以固相微萃取法偵測垃圾污水中揮發性脂肪酸之研究; Determination of Volatile Fatty Acids by Solid-Phase Microextraction from Trash Wastewater
93
0
0
全文
(2) 誌謝 研究所二年半期間,承蒙業師賴俊雄教授、蔡詩偉副教授的耐心指 導,使本論文得以順利完成,在學校課業、實驗分析以及人生經歷上都 令我受益匪淺,在此謹致上最誠摯的謝意。衷心感謝賴以賢教授、陳昭 鈴教授於論文口試時費心指導,提供許多指正及寶貴意見,使得本論文 更趨完善,特致謝忱。 研究所就學期間,在學校時多蒙如淳、峻鳴學長,以及世青、詩婷、 益裕、淑靜、麗梅、文玲、粹文等多位同學的幫忙,在工作職場上,承 蒙台中市環保局張局長皇珍、王副局長進崇、吳技正龍泰、陳課長政良、 白課長玨瑛,以及同事晉燦、裕隆、宏黛、顯鈞、佰昌、富美、婉玲、 文玲、佩宣、莉莉的鼎力相助,使我能在工作百忙中得以撥空,順利完 成研究所課業。 最後,感謝我最親愛的父母,雖然這是一份遲來的學業成績單,仍 然希望已在當天使的您們,能夠收到這份喜訊,還有我的家人,帥氣老 公和二位可愛的兒女、以及所有關心我的親友,感謝多年的照顧與支持, 謹以此論文呈獻給大家,願將畢業的喜悅和榮耀和你們分享。.
(3) 摘要 揮發性脂肪酸是有機物質進行厭氧性分解中最常見的中間產物。本 研究採用頂空固相微萃取具有簡便、快速、不需使用有機溶劑及不受基 質干擾之優點,結合氣相層析儀,測定垃圾污水中揮發性脂肪酸之含量 分析。研究中使用 75 μm Polydimethylsiloxane/Carboxen(PDMS/CAR) 纖維,針對纖維吸附時間、吸附溫度、脫附時間、脫附溫度等因素分別 探討,結果顯示在 1200 rpm 攪拌速率下,於室溫中吸附 20 分鐘後,將 吸附纖維直接插入氣相層析儀,利用儀器注射口之 250℃設定溫度,進行 3 分鐘熱脫附後,再以 GC-FID 進行分析,是為實驗最佳萃取分析條件。 測試酸標準溶液之檢量線及方法偵測極限,在乙酸、丙酸、丁酸、 異丁酸標準品檢量線之相關係數(r 值)均大於 0.995 以上,而方法偵測 極限的結果,乙酸為 9.2μg/ml、丙酸 2.65μg/ml、丁酸 3.11μg/ml、異 丁酸 2.47μg/ml,偵測極限均可達到 ppm 結果;其精密度在 10﹪以下, 準確度之範圍為 88.04-112.32﹪. 固相微萃取之設備簡單,操作容易,非常適合於野外之環境樣品採 樣,本研究選擇垃圾污水做為實際樣品,此法可以簡化採樣及樣品前處 理之繁瑣過程,並能減低經濟分析成本,是為一項實務上應用相當有效 率的樣品前處理方法。 關鍵字:揮發性脂肪酸,固相微萃取,垃圾污水,頂空法,乙酸. I.
(4) Abstract Volatile fatty acids(VFA) are the most common intermediate products generated from anaerobic decomposition of organic substances.. This study. develops a headspace solid phase microextraction procedure for sampling of VFA. This method is simple, convenient, and fast. It does not require organic solvents, and is not interfered by base materials.. It combines the gas. chromatograph to determine the analysis of volatile fatty acids contents in the trash wastewater. In this study, 75 μm PDMS/CAR fiber was used.. The. fiber absorption time, absorption temperature, desorption time and desorption temperature were conditions.. studied to determine the optimal extraction analysis. The results showed that the best absorb time was 20 minutes at. 1200 rpm mixing speed under room temperature. The injection point temperature of the instrument was set at 250℃, with 3 minutes of hot desorption. GC-FID was used for analysis. The linear correlation coefficient of standards acetic acid, propanoic acid, butyric acid, and iso-butyric acid are all greater than 0.995. The detection limits for acetic acid, propanoic acid, butyric acid, and iso-butyric acid were 9.2 μg/ml, 2.65μg/ml, 3.11μg/ml, 2.47μg/ml, respectively. The detection limits were all within ppm range. The precision were lower than 10﹪. The accuracy were in the range of 88.04-112.32﹪. The equipment for solid phase microextraction is simple, easy to operate and is very suitable for field environment sampling.. This research selects trash. wastewater as real sample to test applicability of this microextraction method. II.
(5) This method may simplify the complex procedure of sampling and sample pre-processing, and lower analysis costs. It is a very practical and effective pretreatment method.. Key words:volatile fatty acids ,solid phase microextraction,SPME trash wastewater,headspace,acetic acid. III.
(6) 目錄 頁數 致謝 中文摘要....................................................Ⅰ 英文摘要....................................................Ⅱ 目錄........................................................Ⅳ 表目錄....................................................Ⅷ 圖目錄......................................................Ⅹ. 第一章 前言............................................. 1. 1-1 緣起..................................................... 1 1-2 研究目的................................................. 2. 第二章 文獻回顧....................................... 4. 2-1 揮發性脂肪酸的特性....................................... 4 2-2 揮發性脂肪酸的來源.......................................5 2-3 揮發性脂肪酸的分析應用................................... 6 2-4 揮發性脂肪酸的分析法..................................... 8 2-4-1 管柱色層分析法.................................... 8 2-4-2 直接蒸餾法.....................................10. IV.
(7) 2-4-3 國外揮發性脂肪酸分析參考方法..................... 11 2-5 傳統前處理方法簡介...................................... 11 2-5-1 液相-液相萃取法...................................11 2-5-2 蒸餾法............................................ 12 2-5-3 吹氣捕集法........................................ 13 2-5-4 固相萃取法........................................ 14 2-5-5 固相微萃取法......................................16 2-6 國內惡臭管制概況.......................................... 2. 0. 第三章設備材料與方法................................ 22. 3-1 儀器設備及材料.......................................... 22 3-2 實驗步驟............................................... 23. 3-3 實驗方法............................................... 24. 3-3-1 氣相層析儀最適分析條件測試.......................24 3-3-2 固相微萃取最適分析條件測試....................... 25 3-3-3 標準品檢量線..................................... 26. 3-3-4 儀器偵測極限測試................................. 26. 3-3-5 方法偵測極限測試.................................. 27 3-3-6 精密度與準確度測試................................28. V.
(8) 3-3-7 滯留時窗測試......................................29 3-3-8 樣品儲存穩定度測試................................29 3-4 採樣規劃................................................29 3-4-1 實際樣品採......................................29 3-4-2 採樣點............................................30 3-4-3 實際樣品分析......................................30 3-4-3-1 樣品檢量線..................................30 3-4-3-2 空白樣品測試................................31 3-4-3-3 實際樣品測試................................31 3-5 資料處理及分析..........................................31. 第四章結果與討論.....................................32 4-1 最適分析條件結果........................................32 4-1-1 氣相層析儀實驗條件................................32 4-1-2 固相為萃取法萃取條件..............................32 4-2 標準品分析結果..........................................33 4-2-1 檢量線及圖譜......................................33 4-2-2 儀器偵測極限......................................34 4-2-3 方法偵測極限......................................34 4-2-4 精密度與準確度....................................34. VI.
(9) 4-2-5 滯留時窗..........................................35 4-2-6 樣品儲存穩定度....................................36 4-3 實際樣品分析結果........................................36 4-3-1 檢量線及圖譜......................................36 4-3-2 空白樣品..........................................37 4-3-3 實際樣品分析......................................37. 第五章結論與建議.....................................38 第六章參考文獻........................................41. VII.
(10) 表目錄. 頁數 表 2-1 揮發性脂肪酸的物理常數(C1-C8)....................50 表 2-2 行政院勞工委員會採樣分析建議方法(甲酸).............50 表 2-3 行政院勞工委員會採樣分析建議方法(乙酸).............51 表 2-4 行政院勞工委員會採樣分析建議方法(丙酸).............51 表 2-5 國外揮發性脂肪酸分析參考方法.......................52 表 2-6 固相微萃取靜相纖維種類.............................53 表 2-7 固定污染源空氣污染物排放標準(臭氣)...............54 表 2-8 勞工作業環境空氣中有害物容許濃度標(揮發性脂肪酸)..54 表 3-1 實際樣品採樣點位置紀錄表...........................55 表 4-1 氣相層析儀最適分析條件.............................56 表 4-2 固相微萃取不同吸附時間之回收率(丙酸)..............57 表 4-3 固相微萃取最適萃取條件.............................57 表 4-4 四種酸標準溶液之檢量線.............................58 表 4-5 四種酸標準溶液分析之儀器偵測極限...................58 表 4-6 四種酸標準溶液分析之方法偵測極限(預估值)...........59. VIII.
(11) 表 4-7 四種酸標準溶液分析之方法偵測極限(確認值)...........59 表 4-8 四種酸標準溶液分析之精密度與準確度.................60 表 4-9 混合酸標準溶液分析之精密度與準確度.................61 表 4-10 四種酸標準溶液分析之滯留時間變異情形...............62 表 4-11 四種酸標準溶液之樣品儲存回收率....................63 表 4-12 四種酸標準溶液之檢量線(實際樣品分析之校正曲線)...63 表 4-13 垃圾污水中四種揮發性脂肪酸之濃度值................64 表 4-14 垃圾污水中四種揮發性脂肪酸濃度分析之變異係數......65 表 5-1 相關研究結果比對...............................67. IX.
(12) 圖目錄. 頁數 圖 2-1 厭氣性或好氧性發酵及氧化有機物轉換能量路徑圖.......68 圖 2-2 揮發性脂肪酸產生甲烷之發酵反應.....................68 圖 2-3 複合廢水的甲烷發酵路徑圖...........................69 圖 2-4 固相萃取法的操作流程圖.............................69 圖 2-5 固相微萃取裝置圖...................................70 圖 2-6 固相微萃取法的操作流程圖...........................70 圖 4-1 四種酸標準溶液之分析圖譜...........................71 圖 4-2 固相微萃取不同吸附時間之回收率(丙酸)...............72 圖 4-3 固相微萃取不同脫附時間之脫附效率(四種酸標準溶液)...72 圖 4-4 乙酸標準溶液之檢量線...............................73 圖 4-5 丙酸標準溶液之檢量線...............................73 圖 4-6 丁酸標準溶液之檢量線...............................74 圖 4-7 異丁酸標準溶液之檢量線.............................74 圖 4-8 樣品儲存穩定度之回收率變化圖.......................75 圖 4-9 樣品儲存穩定度之各日平均回收率變化圖...............75. X.
(13) 圖 4-10 乙酸標準溶液之檢量線(實際樣品分析之校正曲線).....76 圖 4-11 丙酸標準溶液之檢量線(實際樣品分析之校正曲線)......76 圖 4-12 丁酸標準溶液之檢量線(實際樣品分析之校正曲線)......77 圖 4-13 異丁酸標準溶液之檢量線(實際樣品分析之校正曲線)...77 圖 4-14 空白樣品去離子純水分析圖譜........................78 圖 4-15 四種酸樣品分析圖譜................................79 圖 4-16 垃圾污水中四種揮發性脂肪酸濃度含量比較............80 圖 4-17 垃圾污水中四種揮發性脂肪酸濃度分析之變異性........80. XI.
