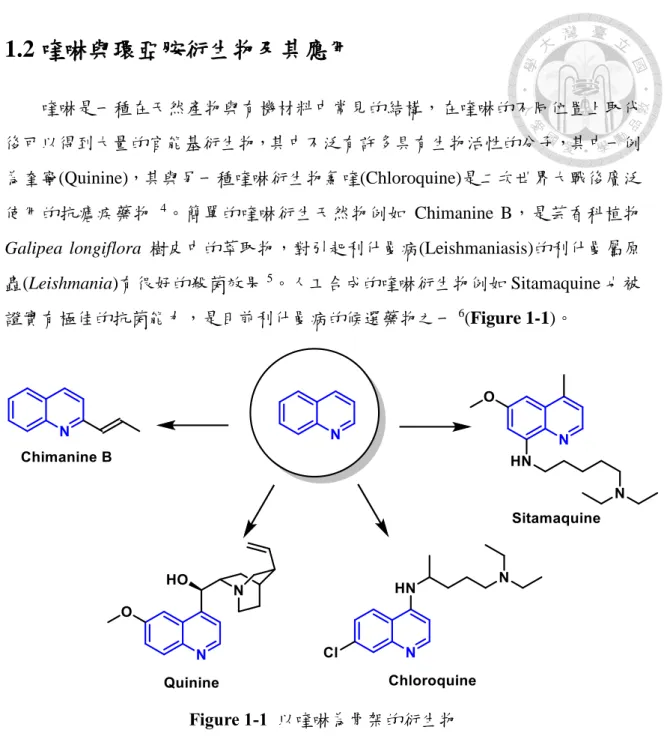
(12) 1.2 喹啉與環亞胺衍生物及其應用 喹啉是一種在天然產物與有機材料中常見的結構,在喹啉的不同位置上取代 後可以得到大量的官能基衍生物,其中不泛有許多具有生物活性的分子,其中一例 為奎寧(Quinine),其與另一種喹啉衍生物氯喹(Chloroquine)是二次世界大戰後廣泛 使用的抗瘧疾藥物 4。簡單的喹啉衍生天然物例如 Chimanine B,是芸香科植物 Galipea longiflora 樹皮中的萃取物,對引起利什曼病(Leishmaniasis)的利什曼屬原 蟲(Leishmania)有很好的殺菌效果 5。人工合成的喹啉衍生物例如 Sitamaquine 也被 證實有極佳的抗菌能力,是目前利什曼病的候選藥物之一 6(Figure 1-1)。. Figure 1-1 以喹啉為骨架的衍生物. 而在喹啉諸多衍生物中,3-羥基喹啉是相對重要但卻較少合成研究的含氮雜環 分子,在自然界中可以找到許多化合物即具有此一結構,例如在黏細菌 Stigmatella aurantiaca 中發現的天然物 Aurachins A 與 Aurachins B (Figure 1-2)即具有 3-羥基 喹啉的骨架,其作為喹啉生物鹼(alkaloids),被證實具有非常好的抗菌效果 7。化合 物 如 Talnetant 則 是 神 經 動 素 Neurokinin-3(NK3) 可 能 的 受 體 拮 抗 劑 (receptor antagonist),可用於治療大腸激躁症 8。PSI-697 則是 P 選擇蛋白(p-selectin)的抑制 劑 9(Figure 1-2)。. 3.
(13) Figure 1-2 以 3-羥基喹啉為骨架的衍生物. 環亞胺一般大多以五員環或六員環及七員環方式存在,其分子骨架中帶有一 碳氮雙鍵,常見於藥物分子的中間合成物以及天然物中 10,例如一種海洋生物毒素 (marine toxin) Portimine,便帶有螺環(spiro)的環亞胺結構 11,列舉部分環亞胺分子 於 Figure 1-3。. Figure 1-3 具有生物活性之環亞胺結構 4.
(14) 1.3 3-羥基喹啉與環亞胺合成方法之文獻回顧 目前文獻中已有許多喹啉的合成方法,但是僅有少數合成研究是同時合成喹 啉的含氮雜環結構並建構三號位的羥基,以下列舉出這些合成方法。. 弗里德蘭德喹啉合成反應(Friedländer quinoline synthesis)12:其反應具有兩種反 應路徑,其一是苯甲酮與另一分子的羰基進行羥醛反應(Aldol reaction)再脫去水, 再經苯胺基與羰基的亞胺化(imium formation),生成喹啉分子;其二為先經苯胺基 與與另一分子的羰基的亞胺化,再經羥醛反應與脫水生成 3-羥基喹啉。此反應的 缺點在於所需要的鄰苯胺基酮、醛類的合成較複雜,且合成的 3-羥基喹啉的 2,4 號 位取代基限制大(Scheme 1-4)。. Scheme 1-4 弗里德蘭德喹啉合成反應. 普菲青格反應(Pfitzinger reaction)13:普菲青格反應可視為弗里德蘭德反應的一 種變化,透過醯胺鍵在鹼中水解,再經過同弗里德蘭德反應的機制,合成 3-羥基喹 啉。反應物靛紅(isatin)可以經由桑德邁爾靛紅反應(Sandmeyer isatin synthesis)由苯 胺製備,相較於弗里德蘭德反應有著容易製備的優點(Scheme 1-5)。. Scheme 1-5 普菲青格反應. 達參反應(Darzen reaction)14:醇鈉與 α-鹵代羧酸酯反應後攻擊酮,生成環氧酸 酯後,再透過連二亞硫酸鈉還原硝基成胺基後,進行分子內苯胺基與羰基的亞胺化 生成 3-羥基喹啉(Scheme 1-6)。. 5.
(15) Scheme 1-6 達參反應. 目前 3-羥基喹啉的合成方法由於需要鄰苯胺基酮、靛紅或 α-鹵代羧酸酯作為 起始物,受限於此三樣物質的衍生物,合成出的 3-羥基喹啉難以引入各式官能基, 尚缺乏有效且易於製備的合成方法,因此在本研究的構想上,是經由 3,4-氫化喹啉 在 3 的-CH2-上進行氧化,合成 3-羥基喹啉,這樣的策略可以使喹啉上可以有更多 基團的引入,亦即合成方法可以容忍更多官能基的變化。 本實驗室與 Youn 的研究團隊於同一時間分別開發出以芳香基重氮氟硼酸鹽、 烯烴與腈類合成 3,4-氫化喹啉之合成 15,不同於 Youn 等人經由氧化或還原生成喹 啉與 1,2,3,4-四氫化喹啉,本研究透過單離出 3,4-氫化喹啉鹽類,進行氧化後得到 3-羥基喹啉(Scheme 1-7)。. Scheme 1-7 經由 3,4-氫化喹啉之喹啉合成方法. 除了直接建構 3-羥基喹啉之外,近年來,也有學者透過喹啉上其他官能基的 轉換,成功合成 3-羥基喹啉。在 2013 年,He 和 Jamison 成功開發出透過連續流合 成(continuous-flow synthesis)將喹啉的格里納試劑(Grignard reagent)轉換為 3-羥基 喹啉 16。在 2017 年,Chauhan 等人透過三號位接上酮基的喹啉,與 N-溴代丁二醯 亞胺(N-bromosuccinimide,NBS)作用後進行羥基化(hydroxylation)生成 3-羥基喹啉 17. (Scheme 1-8)。由於上述兩種合成方法需要先製備帶有取代基的喹啉,再進行官. 能基轉換,因此合成效率較差。. 6.
(16) Scheme 1-8 喹啉官能基轉換 16-17. 在環亞胺之合成中,最為常見是經由環化之含氮雜環,例如環胺(cyclic amine), 透過二級環胺的鹵化並進行脫去反應生成. 18. ,或是使用內醯胺(lactam)與有基鋰試. 劑反應生成 19(Scheme 1-9)。. Scheme 1-9 常見之環亞胺合成方法 18-19. 7.
(17) 1.4. 與腈正離子相關之研究回顧 腈正離子是有機反應中常見的中間體,除了是有機腈類水解的中間體之外,也. 是貝克曼重排反應(Beckmann rearrangement)20、施密特反應(Schmidt reaction)21、里 特反應(Ritter reaction)22 與烏吉反應(Ugi reaction)23 中的中間體(Scheme 1-10)。由於 腈正離子反應性極強,僅有透過六鹵化銻以及其他陰離子穩定的鹽類可在低溫下 製備並保存. 24. ,因此腈正離子的研究大多採用以在反應過程中生成(in situ)的合成. 方式。. Scheme 1-10 具有腈正離子中間體的 Name Reactions20-23. 8.
(18) 自上世紀 70 年代開始,不斷有研究團隊開發以芳香基重氮化物與有機腈類反 應生成腈正離子中間體的分子合成策略. 25-26. ,其中最早是由 Petterson 等人發現在. 芳香基重氮化物以有機腈類作為溶劑的熱分解(pyrolysis)中,成功單離出菲啶 (phenanthridines)以及喹唑啉 25(Scheme 1-11),證實了腈正離子的生成,此一策略使 得腈正離子生成受到控制,並可有效率地合成芳香類含氮雜環。. Scheme 1-11 芳香基重氮化物與有機腈類合成菲啶 25. 9.
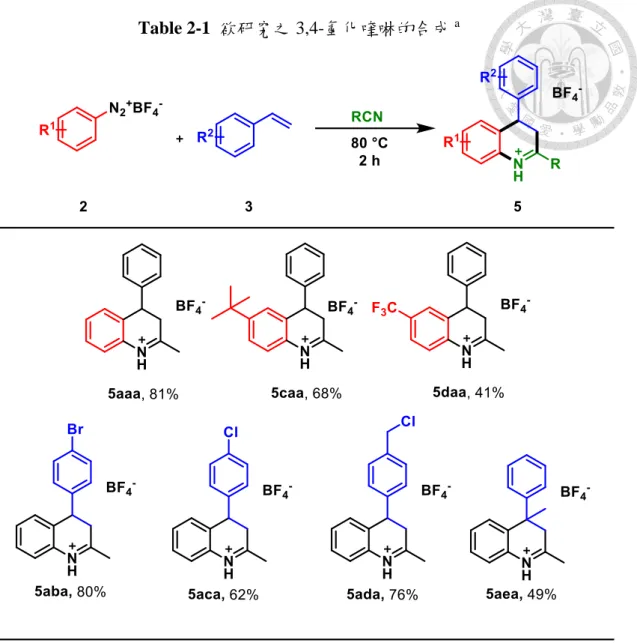
(19) 第二章 3,4-氫化喹啉的合成與純化 在本篇研究中所使用之芳香基重氮四氟硼鹽 2 均是選用市售之苯胺(aniline, 1) 經由席曼反應(Schiemann reaction)製備,各種苯乙烯(styrene 2a)及腈類均為市售之 化學物。. Scheme 2-1 席曼反應. 2.1 3,4-氫化喹啉之合成 根據先前本實驗室合成喹啉的結果 1,並參考 Youn 等人之合成方法. 15. ,由於. 中間產物腈正離子對水分子極為敏感,我們選定在無水乙腈(anhydrous acetonitrile) 4a 作為溶劑,添加苯基重氮四氟硼鹽 2a 一當量與苯乙烯 3a 兩當量在 80 °C 下反 應 2 小時,得到 81%的 2-甲基-4-苯基-3,4-氫化喹啉四氟硼酸鹽(2-methyl-4-phenyl3,4-dihydroquinolinium tetrafluoroborate) 5aaa。除此之外,在含水的乙腈下反應,可 透過薄層層析觀測到乙醯苯胺(N-phenylacetamide)的生成,推測應為腈正離子生成 後水分子進行親核加成所形成,與本實驗欲反應之苯乙烯產生競爭,也因此本反應 需在隔絕水分子的環境下反應。本實驗選用苯基重氮四氟硼鹽為限量試劑,苯乙烯 和乙腈為過量試劑,在反應結束後可觀察到過量的苯乙烯生成聚苯乙烯 (polystyrene),並在純化條件中將其分離。. Scheme 2-2 本篇研究之 5aaa 合成方法 10.
