molecules
ISSN 1420-3049 www.mdpi.com/journal/molecules
Article
Biological Activity of Oleanane Triterpene Derivatives Obtained
by Chemical Derivatization
Shi-Yie Cheng 1,Chao-Min Wang 2, Hsueh-Ling Cheng 3, Hui-Jye Chen 4, Yuan-Man Hsu 5,
Yu-Chi Lin 6 and Chang-Hung Chou 2,6,7,*
1 Department of Life Sciences, National University of Kaohsiung, Kaohsiung 811, Taiwan;
E-Mail: [email protected]
2 Research Center for Biodiversity, China Medical University, Taichung 40402, Taiwan;
E-Mail:[email protected]
3 Department of Biological Science and Technology, National Pingtung University of Science and
Technology, Pingtung 91201, Taiwan; E-Mail:[email protected]
4 Graduate Institute of Molecular Systems Biomedicine, China Medical University, Taichung 40402,
Taiwan; E-Mail: [email protected]
5 Department of Biological Science and Technology, China Medical University, Taichung 40402,
Taiwan; E-Mail: [email protected]
6 Department of Life Sciences, National Cheng Kung University, Tainan 701, Taiwan; E-Mail:
7 Graduate Institute of Ecology and Evolutionary Biology, China Medical University, Taichung
40402, Taiwan
* Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +886-4-2205-3366 (ext.1633); Fax: +886-4-2207-1500.
Received: 5 September 2013; in revised form: 11 October 2013 / Accepted: 14 October 2013 / Published: October 2013
Abstract: Nine new derivatives of oleanane triterpenoids isolated from Fatsia polycarpa Hayata were synthesized through chemical transformations. Acetylation was effected by reaction with acetic anhydride in pyridine to afford compounds 1–5, while compound 6 was obtained using 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC·HCl) in CH2Cl2. The others derivatives 7–9 were obtained in reactions of the
corresponding triterpenoids with EDC·HCl, 4-N,N-dimethylaminopyridine hydrochloride and 4-N,N-dimethylaminopyridine in CH2Cl2. The structures of 1–9 were elucidated from
extensive spectroscopic and HRESIMS data, while the structure of 9 was further confirmed by X-ray diffraction analysis. The cytotoxic, anti-hepatitis B virus (HBV), antibacterial,
hypoglycaemic and Wnt signaling activities of these derivatives were evaluated in vitro. Keywords: oleanane triterpenoids; Fatsia polycarpa Hayata; cytotoxic; anti-hepatitis B virus (HBV); antibacterial; hypoglycaemic; Wnt responsive reporter activities
1. Introduction
Numerous oleananoids have gererated tremendous interest from the standpoint of their biological activities, such as antigiardial, anti-HIV, antihyperglycemia, anti-inflammatory, antimycobacterial, antioxidative, antitumor and cardiovascular properties [1–10]. Some of them are known to have anticarcinogenic activity in experimental animals [11]. Although the bioactivity of oleanolic acid is modest, it has been marketed in China as an oral drug for treating liver disorders in humans. It has been well recognized to possess anti-inflammatory and antihyperlipidemic activities in animals [12].
Structure-activity relationship studies of betulinic acid and dihydrobetulinic acid derivatives have also proved that various structural modifications, for example C-3 ester substitution, may provide analogs with greatly enhanced activity [13,14]. In our previous paper we reported the isolation and characterization of seven oleananoids, named fatsicarpains A–G, with moderate cytotoxic and antibacterial activities [2]. In view of the bioactive potential of these isolated compounds, the goal of this study was to modify the two active portions of them, namely, the C-3 hydroxy group and the carboxyl group at C-28. We have thus prepared nine new derivatives 1–9, as shown in Scheme 1. Compounds 1–5 were synthesized as acetylation products with acetic anhydride in pyridine at 50 °C for 6 h, while 6 was obtained using EDC·HCl in anhydrous CH2Cl2 at 50 °C for 3 h. Derivatives 7–9
were obtained in the reactions of the corresponding triterpenoids with EDC·HCl, DMAP·HCl and DMAP in CH2Cl2 at room temperature overnight. The structure elucidations of 1–9 were performed by
NMR and HR-ESI-MS analyses. The structure of 9 was further confirmed by X-ray diffraction analyses [15].
Compounds 1–9 were evaluated for the in vitro cytotoxicity against cancer cell lines HepG2 2.2.15 (human hepatocellular carcinoma). The anti-hepatitis B virus (HBV) effects of 1–9, in terms of the inhibition of hepatitis B surface antigen (HBsAg) and hepatitis B virus e antigen (HBeAg), were also measured. Moreover, bioassays were conducted for hypoglycaemic activity, and for antibacterial activity against Helicobacter pylori, Bacillus cereus, Enterococcus faecalis, Escherichia coli, Listeria monocytogenes, Salmonella enterica, Staphylococcus aureus and Pseudomonas aeruginosa. Besides, the effects of the tested compounds on the Wnt/β-catenin signaling, a pathway that once it is disregulated will cause many diseases including various types of cancers, were revealed by the Wnt reporter assay [16].