(14) 第一章 前言 1-1 緣起. 揮發性脂肪酸(volatile fatty acids)廣泛存在於環境中各處. (1). ,尤其. 在垃圾、廢水、廢棄物、廚餘中多含有大量有機物質,當有機物質進行 厭氧微生物分解時,會先將有機物質分解為短鏈揮發性脂肪酸,例如: 甲酸、乙酸、丙酸、丁酸、異丁酸等,當環境條件適合時,揮發性脂肪 酸會再分解為甲烷,由於後者分解速率較前者為慢,過多的揮發性脂肪 酸若無法立即分解成甲烷,就會產生大量揮發性脂肪酸累積情形,如此 一來,不僅降低廢水中有機物質轉化成甲烷的分解效率,又因其具有令 人厭惡的氣味,這也是引發空氣刺鼻味道為人們所詬病的惡臭因素之一。 惡臭氣味本屬人類嗅覺感受不愉快之主觀判斷,許多臭味的問題, 並非單一物質所致,而是源自多種物質相互作用與影響的結果,且惡臭 之組成多為沸點較低的揮發性或氣態物質,例如揮發性有機酸、有機胺 及有機硫化物等,而人類感官對於這些化合物的閾值,往往遠低於儀器 所能偵測之極限. (2). ,所以,對於臭味成分的精確分析實屬一種挑戰,本. 研究以揮發性脂肪酸之惡臭濃度做為主要探討的分析物,利用儀器分析 獲得客觀的數據,希望協助瞭解有機酸臭味來源的污染程度。 過去對於分析樣品前處理技術的傳統方法,大多需要使用大量,且. 1.
(15) 昂貴的有機溶劑來萃取分析物,不僅相當耗時費力,同時容易造成二次 環境污染問題,直到 1990 年,加拿大 Pawliszyn 教授實驗室團隊發展出 固相微萃取技術(solid-phase microextraction,SPME)3 ,係為一種免溶 ( ). 劑技術,由於其整合取樣、萃取、濃縮及注入於同一個步驟中,而且擁 有裝置簡單、攜帶方便、採樣時間短、偵測極限佳等優點,加上並沒有 使用有機溶劑造成環境污染的困擾,因此將固相微萃取方法應用於環境 樣品檢測之研究,已經日益受到分析者的重視及推廣。 所以本研究藉由固相微萃取技術適用於野外採樣之優點,選擇臺中 市垃圾車所殘留之垃圾污水,做為實際樣品分析,建立垃圾污水中揮發 性脂肪酸濃度分析的檢測機制,發展一個簡單、快速之臭味成分分析的 方法,可以即時提供揮發性脂肪酸污染濃度值,做為惡臭污染源防制改 善方法的參考。. 1-2 研究目的. 1.參考行政院環保署環檢所相關公告方法以及行政院勞工委員會採樣分 析建議方法的採樣及分析步驟,建立本研究之適用方法。 2.評估固相微萃取技術應用於揮發性脂肪酸檢測之可行性。 3.應用固相微萃取野外採樣的優點,確立環境實際樣品檢測之適用性。 4.經由揮發性脂肪酸檢測值,進而瞭解垃圾污水中惡臭物質的污染源及濃 2.
(16) 度值,做為研擬改善方案的參考。 5.迅速提供揮發性脂肪酸初步檢測值,做為有機廢水厭氧處理程序控管條 件的參考數據。 6.利用儀器分析測得科學數據,瞭解有機酸惡臭污染源的濃度變化,提供 處理惡臭污染問題的參考。. 3.
(17) 第二章 文獻回顧 2–1 揮發性脂肪酸的特性. 揮發性脂肪酸(volatile fatty acids),主要為含有一至八個碳數的低 分子量短鏈脂肪酸,包括甲酸(formic acid)、乙酸(acetic acid)、丙酸 (propanoic acid) 、丁酸(butanoic acid) 、戊酸(valeric acid) 、己酸(hexanoic acid) 、庚酸(heptanoic acid) 、辛酸(caprylic acid)等,由於這些短鏈脂 肪酸在常壓下可被蒸餾,故將其稱為揮發性脂肪酸(VFA)4 。 ( ). 揮發性脂肪酸分子具有羧基(–COOH)和碳氫基(–R),前者是極 性基,後者是非極性基,因此具有親水性和疏水性兩種不同性質,對水 的溶解度依碳氫鏈的長短而異,四個碳數以下的低級短鏈脂肪酸易溶於 水而呈弱酸性,隨著碳數的增加,對水的溶解度降低,碳數一至八個的 脂肪酸在室溫時為液狀,甲酸、乙酸、丙酸具有強烈刺激味,其餘的液 態酸均有令人作噁的難聞氣味,特別是丁酸與戊酸,而腐敗的奶油味道, 即是丁酸所造成. (5). 。. 揮發性脂肪酸的化學性質以鹽及酯的生成最常被應用,揮發性脂肪 酸的羧基易與 Li、Na、K 等鹼金屬反應,形成鹽類,亦可使用硫酸、鹽 酸等強酸做為觸媒,形成酯化反應,脂肪酸酯一般是中性、對熱安定, 可以蒸餾法蒸餾,不致發生分解現象. 4. (4). 。.
(18) 揮發性脂肪酸的名稱、分子式與物理常數分列於表 2-1. (5). 。. 2–2 揮發性脂肪酸的來源. 揮發性脂肪酸為低分子量有機酸,是碳水化合物、蛋白質、脂類等 有機物質分解時的中間產物,有關有機物氧化及發酵轉換能量路徑圖, 如圖 2-1 6 ,圖中這些揮發性脂肪酸的中間產物會在無氧發酵環境下,繼 ( ). 續轉化成最終產物甲烷及二氧化碳,而整個厭氧作用的轉換過程,是二 個或二個以上的反應結果,如圖 2-2. (6). 。. 揮發性脂肪酸廣泛存在於環境廢水、廢棄物、活性污泥 出水. (8). (7). 及垃圾滲. 中,當有機物質被生物分解時,是由兩種族群的微生物共同作用. 使反應順利進行,此兩種族群分別為使有機物分解成揮發性脂肪酸的水 解酸化菌及使揮發性脂肪酸轉變成甲烷及二氧化碳的甲烷生成菌。 近年來,廢水中揮發性脂肪酸濃度的測定,漸受環境工程研究者重 視,主要是應用於廢水厭氧消化處理程序的控制. (9). ,因為揮發性脂肪酸. 存在量的多寡,會直接影響廢水處理系統的運作成效,例如:生物處理 法. (10). 、化學沉澱去磷酸鹽. (11). 、脫硝及氨氣提法. ( 12). 等,如在廢水處理過. 程中有大量揮發性脂肪酸累積,將無法順利分解成甲烷,不僅會破壞廢 水處理系統的最終穩定性功能,同時也會產生嚴重惡臭的空氣污染問題。 圖 2-3 顯示,一複合廢水. (6). ,揮發性脂肪酸可經由數種階段轉變成 5.
(19) 甲烷。在各種有機物的厭氧過程中,約有 30﹪的複合廢水,先轉變為丙 酸,再變為甲烷,而乙酸則為最多的中間產物,約有 72﹪的有機物會先 轉變為乙酸,再轉化為甲烷,根據圖 2-3 複合廢水的甲烷化路徑圖中所 示,約有 85﹪的廢水,是先轉變為乙酸與丙酸的中間產物,由此可見這 兩種揮發性脂肪酸的重要性,而其餘 15﹪的甲烷,則是由其他揮發性脂 肪酸(甲酸或丁酸等)發酵而得,因此,本研究將選擇污水樣品中所含 乙酸、丙酸、丁酸、異丁酸進行濃度分析,做為本研究探討的主要標的 分析物,藉以建立固相微萃取技術適合於揮發性脂肪酸偵測的檢驗模式。. 2-3 揮發性脂肪酸的分析應用. 現行廢水三級處理系統的處理方法,依處理操作之不同可分為物理 、化學及生物處理三種. (13). ,其中生物處理法,可依氧氣之有無分為好氧. 性及厭氧性處理。好氧性處理是將廢水中的有機物,利用好氧性微生物 所分解,產生二氧化碳及水,如活性污泥法、滴濾池法、生物旋轉圓盤 法均屬之,厭氧性處理是利用微生物在缺氧情況下,分解廢水中有機物 形成甲烷、氮及硫化氫等,如厭氣消化法、厭氣濾床法、厭氣流體化床 等均屬之,一般常用方法多為好氧性處理,但主要缺點是需要較高電費 以提供所須之氧氣,且不能回收有用的副產品,因此,厭氧性處理技術 近年來為節省廢水處理之動力費,減少污泥產生量的特點,而為小規模 6.
(20) 處理設施所重視採用,而且厭氣消化結果所產生的沼氣亦可用來發電, 或經純化可充作燃料或製成其他石化產品。 在穩定運轉的厭氧消化設備中,有二種細菌必須和諧的作用,才得 以完成有機物的分解,一為水解酸化菌,另一為甲烷生成菌,其反應式 如下. (14). :. 有機物 + 水解酸化菌→ 有機酸 + 甲烷生成菌 → CH4 + CO2 廢污水在厭氧系統中生物分解,會產生大量有機酸,當環境條件有 利於生長時,由水解酸化過程中所產生的揮發性脂肪酸,便會很快的為 甲烷生成菌所分解,因此揮發性脂肪酸將不會造成累積情形,若這些短 鏈有機酸無法及時移除或形成甲烷,此有機酸濃度便會增加,而降低系 統之 PH,在 PH 小於 6.5 時. (6). ,甲烷菌會受到抑制而使活性減弱,在此. 不平衡的情況下,將對甲烷消化系統造成不利的影響,因此,揮發性脂 肪酸測定的實務應用價值,對於及時偵測廢水厭氧消化反應槽中,甲烷 生成的不平衡情況有極大的幫助,一般於消化程序處理時,監測甲烷發 酵最佳的環境條件為 PH 範圍是 6.8 ~ 7.4,揮發性脂肪酸含量約為 50~300 (10). mg/l(用乙酸表示). ,另一方面,也可以藉由揮發性脂肪酸的濃度測. 定,初步瞭解環境中廢污水惡臭物質的污染來源,做為空氣污染防制策 略的參考依據。. 7.
(21) 2-4 揮發性脂肪酸的分析法. 環保署環檢所已公告之標準檢測方法中,尚未建立廢水中揮發性脂 肪酸之檢測方法,但有許多實驗室,已利用氣相層析及離子層析法來分 析揮發性脂肪酸的濃度. (15). ,儘管這些方法尚未被正式認可為標準方法,. 但將來了解到揮發性脂肪酸,在廢水處理程序上的重要性時,便可能成 為標準方法。 依據行政院勞工委員會採樣分析建議方法. (16). ,分列出空氣樣品中甲. 酸、乙酸、丙酸採樣分析建議方法,如表 2-2、表 2-3、表 2-4,其中甲酸 以離子層析法(IC/ECD)17 ,乙酸及丙酸以氣相層析法(GC/FID)18-19 。 (. ). (. ). 目前實驗室常用於測定消化污泥中揮發性脂肪酸濃度的方法有二 種。一種是利用管柱色層分析法. (20). (column partition chromatography),. 另一種是直接蒸餾法(direct distillation)5 。 ( ). 2-4-1 管柱色層分析法. (20). 本法中包含有兩種溶劑,一為固定於固態吸附劑表面(固定相) ,例 如矽酸(silicic acid) ,並置於管柱中,需分析監測的水樣則置於管柱的頂 部。第二種溶劑則為攜帶溶劑(移動相) ,將其加入於管柱中時,便可將 欲分析的物質帶進管柱內進行分離。在管柱中,分析物不斷受到固定溶 8.