(20) 2.2 3,4-氫化喹啉的純化與氧化產物的鑑定 在處理化合物 5aaa 的純化時,首先嘗試了經鹼性水溶液中和後,透過傳統之 管柱層析(column chromatography)分離,我們意外發現在純化過程中儲存的試管內 不斷有白色固體析出,經核磁共振(nuclear magnetic resonance,NMR)與質譜(mass spectrometry)實驗後,根據所得到的數據,鑑定出該白色固體為 5aaa 的氧化產物 2-甲基-4-苯基-3-羥基喹啉(2-methyl-4-phenyl-3-hydroxyquinoline) 6aaa,化合物 6aaa 的高解析電灑游離質譜(ESI-mass)顯示 6aaa 之分子質量荷值比(m/z) = 236.1074 之 訊號,符合[M-BF4-]的陽離子片段,6aaa 之核磁共振圖譜資訊也與文獻值一致 17。. Figure 2-1 2-甲基-4-苯基-3-羥基喹啉質譜訊號(ESI-TOF). 根據此一現象,我們推論 3,4-氫化喹啉本身極易氧化,其反應驅動力(driving force)應來自於氧化生成具有芳香性的喹啉分子的共振能,造成在空氣下即可氧化 生成產物。為了能有效分離出高純度之 3,4-氫化喹啉,我們在反應結束後將溶液緩 慢倒入乙醚(diethyl ether)中,由於 3,4-氫化喹啉本身會捕捉經由芳香親電加成生成 的氫離子,在乙醚中會和四氟化硼陰離子生成穩定的 3,4-氫化喹啉四氟硼酸鹽,供 11.
(21) 後續之研究。. Scheme 2-3 芳香親電加成反應機制. 此一 3,4-氫化喹啉氧化生成 3-羥基喹啉之反應在文獻中從未有學者報導過, 在第三章將會對其氧化條件與反應機制進行探討。. 2.3 多取代 3,4-氫化喹啉之合成 藉由 Scheme 2-2 的合成方法,更換不同的芳香基重氮氟硼酸鹽、烯烴與有機 腈類的組合,可大量合成不同排列組合的 3,4-氫化喹啉 5,本篇論文將使用 5aaa5aea 等數個分子進行後續研究(Table 2-1)。. 我們也嘗試在苯乙烯之 α 位接上取代基,如此一來可製備出在四號位接有雙 取代基的氫化喹啉,如 Table 2-1,5afa,選用 α-溴代苯乙烯 3l,則發現會生成 2甲基-4-苯基喹啉(2-methyl-4-phenylquinoline) 6,推測其反應機制是生成 5ala 分子 後經由脫去反應(elimination)生成 6 (Scheme 2-4)。. Scheme 2-4 α-溴代苯乙烯生成喹啉. 12.
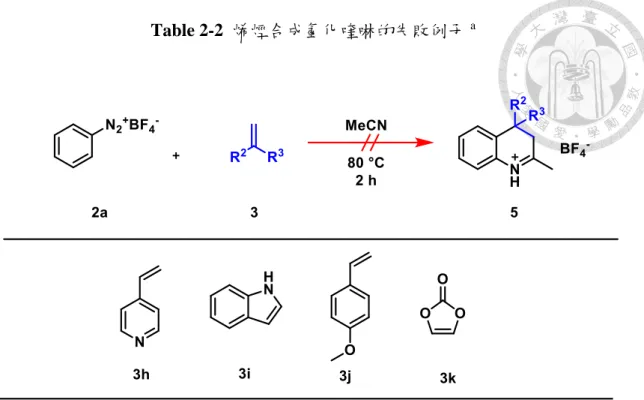
(22) Table 2-1 欲研究之 3,4-氫化喹啉的合成 a. 然而,也有一些烯烴衍生物無法在標準條件下生成 3,4-氫化喹啉,例如具有親 核性的氮原子的吡啶(pyridine)衍生物、吲哚(indole)以及電子密度較高的苯乙烯衍 生物以及碳酸亞乙烯酯(vinylene carbonate)都完全無法得到相對產物(Table 2-2,3h3k),推測是由於反應物的親核性較高時,易與重氮鹽或腈正離子發生副反應。. 13.
(23) Table 2-2 烯烴合成氫化喹啉的失敗例子 a. 14.
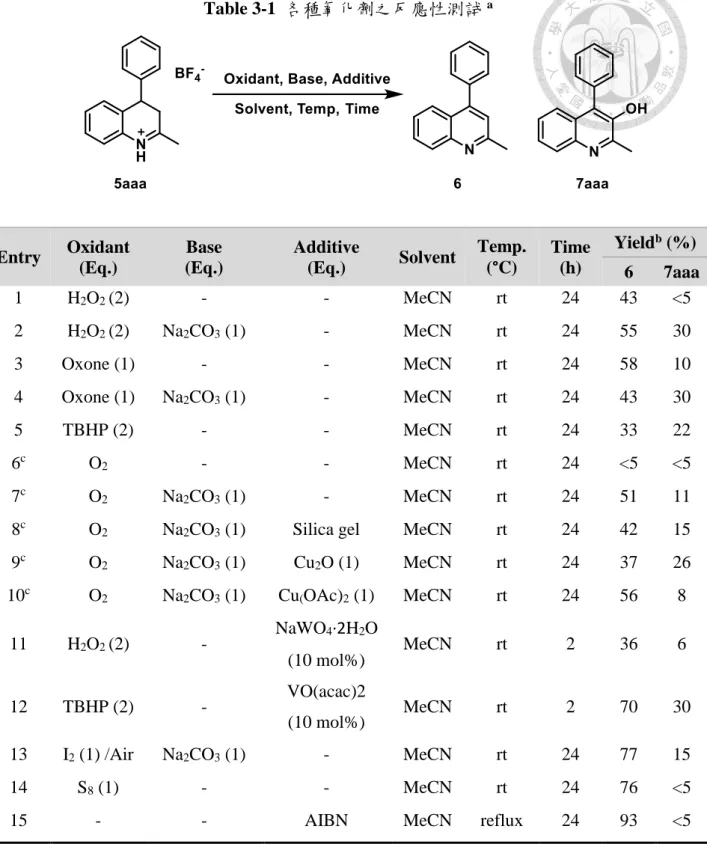
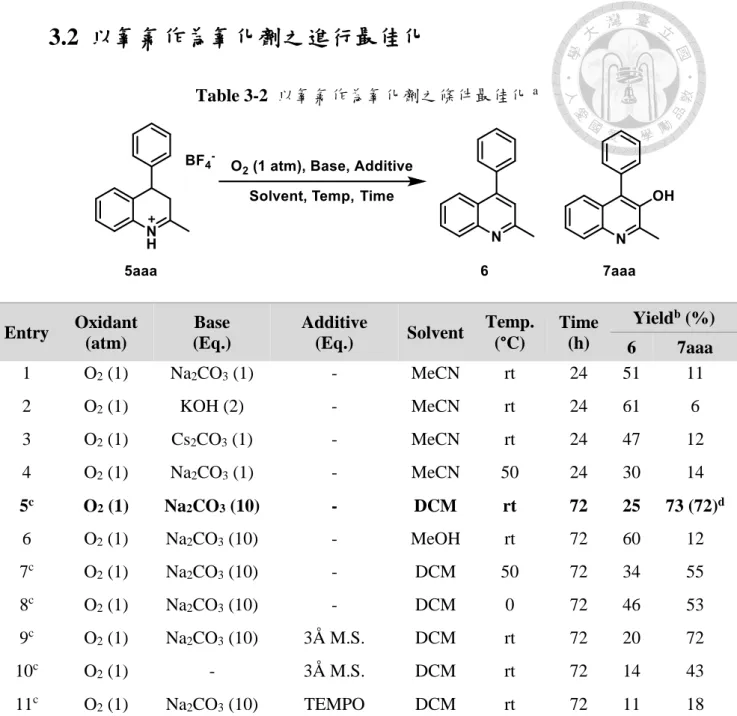
(24) 第三章 3-羥基喹啉之合成 在第二章有提及在純化 3,4-氫化喹啉中發現了 3-羥基喹啉的生成,其結果十 分特別,且在文獻中從未有學者報導此一發現,於是本研究接續 3,4-氫化喹啉的合 成,嘗試找到 3,4-氫化喹啉氧化到 3-羥基喹啉的最佳化條件,得到以芳香基重氮氟 硼酸鹽、烯烴與有機腈類的三成分一鍋化合成衍生應用。. 3.1 各種氧化劑之反應性測試 在氧化條件的測試之初,首先測試的是常見的有機化合物氧化試劑,例如:過 氧化氫(hydrogen peroxide)、Oxone、過氧化叔丁醇(tert-butyl hydroperoxide,TBHP)、 氧氣,均無法有效氧化成 3-羥基喹啉,而是得到喹啉為主的氧化產物(entry 1, 3, 5), 在氧氣下化合物 5aaa 幾乎無法進行氧化(entry 6)。添加碳酸納(sodium carbonate)作 為鹼源,發現氧化效率相對不添加鹼有顯著提升,雖然主要產物仍然以 6 為主要 產物,可觀察到 10-30%不等的 7aaa 生成(entry 2, 4, 7)。 由於最早觀察到的 3-羥基喹啉是在管柱層析後生成之沉澱物,我們懷疑矽膠 (silical gel)可能促進此氧化反應,但經過實驗結果排除了這一可能(entry 8)。參考文 獻中已知氧氣氧化反應 27,在氧氣條件下,加入一價及二價銅催化劑均無法有效提 升 7aaa 之產率(entry 9-10)。由於懷疑產物 7aaa 可能是是經由 6 生成,在烯烴的環 氧化條件下進行反應測試,仍然得到以 6 為主的產物(entry 11-12),將 entry 12 的 反應時間加長到 24 小時也無明顯產率變化。 除此之外,在進行非氧原子氧化劑的測試時,發現碘的添加會促進 6 的生成 (entry 13),添加偶氮二異丁腈(azobisisobutyronitrile,AIBN)作為自由基的測試中, 發現加入一當量的 AIBN 並以加熱起始自由基生成,可以得到將近等量的 6 生成 (entry 15),推測為 7aaa 生成二苯基自由基後經脫去反應得到。. 15.