Scheme 1. Outline of the present work (Preparation of compounds 1–9).
2. Results and Discussion
Compound 1 was prepared in the overnight reaction of fatsicarpain D [2] and acetic anhydride in anhydrous pyridine at 50 °C. The HRESIMS of 1 exhibited a pseudomolecular ion peak at m/z 519.3445 [M + Na]+, consistent with the molecular formula of C
32H48O4, requiring nine degrees of
unsaturation. The IR spectrum of 1 showed the diagnostic absorption band of an acetoxy functionality at 1,733 cm−1, which was further supported by the 1H-NMR signals at δ
H 2.04 (3H, s) and at δH 4.64
ppm for the H-3 (situated downfield from the resonance of the corresponding proton in the starting compound (δH 3.42 ppm) [2], and 13C-NMR signals at δC 170.8 (qC) and 21.3 (CH3). Additionally,
acetylation induced significant downfield shifts of H-3 (ΔδH = 1.22 ppm) [2]. The carbonyl signal was
attributed to the acetate moiety linked to C-3, as further confirmed through the crucial HMBC correlation from H-3 to the carbonyl carbon of 3-OAc. Similarly, fatsicarpain C, 3α-hydroxyolean-11-en-28,13β-olide, fatsicarpain F and 3α-hydroxyolean-11,13(18)-dien-28-oic acid were submitted to acetylation with Ac2O in pyridine at room temperature overnight to yield 2–5, respectively. The NMR
data revealed the presence of characteristic O-acetyl group signals (see Experimental). Furthermore, the structures of 2–5 were definitely confirmed by the crucial HMBC correlations of the acetyl carbonyl carbon signals in each case with H-3.
N-(3-(Dimethylamino)propyl)-N-(ethylcarbamoyl)-3α,23-dihydroxyolean-11,13(18)-dien-28-amide (6), synthesized from fatsicarpain A and EDC·HCl in dry CH2Cl2 (at 50 °C for 3 h), was obtained as a
white amorphous powder. Compound 6 was analyzed for the molecular formula of C38H64O4N3 by the
Experimental). The NMR features of 6 were analogous to those of fatsicarpain A except that the resonances of the carboxylic acid at C-28 were replaced by those of N-(3-(dimethylamino)propyl)-N-(ethylcarbamoyl)formamide group. By interpretation of 1H-1H COSY correlations, it was possible to
establish two partial structures of consecutive proton systems extending from H2-1' to H2-3' through
H2-2', and from H2-6' to H2-7'. The crucial HMBC correlations from H2-16 to C-28 and from H2-6' to
C-8' revealed the connectivity of the above partial structures (Figure 1). Consequently, the structure of 6 was unambiguously established. Moreover, the suggested pathway involves the rearrangement of the O-acylisourea 6a to the stable N-acylurea 6, as illustrated in Figure 2. O-acylisourea 6a is unstable in anhydrous CH2Cl2 and can undergo cyclic electronic displacement (O→N acyl migration), producing
the thermodynamically more stable N-acylurea 6 [17].
Figure 1. Selected 1H-1H COSY (▬) and HMBC (→) correlations of 6.
Figure 2. Suggested pathway for the conversion of fatsicarpain A to 6.
Compound 7, prepared by overnight reaction of fatsicarpain A with EDC·HCl, DMAP and DMAP·HCl in CH2Cl2 at room temperature, was obtained as a white amorphous powder. It was
analyzed for the molecular formula of C60H90O7 by the positive HRESIMS (m/z 945.6595, [M+Na]+)
coupled with its 13C-NMR spectroscopic data (see Experimental). The characteristic pattern for a
noncyclic and saturated anhydride in the IR spectrum of 7 is the appearance of two strong bands at 1,793 and 1,772 cm−1, and this was further supported by the 13C-NMR signals at δ
C 172.7 (qC).
Anhydride coupling induced significant upfield shifts of C-28 (ΔδC = 7.3 ppm) [2]. The position of the
compounds 8 and 9 were prepared by anhydride coupling of 3α-hydroxyolean-11,13(18)-dien-28-oic acid and 5, respectively. The IR spectra and 13C-NMR data revealed the presence of characteristic
anhydride group signals (see Experimental). In addition, compound 9 was fully characterized by the X-ray analysis (Figure 3) which confirmed the structure and presence of anhydride group.