(22) 劑與流動溶劑的分離,因而產生不同的移動現象,分析物在移動相的溶 解度大於固定相的溶解度時,此分析物將會從管柱中先行分離出來,此 原理與氣相層析法相同,不同處僅在於移動相的不同而已。 本法分析前須先藉由過濾或離心的方法,將懸浮固體物予以分離, 再以硫酸類的無機酸將水樣酸化,以轉變離子化的揮發性脂肪酸成為分 子化形式,反應式如右所示:. R-COO + H+ → R-COOH -. 此形成分子化的酸化生成物,在氯仿丁醇溶劑中具有高溶解度。將酸化 後的水樣均等的注入於含有預乾燥處理矽酸的管柱內,以預乾燥處理的 矽酸當做固定相溶劑,用以吸收水樣中的水分,再將定量的氯仿丁醇混 合液通入管柱中,因分子形式存在的揮發性脂肪酸,在移動相的溶解度 較高,故先被分離出來,硫酸及離子化之鹽類則在矽酸所吸收的水中, 有較高的溶解度,所以在揮發性脂肪酸之後出現。為使揮發性脂肪酸完 全被萃取,必須加入充分量的攜帶溶劑混合液,但是加量不宜過高,以 免將無機酸一起攜出。 萃取出來的揮發性脂肪酸,以酚太為指示劑,採用 NaOH 滴定法滴 定至酚太終點以求出揮發性脂肪酸的濃度,其反應式如下: -. R-COOH + NaOH → R-COO + Na. +. + H2O. 滴定用 NaOH 必須加入甲醇,因水不溶於有機溶劑,所以此溶液並不穩 定,必須經常標定或每日配置,通常用甲醇稀釋 1.00 N 液態 NaOH 備用。. 9.
(23) 2-4-2 直接蒸餾法. (5). 本法分析原理為低分子量脂肪酸,在水的沸點 100℃時,均有可觀的 蒸氣壓. (21). ,如表 2-1,所以蒸餾液中應含有大量的揮發性脂肪酸,因此,. 可以使用蒸餾法來萃取分析物。。 消化污泥的 PH 值約在 6.5-7.5 間,在這範圍內,有機酸多呈游離態, 無法蒸餾,藉由加入非揮發性強酸,例如:硫酸,使 PH 值降至 1.0 以下, 可將有機酸轉變成非離子形式,以醋酸表示如下: -. +. CH3 COO + H. → CH3 COOH. 若由表 2-1 所示,低分子量的揮發性脂肪酸由於具有很高的蒸氣壓,應很 容易分離,但事實上,甲酸、乙酸、丙酸的稀釋溶液,卻不易使用蒸餾 法予以分離. (22). ,其原因主要為稀釋水溶液中,酸與水分子發生結合,已. 與原來的酸不一樣,形成不是理想溶液的性質所致,而酸和水形成的混 合液,其氣相中的主要物質為水,將其精餾,則可得到純水蒸餾液,但 有機酸卻仍殘留在未蒸餾液中,因此,在本法操作時,精餾的氣體應保 持最低量,避免僅得到純水蒸餾液的困擾結果,由此可見,蒸餾的速率, 對揮發性脂肪酸的測定,有很大的影響,必須嚴格控制,蒸餾出來的酸, 用標準 NaOH 滴定,以酚太為指示劑,滴定至終點,計算揮發性脂肪酸 的濃度。. 10.
(24) 2-4-3 國外揮發性脂肪酸分析參考方法. 蒐集自美國紐約社會圖書館內 “ A Wiley- Interscience Publication”藏 書,書中所載有機酸混合物的定性定量分析方法,包括離子交換樹脂法 (23). 、氣相層析法. 層析法. (24-25). 、衍生化氣相層析法. (29-30). 、薄層層析法. (31). (26). 、液相層析法. (27-28). 、濾紙. 等,如表 2-5。. 2-5 傳統前處理方法簡介. 以往常用於環境樣品中揮發性脂肪酸濃度分析的前處理法,包括液 相-液相萃取法. (32). (liquid-liquid extraction method , LLE)、蒸餾法. (distillation) 、吹氣捕集法. (33). (34). (purge and trap method) ,實驗者多以這三. 種前處理法結合 GC、GC-MS、LC 等儀器進行分析。. 2-5-1 液相-液相萃取法(liquid-liquid extraction method , LLE)32 (. ). 液相-液相萃取法是一種傳統且被廣泛使用的萃取方法,其原理是根 據 Nernst 所提出的分配率(partition)理論. (35). ,主要是於固定溫度下,. 利用分析物於水溶液和有機溶液之間分配不同,而將欲分析物由水層萃 取至有機層中,即在常溫下,當達成平衡狀態時,溶質在兩不互溶的兩. 11.
(25) 溶劑中,其濃度比值為一特定的常數,稱為分配係數 K(distribution coefficient) ,任一溶液中分析物的分配係數越大,表示萃取效果較好,如 下列公式所示: [ A ]0 K = —————— [A] [ A ]0:為溶質在有機溶劑之莫耳濃度 [ A ]:為溶質在水溶液中之莫耳濃度 K:. 為溶質之分配係數或分配常數 液相-液相萃取法的優點在於適用的樣品範圍極廣,任何複雜的基質. 均可萃取,例如:廢污水. ( 36). 、垃圾污水. ( 76). 、海水. ( 38). 等水樣中揮發性脂. 肪酸的萃取處理,此法雖然操作不難,但其缺點為萃取過程中必須使用 大量有機溶劑,不但危害身體健康,且容易造成二次環境污染,加上昂 貴的高純度有機溶劑及實驗廢液的處理費用,使得分析成本相當不符合 經濟實惠原則。. 2-5-2 蒸餾法(distillation)33 (. 蒸餾法適用於消化污泥. ). (22). 或垃圾滲出水中. (39). 揮發性脂肪酸萃取前. 處理,揮發性脂肪酸在中性或鹼性溶液中,會有解離及鹽化現象,所以 在蒸餾時,必須加入強硫酸,使其有機酸呈現非游離態,才得以被蒸餾 12.
(26) 出來,此法的缺點為所加入的強硫酸,會在污泥分解成有機酸的存在下, 還原成 SO2,SO2 又會溶於蒸餾液中形成亞硫酸而造成嚴重分析誤差。 蒸餾法必須嚴格控制蒸餾的速率,如在低蒸餾速率下,位於燒瓶瓶 頸處,會有多量精餾現象發生,這種現象將造成揮發性脂肪酸不易為蒸 餾液所帶出,另由於酸的濃度會隨蒸餾程度增加而增加,若蒸餾程度過 高時,反使硫酸將污泥分解並產生還原現象,因此,可採用電熱器加熱 蒸餾,避免加熱控制不當,造成蒸餾速率不穩定,而影響分析結果。蒸 餾後的酸,用標準 NaOH 滴定,以酚太為指示劑,滴定至終點以求揮發 性脂肪酸的濃度。. 2-5-3 吹氣捕集法(purge and trap method)34 (. ). 1996 年 Takanobu 等人利用吹氣捕集法來分析水樣中 23 種揮發性有 機化合物. (40). ,此法廣泛應用於水中揮發性及半揮發性有機物分析的萃取. 處理。其原理是利用高純度的惰性氣體,如高純度氮氣或高純度氦氣, 通入欲分析的水樣中,使產生氣泡,分析物經一平衡過程,會從液相分 配至氣相. (41). ,含有分析物的氣體,通過一填充吸附物質的吸附管,直到. 吸附完成後再利用熱脫附儀器,將分析物脫附出來,美國環保署(USEPA) 524.2 分析方法. (42). 及我國行政院環保署環檢所公告 NIEA W784.51C 和. NIEA W785.53B 檢測方法. (43). ,即是使用此方法做為樣品前處理方法,以 13.
(27) 分析水樣中揮發性有機化合物。 吹氣捕集法所使用的吸附劑大多為多孔隙固體物質,如:Tenax-TA 、Tenax-GC、XAD-2、分子篩等. (44). ,還有具多重性能的吸附劑,如:. Tenax-GC-silica gel-Charcoal 及 Tenax-TA-Chromsorb 100-Spherorab 等三 段式填充吸附劑,這些吸附劑具有不同的吸附特性,必須針對分析物選 擇一適當的吸附劑,以達最佳的分析結果。在吸附劑中,以 Tenax-TA 最 被廣泛使用,因其具有熱穩定性、適用性廣,以及吸附時並不受水氣影 響的優點. (45). 。. 吹氣捕集法是一種免溶劑的萃取濃縮方法,也有不錯的靈敏度,但 其設備成本過高,而且吹捕系統的洩漏(Leak)及分析物殘留的問題, 常會造成分析物損失,使得分析結果產生負誤差的影響. 2-5-4 固相萃取法. (46). 。. (49). (solid phase extraction method , SPE). 固相萃取法是利用固相萃取管柱內,填充高吸附能力的正相或逆相 (47). 吸附劑當作靜相,例如 C8、C18. 等不同化性物質,這些吸附劑與分析. 物經由分配原理,將分析物吸附於靜相中,再以適當溶劑將分析物沖提 出來. (48). ,連結 GC 或 HPLC 進行分析。. 固相萃取法的操作流程如圖 2-4 所示. (49). :. (1) 選擇適當的固相萃取管柱(SPE cartridge) 14.
(28) 依據欲分析物的物化特性選擇一適當的固相萃取管,因其性質區分 為四類:1.正相萃取管是極性較強的填充物,萃取非極性至高極性 的樣品。2.逆相萃取管是極性較弱的填充物,萃取水中低極性至中 極性的樣品。3.離子交換相是藉由離子吸引力滯留樣品。4.吸附相 通常是萃取非極性樣品中的極性物質。 (2) 管柱活化(column condition) 選擇適當的有機溶劑通過固相萃取管,活化的目的是去除吸附管柱 中的不純物,可加入緩衝溶液調整至適當 PH 值,以提高吸附能力。 (3) 載入樣品(sample loading) 將樣品緩慢的注入固相萃取管中,使分析物與靜相有足夠時間產生 分配平衡作用,使分析物滯留於吸附劑上。 (4) 沖洗(washing) 選擇適當極性較大的溶劑將吸附劑所吸附的雜質沖洗出來,此步驟 必須注意,勿使分析物也被沖提出來,而造成負誤差的分析結果。 (5)分析物沖提(sample eluting) 選擇與分析物極性相近的適當溶劑將分析物沖提出來,沖提液可分 數次沖提,可得較純淨分析溶液,其回收率較佳。 固相萃取法具有純化、濃縮、操作簡單、有機溶劑少的優點,其干 擾物較液相-液相萃取法少,可提高分析物濃度及回收率,但對於較複雜. 15.
(29) 或具固態懸浮物的樣品,並不適合採用此法萃取,因其固體雜質會阻塞 固相萃取管柱或造成吸附劑的孔隙被堵住,而影響分析結果。. 2-5-5 固相微萃取法. (50-55). (solid phase microextraction,SPME). 固相微萃取法是 1990 年由加拿大 Pawliszyn 實驗室所提出的新萃取 (50). 技術. ,近年來廣泛受到分析研究者的重視應用. (51-55). ,其主要特點是整. 合樣品採樣、萃取、濃縮及導入分析物於一個單一步驟中,具有萃取時 間短、設備操作簡單、不需使用有機溶劑、高靈敏度及再現性佳等優點, (56-57). 可以直接結合 GC 或 GC/MS 儀器進行定性定量分析. ,偵測極限可達. ppt 程度。 固相微萃取法的裝置如圖 2-5 所示. (58). ,主要分為兩部份,一為固定. 器(holder),另一為塗覆高分子聚合吸附物質的熔矽纖維(fused silica fiber)。固定器主要功用是固定及保護纖維,而纖維是固相微萃取法中最 重要的心臟部份,攸關整個樣品前處理技術的萃取成效。 固相微萃取法的操作流程如圖 2-6 所示. (59). :. (1) 先將固相微萃取裝置之不銹鋼針插進樣品瓶中,伸出塗覆纖維。 (2) 待一段吸附時間使分析物於塗覆纖維與樣品間達分配平衡,縮回纖 維,取出固相微萃取裝置。 (3) 將固相微萃取裝置插進 GC 或 GC/MS 儀器注射口部,伸出塗覆纖 16.