(25) Table 3-1 各種氧化劑之反應性測試 a. Entry. Oxidant (Eq.). Base (Eq.). Additive (Eq.). Solvent. Temp. (°C). Time (h). 1. H2O2 (2). -. -. MeCN. rt. 2. H2O2 (2). Na2CO3 (1). -. MeCN. 3. Oxone (1). -. -. 4. Oxone (1). Na2CO3 (1). 5. TBHP (2). 6c. Yieldb (%). 24. 6 43. 7aaa <5. rt. 24. 55. 30. MeCN. rt. 24. 58. 10. -. MeCN. rt. 24. 43. 30. -. -. MeCN. rt. 24. 33. 22. O2. -. -. MeCN. rt. 24. <5. <5. 7c. O2. Na2CO3 (1). -. MeCN. rt. 24. 51. 11. 8c. O2. Na2CO3 (1). Silica gel. MeCN. rt. 24. 42. 15. 9c. O2. Na2CO3 (1). Cu2O (1). MeCN. rt. 24. 37. 26. 10c. O2. Na2CO3 (1). Cu(OAc)2 (1). MeCN. rt. 24. 56. 8. 11. H2O2 (2). -. MeCN. rt. 2. 36. 6. MeCN. rt. 2. 70. 30. NaWO4·2H2O (10 mol%). a. VO(acac)2. 12. TBHP (2). -. 13. I2 (1) /Air. Na2CO3 (1). -. MeCN. rt. 24. 77. 15. 14. S8 (1). -. -. MeCN. rt. 24. 76. <5. 15. -. -. AIBN. MeCN. reflux. 24. 93. <5. (10 mol%). Reaction conditions: a mixture of 5aaa (0.16 mmol) in the solvent (2 mL) was treated with oxidant as indicated in each entry. b NMR yield: 1,3,5-trimethoxybenzene was used as internal standard. c O2 (1 atm) balloon was used.. 鑒於諸多氧化條件均未能得到 7aaa 為主的產物,重新思考最初得到的 7aaa 生 成條件,僅有氧氣即可進行氧化,同時在其他氧化劑下,經薄層層析大多可觀察到 大量的副產物生成,因此選用氧氣作為氧化劑,進行後續的最佳化測試。 16.
(26) 3.2 以氧氣作為氧化劑之進行最佳化 Table 3-2 以氧氣作為氧化劑之條件最佳化 a. Solvent. Temp. (°C). Time (h). -. MeCN. rt. KOH (2). -. MeCN. O2 (1). Cs2CO3 (1). -. 4. O2 (1). Na2CO3 (1). 5c. O2 (1). 6. Entry. Oxidant (atm). Base (Eq.). Additive (Eq.). 1. O2 (1). Na2CO3 (1). 2. O2 (1). 3. a. Yieldb (%). 24. 6 51. 7aaa 11. rt. 24. 61. 6. MeCN. rt. 24. 47. 12. -. MeCN. 50. 24. 30. 14. Na2CO3 (10). -. DCM. rt. 72. 25. 73 (72)d. O2 (1). Na2CO3 (10). -. MeOH. rt. 72. 60. 12. 7c. O2 (1). Na2CO3 (10). -. DCM. 50. 72. 34. 55. 8c. O2 (1). Na2CO3 (10). -. DCM. 0. 72. 46. 53. 9c. O2 (1). Na2CO3 (10). 3Å M.S.. DCM. rt. 72. 20. 72. 10c. O2 (1). -. 3Å M.S.. DCM. rt. 72. 14. 43. 11c. O2 (1). Na2CO3 (10). TEMPO. DCM. rt. 72. 11. 18. Reaction conditions: a mixture of 5aaa (0.16 mmol) in the solvent (2 mL) was treated with oxidant as indicated in each. entry. b NMR yield: 1,3,5-trimethoxybenzene was used as internal standard. c Neutralized with NaHCO3 aqueous solution (5 mL). d Isolated yield.. 基於 Table 3-1, entry 7 的條件,將鹼換為氫氧化鉀與碳酸銫對產率無明顯影響 (Table 3-2, entry 1-3 ),將溫度升高到 50°C 也無明顯影響(entry 4)。由於 5aaa 鹽類 在 DCM 中溶解度較差,將鹽類以飽和碳酸氫鈉水溶液沖洗過後以 DCM 萃取,並 增加碳酸鈉到十當量除去多餘水分,可觀察到 73%的 7aaa 生成(entry 5),將反應 溶液換為甲醇則產率有顯著下降(entry 6)。維持 DCM 為反應溶劑,若將反應溫度 增高到 50 °C 或降低到 0 °C,可以發現 7aaa 產率都會降低,而 6 產率則上升(entry 7-8)。若在反應系統中加入活化過之 3Å 分子篩(molecular sieve,M.S.)產率無明顯 17.
(27) 變化,若不添加碳酸鈉而只加入分子篩,則產率會降低到 43%,反應轉化率也會降 低至約 60%,此一現象代表在反應當中碳酸鈉除了扮演鹼的角色之外,也有可能 是好的吸水劑,移除反應當中多餘的水分子。而若在標準條件下加入自由基淬滅劑 —四甲基哌啶氧化物(2,2,6,6-tetramethylpiperidine-1-oxy,TEMPO),則會發現無論 是 3-羥基喹啉或是喹啉的生成均會被抑制。. 3.2 3-羥基喹啉官能基耐受性測試 選用第二章中合成的 3,4-氫化喹啉添加 10 當量的碳酸鈉以及一大氣壓的氧氣 下,使用 DCM 作為溶劑,在室溫下經 72 小時的反應時間,比較經過後處理後的 3-羥基喹啉(7)產率(Table 3-3)。 更改喹啉上六號位的取代基,可以觀察到接有強拉電子基的-CF3 產率(7daa)與 接有推電子的甲基的產率(7baa)相比有明顯下降,但是甲基同樣比沒有取代基 (7aaa)的產率低。更換 2 號位的取代基,發現換成環丙烷以及苯環取代基,產率有 稍微下降(7aab, 7aac),更進一步換成立體障礙更大的三級丁基取代基,更是可觀 察到該 3,4-氫化喹啉不但在標準環境下無法完全氧化,在七天後得到起始物與少許 2-第三丁基-4-苯基喹啉 6aad 生成。這一結果證實了在涉及氧氣氧化生成 3-羥基喹 啉時,氧氣是否能觸及喹啉骨架之三號位影響了生成產物的比例與反應速率。除此 之外,若選用在喹啉骨架之四號位是雙取代的起始物,可以得到與無取代基接近產 率的 8afa 除了證實了喹啉骨架之三號位是氧化關鍵外,根據此項實驗證據可推論 3-羥基喹啉 7 的生成是經由 8,當 R3 是氫原子時再進行分子內異構化所生成。從 實驗結果亦可推論此氧化反應對喹啉二號位的取代基的影響較為明顯,帶有立障 越大的基團,氧化反應速率越慢。. 18.
(28) Table 3-3 3,4-氫化喹啉氧化至 3-羥基喹啉的官能基耐受性. 19.
(29) 3.3 一鍋化合成 3-羥基喹啉及反應機制探討 得到標準氧化條件後,我們試圖將 3,4-氫化喹啉與 3-羥基喹啉的合成合而為 一,從芳香基重氮氟硼酸鹽與腈與苯乙烯一鍋化合成出各式取代基的 3-羥基喹啉, 如此一來就能透過便宜、易於製備的化合物製備各式 3-羥基喹啉,而反應當中不 需要純化的方式也使之更便於操作。 其產率大致在 30 - 40%之間,得到的產率比 3,4-氫化喹啉合成產率乘上氧化產 率低一點(以無取代基為例,81% x 73% = 59%,而一鍋化合成僅僅得到 36%)。. Table 3-4 一鍋化合成 3-羥基喹啉的官能基耐受性測試 a.b. 整合文獻以及我們在氧化條件中針對立體障礙的討論、自由基的淬滅劑同時 抑制兩種氧化產物的結果,可能的反應機制是芳香基重氮鹽 2a 與乙腈產生腈正離 子後,經由分子間的親核加成,與分子內之芳香親電子取代,生成 3,4-氫化喹啉 5aaa,3,4-氫化喹啉分子上與自由基反應拔去一個氫原子時,有兩個可生成自由基 的位點,分別為 azaallylic 和 bisbenzylic。azaallylic 的自由基會傾向與氧氣氧化生 成 3-oxoquinoline,再經由分子異構化,生成有芳香性的 3-羥基喹啉 7aaa。bisbenzylic 位點的自由基則傾向經由脫去反應得到生成喹啉 6。 20.
(30) Scheme 3-1 生成 3,4-氫化喹啉與 3-羥基喹啉的反應機制. 第一部分之研究成果已於 2018 年發表於國際期刊,本篇學士論文作者為共同 第一作者 28。. 21.
(31) 第四章 里特反應衍生之腈正離子-烯烴環化反應 4.1 里特反應衍生環化反應之反應設計 回顧近年來多個團隊透過芳香基重氮化物與有機腈類反應生成腈正離子中間 體的分子合成策略. 15, 29. ,可以發現此一方法雖然有易於製備以及起始物價格低廉. 等等優點,但是芳香基重氮化物的使用讓分子結構局限於芳香性的骨架,於是瀏覽 許多有腈正離子中間體的人名反應並整理反應中腈正離子的生成方法,試圖透過 已知的腈正離子生成方法進行環化。其中最適宜的方法是參考里特反應 30-31。里特 反應多是經 SN1 反應,透過強酸或路易士酸(Lewis acid)產生碳陽離子,腈類上的 氮對碳陽離子進行親核加成,水解後生成醯胺。參考其使用三級醇類產生腈正離子 的機制,加入雙鍵烯烴進行環化反應,使用與第一部分類似的策略,進行另一種類 之腈正離子-烯烴環化反應,生成環亞胺結構,但此一策略會留下一個碳陽離子, 要如何淬息(quench)此碳陽離子是將會面臨的難題(Scheme 4-1)。. Scheme 4-1 里特反應與本研究之合成策略比較 22.