Figure 3. X-ray ORTEP diagram of 9.
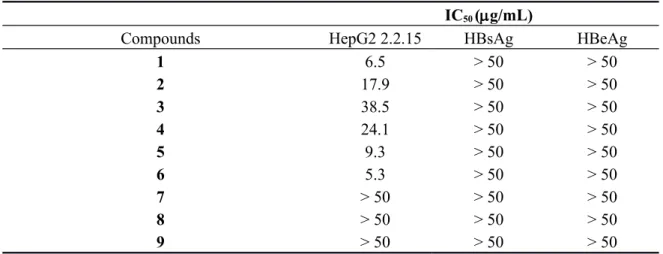
Of four million patients worldwide infected with hepatitis B virus (HBV), about 20% are expected to develop chronic hepatitis, liver cirrhosis, or hepatocarcinoma [18]. Anti-HBV effects of compounds 1–9 were screened in vitro using the human hepatocellular carcinoma (HepG2 2.2.15) cell model system with fluorouracil as a positive control (Table 1). Preliminary cytotoxicity screening revealed that 1–6 exhibited inhibition effects on HBV replicated DNA level in the IC50 values of 6.5,17.9,38.5,
24.1, 9.3 and 5.3 μM, respectively. The esterification of the hydroxy group at C-3 increased the resultant cytotoxicity against HepG2 2.2.15, as shown for the acetylated derivatives 1–5. By far, compound 6 exhibited the highest potency against HepG2 2.2.15. Particularly, compounds 7–9, possessing an anhydride group, showed no significant activity against HepG2 2.2.15 (IC50 > 50 μM),
which confirmed that the carboxylic acid at C-28 is essential to their cytotoxicity. After varying its structure at C-3 and C-28 positions, we found 1–6 to be more potent than the parent compounds (Table 1), and these findings are in accordance with the available literature data concering structure−activity relationship [13,14]. However, hepatitis B surface antigen (HBsAg) and hepatitis B virus e antigen (HBeAg) in HepG2 2.2.15 cells were not significantly inhibited by the tested compounds.
Table 1. Cytotoxicity data of compounds 1–9, fatsicarpains A, C, D, F, 3α-hydroxyolean-11-en-28,13β-olide and 3α-hydroxyolean-11,13(18)-dien-28-oic acid.
IC50 (g/mL)
Compounds HepG2 2.2.15 HBsAg HBeAg
1 6.5 > 50 > 50 2 17.9 > 50 > 50 3 38.5 > 50 > 50 4 24.1 > 50 > 50 5 9.3 > 50 > 50 6 5.3 > 50 > 50 7 > 50 > 50 > 50 8 > 50 > 50 > 50 9 > 50 > 50 > 50
Table 1. Cont.
IC50 (g/mL)
Compounds HepG2 2.2.15 HBsAg HBeAg
Fatsicarpain A 18.9 > 50 > 50 Fatsicarpain C 16.7 > 50 > 50 Fatsicarpain D 28.8 > 50 > 50 Fatsicarpain F 23.9 > 50 > 50 3á-Hydroxyolean-11-en-28,13â-olide > 50 > 50 > 50 3á-Hydroxyolean-11,13(18)-dien-28-oic acid > 50 > 50 > 50
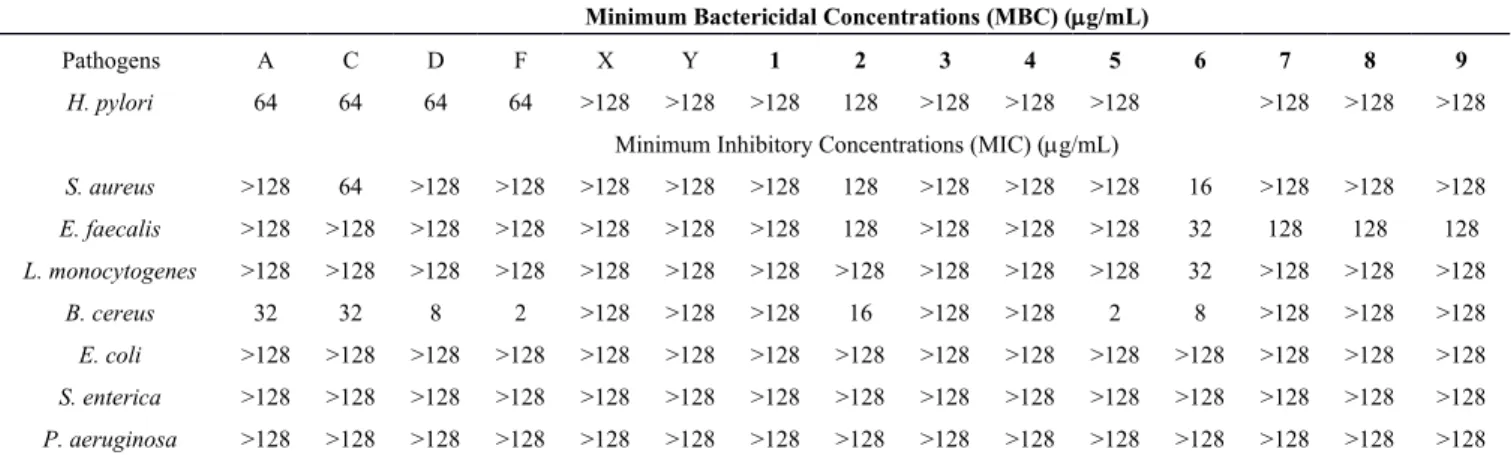
Helicobacter pylori infection is associated with an increased risk for development of duodenal ulcers, gastric ulcers, gastric adenocarcinomas and gastric lymphomas. However, as other bacterial pathogens, antibiotic resistance to H. pylori is an increasing problem for eradicating infection [19]. Therefore, finding a safe and efficient treatment to decrease the need or even replace antibiotics for eradicating H. pylori infection in human becomes necessary and an important task. Preliminary anti-H. pylori activity revealed that compound 2 exhibited moderate antibacterial activity with a minimum bactericidal concentrations (MBC) of 128 μg/mL. With the exception of the above observations, the obtained negative results showed that compounds 1, 3–5 and 7–9 exhibited no discernible activity (MBC > 128 μg/mL) (Table 2).