(30) 維,以熱脫附方式使分析物自纖維上脫附出來,進入儀器分析。 固相微萃取法可區分為直接固相微萃取(direct SPME)及頂空固相 微萃取(headspace SPME) ,此兩種技術的萃取原理,主要為基質中標的 分析物與塗覆纖維進行平衡分配(equilibrium partition)的作用. (60-61). 。. (1) 直接固相微萃取(direct SPME) 萃取系統中的水相與纖維靜相達分配平衡,其二相間(水相、靜相) 的濃度關係可用下列方程式表之. (53). :. n=C0V1V2K/(KV1+V2). (公式 1). n: 分析物被纖維靜相所吸附量 C0:分析物在基質水樣品中的初始濃度 V1:纖維靜相的體積 V2:基質水樣品的體積 K: 分析物在基質水樣品與纖維靜相的分配係數 當基質水樣品的體積遠大於纖維靜相的體積(V2>> KV1),則上式 方程式可簡化為: n=KV1C0. (公式 2). 由公式(2)可見分析物被纖維靜相所吸附的量與分析物初始濃度 呈線性正比關係,另外纖維靜相吸附分析物的量是與基質水樣品的 體積大小無關,因此固相微萃取法非常適合野外樣品的採樣分析。. 17.
(31) (2) 頂空固相微萃取(headspace SPME) 分析固態樣品、複雜污穢樣品及揮發性化合物樣品,可以利用頂空 氣相介入萃取系統,先使分析物揮發至氣相,由於氣相的擴散係數較大, 藉此提昇分析物的萃取效率,其三相間(水相、氣相、靜相)的濃度關 係可用下列方程式表之. (53). :. n=C0V1V2K1K2/(K1K2V1+K2V3+V2) (公式 3) n: 分析物被纖維靜相所吸附量 C0:分析物在基質水樣品中的初始濃度 V1:纖維靜相的體積 V2:基質水樣品的體積 V3:頂空氣相的體積 K1:分析物在纖維靜相與頂空氣相的分配係數 K2:分析物在頂空氣相與基質水相的分配係數 由於平衡時分析物於三相中的化學位能均相等,根據亨利定律及理 想氣體定律的推演可得方程式: K=KH/KF=K1K2. (公式 4). KH 及 KF 分別為分析物於纖維靜相及基質水相的亨利常數。 將公式(4)代入公式(3),可得公式(5)如下: n=C0V1V2K/(KV1+K2V3+V2) (公式 5). 18.
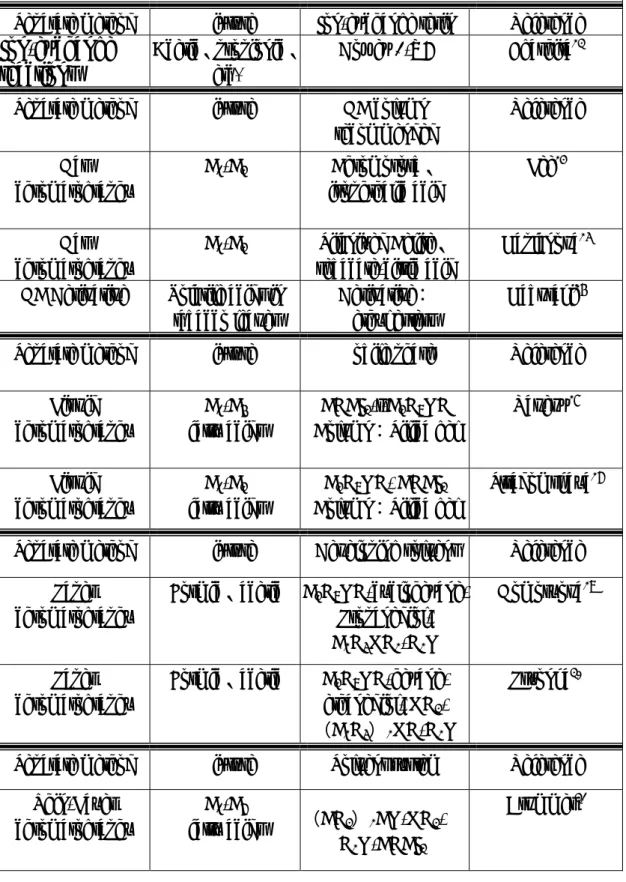
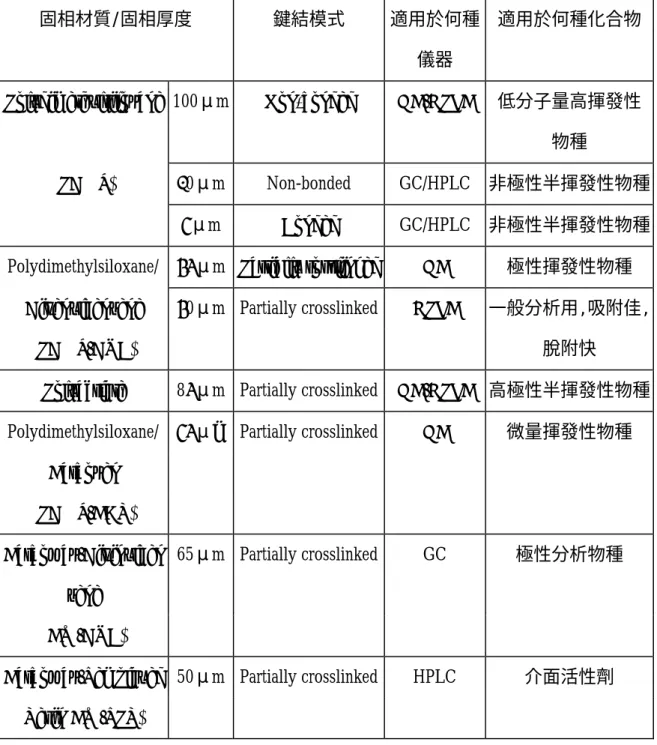
(32) 比較公式(1)及公式(5) ,發現三相萃取系統較兩相萃取系統於分母多 一個 K2V3 的項目,由於對大多數化合物而言 K2 相對較小於 K(K , 1 2<K1) 如果頂空氣相的體積遠小於水樣品體積(V3<<V2),則 K2V3 的項目幾 乎可被省略,換句話說,分析物被纖維靜相所吸附的量,並未因頂空氣 相的介入而有所改變。 1990 年發展初期,固相微萃取技術的纖維,並沒有塗覆上一層高分 子吸附材質,而是直接以熔融矽纖維(fused silica fiber)本身就當作吸附 物質,所以早期固相微萃取法的萃取效率及濃度分析的線性範圍,都不 能得到理想的分析結果,後來分析研究者開始發展使用非極性高分子塗 覆物質,例如 poly-dimethlysiloxane. (62). ,以及極性高分子塗覆物質,例如. (63). poly-acrylate. ,使用這些塗覆材質後,不僅增加分析物的萃取效率,更. 提昇固相微萃取法多元化分析的發展趨勢,所以說使用固相微萃取法, 其萃取效率的優劣評價,首重條件是塗覆纖維種類的正確選擇,目前市 面上可供選擇的商業產品種類眾多,一般常用者可分為六種:如表 2-6 (64). ,由於不同的塗覆纖維,其材質特性各異,所適用於分析物的種類也. 就不同,一般的選擇基準,是依據“極性溶於極性;非極性溶於非極性” 的基本原則,如具非極性、高揮發性之碳氫化合物可選擇 PDMS (poly-dimethlysiloxane)塗覆材質做為吸附纖維. (65). ,而具極性及揮發性. 之酚類化合物則選用 PA(poly-acrylate)塗覆材質做為吸附纖維. 19. (66). ,由.
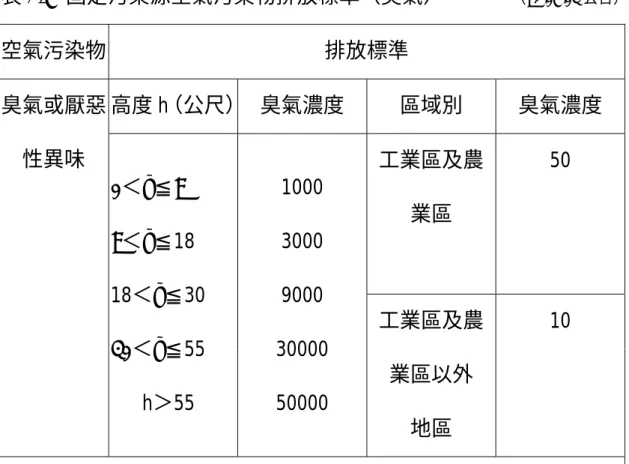
(33) 於塗覆纖維材質具多樣化的選擇,這也是固相微萃取技術廣受分析者喜 愛選用的原因之一。 現今固相微萃取技術之應用已相當廣泛,除環境分析樣品外. (67). ,其. (68-71). 他在食品、藥物、有機農藥、刑事鑑定等方面,都有許多文獻的發表 而其中以揮發性脂肪酸為分析標的物的報告. ,. (72-81). ,更是不勝枚舉,所以. 建立固相微萃取法測定垃圾污水中揮發性脂肪酸之研究,理論上是合理 可行的,但評估能適用於環境實際樣品的分析成效,就是本研究主要尋 求的研究目標。. 2-6 國內惡臭管制概況. 空氣污染防制法施行細則第二十六條指出,所稱惡臭是足以引起厭 惡或其他不良情緒反應之氣味,同法第三十三條規定,惡臭測定指檢查 人員以嗅覺進行氣味之判定,現行國內對於空氣中臭味之測定方法,是 以臭氣及異味官能測定法-三點比較式嗅袋法為公告標準方法,係將試 樣氣體以純淨空氣適當稀釋後,置於三個嗅袋中的一個,由六名合格嗅 覺判定員判別可聞出之稀釋倍數,以臭氣濃度表示之。 引起臭味的成分有很多種類,其中以硫化物、有機胺、有機酸、Indols、 Phenols 最為主要原因. (2). ,由於惡臭對人類的健康有害,在高濃度時會引. 起呼吸障礙、肺水腫、死亡等中毒現象,因此政府訂有空氣污染防制法 20.
(34) 及勞工安全衛生法,規定空氣污染物排放標準及勞工作業環境空氣中有 害物質容許濃度標準,做為臭氣污染源的管控指標,如表 2-7、表 2-8。 法定之人為官能檢查法,並非測定單一物質之臭氣,而是多種成分 之複合臭氣,基於此因素,本研究為發展固相微萃取法測定廢水中揮發 性脂肪酸之檢測機制,嘗試建立一個簡單方便測試單一臭味成分的分析 方法,如同行政院環境保護署環境檢驗所於九十二年八月五日公告,水 中 Geosmin 及 2-Methylisoborneol 檢測方法,即是以固相微萃取/氣相層析 質譜儀法,測定飲用水中二種土霉味道成分的檢測方法,這也是首件以 固相微萃取法測定臭味成分的公告方法,可見未來,使用固相微萃取法 做為臭味成分的監測分析工具,是有相當寬廣的發展研究空間,同時可 以將實驗分析結果進一步與官能檢查法應用比對,一併探討惡臭污染的 成分來源,提供適當的除臭方案參考。. 21.