(32) 4.2 高烯丙基醇與多取代松油醇之合成 欲研究的反應所需之分子骨架為高烯丙基醇(homoallylic alcohol),目前文獻中 已有許多高烯丙基醇的合成方法,其中有兩種合成方法最為適宜,分別是格里納反 應和野崎—檜山—岸反應(Nozaki–Hiyama–Kishi reaction)32,透過烯丙基鹵烷與鎂 或鉻/鎳的反應系統以及適當的酮或醛類,便可以有效的合成高烯丙基醇。. Scheme 4-2 高烯丙基醇 11 之合成方法. 除此之外,選用同時具有烯烴與羥基的天然物 α-松油醇(terpineol)16a 進行合 成測試,也可透過狄耳士–阿爾德反應(Diels–Alder reaction)與格里納反應,合成 帶有不同取代基的松油醇衍生物 16。. Scheme 4-3 松油醇衍生物 16a 之合成方法. 23.
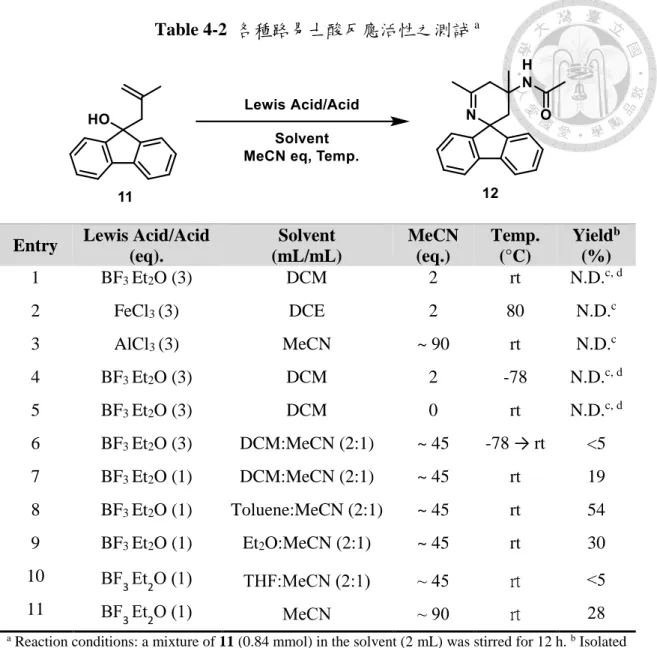
(33) 4.3 高烯丙基醇與有機腈類環化反應條件之最佳化 根據傳統的里特反應操作環境 22,以乙腈作為反應溶劑,加入硫酸進行測試, 其結果統整於 Table 4-1,在加入 5 當量的硫酸後,反應溶液的薄層層析得到非常 雜亂的結果(entry 1),而若降低酸的用量至 1 當量酸(相當於 0.5 當量硫酸,因為硫 酸是二質子酸),則可以收集到少於 5%的固體生成(entry 2),經質譜分析後確認環 亞胺產物 12 的生成,提高當量到添加 2.5 當量的硫酸,可以得到 20%的產率,若 降低溫度至-10°C,則可提高產率至 25%,選用 1 當量的氟硼酸,只能得到 12%的 產率。. Table 4-1 質子酸反應活性之測試 a. a. MeCN. MeCN (eq.) ~ 90. Temp. (°C) rt. Yieldb,c (%) N.D.. 1. Lewis Acid/Acid (eq.) H2SO4 (5). 2. H2SO4 (0.5). MeCN. ~ 90. rt. <5. 3. H2SO4 (2.5). MeCN. ~ 90. rt. 20. 4. H2SO4 (2.5). MeCN. ~ 90. -10. 25. 5. HBF4 (1). MeCN. ~ 90. rt. 12. Entry. Solvent. Reaction conditions: a mixture of 11 (0.84 mmol) in the solvent (2 mL) was stirred for 12 h. b Isolated. yield. c N. D. = Not detected.. 由於 11 分子結構中的雙鍵在酸性環境下有可能會被質子化,造成反應系統的 複雜化,也會導致產率偏低,副產物相當複雜,為了減少反應副產物以及增加官能 基的耐受度,於是我們捨棄了質子酸的使用,而開始對路易斯酸進行測試,並透過 更改溶劑比例測試反應性(Table 4-2)。. 24.
(34) Table 4-2 各種路易士酸反應活性之測試 a. 1. Lewis Acid/Acid (eq). BF3 Et2O (3). Solvent (mL/mL) DCM. MeCN (eq.) 2. Temp. (°C) rt. Yieldb (%) N.D.c, d. 2. FeCl3 (3). DCE. 2. 80. N.D.c. 3. AlCl3 (3). MeCN. ~ 90. rt. N.D.c. 4. BF3 Et2O (3). DCM. 2. -78. N.D.c, d. 5. BF3 Et2O (3). DCM. 0. rt. N.D.c, d. 6. BF3 Et2O (3). DCM:MeCN (2:1). ~ 45. -78 → rt. <5. 7. BF3 Et2O (1). DCM:MeCN (2:1). ~ 45. rt. 19. 8. BF3 Et2O (1). Toluene:MeCN (2:1). ~ 45. rt. 54. 9. BF3 Et2O (1). Et2O:MeCN (2:1). ~ 45. rt. 30. 10. BF3 Et2O (1). THF:MeCN (2:1). ~ 45. rt. <5. 11. BF3 Et2O (1). MeCN. ~ 90. rt. 28. Entry. a. Reaction conditions: a mixture of 11 (0.84 mmol) in the solvent (2 mL) was stirred for 12 h. b Isolated. yield. c N. D. = Not detected. d Solution turns into dark purple color right after adding lewis acid.. 降 低 乙 腈 用 量 並 選 用 不 具 有 配 位 能 力 的 溶 劑 如 DCM 及 二 氯 乙 烷 (dichloroethane, DCE),對三氟化硼-乙醚絡合物(boron trifluoride-diethyl etherate)、 三氯化鐵(iron (III) trichloride)和三氯化鋁(aluminum (III) trichloride)三種路易斯酸進 行測試後,均得到難以分析的混合溶液(entry 1-3),於是使用三氟化硼-乙醚絡合物 繼續測試其他條件。由於懷疑是在室溫下產生副反應分解產物,將溫度降低至-78 °C,卻得到同樣複雜的產物(entry 4),並且溶液在加入路易士酸的一瞬間就會產生 明顯的顏色變化,為了確認該顏色變化與腈正離子的生成有無關係,在不添加乙腈 的狀態下,會得到與 entry 1 接近的 NMR 光譜、薄層分析,推測這是由於過量的 路易士酸所造成的副反應。增加乙腈當量數為約 45 當量時,在-78°C 可明顯觀察 到經過一小時後溶液顏色無明顯變化,在升至室溫時溶液顏色漸漸轉棕,可收集到 25.
(35) 不到 5%之 12 生成,將三氟化硼-乙醚絡合物的用量降至 1 當量後得到 19%的產物 12(entry 7),而此產物經後續分析是 8a 與三氟化硼的絡合物,在更改各種溶劑後 (entry 7-11),我們發現在甲苯與乙腈的混合溶劑中得到最高的產率(54%,entry 8)。. Table 4-3 各種路易士酸在甲苯與乙腈混合溶液下反應活性之測試 a. 1. Lewis Acid/Acid (eq). FeCl3 (1). Solvent (mL/mL) Toluene:MeCN (2:1). MeCN (eq.) ~ 45. Temp. (°C) rt. Yieldb (%) <5. 2. Zn(OTf)2 (1). Toluene:MeCN (2:1). ~ 45. rt. N.R.c. 3. Zn(OTf)2 (1). Toluene:MeCN (2:1). ~ 45. 50. N.R.c. 4. Cu(OTf)2 (1). Toluene:MeCN (2:1). ~ 45. rt. N.D.d. 5. TMSOTf (1). Toluene:MeCN (2:1). ~ 45. rt. N.D.d. 6. Tf2O (1). Toluene:MeCN (2:1). ~ 45. rt. N.D.d. 7. FeCl3 (1). Toluene:MeCN (2:1). ~ 45. rt. <5. 8. Al(O-i-Pr)3 (1). Toluene:MeCN (2:1). ~ 45. rt. N.R.c. Entry. a. Reaction conditions: a mixture of 11 (0.84 mmol) in the solvent (2 mL) was stirred for 12 h. b Isolated. yield. c N. R. = No reaction. d N. D. = Not detected.. 以 Table 4-2 得到的溶劑比例下對更多路易斯酸進行測試(Table 4-3,entry 18),但是均無法得到可分析的 12 產物,其中在三氯化鐵均可觀察到及少量的產物 生成,若選用三氟甲磺酸鋅 Zn(OTf)2,和異丙醇鋁 Al(O-i-Pr)3,則起始物 10 無法 進行反應。在三氟甲磺酸銅 Cu(OTf)2 與三氟甲磺酸三甲基硅酯 TMSOTf 和三氟甲 磺酸酐 Tf2O 等酸性環境下均得到複雜的生成物。. 26.
(36) Table 4-4 三氟化硼-乙醚絡合物當量數對反應活性之測試 a. 1. Lewis Acid/Acid (eq). BF3 Et2O (0.75). Solvent (mL/mL) Toluene:MeCN (2:1). MeCN (eq.) ~ 45. Temp. (°C) rt. Yieldb (%) 19. 2. BF3 Et2O (1.0). Toluene:MeCN (2:1). ~ 45. rt. 54. 3. BF3 Et2O (1.5). Toluene:MeCN (2:1). ~ 45. rt. 43. 4. BF3 Et2O (2.0). Toluene:MeCN (2:1). ~ 45. rt. 24. 5. BF3 Et2O (1+0.5). Toluene:MeCN (2:1). ~ 45. rt. 37. 6. BF3 Et2O (1.0). Toluene:MeCN (2:1). ~ 45. -78. 33. 7. BF3 Et2O (1.0). Toluene:MeCN (2:1). ~ 45. -20. 36. 8. BF3 Et2O (1.0). Toluene:MeCN (2:1). ~ 45. 0. 43. Entry. a. Reaction conditions: a mixture of 11 (0.84 mmol) in the solvent (2 mL) was stirred for 12 h. b Isolated. yield.. 針對三氟化硼-乙醚絡合物進行當量數與溫度的測試後(Table 4-4),發現約等 當量的三氟化硼得到的產率最高,而若降低反應溫度,則產率降低,甚至有不反應 的狀況,其中 entry 6-7 直到回復到室溫後才開始反應,配合在混合低極性的甲苯 後反應產率上升,可以推測反應三氟化硼-乙醚絡合物需要大量釋放出三氟化硼後 將羥基轉為好的離去基反應才能進行的順利,而過量的三氟化硼又會造成副產物 的生成,而在分子內環化後生成的三級碳陽離子會與另一分子的乙腈進行里特反 應才能得到對應的 12 產物。選定產率最高的 entry 2 作為最佳化條件,進行後續合 成研究。. 27.
(37) 4.4 環亞胺產物之結構鑑定 根據 Table 4-1 entry 5 的條件,將 11 加入含有一當量氟硼酸,在反應結束後 將乙腈溶液滴入乙醚中即可收集 12’之固體沉澱。. Scheme 4-4 環亞胺酸鹽 12’之合成方法. 最初 12’在合成之時,無法確認其結構,且不確定其反應最後的碳陽離子會如 何被淬息,因此透過一維與二維 NMR 光譜進行結構解析,由 1H 積分值與分裂 (splitting)可知該 12’應有三個-CH3,並且由於無明顯 3J 耦合訊號,三個-CH3 應連 接著四級碳或異原子,而在芳香族訊號部分,可以觀察到芴(fluorene)的訊號分成了 八個不同的化學環境,表示其應具有不對稱的結構,並至少有一個手性中心。. Figure 4-1 環亞胺鹽 12’之 1H NMR 解析 28.
(38) Figure 4-2 環亞胺鹽 12’之 13C NMR 解析. Figure 4-3 環亞胺鹽 12’之 13C + DEPT NMR 解析. 29.
(39) 從 13C+DEPT 光譜結果可清楚發現這個 12’分子中有兩個-CH2-,且在芳香區 (120-140 ppm)之後往低場(downfield)的部分有兩根四級碳訊號,代表除了環亞胺 上氮旁邊的四級碳之外,分子結構中可能還有一個羰基或亞胺存在。. Figure 4-4 環亞胺鹽 12’之 HMQC 光譜解析 30.
(40) Figure 4-5 環亞胺鹽 12’之 HMBC 光譜解析. 12’之核磁共振光譜如 Figure 4-1~5 所示,透過二維的異核多量子相關光譜 (heteronuclear multiple quantum coherence, HMQC)中證實於 2.2 ppm 及 2.9 ppm 的 兩個氫是於同一個碳上,屬於 AB pattern,由於兩個氫旁有一光學中心,兩個氫的 化學環境並不對等且並相互產生 2J 耦合所導致。而 8.1 ppm 有一個氫並不是接於 碳上,可能是接在氮上的-NH,同時在 13.3 ppm 之訊號很明顯是亞胺離子的訊號, 因此可以推論 12’應至少有兩個氮原子。從異核多鍵相關光譜 heteronuclear multiple bond correlation, HMBC)可以得知 1H 圖譜中位於 1.83 ppm 的-CH3,只有連接著一 個位於 169.9 ppm 之四級碳,該四級碳另一端連接的是-NH,且無其他相近的訊號, 因此推論這是一個醯胺官能基。1H 圖譜中 2.53 ppm 之-CH3,與另一個應為亞胺四 級碳的 189.4 ppm 有空間相關,且與一-CH2-訊號接近,可推測這是經由腈正離子 生成之環亞胺旁的甲基訊號。從 1H 圖譜中 1.44 ppm 的-CH3 的附近有一個位於 47.9 之四級碳,以及這個-CH3 的氫與醯胺的四級碳有微弱之 4J 耦合訊號,可以推論應 為成環後生成與氮連接之四級碳。. 31.
(41) 化合物 12’的高解析電灑游離質譜顯示 12’之分子質量荷值比(m/z) = 319.1812 之訊號,符合[M] +的陽離子片段。. Figure 4-4 12’質譜訊號(ESI-TOF). 從核磁共振光譜與質譜的鑑定中,可以推論 12’應帶有一個螺環,並且除了反 應合成策略中設想的經由腈正離子-烯烴環化接上一個乙腈外,應有另一分子的乙 腈進行里特反應,生成醯胺。此一合成方法透過腈正離子環化後又與腈作用生成醯 胺,可稱為腈正離子-烯烴里特反應(nitrilium-ene Ritter reaction)。. 32.
(42) 4.5 環亞胺之生成機制 根據 12’的結構,可以推論此一腈正離子-烯烴環化反應機制應是經由路 易士酸活化羥基後生成碳陽離子,由乙腈氮上的孤對電子進行親核攻擊生成 C-N 鍵,生成腈正離子,由分子內之烯烴進行第二次親核攻擊生成 C-C 鍵, 生成碳陽離子後在與另一分子之乙腈生成腈正離子,經由里特反應之機制生 成醯胺。. Scheme 4-5 高烯丙基醇與腈生成環亞胺之反應機制. 33.
(43) 第五章 腈正離子-烯烴環化的合成應用 5.1 Aristoteline 的簡潔合成策略探討 Aritoteline 是在馬基果(maqui,學名為 Aristotelia chilensis)中被發現的一種 aristotelia-type 生 物 鹼 , 具 有 一 個 吲 哚 與 3- 氮 雜 雙 環 [3.3.1] 壬 烷 (3azabicyclo[3.3.1]nonane)的分子骨架(Figure 5-1)。Aritoteline 被證實有相當好的抗炎 性 33-34,但由於 Aritoteline 在馬基果與樹皮中的濃度低(約為 0.00001%)35,無法經 由萃取大量製備,因此經由有機合成 Aritoteline 是相對可行的製備方案,於 1980 年代已有多個研究團隊以不同合成策略進行合成,以下介紹兩個 Aristoteline 的全 合成方案。. Figure 5-1 Aritoteline 與其他結構相近之 aristotelia-type 生物鹼. Scheme 5-1 Stevens 和 Kenney 的 Aritoteline 全合成 34.
(44) Stevens 和 Kenney 於 1983 年提出以二價汞試劑成功將(-)-β-蒎烯(pinene)的四 員環開環,並與吲哚衍生之腈類反應,合成帶有環亞胺之雙環系統,他們透過該雙 環的立體選擇性還原,成功合成 Aritoteline(Scheme 5-1)36。但此一方法需要添加等 當量以上之二價汞試劑,基於重金屬之毒性,此一合成方法並不能提供理想的合成 製程。. Scheme 5-2 Darbre 等人的 Aritoteline 全合成. Darbre 等人於 1984 年提出 Aritoteline 的生物仿生合成(biomimetic synthesis), 參考 Aritoteline 的生物合成機制,以(-)-α-松油醇為起始物,將羥基轉為胺基後與 吲哚衍生的醛類經縮合,透過在甲酸下進行環化反應後合成 Aritoteline (Scheme 52)35。此一合成雖然是目前相當有效的 Aritoteline 合成方法,但由於其需經由胺基 化,造成其合成上略顯過於繁複。. 35.
(45) 在了解腈正離子-烯烴環化反應之機制後,並根據 Stevens 和 Kenney 所提出氫 化之立體選擇性,我們提出 Aristoteline 的逆合成分析如下。. Scheme 5-3 腈正離子-烯烴環化之 Aristoteline 合成策略. 根據第四章所展示的合成機制,若在反應系統之中,透過芳香親電加成,淬息 碳陽離子並建構 C-C 鍵,便可提供一個有效且簡潔的 Aristoteline 合成方法。. 36.
(46) 5.2 以苯乙腈為模板的 Aristoteline 合成測試 鑒於 3-吲哚乙腈 17 價格較昂貴,且為了方便試驗控制條件的更動,於是選用 苯乙腈為反應模板,若能透過苯乙腈根據相同的機制進行腈正離子-烯烴環化反應, 進行反應最佳化篩選後,再與 3-吲哚乙腈 17 進行 Aristoteline 之合成。. ’ Scheme 5-4 苯乙腈之腈正離子-烯烴環化構想. 使用 Table 4-2 entry 11 之條件進行測試,發現並無生成所預想之產物 20,而 會觀察到有 73%之 21 產物生成。. Scheme 5-5 3-氮雜雙環[3.3.1]的建構與 21 的生成. 37.
(47) 5.3 松油醇之腈正離子-烯烴環化反應條件最佳化 Table 5-1 不同溶劑、比例與溫度下環化反應的最佳化 a. 1. 19 (eq.) ~ 18. Temp. (°C) rt. 2. Benzyl cyanide 19 (2). ~ 18. 80. N.D.. 68. 5. rt. N.D.. -. 3. a. Yieldb,c (%) 20 21 N.D. 73. Solvent (mL:mL) Benzyl cyanide 19 (2). Entry. Toluene: Benzyl cyanide 19 (1.6:0.4). 4. Toluene (2). 1. rt. N.D.d. -. 5. Toluene (2). 1. reflux. N.D.d. -. 6. DCE (2). 5. 80. N.D.. -. 7. DCE (4). 2. rt. N.D.. -. 8. Toluene (4). 2. rt. N.D.. -. Reaction conditions: a mixture of 16 (0.65 mmol) and BF3·Et2O (0.65mmol) in the solvent was stirred. for 12 h. b Isolated yield. c N.D. = Not detected. d 16 decomposed.. 首先將反應溫度升高至 80 °C,只能得到與 21 生成,產率與室溫下接近(entry 2)。由於懷疑是過量的苯乙腈 19 與芳香親電加成互相競爭,於是將 19 的當量減少 至五當量,仍然只能觀察到 21 的生成(entry 3),進一步降低當量至一當量時可發現 溶液在加入三氟化硼-乙醚絡合物後又呈現起始物分解、溶液轉為棕紫色的現象 (entry 4-5),更換溶劑為高極性之 DCE,並將苯乙腈 19 修改為五當量,並加熱至 80 °C 也無法觀察到 20(entry 6),嘗試將溶液稀釋兩倍,在室溫下反應,也不能得 到好的條件合成出化合物 20(entry 7-8)。. 38.