(35) 第三章設備材料與方法 3-1 儀器設備及材料. 1. 固相微萃取裝置固定器(SPME manual holder)(Supelco 公司 No.57330-U) 2. 固相微萃取吸附纖維(SPME fiber)(Supelco 公司 75 μm polydimethylsiloxane-carboxen fiber;PDMS-CAR fiber) 3. 固相微萃取採樣裝置(SPME sampling stand stand / 4mL vial puck) (Supelco 公司 57333-U 含電磁攪拌器 stir plate) 4. 氣相層析儀 GC:PE-AutoSystem XL (1)偵測器:火焰游離偵測器(Flame Ionization Deterctor;FID) (2)層析管柱:J&W Scientific DB-5 30m×0.25mm ID(SUPELCO) (3)高壓氣體:用於 GC-FID(國產三福化工) (4)高壓氫氣:用於 GC-FID(國產三福化工) (5)高壓氮氣:用於 GC 之攜行氣體(純度為 99.995﹪) 5. 取樣瓶 vial:4mL,glass,KIMAX,Mexico(含 PTFE 中空螺蓋與墊片) 6. 定量瓶 volumetric flask:10、25、200 mL, glass, IWAKI, Japan 7. 計時器 8. 藥品:. 22.
(36) (1)乙酸 (Acetic Acid):99.5﹪、丙酸 (Propionic Acid):99.5 ﹪、丁酸 (Butyric Acid):99.5﹪、異丁酸 (Isobutyric Acid): 99.5﹪(CHEM SERVIEE)分析級 (2)Volatile Acid Standard Mix:10mM(UPELCO-46975-U)分析級. 3-2 實驗步驟. 1.先配製不同濃度之酸標準溶液注入氣相層析儀建立初步圖譜。 2.以固相微量萃取裝置之不銹鋼針插入樣品瓶中, (內含已知濃度之 酸標準液)伸出塗覆纖維待一段時間,使分析物於塗覆纖維與樣品間 達到分配平衡後縮回纖維,再將固相微量萃取裝置插入氣相層析儀 注射口內,利用注射口的溫度將分析物自纖維上脫附下來,進入儀 器中分析。 3.以標準混合酸,建立氣相層析儀最適分析條件參數。 4.建立酸標準溶液固相微萃取分析之檢量線。 5.建立酸標準溶液固相微萃取之偵測極限。 6.計算分析方法精密度與準確度。 7.樣品滯留時窗測試。 8.樣品儲存穩定度測試。. 23.
(37) 9.實際樣品採樣分析。 3-3 實驗方法. 3-3-1 氣相層析儀最適分析條件測試. 本研究參考行政院環境保護署環境檢驗所制定之「環境檢測標準方 法驗證程序準則」82 做為分析方法判定之依據,同時參考顏氏 (. (39). ). 研究指. 出,使用 GC-FID 法,對於垃圾水中甲酸之濃度值,無法獲得靈敏的偵測 結果,而在垃圾污水中甲酸的量相當微少,因此本實驗將針對垃圾污水 中含量較多之乙酸、丙酸、丁酸、異丁酸,做為實驗標的分析物。 1.標準溶液製備 乙酸、丙酸、丁酸、異丁酸、揮發性混合酸等分析物,均易溶於水, 所以本實驗將酸標準溶液,直接以去離子水配製成不同濃度之儲備標 準溶液備用。 2.標準品檢測 取一固定量儲備標準溶液,經固相微萃取/頂空法吸附樣品後,以 GC/FID 進行分析,設定注射口溫度 250℃,火焰離子偵測器溫度 250 ℃條件下,探討管柱中不同流速和升溫程式,尋求最佳分離條件。. 24.
(38) 3-3-2 固相微萃取最適分析條件測試. 1.固相微萃取纖維選擇 揮發性脂肪酸為具兩極性的揮發性物質,依據「分析物質極性吸附極 性,非極性吸附非極性」原則,參考相關文獻. (73,80). 指出,選擇兩極性. 用途之 polydimethylsiloxane/carboxen(PDMS/CAR)纖維,能夠得到最 佳的分析結果,因此,本實驗採用 PDMS/CAR 75μm 做為固相微萃取 吸附纖維。 2.固相微萃取吸附時間探討 樣品吸附時間的長短,對分析物是否達最大吸附量的平衡狀態有關,以 丙酸標準溶液配製成 10μg/ml , 20μg/ml 二種濃度,吸附時間分別以 5,10,15,20,25,30 分鐘進行吸附量評估測試。 3.固相微萃取吸附溫度探討 固相微萃取為適合於野外採樣的前處理技術,所以選擇以室溫(約 25±1 ℃)做為吸附溫度條件。 4.固相微萃取脫附溫度探討 樣品吸附量受萃取時溫度所影響,參考固相微萃取設備購買廠商所提供 的資料,PDMS/CAR 適用溫度範圍為 250-310℃,為防止高溫條件下, 容易造成揮發性分析物流失,所以本實驗選擇 250-℃做為分析條件。. 25.
(39) 5.固相微萃取脫附時間探討 將樣品進行第一次脫附分析完成後,再進行相同條件第二次脫附,選擇 脫附效率接近 100﹪的最適脫附時間。 脫附效率的計算式如下:. 第一次化合物脫附積分面積 脫附效率(﹪)=. ×100﹪ 第一次+第二次化合物積分面積. 3-3-3 標準品檢量線. 以儲備標準溶液配製至少 5 種不同濃度之校正用標準溶液,其中一 個濃度需接近但稍高於儀器偵測極限,其餘濃度可為偵測器工作濃度範 圍內之濃度,研定檢量線線性相關係數(correlation coefficient , r)必須 大於 0.995 以上。 3-3-4 儀器偵測極限測試(Instrumental Detection Limit ;IDL). 先將儀器感度調至最靈敏程度,直接以去離子水分別測定 7 次,計 算所得儀器訊號之平均值及標準偏差,儀器偵測極限為平均值加上 3 倍 標準偏差相對應之濃度。. 26.
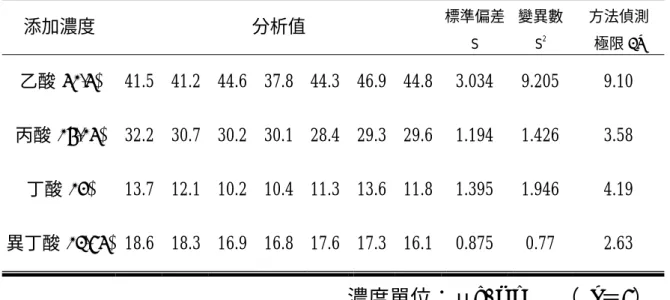
(40) 3-3-5 方法偵測極限測試. (82). (Method Detection Limit;MDL). 建立方法偵測極限測定步驟如下: (1) 預估偵測極限:採用已知儀器極限濃度值為預估值。 (2) 測定分析物之方法偵測極限. (82). :. a. 準備試劑水,試劑水中不得分析物或干擾物之干擾。 b. 於試劑水中之分析物,其濃度為預估偵測極限之 1-5 倍。 c. 重覆分析水樣 7 次,並將測得結果換算求淂濃度。 d. 計算 7 次測定值之標準偏差 S。 e. 方法偵測極限 MDL= t(n-1,1-α=0.99)(S) ,當 n=7,在 99 ﹪可信度,由 Students΄t 值表中查得 ( t n-1, 1-α=0.99)=3.143, 故方法偵測極限定為 3S。 (3)方法偵測極限之確認. (82). a. 將 MDL 濃度之分析物添加於試劑水中,重覆分析 7 次。 b. 若 a.之測定不能得到分析物之定性測定,則 MDL 應介於目前 之添加值及前次可測得定性測定之添加值之間。 c. 若 a.之添加測定能得到定性測定結果,計算標準偏差的平方 SB2 。若 SA2 / SB2 小於 3.05,依下列計算: Spooled =【(6 SA2 +6 SB2 / 12)】1/2. 27.
(41) d. 若 SA2 / SB2 大於 3.05,重覆步驟(3)a. MDL 之測定。 e. MDL=2.681(Spooled),t(12, 1-α=0.99)=2.681 (4)分別添加乙酸 52.5 μg/ml,丙酸 26.25μg/ml,丁酸 24μg/ml, 異丁酸 23.75μg/ml 濃度的標準溶液各進行 7 次重覆分析,計算平 均值及標準偏差,以求方法偵測極限。 (5)方法偵測極限確認之添加濃度,乙酸 28μg/ml,丙酸 13.44μg/ml, 丁酸 21.04μg/ml,異丁酸 13.18μg/ml 濃度的標準溶液各進行 7 次 重覆分析,計算平均值及標準偏差,以共同的標準偏差(pooled standard deviation,Spooled)確認方法偵測極限值。. 3-3-6 精密度與準確度測試. (82). (1)取不同濃度之標準酸儲備溶液,乙酸 15μg/ml、丙酸 2.5μg/ml、 丁酸 2μg/ml、異丁酸 4.75μg/ml,分別進行 7 次重覆分析,依 USEPA SW-846 方法. (83). ,計算相對標準偏差以求精密度,以及百分回收率. 以求準確度。 (2)取定量之混合酸儲備溶液,內含乙酸 60μg/ml,丙酸 74μg/ml,丁 酸 88μg/ml,異丁酸 88μg/ml,進行 7 次重覆分析,計算相對標準 偏差以求精密度,以及百分回收率以求準確度。. 28.
(42) 3-3-7 滯留時窗測試. (82). 利用乙酸、丙酸、丁酸、異丁酸標準溶液,檢量線中間點濃度連續 三天每天分析一次,由 3 次的滯留時間統計得到平均滯留時間及其標準 偏差,可接受的滯留時窗即為平均滯留時間±3S。. 3-3-8 樣品儲存穩定度測試. (82). 取不同濃度之標準酸儲備溶液,乙酸濃度為 146.16μg/ml,丙酸 159.28μg/ml,丁酸 220.40μg/ml,異丁酸 152.22μg/ml,將樣品放置於 4℃冷藏冰箱中保存,分別依第 1 天、第 2 天、第 3 天、第 4 天、第 5 天、 第 7 天、第 9 天保存後,各日將樣品各做 3 次重覆分析,計算平均濃度 值及標準偏差,以分析濃度結果相對於第 1 天檢測之濃度,並以回收率 計算樣品之各日儲存穩定度變化情形。. 3-4 採樣規劃. 3-4-1 實際樣品採樣. 本研究選擇垃圾污水做為實際樣品採樣分析,並以固相微萃取前處. 29.
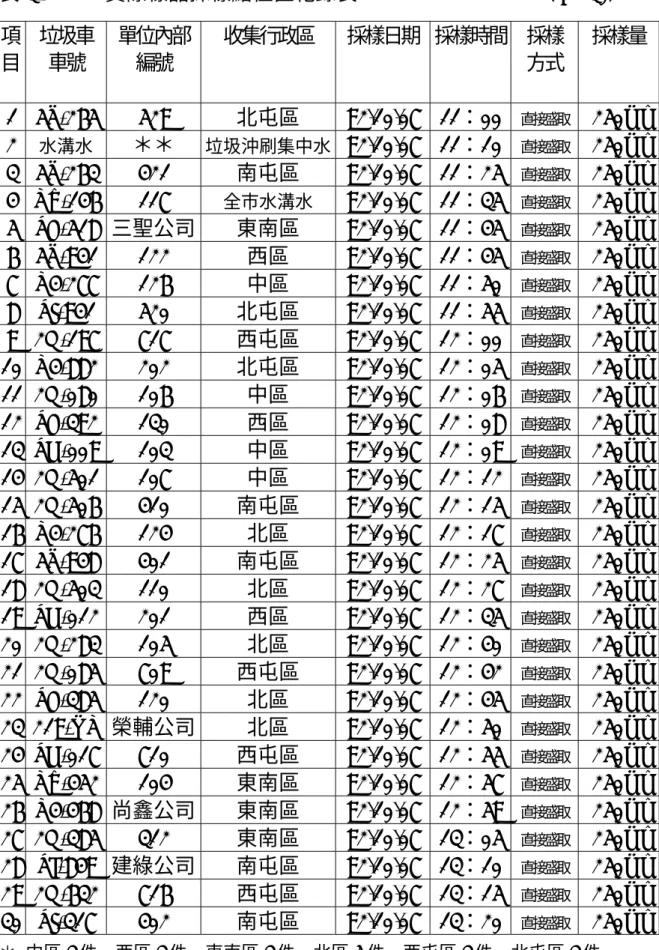
(43) 理技術結合氣相層析儀進行定性定量分析,為建立固相微萃取適用於野 外採樣分析之檢測機制,並能瞭解垃圾污水中揮發性脂肪酸含量濃度與 惡臭污染源的相關聯性,做為即時掌握污染源訊息的參考。. 3-4-2 採樣點. 以台中市垃圾車所殘留之垃圾污水做為實際樣品,共計採樣 30 點 次,採樣時間為九十二年十月七日,採樣車種為台中市八個行政區之垃 圾車輛,係定期收集台中市民之一般廢物及廚餘,組成物包括可燃物: 紙、木材、塑膠、橡膠、皮革類等,以及烹調、膳後之廢棄食物,產生 源為家庭、餐廳、市場、機關、學校、商店等;隨機採樣分為中區 4 件、 西區 3 件、東南區 4 件、北區 5 件、西屯區 4 件、北屯區 3 件、南屯區 5 件、垃圾水溝水 2 件,採樣方式是以採樣塑膠瓶直接盛取垃圾污水出水 口水樣,採樣量為 250 ml,樣品採集完成後,立即密封瓶蓋,並清楚標 明採樣時間、車輛編號等資料,放置於攜帶型冷藏箱內,並於 2 小時內 送至實驗室,放入 4℃之冰箱冷藏管理,以利進行後續分析。採樣地點紀 錄表及採樣圖,如表 3-1。. 3-4-3 實際樣品分析 3-4-3-1 樣品檢量線 30.
(44) (1) 配製前參考相關文獻. (39,81). ,決定檢量線濃度範圍。. (2) 以儲備標準溶液配製至少 5 種不同濃度之校正用標準溶液,濃 度範圍以能涵蓋樣品濃度範圍為原則,其可接受的檢量線線性 相關係數(correlation coefficient , r)必須大於 0.995 以上。. 3-4-3-2 空白樣品測試. 使用去離子水,依同分析物樣品分析步驟檢測,每批次至多 10 個樣 品時,需製備一個空白樣品,判別分析過程或儀器方面是否遭受污染, 空白樣品的分析結果,須低於分析物的方法偵測極限的 2 倍。. 3-4-3-3 實際樣品測試. 30 個採樣樣本,依乙酸、丙酸、丁酸、異丁酸濃度,各進行重覆分 析 3 次,計算平均值、標準偏差及各揮發酸之含量濃度值。. 3-5 資料處理及分析. 原始的分析數據須加以處理,以便進行定量工作,本實驗分析之所 有數據,包括標準品測試、實際樣品分析等數據,均以 Excel 2000 應用 軟體處理建檔,輔以 SAS 套裝軟體進行統計分析。 31.
(45) 第四章結果與討論 4-1 最適分析條件結果. 4-1-1 氣相層析儀實驗條件. 氣相層析法適用於在分析條件下,不易被裂解或發生化學結構改變 的可揮發性有機物,本實驗以揮發性脂肪酸為分析物,其最適氣相層析 儀器分析條件,GC 注射口溫度 250℃、偵測器溫度 250℃,載流氣體流 速 1.33ml/min,管柱起始溫度 80℃維持 1 分鐘,以每分鐘上升 5℃升至 85℃維持 3 分鐘,再以每分鐘 20℃上升至 150℃維持 1 分鐘,結果如表 4-1,以此條件可以有效的將分析物分開,所得分析層析圖,如圖 4-1。. 4-1-2 固相微萃取法萃取條件. 1.吸附時間 以丙酸標準溶液配製成 10μg/ml , 20μg/ml 二種濃度,由表 4-2、圖 4-2 結果顯示,吸附 10 分鐘後,回收率分別達到 79.1%(10μg/ml) 及 103.2 %(20μg/ml),吸附 20 分鐘後,回收率達到 85.5%(10μ g/ml)及 104.1%(20μg/ml),為確保分析準確度,本實驗將吸附時 間設定為 20 分鐘。. 32.
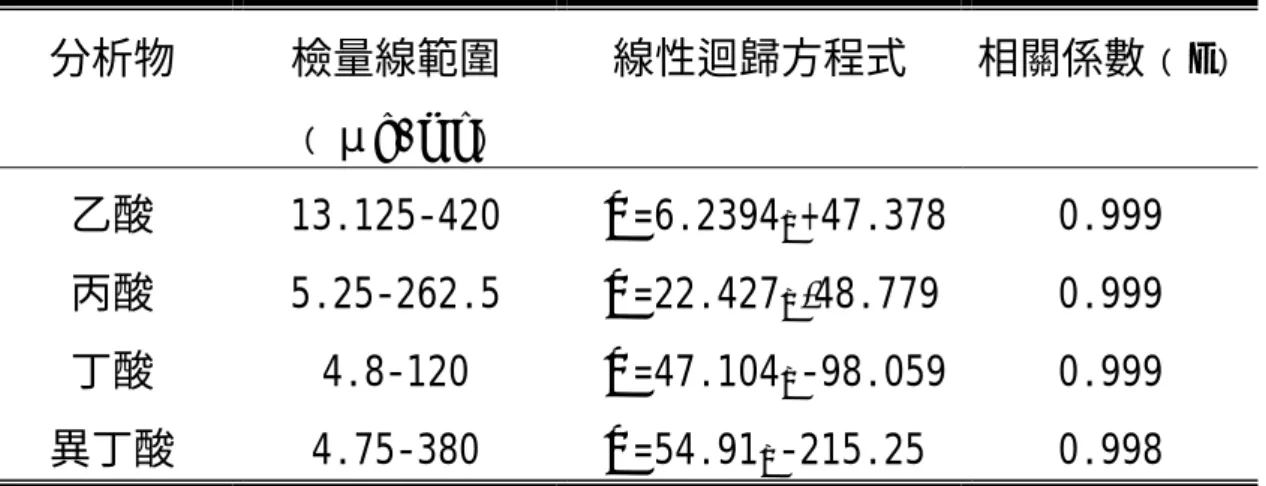
(46) 2.脫附時間 脫附時間設定為 1,2,3,5 分鐘,計算脫附效率,如圖 4-3 的結果,在 脫附 3 分鐘時,乙酸達 100﹪、丙酸達 99.6﹪、丁酸達 99.9﹪、異 丁酸達 99.8﹪,其脫率效率極佳,因此,本實驗採用 3 分鐘為脫附 時間。 3.固相微萃取最適萃取條件 採用 PDMS/CAR 75μm 吸附纖維,以頂空方式萃取,吸附溫度為室 溫 25±1℃,吸附時間 20 分鐘,脫附溫度為 250℃,脫附時間為 3 分鐘, 如表 4-3 之參數條件。. 4-2 標準品分析結果. 4-2-1 檢量線及圖譜. 配製不同濃度之酸標準儲備溶液,建立標準品之檢量線及 GC 圖 譜,如圖 4-4 至圖 4-7 及圖 4-1,檢量線之線性範圍,乙酸 13.125-420 μg/ml、丙酸 5.25-262.5μg/ml、丁酸 4.8-120μg/ml、異丁酸 4.75-380 μg/ml,其線性相關係數(r),乙酸 0.999、丙酸 0.999、丁酸 0.999、 異丁酸 0.998,均大於 0.995 以上,如表 4-4。. 33.
(47) 4-2-2 儀器偵測極限 以去離子水分別重覆測定 7 次,計算所得儀器訊號之平均值及標準 偏差,儀器偵測極限為平均值加上 3 倍標準偏差相對應之濃度。進行四 次測得結果分別為 4.3μg/ml、2.58μg/ml、1.01μg/ml、0.47μg/ml,各 分析值、平均值及標準偏差結果,如表 4-5。. 4-2-3 方法偵測極限. 本實驗採用行政院環境保護署環境檢驗所所制定之「方法偵測極限 之定義及測定步驟」82 ,以不同濃度之酸標準溶液各進行重覆分析 7 次, (. ). 測得方法偵測極限預估值(3S) ,乙酸 9.1μg/ml、丙酸 3.58μg/ml、丁酸 4.19μg/ml、異丁酸 2.63μg/ml,並經由確認步驟後,所得方法偵測極限 結果,乙酸 9.2μg/ml、丙酸 2.65μg/ml、丁酸 3.11μg/ml、異丁酸 2.47 μg/ml,定量偵測極限結果(10 Spooled) ,乙酸 34.28μg/ml、丙酸 9.86 μg/ml、丁酸 11.61μg/ml、異丁酸 9.21μg/ml,各分析值及結果如表 4-6、表 4-7。. 4-2-4 精密度與準確度. (82-83). 1.配製不同濃度之酸標準儲備溶液,分別進行 5 次重覆分析,以相對. 34.
(48) 標準偏差(RSD﹪)表示精密度,四種酸分析物的精密度結果,乙 酸 6.04﹪、丙酸 5.19﹪、丁酸 7.29﹪、異丁酸 6.15﹪,精密度皆小 於 10﹪;以百分回收率表示準確度,結果為乙酸 105.32±12.72﹪、 丙酸 97.68±10.14﹪、丁酸 102.40 ±14.94﹪、異丁酸 97.09 ±11.94﹪, 所有分析物的準確度結果皆位於 120-80﹪的範圍,所測得之精密度 及準確度結果,如表 4-8。 2.取定量之混合酸儲備溶液,內含乙酸 60μg/ml、丙酸 74μg/ml、丁 酸 88μg/ml、異丁酸 88μg/ml,進行 7 次重覆分析,所得四種揮發 酸精密度之相對標準偏差結果為乙酸 4.51﹪、丙酸 3.19﹪、丁酸 6.06﹪、異丁酸 5.02﹪,精密度皆小於 10﹪;以百分回收率表示準 確度,結果為乙酸 98.94±8.93﹪、丙酸 100.86±6.44﹪、丁酸 100.18 ±12.14﹪、異丁酸 98.73 ±9.91﹪,所有分析物的準確度結果皆位於 115-85﹪的範圍,所測得之精密度及準確度結果,如表 4-9。. 4-2-5 滯留時窗. (82). 利用乙酸、丙酸、丁酸、異丁酸標準品檢量線中間點濃度之儲備標 準溶液,連續三天每天分析一次,由三次的滯留時間作統計,結果如表 4-10 所示,滯留時間的變異係數 CV﹪,乙酸 3.04﹪、丙酸 2.4﹪、丁酸 2.77﹪、異丁酸 3.08﹪,皆小於 5﹪,表示分析儀器具有良好的再現性 35.