(48) 5.3 松油醇之腈正離子-烯烴環化的機制討論 目前經過一系列之測試,均無法得到化合物 20,由於本反應是透過與第一個 苯乙腈生成腈正離子後合環,產生出的碳陽離子由原本所設計之芳香親電取代反 應生成多環產物 20,最後一步的合環反應是一個分子內的反應,其反應速率應快 於而與另一苯乙腈的分子間反應 (Scheme 5-6)。. Scheme 5-6 化合物 21 的生成機制. 但是在 Table 5-1 的所有 entry 中,都只能觀察到 21 的生成。因此,根據此一 現象進行討論,由於使用苯乙腈作為模板測試,在芳香親電取代反應中過度態 (transition state,TS)需打破之芳香共振能(conjugation energy)相較於吲哚要來得高, 也因此改成使用 3-吲哚乙腈將有可能可以增進環化產物的生成,除此之外,將本 篇研究的內容與已知的 Aristoteline 方法進行比較,前人的策略是先還原[3,3,1]壬 烯至[3,3,1]壬烷,最後才在酸性環境下封閉最後一個環,因此有可能是[3,3,1]壬烯 的結構因為環張力(ring strain)而不傾向生成環,並生成 21(Scheme 5-7)。 39.
(49) Scheme 5-7 閉環機制的比較. 在本反應系統中,目前已觀察到腈正離子-烯烴環化反應的發生,但仍無法達 成目標分子 Aristoteline 的合成,未來仍然需要繼續嘗試。除此之外,此一方向之 研究也需要開發更多能應用腈正離子-烯烴環化反應的分子骨架設計,除了在本研 究中所提出的喹啉、螺環與[3,3,1]壬烯產物之外,應仍有可合成之結構,未來還有 很長的路要走。. 40.
(50) 第六章 結論 在本篇論文中,使用了芳香基重氮氟硼酸鹽、腈與烯烴之三成分一鍋化反應合 成出 3,4-氫化喹啉,並成功透過 3,4-氫化喹啉易於氧化之特性,經由對氧化劑、酸 鹼性環境、溶劑與溫度的測試,開發出在氧氣下氧化 3,4-氫化喹啉生成 3-羥基喹啉 之反應。此氧化方法對一般的鹵素取代以及推拉電子基例如甲基以及三氟甲基均 有不錯的產率,惟在 3,4-氫化喹啉之二號位有立障較大的基團,例如叔丁基會不利 於 3-羥基喹啉的生成。本方法可以在一大氣壓之氧氣與室溫下,進行一個相當溫 和的氧化反應。 透過對此一氧化反應進行大範圍的化學環境測試,我們發現在此氧化反應中 添加自由基淬滅劑 TEMPO 後,3-羥基喹啉的生成被抑制,與添加自由基狀態下生 成喹啉之結果綜合比較,可以發現此氧化反應中很有可能涉及自由基的生成,以及 氮烯丙基自由基與芐位自由基的競爭。結合一鍋化生成 3,4-氫化喹啉之研究,我們 提出以芳香基重氮氟硼酸鹽、腈與烯烴一鍋化合成 3-羥基喹啉的合成方法。透過 易於合成之起始物,以及三成分均可變換之特性,此一反應可望能應用於帶有多取 代基之 3-羥基喹啉合成方法。. 基於腈正離子合成 3,4-氫化喹啉所展示出的高活性,以及里特反應的過往研 究,本篇論文成功設計出以羥基在路易士酸下,產生碳陽離子,與腈反應後轉化為 腈正離子的 in situ 生成方法,並與烯烴進行合環之新穎環亞胺合成策略,並發現 以三氟化硼-乙醚絡合物做為路易士酸有最佳的反應活性。此一反應成功串聯腈正 離子、烯烴合環與里特反應,於一步合成反應中建構三個化學鍵。並透過二維核磁 共振實驗與質譜解析合成的未知結構。. 41.
(51) 根據腈正離子-烯烴環化反應,本研究中也提出合成天然物的應用,雖然到目 前為止在測試中僅能得到副產物,但在反應過程中,確實能觀察到腈正離子與烯烴 的環化,此結果展現出此一策略具有可行性,除了此一應用外,也期待未來能將腈 正離子-烯烴環化合成策略應用於其他分子骨架的合成。. 42.
(52) 第七章 實驗部分 7.1. 實驗儀器. 1. 核磁共振光譜(nuclear magnetic resonance spectroscopy) 本 篇 論 文 中 所 提 及 的 核 磁 共 振 氫 譜 (1H NMR) 是 以 Bruker AVANCE-400 (400.13MHz)所測得,化學位移(chemical shift, δ)以 ppm 為單位,以溶劑訊號或 四甲基矽烷(tetramethylsilane, TMS)之δ= 0 作為核磁共振儀的內標值;核磁共 振碳譜(13C NMR),是以 Bruker AVANCE-400 (100.62 MHz)所測得,化學位移 以 ppm 為單位,耦合常數(coupling constant)以 J 來表示,其單位為 Hz,而分裂 形式(splitting pattern)定義如下:s,單峰(singlet);d,雙峰(doublet);t,三重峰 (triplet);q,四重峰(quartet);m,多重峰(multiplet);br,寬峰(broad)。 2. 質譜分析(mass spectrum analysis) ESI-Mass 之機型為 Water LCT Premier XE,將待測樣品溶於乙腈,注入量測管 柱內鑑定有機物之荷質比。 3. 管柱色層分析(column chromatography) 一般有機物的重力管柱層析以 Merck Silica gel 60 (40 – 63 μM)填充。 4. 薄層色層分析(thin layer chromatography, TLC) 將樣品以毛細管點於 Merck Art. 5544 precoated sheet 薄片後至於展開液中,再 以 254 nm 紫外光燈檢視。. 7.2. 試劑來源與前處理. 1. 溶劑之純化 (a) 無水乙腈:加入氫化鈣(calcium hydride),加熱回流數小時後於氮氣下蒸餾 純化,並保存於活化過之 3Å 分子篩中。 (b) 無水二氯甲烷:取自 MBRAUN Solvent Purification Systems (MB-SPS-800) , 並保存於活化過之 3Å 分子篩中。 (c) 無水甲苯:取自 MBRAUN Solvent Purification Systems (MB-SPS-800),並保 存於活化過之 3Å 分子篩中。 43.
(53) 2. 試劑來源 其他溶劑與試劑一般是直接由藥品供應商(Sigma-Aldrich Co., Acros Organic Co., Alfa Aesar Co., Merck Co. and TCI Co.)所供應之試藥級藥品、溶劑級藥品,如藥 品需要進一步純化或乾燥,則依標準方式處理純化。. 7.3. 實驗步驟與數據. 7.3.1 3,4-氫化喹啉之合成與鑑定 3,4-氫化喹啉的三成分一鍋化合成之反應步驟; 將 aryldiazonium tetrafluoroborate 2 (0.52 mmol)、styrene derivative 3 置於 10 mL 之密封管內,打入無水 nitrile 4 (2 mL),在 80°C 下反應 2 小時,反應結束後冷卻置 室溫,將溶液逐滴加入乙醚(30 mL)中,產生沉澱物,經抽氣過濾並乙乙醚沖洗數 次後,置於高真空下烘乾,可得 3,4-氫化喹啉之衍生物 5。. 2-Methyl-4-phenyl-3,4-dihydroquinolinium tetrafluoroborate (5aaa). 得白色固體 130 mg,產率 81%。mp: 185-186 °C 1H. NMR (400 MHz, CD3CN): δ 12.46 (br, 1H, N-H), 7.52 – 7.32 (m, 6H, Ar-H), 7.22 (d,. J = 7.2 Hz, 2H, Ar-H), 7.02 (d, J = 7.2 Hz, 1H, Ar-H), 4.50 (t, J = 8.9 Hz, 1H, -CH), 3.48 – 3.33 (m, 2H, -CH2), 2.66 (s, 3H, -CH3) ppm. 13C. NMR (100 MHz, CD3CN): 189.25, 141.84, 133.20, 132.66, 131.39, 130.51, 130.34,. 130.11, 129.28, 129.16, 121.80, 38.72, 38.67, 25.72 ppm. HRMS (ESI-TOF) calcd. for C16H16N (M - BF4) m/z = 222.1283; found: 222.1281.. 44.
(54) 6-(tert-butyl)-2-methyl-4-phenyl-3,4-dihydroquinolinium tetrafluoroborate (5caa). 得黃色固體 101 mg,產率 68%。mp: 190–191 °C 1H. NMR (400 MHz, CD3CN): δ 7.53 – 7.50 (m, 1H, Ar-H), 7.44 – 7.37 (m, 3H, Ar-H),. 7.20 – 7.18 (m, 2H, Ar-H), 7.14 (s, 1H, Ar-H), 4.48 (t, J = 8.4 Hz, 1H, -CH), 3.47 – 3.30 (m, 2H, -CH2), 2.62 (s, 3H, -CH3), 1.22 (s, 9H, -C(CH3)3) ppm. 13C. NMR (100 MHz, CD3CN): 187.54, 158.18, 141.93, 130.74, 130.59, 130.24, 128.83,. 127.43, 126.82, 121.20, 38.68, 38.59, 35.86, 31.21, 25.40 ppm. HRMS (ESI-TOF) calcd. for C20H24N (M - BF4) m/z = 278.1903; found 278.1914.. 2-methyl-4-phenyl-6-(trifluoromethyl)-3,4-dihydroquinolinium tetrafluoroborate (5daa). 得白色固體 81 mg,產率 41%。 1H. NMR (400 MHz, CD3CN): δ 7.79 (d, J = 8.2 Hz, 1H, Ar-H), 7.67 (d, J = 8.2 Hz, 1H,. Ar-H), 7.46 – 7.37 (m, 3H, Ar-H), 7.32 (s, 1H, Ar-H), 7.24 – 7.22 (m, 2H, Ar-H), 4.57 (t, J = 8.9 Hz, 1H, -CH), 3.56 – 3.39 (m, 2H, -CH2), 2.70 (s, 3H, -CH3) ppm. 13C. NMR (100 MHz, CD3CN): 192.42, 140.76, 135.59, 133.20, 132.87, 132.59, 130.48,. 129.29, 129.13, 127.18, 127.13, 127.11, 127.07, 125.80, 123.10, 122.48, 38.50, 38.36, 25.89 ppm.. 45.
(55) 4-(4-Bromophenyl)-2-methyl-3,4-dihydroquinolinium tetrafluoroborate (5aba). 得橘色固體 162 mg,產率 80%。mp: 145-147 °C 1H. NMR (400 MHz, CD3CN): δ 12.44 (br, 1H, N-H), 7.56 – 7.42 (m, 5H, Ar-H), 7.16-. 7.13 (m, 2H, Ar-H), 7.02 (d, J = 7.4 Hz, 1H, Ar-H), 4.50 (t, J = 8.8 Hz, 1H, -CH), 3.47 – 3.29 (m, 2H, -CH2), 2.65 (s, 3H, -CH3) ppm. 13C. NMR (100 MHz, CD3CN): 188.70, 140.77, 133.01, 132.77, 132.31, 131.03, 130.32,. 129.87, 122.06, 121.45, 38.06, 37.76, 25.31 ppm. HRMS (ESI-TOF) calcd. for C16H15N79Br (M-BF4): m/z = 300.0388; found: 300.0379. HRMS (ESI-TOF) calcd. for C16H15N81Br (M-BF4): m/z = 302.0368; found: 302.0359.. 4-(4-Chlorophenyl)-2-methyl-3,4-dihydroquinolinium tetrafluoroborate (5aca). 得黃色固體 111 mg,產率 62%。 mp: 161-162 °C 1H. NMR (400 MHz, CD3CN): δ 7.51 – 7.39 (m, 5H, Ar-H), 7.22 – 7.19 (m, 2H, Ar-H),. 7.03 (d, J = 7.2 Hz, 1H, Ar-H), 4.51 (t, J = 8.4 Hz, 1H, -CH), 3.48 – 3.29 (m, 2H, -CH2), 2.65 (s, 3H, -CH3) ppm. 13C. NMR (100 MHz, CD3CN): 188.66, 140.30, 134.00, 132.82, 132.34, 130.72, 130.43,. 130.04, 129.92, 129.46, 121.46, 38.16, 37.71, 25.31 ppm. HRMS (ESI-TOF) calcd. for C16H15N35Cl (M - BF4) m/z = 256.0893; found: 256.0897. HRMS (ESI-TOF) calcd. for C16H15N37Cl (M - BF4) m/z = 302.0367; found: 302.0369.. 46.
(56) 4-(4-(chloromethyl)phenyl)-2-methyl-3,4-dihydroquinolinium tetrafluoroborate (5ada). 得橘色固體 120 mg,產率 64%。mp: 146-147 °C 1H. NMR (400 MHz, CD3CN): δ 12.44 (br, 1H, N-H), 7.53 – 7.42 (m, 5H, Ar-H), 7.23 –. 7.21 (m, 2H, Ar-H), 7.04 – 7.02 (d, J = 7.2 Hz, 1H, Ar-H), 4.67 (s, 2H, -CH2Cl), 4.54 – 4.50 (t, J = 8.8 Hz, 1H, -CH), 3.49 – 3.32 (m, 2H, -CH2), 2.66 (s, 3H, -CH3) ppm. 13C. NMR (100 MHz, CD3CN): 188.71, 141.75, 138.49, 132.73, 132.29, 130.63, 130.42,. 129.92, 129.30, 121.42, 46.53, 38.13, 37.99, 25.30 ppm. HRMS (ESI-TOF) calcd. for C17H17N35Cl ((M - BF4): = 270.1050; found: 270.1050. HRMS (ESI-TOF) calcd. for C17H17N37Cl ((M - BF4): = 272.1015; found: 272.1058.. 2,4-dimethyl-4-phenyl-3,4-dihydroquinolinium tetrafluoroborate (5aea). 得白色固體 white solid (60 mg, 49%). 1H. NMR (400 MHz, CD3CN): δ 7.55 – 7.51 (m, 3H, Ar-H), 7.37 – 7.29 (m, 4H, Ar-H),. 7.21 – 7.19 (m, 2H, Ar-H), 3.71 – 3.66 (d, J = 18.8 Hz, 1H, -CHa(Hb)), 3.23 – 3.19 (d, J = 18.8 Hz, 1H, -C(Ha)Hb), 2.63 (s, 3H, -CH3), 1.72 (s, 3H, -CH3) ppm. 13C. NMR (100 MHz, CD3CN): 188.33, 144.81, 135.89, 132.53, 132.44, 129.74, 129.68,. 128.65, 128.39, 127.36, 121.70, 44.85, 40.02, 27.39, 25.47 ppm. mp: 171-172°C HRMS (ESI-TOF) calcd. for C17H18N ((M - BF4): = 236.1439; found: 236.1442.. 47.
(57) 7.3.2 3-羥基喹啉之合成與鑑定 3,4-氫化喹啉氧化生成 3-羥基喹啉或喹啉酮之反應步驟; 將 3,4-dihydroquinolinium tetrafluoroborate 5 (0.3 mmol)加入飽和碳酸氫鈉水溶 液(5 mL),以 DCM(5 mL x 3)萃取,合併有機層並加入過量無水硫酸鎂除去有機層 的水分,加入 sodium carbonate (3.0 mmol, 318 mg)在室溫下反應 72 小時,反應結 束後加入飽和 NH4Cl(aq)(5 mL),以 DCM(10 mL x 2)萃取,合併有機層並加入過量 無水硫酸鎂除去有機層的水分,將溶液旋乾後以管柱層析分離純化(silica gel, 2040% EtOAc in hexane),可得相對應的 3-羥基喹啉產物 7 或喹啉酮 8。. 3-羥基喹啉的三成分一鍋化合成之反應步驟; 將 aryldiazonium tetrafluoroborate 2 (0.52 mmol)、styrene derivative 3 置於 10 mL 之密封管內,打入無水 nitrile 4 (2 mL),在 80°C 下反應 2 小時,反應結束後冷卻 置室溫,加入飽和碳酸氫鈉水溶液(5 mL),以 DCM(5 mL x 3)萃取,合併有機層並 加入過量無水硫酸鎂除去有機層的水分,加入 sodium carbonate (3.0 mmol, 318 mg) 在室溫下反應 72 小時,反應結束後加入飽和 NH4Cl(aq)(5 mL),以 DCM(10 mL x 2) 萃取,合併有機層,加入過量無水硫酸鎂除水,將溶液旋乾後以管柱層析分離純化 (silica gel, 20-40% EtOAc in hexane),可得相對應的 3-羥基喹啉產物 7。. 2-methyl-4-phenylquinolin-3-ol (7aaa). 得白色固體 52 mg,產率 73%。 mp: 238-239 °C 1H. NMR (400 MHz, DMSO-d6): δ 8.88 (br, 1H, O-H), 7.87 (d, J = 8.0 Hz, 1H, Ar-H),. 7.56 – 7.46 (m, 4H, Ar-H), 7.36 – 7.32 (m, 3H, Ar-H), 7.24 (d, J = 8.0 Hz, 1H, Ar-H), 2.63 (s, 3H, -CH3) ppm. 13C. NMR (100 MHz, DMSO-d6): δ 152.52, 145.41, 142.10, 133.79, 130.33, 128.70,. 128.50, 128.21, 127.85, 127.50, 125.79, 125.74, 124.13, 21.24 ppm. HRMS (ESI-TOF) calcd. for C16H13NO (M+H+): = 236.1075; found: 236.1074. 48.
(58) 4-(4-chlorophenyl)-2-methylquinolin-3-ol (7aca). 得亮黃色固體 41 mg,產率 50%。mp: 272-274 °C 1H. NMR (400 MHz, DMSO-d6): δ 7.87 (d, J = 8.4 Hz, 1H, Ar-H), 7.59 (d, J = 8.4 Hz,. 2H, Ar-H), 7.50 – 7.46 (m, 1H, Ar-H), 7.38 – 7.33 (m, 3H, Ar-H), 7.23 (d, J = 8.0 Hz, 1H, Ar-H), 2.61 (s, 3H, -CH3) ppm. 13C. NMR (125 MHz, DMSO-d6): δ 152.61, 145.68, 141.96, 132.71, 132.36, 128.64,. 128.29, 127.28, 127.19, 125.96, 125.85, 123.88, 21.24 ppm. HRMS (ESI-TOF) calcd. for C16H1335ClNO (M+H+): = 270.0680; found: 270.0695. HRMS (ESI-TOF) calcd. for C16H1337ClNO (M+H+): = 272.0651; found: 272.0692.. 4-(4-(chloromethyl)phenyl)-2-methylquinolin-3-ol (7ada). 得亮黃色固體 59mg,產率 69%。 1H. NMR (400 MHz, DMSO-d6): δ 8.98 (br, 1H, O-H), 7.88 (d, J = 8.4 Hz, 1H, Ar-H),. 7.60 (d, J = 8.0 Hz, 2H, Ar-H), 7.50 – 7.46 (m, 1H, Ar-H), 7.37 – 7.33 (m, 3H, Ar-H), 7.23 (d, J = 8.4 Hz, 1H, Ar-H), 4.88 (s, 2H, -CH2Cl), 2.63 (s, 3H, -CH3) ppm. 13C. NMR (100 MHz, DMSO-d6): δ 152.56, 145.48, 142.08, 137.11, 133.90, 130.68,. 128.99, 128.25, 128.19, 127.38, 125.84, 124.02, 46.00, 21.25 ppm. HRMS (ESI-TOF) calcd. for C17H1435ClNO (M+H+): = 284.0842; found: 284.0840. HRMS (ESI-TOF) calcd. for C17H1437ClNO (M+H+): = 286.0813; found: 286.0807.. 49.
(59) 2,4-diphenylquinolin-3-ol (7aab). 得白色固體 68 mg,,產率 46%。mp: 123-124 °C 1H. NMR (400 MHz, CDCl3): δ 8.15 (d, J =8 .4 Hz, 1H), 8.01 – 7.93 (m, 2H), 7.62 – 7.58. (m, 2H), 7.58 – 7.53 (m, 2H), 7.52 – 7.48 (m, 2H), 7.48 – 7.42 (m, 3H), 7.42 – 7.37 (m, 2H), 5.45 (brs, 1H) ppm. 13C. NMR (100 MHz, CDCl3): δ 150.0, 143.9, 143.3, 137.2, 132.2, 130.3,129.5, 129.4,. 129.1, 128.4, 127.6, 126.8, 124.5 ppm. HRMS (ESI-TOF) calcd. for C21H16NO (M+H+): = 298.1232; found: 298.1242.. 2-cyclopropyl-4-phenylquinolin-3-ol (7aac). 得白色固體 57 mg,,產率 42%。mp: 127-128 °C 1H. NMR (400 MHz, CDCl3): δ = 7.97 (d, J = 8.4 Hz, 1H), 7.58 (t, J = 7.6 Hz, 2H), 7.54. – 7.44 (m, 2H), 7.41 – 7.39 (m, 2H), 7.31 – 7.26 (m, 2H), 5.35 (brs, 1H), 2.63 (m, 1H), 1.36 – 1.35 (m, 2H), 1.10 – 1.06 (m, 2H)ppm. 13C. NMR (100 MHz, CDCl3): δ 154.5, 144.4, 132.2, 130.3, 129.9 (2C), 129.6, 129.0,. 128.4, 126.6, 126.3, 125.6, 124.3, 11.9, 9.5 ppm. HRMS (ESI-TOF) calcd. for C18H16NO (M+H+): = 262.1232; found 262.1232.. 50.