(49) 及可靠性。 4-2-6 樣品儲存穩定度. 如表 4-11、圖 4-8、圖 4-9 結果顯示,樣品在 4℃下儲存至第 7 天, 平均回收率為乙酸 98.1﹪、丙酸 97.07﹪、丁酸 94.24﹪、異丁酸 100.11 ﹪,第 7 天起回收率開始呈現明顯下降趨勢,儲存至第 9 天之平均回收 率為乙酸 96.33﹪、丙酸 95.69﹪、丁酸 91.95﹪、異丁酸 98.08﹪,因此 採用本法分析,應於 7 天內完成分析進度,才能獲得有效性的數據結果, 避免產生分析誤差。. 4-3 實際樣品分析結果. 4-3-1 檢量線及圖譜 配製不同濃度之酸標準儲備溶液,建立樣品校正用之檢量線,如圖 4-10 至圖 4-13,檢量線之線性範圍,乙酸 60-900μg/ml、丙酸 74-740 μg/ml、丁酸 44-660μg/ml、異丁酸 10-660μg/ml,其線性相關係數(r) , 乙酸 0.997、丙酸 0.996、丁酸 0.997、異丁酸 0.999,均大於 0.995 以上, 如表 4-12。. 36.
(50) 4-3-2 空白樣品. 實際樣品分析時必須每批次同時進行一個空白樣品分析,以去離子 水當做空白樣品分析,如圖 4-14。. 4-3-3 實際樣品分析. 30 個實際樣品分析,垃圾污水中四種揮發性脂肪酸分析濃度結 果,如圖 4-15 及表 4-13;平均濃度為乙酸 755.30μg/ml、丙酸 115.02 μg/ml、丁酸 146.93μg/ml、異丁酸 13.08μg/ml,如圖 4-16;各揮發酸 分別進行 3 次重覆分析,其積分面積之變異係數(CV﹪),除丁酸、異 丁酸變異情形較大外,乙酸、丙酸之變異係數(CV﹪)均小於 30﹪,結 果如表 4-14 及圖 4-17。由以上綜合結果得知,垃圾污水中所含短鏈揮發 性脂肪酸,以乙酸含量最多,在 30 個實際樣品分析結果中顯示,乙酸濃 度分析之變異係數均小於 20﹪,因其廢水厭氧處理程序中,是以乙酸的 濃度值當做偵察指標值(50-300mg/l)10 ,所以選擇固相微萃取法檢測揮 (. ). 發性脂肪酸之濃度分析,應用於環保實務上的廢水監測管理,是一項合 理可行的樣品前處理技術。. 37.
(51) 第五章結論與建議 結論 1. 固相微萃取之設備簡單,操作容易,非常適合於野外之環境樣品採樣, 本研究選擇垃圾污水做為實際樣品,固相微萃取法可以簡化採樣及樣 品處理之繁瑣過程,並能減低經濟分析成本,是為一項實務應用相當 有效的樣品前處理方法。 2. 垃圾污水中所含的成份相當繁多複雜,本研究採用固相微萃取法,進 行揮發性脂肪酸樣品前處理萃取步驟,因其對分析物的萃取濃縮效率 ,並不受樣品體積大小及複雜基質干擾的因素所限制,這就是選擇固 相微萃取法具備優勢條件的原因,也必能獲得事半功倍的分析效益。 3. 固相微萃取法經由檢測方法驗證,四種揮發性脂肪酸檢量線之線性相 關係數均可達 0.995 以上,其偵測極限可達 ppm 之靈敏度,惟對甲酸 偵測的靈敏度較低,無法偵測得知甲酸含量,但對於垃圾污水中高濃 度含量的乙酸、丙酸、丁酸、異丁酸,均可快速檢出,可以即時提供 惡臭污染源的濃度資料,具有相當實用的參考價值。 (39). 4. 回觀過去國內針對垃圾滲出水中揮發性脂肪酸之測定研究,僅顏氏. 一篇研究結果,現與本研究比對結果,如表 5-1,可以說明,本研究確 實可以應用於垃圾污水之檢測模式。 5. 本研究之方法偵測極限,乙酸 9.2μg/ml、丙酸 2.65μg/ml、丁酸 3.11. 38.
(52) μg/ml、異丁酸 2.47μg/ml,定量偵測極限結果,乙酸 34.28 μg/ml、 丙酸 9.86μg/ml、丁酸 11.61μg/ml、異丁酸 9.21μg/ml,而儀器偵測 極限結果,乙酸 4.3μg/ml、丙酸 2.58μg/ml、丁酸 1.01μg/ml、異丁 酸 0.47μg/ml。 6. 本研究之精密度與準確度,精密度結果,乙酸 4.51﹪、丙酸 3.19﹪、 丁酸 6.06﹪、異丁酸 5.02﹪,精密度皆小於 10﹪;準確度結果,乙酸 98.94±8.93﹪、丙酸 100.86±6.44﹪、丁酸 100.18 ±12.14﹪、異丁酸 98.73 ±9.91﹪,準確度的範圍在 112.32-88.04﹪。 7. 樣品儲存穩定度測試結果,顯示樣品儲存至第 7 天起,回收率濃度開 始呈現明顯下降趨勢,採用本法分析的進度,應於一星期內完成分析, 才能獲得有效性的數據結果,避免產生回收率降低的誤差。 8. 30 個實際樣品分析,測定垃圾污水中揮發性脂肪酸含量,平均濃度為 乙酸 755.30μg/ml、丙酸 115.02μg/ml、丁酸 146.93μg/ml、異丁酸 13.08μg/ml,其中以乙酸含量最多,而乙酸重覆分析之變異係數均小 於 20﹪,表示本法確有可接受程度的再現性結果,所以應用於實務分 析,將是一項合理可行的技術。. 39.
(53) 建議 1. 環保署環檢所已公告之標準檢測方法中,尚未建立廢水中揮發性脂肪 酸之檢測方法,但揮發性脂肪酸的濃度控管,在廢水厭氧處理程序上 是相當重要的,建議以固相微萃取法應用於廢水中揮發性脂肪酸之檢 測,做為未來標準方法制定的參考。. 2. 硫化物、氨氮、揮發性脂肪酸等三種物質是環境中產生惡臭之主要物 質,目前針對惡臭之偵測方法,多以人為嗅覺測定法為主,缺點是較 主觀且影響因素多,容易造成誤差,建議採用快速的分析數據結果, 對應惡臭污染源濃度的含量,積極發展防制惡臭污染的因應對策,. 3. 本研究採用固相微萃取之最適條件,部份以參考文獻為主,對於提高 吸附效率的促進條件,例如:使分析物更容易揮發至氣相、樣品加熱、 加入鹽類、使用不同吸附纖維等因素,建議未來可以配合使用氣相層 析質譜儀做進一步定性定量分析,將可獲得更多精密分析的訊息。. 40.
(54) 第六章. 參考文獻. 1.N. Masque, R.M. Marce, F.Borrull, Anal. Chem.,17(6),384(1998). 2.羅中恆、劉惠群、吳繼芳、郭偉明,“豬場空氣中臭味污染物質的特性 研究”,中華農學會報,新第 172 期(1995). 3. J. Pawliszyn,“Inexpensive Fluid Delivery System For Supercritical Fluid Extraction”J. High Resolut. Chromatogr.,13,199-202(1990). 4.賴滋漢,黃卓治,“基礎食品化學”,富林出版社(1999). 5. Clair N. Sawyer / Perry L. McCarty / Gene F. Parkin (原著)、蕭蘊華、 傅崇德、許鼎居(譯述),“環境工程化學”,美商麥格羅、希爾國際股 份有限公司(1999). 6. Sawyer/McCarty(原著) 、謝立生、黃建業(譯述),“環境工程化學”, 乾泰圖書有限公司(1986). 7. E. Lie, T. Welander, Water Res.,31, 1269(1997). 8. G. Manni, F. Caron, J., Chromatogr. A ,690, 237(1995). 9. A.Wheatley (Editor),“Anaerobic Digestion: A Waste Treatment Technology”, Elsevier Applied Science , London(1990). 10.周志儒、林健三、楊惠嘉,“環境污染防治”,台灣復文興業股份有限公 司(1999). 41.
(55) 11. A.A. Randall, L. D. Benefield, W. E. Hill, J. P. Nicol, G. K. Boman, S. R. Jing, Water Sci. Technol. 35, 153(1997). 12.A. M. Eilersen, M. Henze, L. Kloft, Water Res. 29, 1259(1995). 13.工業污染防治手冊之二十五,“廢水處理廠操作管理”,經濟部工業污 染防治技術服務團(1991). 14.張漢昌,“環境污染防治”,文京圖書有限公司(1996). 15.H. H.Willard, L. I. Merritt and J. A. Dean:,“Instrumental Methods of al Analysis.”,5th ed..D. Van Nostrand. New York. (1974). 16.行政院勞工委員會,“勞工作業環境空氣中有害物採樣分析建議方法 通則篇”(1995). 17.“Formic Acid in Workplase Atmospheres”, Method ID-112(1985). Inorganic Section, OSHA Analytical Laboratory, Salt Lake City, Utah. 18.Backup Date Report for Acetic Acid, prepared under NIOSH Contract No. 210-76-0123, available as “Ten NIOSH Analytical Methods”, Order No. PB 275-834 from NTIS, Springfield, VA 22161. 19.NIOSH Research Report,“Development and Validation of Methods for Sampling and Analysis of Workplace Toxic Substances”, U. S. Department of Health and Human Services, Publ.( NIOSH)80-133(1980). 20.H. F. Mueller, A. M. Buswell, and T. E. Larson, Chromatographic Determination of Volatile Acids, Sewage and Ind. Wastes , 28:255(1956).. 42.
(56) 21.“International Critical Tables”McGraw-Hill, New York. 22.A. M. Buswell and S. L. Neave,, Laboratory Studies on Sludge Digestion, Iillinois State Water Survey Bull., 30,76(1930). 23.S.Egashira, Jap. Anal.,15,1356(1966). 24.W. K. Lee and R. M. Bethea, J. Gas Chromatogr. , 5 , 99(1967) . 25.B. Kaplanova and J. Janak, Mikrochim. Acta , 119(1966). 26.Y. Kaburaki and Y. Sato , J. Agric. Chem. Soc. Jap. , 36. 865(1962). 27.R. Raveux and J. Bove, Bull. Soc. Chim. Fr. ,369(1967). 28.A.G.Stradomskaya and I.A.Goncharova, Gidrokhim.Mater.,39.169(1965). 29.A. Gomoryova , Acta Chim. Acad. Sci. Hung.,33.251(1962). 30.B. Pszonka , Chem. Anal. ( Warsaw) , 13 , 83(1968) . 31.J. M. Brot Gebeck , 19 , 238 (1965). 32.K.J. Hauser, R. J. Zabransky, J. Clin. Microb. 2,1(1975). 33.A. E. Greenbrg, R. R. Trussell, L. S. Clesceri(Eds.),Standard Methods for the Examination of Water and Wastewater, American Public Haelth Association, Washington, DC, 16th ed., Section 504, A, B. 34.T. J. Clark, J. E. Bunch, J. Chromatogr. Sci. 35, 206(1997). 35.D. A. Skoog, D. M. West, and F. J. Holler, Fundamental of Analytical Chemistry, 7th ed., Saunders Collwege Publishung, Philadolphia(1996). 36.Giulio Manni, Francois Caron, “Calibration and determination of volatile fatty acids in waste leachates by gas chromatography”,Journal of 43.
(57) Chromatography A, 690 ,237-242(1995). 37.P. J. Hutchinson,The Geology of Landfills ,Env. Geosci.,2, pp.2-14(1995). 38.X.Yang , C. Lee and M.Scranton I.,.“Determination of nanomolar amounts of individual dissolved low molecular weight amines and organic acids in sea water” Anal. Chem., 65: 572-576(1993). 39.顏慶堂、鄭政峰,“以層析法測定垃圾滲出水中揮發性脂肪酸之研究”, 國立中興大學化學研究所,碩士論文(1990). 40.T. Hino, S. Nakanishi, T. Hobo, “Development of a whole headspace injection method for the determination of volatile organic compounds in water”, J. of Chromatography A, 746, 83-90(1996). 41.G. Baykut and A. Voigt, “Spary Agueous into the Gas Phase for Gas Chromatography/Mass Spectrometry” , Anal. Chem.,64,677(1992). 42.H. Kessels, W. Hoogerwerf and J. Lips, “The Determination of Volatile Organic Compounds from EPA 524.4 Using Purge and Trap Capillary Gas Chromatogr”. Sci., 30, 207(1992). 43.行政院環保署環境檢驗所九十一年十一月二十八日環署檢字第 0910083743 A 號公告“水中揮發性有機化合物檢測方法”. 44.T. Class and K. Ballschmiter, “Chemistry of Organic Traces in Air V. Determination of Halogenated C1-C2 Hydrocarbons Clean Marine Air and Amibient Continental Air and Rain by High Resolution Gas Chromato -graphy Using Different Stationary Phase ” , Anal. Chem.,325,1(1986). 45.C.Leuenberger, M. P. Ligocki and J. F. Pankow,“Trace Organic 44.
(58) Compounds in Rain. 4. Identities Concentrations, and Scavenging Mechanisms for Phenols in Urban Air and Rain”,Environ. Sci. Technol.,19, 1053(1985). 46.N. Ragunathan, K. Krock, C. Klawun, T. A. Sasaki, and C. L. Wilkins, J. Chromatogr. A, 703,277(1995). 47.Janusz Pempkowiak*,“C18 Reversed-Phase Trace Enrichment of Shortand Long- Chain (C2-C8-C20) Fatty Acids from Dilute Aqueous Solutions and Sea Water”, Journal of Chromatography, 258, 93-102(1983). 48.黃寶慧、李茂榮,“固相萃取技術”,Chemistry Dec., Vol. 56 No. 4. pp. 319-326(1998). 49.SUPELCO Chromatography Products, 33(1998). 50.C.L. Arthur,. J. Pawliszyn, “Solid phase microextraction with thermal desorption using fused silica optical fiber” Anal. Chem. Vol.62, No.19 , pp.2145-2148(1990). 51.黃賢達, “氣相層析及固相微萃取技術” Chemistry Sept. Vol. 54. No.3, pp. 119-122(1996). 52.黃寶慧、李茂榮, “固相微量萃取法” 科儀新知第十八卷二期(1996). 53.黃敬德、謝有容,“固相微萃取技術之原理及其應用” Chemistry Dec. Vol. 56. No.4, pp. 311-318(1998). 54.黃敬德、謝有容, “固相微萃取法” 科儀新知第十八卷四期(1997). 55.李德仁, “固相微萃取樣品前處理技術應用於揮發或半揮發有機物質之 45.
(59) GC 分析” 科儀新知第十九卷六期(1998). 56.吳鎧光、蔡詩偉, “以固相微萃取技術發展空氣中環氧乙烷”中國醫藥學 院環境醫學研究所,碩士論文(2002). 57.張峻鳴、蔡詩偉, “水中醛類檢測方法之改進”中國醫藥學院環境醫學研 究所, 碩士論文(2002). 58.Z. Zhang, M. J. Yang and J. Pawliszyn,Anal. Chem. 66 844A(1994). 59.SUPELCO Chromatography Products, 373(1995). 60.Z: Zhang, J. Pawliszyn, “Headspace Solid phase microextraction ” Anal. Chem. Vol.65, No.14, pp.1843-1852(1993). 61.J. Pawliszyn,1997, Solid- Phase Microextraction: Theory and Practice , Wiley- VCH, New York(1997). 62.M. Chai, C. L. Arthur and J. Pawliszyn , “Determination of Volatile Chlorinated Hydrocarbons in Air and Water with Solid-Phase Microextraction”, Analyst, 118, 1501(1993). 63.K. D. Bochholz and J. Pawliszyn ,“Optimization of Solid-Phase Microextraction for Determination of Phenols”,Anal. Chem.,66,160 (1994). 64.項維欣、李茂榮, “固相微萃取/脈衝噴霧捕捉技術於水中氯酚之偵測” 國立中興大學化學研究所,碩士論文(1998). 65.許麗秋、郭錦堂, “利用固相微萃取量測尿中 BTEX 之技術” 中國醫藥 學院環境醫學研究所,碩士論文(2000). 46.
(60) 66.葉曜嘉、李茂榮, “固相微萃取法於微量酚之萃取偵測” 國立中興大學 化學研究所,碩士論文(1996). 67.Maria de Fatima Alpendurada, “Solid-phase microextraction: a promising technique for sample preparation in environmental analysis -Review ” Journal of Chromatography A , 889, 3-14(2000). 68.H.Kataoka, H. Lord and J. Pawliszyn, “Applications of SPME in Food Analysis” J. Chromatogr. 880, 35-62(2000). 69.H.Lord, H. Kataoka, J. Wu and J. Pawliszyn,“Drug Analysis in Biological Matrices Using Fiber and In- tube SPME/LC/MS” ExTech’99, International Symposium on Advances in Extraction Technologies, Waterloo, ON, May(1999). 70.H..Lord, Y. Guo and J. Pawliszyn, “SPME for Analysis of Pesticides” 31th Eastern Canada Pesticide and environmental Contaminants Workshop, Niagata on the Lake, Ontario,May(1999). 71.X. Yu, H. Yuan, T. Gorecki and J. Pawliszyn ,“Determination of Blood and Urine by SPME”The Pittsburgh Conference,Orlando,Florida, March (1999). 72.L. Pan, J. Pawliszyn, “Determination of Short Fatty Acids in Aqueous Sample by Using Solid-Phase Microextraction” The Pittsburgh Conference, Chicago, Illinois, March(1994). 73.M. Abalos. J. Bayona and J. Pawliszyn,“ Development of a Headspace SPME procedure for the Determination of Free Volatile Fatty Acids in. 47.
(61) Waste Waters” J. Chromatogr. 873, 107-115(2000). 74.H.Kim, C.Nochetto, L.L. McConnell,“Gas-Phase Analysis of Trimethylamine,Propionic and Butyric Acids,and Sulfur Compounds Using SolidPhase Microextraction” Anal. Chem.; (Article), 74(5); 1054-1060(2002). 75. A. F.Gonzalez-Cordova; B. Vallejo-Cordoba,“Quantitative Determination of Short-Chain Free Fatty Acids in Milk Using Solid-Phase Microextraction and Gas Chromatography ”J. Agric. Food Chem.; (Article); 49(10);4603-4608(2001). 76. O.Pinho, I. M. P. L. V. Ferreira, M. A. Ferreira,“Solid-Phase Microextraction in Combination with GC/MS for Quantification of the Major Volatile Free Fatty Avids in Ewe Cheese” Anal. Chem.; (Article);74 (20), 5199-5204(2002). 77. R. M.Tomqino, J. D. Parker, D. K. Larick,“Analysis of Free Fatty Acids in Whey Products by Solid-Phase Microextraction” J. Agric. Food Chem.; (Article); 49(8);3993-3998(2001). 78. J.A. Cruwys, R. M. Dinsdale, F. R. Hawkes, D.L. Hawkes, “Development of a static headspace gas chromatographic procedure for the routine analysis of volatile fatty acids in wastewaters” Journal of Chromatography A , 945 , 195-209(2002). 79.Gy. Wittmann, H. Van Langenhove, J. Dewulf, “Determination of acetic acid in aqueous samples, by water-phase derivatisation, solid-phase microextraction and gas chromatography” Journal of Chromatography A , 874, 225-234(2000).. 48.
(62) 80.M. Abalos, J. M. Bayona ,“Application of gas chromatography coupled to chemical ionization mass spectrometry following headspace solid-phase microextraction for the determination of free volatile fatty acids in aqueous samples ” Journal of Chromatography A , 891, 287-294(2000). 81.Shao-Pin Yo.,“Analysis of volatile fatty acids in wastewater collected from a pig farm by a solid-phase microextraction method” Chemosphere Vol:38, Issue: 4, February, pp. 823-834(1999). 82.行政院環境保護署環境檢驗所環境檢測方法驗證程序準則,凌永健、 陳秋雲,“化學分析的偵測極限(上)”,科儀新知第十六卷第一期,8 (1994). 83.USEPA SW-846,“Test Methods for Evaluating Solid Waste”,Quality Control,(1994).. 49.
數據
![表 2-1 揮發性脂肪酸的物理常數(C 1 -C 8 ) ( 5,21 ) 表 2-2 行政院勞工委員會採樣分析建議方法(甲酸)( 16 ) 採樣 分析 採樣介質: 0.01 N NaOH 10mL 於標準小 型多細孔噴頭衝擊瓶內 流速:1000 mL / min 採樣量:120 L 樣品運送:密封 樣品穩定性:未測定 現場空白樣品:樣品數的 10﹪ 準確度 範圍:4.17~12.5 mg / m 3 偏差:不顯著[ 1 ] 全精密度偏差(CV T ) :0.111[ 1 , 4 ]](https://thumb-ap.123doks.com/thumbv2/9libinfo/8979366.281192/63.892.124.794.173.491/行政院採樣N於標準小型多細孔噴頭衝擊流速採樣量準確度不顯著.webp)
![表 2-3 行政院勞工委員會採樣分析建議方法(乙酸) ( 16 ) 採樣 分析 採樣介質:活性碳管(100 mg / 50 mg ) 流率:10~1000 mL / min 採樣體積:最小 20 L 最大 300 L 樣品運送:例行性 樣品穩定性:大於 7 天 @25℃ 現場空白樣品:每批樣品數的 10﹪,至 少二個以上 準確度[ 1 ] 範圍: 12.5~50 mg / m 3 (173<空氣樣品) 偏差:5.4﹪ 總變異係數(CV T ) :5.8﹪ 準確度:15.5﹪ 方法](https://thumb-ap.123doks.com/thumbv2/9libinfo/8979366.281192/64.892.126.782.138.589/行政院體積L最大L樣品運送例行樣品穩定準確度空氣樣品準確度.webp)
+7


相關文件
Finally, we want to point out that the global uniqueness of determining the Hartree po- tential (Theorem 2.5) and the determination of the nonlinear potential in the
Vessella, Quantitative estimates of unique continuation for parabolic equa- tions, determination of unknown time-varying boundaries and optimal stability estimates, Inverse
Attributes characterized in this campaign for the turboshaft engine emissions include, for particulate phase, engine smoke number (SN) (which currently is the only accepted method
Recommendation 14: Subject to the availability of resources and the proposed parameters, we recommend that the Government should consider extending the Financial Assistance
stating clearly the important learning concepts to strengthen the coverage of knowledge, so as to build a solid knowledge base for students; reorganising and
• • Linear Mode : Provide Wild Mass Range Linear Mode : Provide Wild Mass Range – – Protein MW determination Protein MW
• Non-vanishing Berry phase results from a non-analyticity in the electronic wave function as function of R.. • Non-vanishing Berry phase results from a non-analyticity in
Let T ⇤ be the temperature at which the GWs are produced from the cosmological phase transition. Without significant reheating, this temperature can be approximated by the