(60) 2,6-dimethyl-4-phenylquinolin-3-ol (7baa). 得黃色固體 40 mg,53%。mp: 205-206 °C 1H. NMR (400 MHz, DMSO-d6): δ 8.78 (br, 1H, O-H), 7.76 (d, J = 8.4 Hz, 1H, Ar-H),. 7.56 – 7.52 (m, 2H, Ar-H), 7.48 – 7.45 (m, 1H, Ar-H), 7.33 – 7.29 (m, 3H, Ar-H), 6.99(s, 1H, Ar-H), 2.59 (s, 3H, -CH3), 2.30 (s, 3H, -CH3) ppm. 13C. NMR (100 MHz, DMSO-d6): δ 151.37, 145.51, 140.70, 134.89, 133.94, 130.32,. 128.50, 128.28, 127.86, 127.79, 127.45, 122.93, 21.34, 21.10 ppm. HRMS (ESI-TOF) calcd. for C17H16NO (M+H+): = 250.1232; found 250.1239.. 2-methyl-4-phenyl-6-(trifluoromethyl)quinolin-3-ol (7daa). 得黃色固體 30 mg,32%。 1H. NMR (400 MHz, DMSO-d6): δ 9.28 (br, 1H, O-H), 8.10 (d, J = 8.8 Hz, 1H, Ar-H),. 7.76 (d, J = 8.8 Hz, 1H, Ar-H), 7.63 – 7.54 (m, 4H, Ar-H), 7.41 – 7.39 (m, 2H, Ar-H), 2.70 (s, 3H, -CH3) ppm. 19F. NMR (376 MHz, DMSO-d6): δ -61.71 ppm.. 51.
(61) 2,4-dimethyl-4-phenylquinolin-3(4H)-one (8aea). 得黃色油狀物 52 mg,70%。 1H. NMR (400 MHz, CDCl3): δ 7.60 (d, J = 7.2 Hz, 1H, Ar-H), 7.46 – 7.42 (m, 1H, Ar-. H), 7.46 (m, 1H, Ar-H), 7.37 – 7.33 (m, 1H, Ar-H), 7.29 – 7.23(m, 4H, Ar-H), 7.03 – 7.01 (m, 2H, - Ar-H), 2.28 (s, 3H, -CH3), 1.89 (s, 3H, -CH3) ppm. 13C. NMR (100 MHz, CDCl3): δ 196.44, 160.66, 140.69, 140.58, 136.82, 129.40, 128.77,. 128.73, 127.85, 127.59, 127.17, 53.56, 23.26, 20.01 ppm. HRMS (ESI-TOF) calcd. for C17H14NO (M+H+): = 250.1232; found: 250.1243. 7.3.3 高烯丙基醇與松油醇的合成 9-(2-methylallyl)-9H-fluoren-9-ol (11). 將 magnesium turnings (1.5 g, 60 mmol)置於經烘箱除水的 100 mL 雙頸瓶中, 抽灌氮氣數次後,打入無水乙醚(10 mL)。在 0 °C 下逐滴加入 methylallyl chloride (5.54 g, 60 mmol),慢慢以熱水加熱直到 Grignard 試劑被起始為止,將反應溶液立 即以冰浴降至 0 °C,逐滴加入 fluorenone (3.6 g, 20 mmol)的無水乙醚(30 mL)溶液, 移除冰浴,在室溫下反應 12 小時後,逐滴加入 NH4Cl(aq) (5 mL)淬息反應,用乙醚 (40 mL x 2)萃取後,合併有機層並以無水硫酸鎂乾燥,將溶液旋乾後以管柱層析分 離純化(silica gel, 10% EtOAc in hexane)即可得白色固體 11(4.7 g, 98%)。 1H. NMR (400 MHz, CDCl3): δ7.61 (d, J = 7.2 Hz, 2H, Ar-H), 7.52 (d, J = 7.2 Hz, 2H,. Ar-H), 7.37 (ddd, J = 7.2, 7.2, 1.2 Hz, 2H, Ar-H), 7.31 (ddd, J = 7.2, 7.2, 1.2 Hz, 2H, ArH), 4.65 (m, 1H, allyl-H), 4.42 (m, 1H, allyl-H), 2.86 (s, 2H, -C-CH2-C-), 2.32 (br, 1H, OH), 1.41 (s, 3H, -CH3) ppm. 13C. NMR (100 MHz, CDCl3): δ 148.4, 140.8, 139.4, 128.8, 127.6, 124.1, 119.8, 115.3, 52.
(62) 82.0, 46.9, 24.0 ppm.. methyl 4-methylcyclohex-3-ene-1-carboxylate (15). 取 3-methyl-1,2-butadiene (6.0 mL, 60 mmol)與 methyl acrylate(2.7 mL,30 mmol) 置入 50 mL 單頸瓶中,加入 AlCl3 (402 mg, 3 mmol)在 0 °C 下反應 6 小時後,以二 氯甲烷(20 mL)稀釋反應溶液,逐滴加入水淬息反應,用飽和食鹽水(10 mL)萃取後, 以無水硫酸鎂乾燥,將溶液旋乾後即可得黃色液體 15 (3.86 g, 83%)。 1H. NMR (400 MHz, CDCl3): δ 5.32 (s, 1H), 3.62 (d, J = 1.2 Hz, 3H, -CH3), 2.47 – 2.40. (m, 1H), 2.17 – 2.16 (m, 2H), 1.94 (br, 1H), 1.55 – 1.45 (m, 5H) ppm. 13C. NMR (100 MHz, CDCl3): δ 176.1, 133.4, 119.1, 51.3, 38.9, 29.1, 27.5, 25.3, 23.2. ppm. α-terpineol (16). 取 mthyl 4-methylcyclohex-3-ene-1-carboxylate 15 (2.3 g, 15 mmol) 置入 50 mL 單 頸 瓶 中 , 抽灌 氮 氣數 次 後 , 打 入無 水 乙醚 (10 mL) , 在 0 °C 下 逐 滴 加 入 methylmagnesium bromide solution 3.0 M in dibutyl ether (10 mL, 30 mmol),在室溫 下反應 12 小時後,逐滴加入 NH4Cl(aq) (5 mL)淬息反應,用乙醚(40 mL x 2)萃取後, 合併有機層並以無水硫酸鎂乾燥,將溶液旋乾後以管柱層析分離純化(silica gel, 20% EtOAc in hexane)即可得無色溶液 16(2.1 g, 91%)。 1H. NMR (400 MHz, CDCl3): δ 5.32 (s, 1H), 3.62 (d, J = 1.2 Hz, 3H, -CH3), 2.47 – 2.40. (m, 1H), 2.17 – 2.16 (m, 2H), 1.94 (br, 1H), 1.5 – 1.55 (m, 5H) ppm. 13C. NMR (100 MHz, CDCl3): δ 134.0, 120.5, 72.7, 45.0, 31.0, 27.4, 26.8, 26.2, 23.9,. 23.3 ppm.. 53.
(63) 7.3.4 高烯丙基醇與松油醇之腈正離子-烯烴環化產物. N-(4',6'-dimethyl-4',5'-dihydro-3'H-spiro[fluorene-9,2'-pyridin]-4'-yl)acetamide (12). 將 9-(2-methylallyl)-9H-fluoren-9-ol 11 (100 mg, 0.42 mmol) 置入 4 mL 反應管 中,抽灌氮氣數次後,打入無水乙腈(1 mL)和無水甲苯(1 mL)。將 trifluoroborate etherate(60 mg, 0.42 mmol)於無水甲苯(1 mL)的溶液緩慢滴入反應管中。在室溫下 反應 12 小時後,滴入數滴甲醇淬息反應,以 DCM (10 mL x 2)與 NaHCO3 (2 mL) 萃取,合併有機層並以無水硫酸鎂乾燥,將溶液旋乾後以管柱層析分離純化(silica gel, EtOAc:hexane:triethylamine = 50:49:1)得到白色固體 (72.8 mg, 54%). 1H. NMR (400 MHz, CDCl3): δ 7.73 - 7.69 (m, 2H, Ar-H), 7.38 – 7.32 (m, 4H, Ar-H),. 7.26 – 7.22 (m, 2H, Ar-H), 5.60 (br, 1H, N-H), 3.63 - 3.58 (m, 1H, AB-H), 2.36 - 2.32 (m, 1H, AB-H), 2.14 (s, 3H, -CH3), 2.08 – 2.02 (m, 2H, AB-H), 1.96 (s, 3H, -CH3), 1.49 (s, 3H, -CH3) ppm. 13C. NMR (100 MHz, CDCl3): δ 169.7, 168.7, 152.3, 149.7, 140.8, 139.6, 128.4, 128.2,. 128.0, 126.9, 125.0, 123.6, 120.5, 120.0, 70.6, 50.5, 43.3, 38.8, 28.7, 28.2, 24.6 ppm. HRMS (ESI-TOF) calcd. for C21H23N2O (M+H+): = 319.1805; found: 319.1812.. N-((1S,5S)-4-benzyl-2,2,6-trimethyl-3-azabicyclo[3.3.1]non-3-en-6-yl)-2phenylacetamide (21). 54.
數據

+7



相關文件
1、取一10mL的量筒量取5mL 1M的氫氧化鈉溶液置 於250mL錐形瓶中。再取一10mL的量筒量5mL 1M
雖然水是電中性分子,然其具正極區域(氫 原子)和負極區域(氧原子),因此 水是一種極 性溶劑
而在後續甲烷化反應試驗方面,以前段經厭氧醱酵產氫後之出流水為進流基 質。在厭氧光合產氫微生物方面,以光合作用產氫細菌中產氫能力最好的菌株 Rhodopseudomonas palustris
簡報第 40 頁:甲烷 結構式 University of Illinois
探討燃燒所得的碳簇、活性碳及二氧化錳對 雙氧水分解的影響。將 3個100 mL量筒中各加 入 5 mL雙氧水和1 mL的清潔劑水溶液,分別 加入 0 .2 g碳黑、0.2
溶液中離子沈澱反應的化學反應式表示法:以氯化鈉和硝酸銀的反應為例(如上圖)
酸性氣體(二氧化硫、二氧化氮)可以飄浮到離源頭很遠的地
1、取一10mL的量筒量取5mL 1M的氫氧化鈉溶液置 於250mL錐形瓶中。再取一10mL的量筒量5mL 1M
![HPSH [ 氧化數平衡反應式係數 ]](data:image/gif;base64,R0lGODlhAQABAIAAAP///wAAACH5BAEAAAAALAAAAAABAAEAAAICRAEAOw==)