In the past decades, bacterial resistance to the antibiotics has emerged a serious global problem in human and veterinary medicine. The abuse of antibiotics for non-perscription application has accelerated the generation of superbacteria which makes a critical issue. According to a previous report from 1991 to 2000, the Bacillus cereus played the leading role of outbreak case of food-borne pathogens (41.2%, 113 of 171 outbreaks), followed by Staphylococcus aureus (17.9%) and Vibrio parahaemolyticus (15.7%) in central Taiwan [20]. Also, the modifiction at C-3 position of betulinic acid, oleanolic acid and ursolic acid increased antimycobacterial activity aganist Mycobacterium tubereulosis [21]. For that reason the antibacterial activities of new modified oleanane-type derivatives 1–9 were evalutaed against seven bacteria: B. cereus, E. faecalis, E. coli, L. monocytogenes, S. enterica, S. aureus and P. aeruginosa and compared with the activity of parent compounds (Table 2) [2]. As expected, the C-3 acetylated derivatives 2, 5 and 6 exhibited more potent than the parent compounds. Additionally, compounds 2, 5 and 6 revealed greater antibacterial potential than the positive control (ampicillin) against B. cereus. Particularly, compounds 5 and 6 showed significant antibacterial activity against B. cereus with MIC values at 2 and 8 µg/mL, respectively. Only 6 showed specific antibacterial activity against S. aureus and L. monocytogenes with MIC values at 16 and 32 µg/mL, respectively. It was noteworthy to mention that modification of the functional group at C-28 from carboxylic acid to N-(3-(dimethylamino)propyl)-N-(ethylcarbamoyl)formamide moiety increased the antibacterial activity against L. monocytogenes significantly. Moreover, it is thought that coupling of two active compounds would generate more activity, but anhydride derivatives 7–9 did not exhibit significant antibacterial activity against all tested pathogens. The present result suggested that the presence of the C-28 carboxylic acid moiety is important for significant activity against B. cereus. However, none of the tested compounds had significant activity against Gram-negative pathogens, E. coli, S. enterica and P. aeruginosa.
Table 2. Antibacterial activity of 1–9, fatsicarpains A, C, D, F, 3α-hydroxyolean-11-en-28,13β-olide (X) and 3α-hydroxyolean-11,13(18)-dien-28-oic acid (Y).
Minimum Bactericidal Concentrations (MBC) (g/mL)
Pathogens A C D F X Y 1 2 3 4 5 6 7 8 9
H. pylori 64 64 64 64 >128 >128 >128 128 >128 >128 >128 >128 >128 >128
Minimum Inhibitory Concentrations (MIC) (g/mL)
S. aureus >128 64 >128 >128 >128 >128 >128 128 >128 >128 >128 16 >128 >128 >128 E. faecalis >128 >128 >128 >128 >128 >128 >128 128 >128 >128 >128 32 128 128 128 L. monocytogenes >128 >128 >128 >128 >128 >128 >128 >128 >128 >128 >128 32 >128 >128 >128 B. cereus 32 32 8 2 >128 >128 >128 16 >128 >128 2 8 >128 >128 >128 E. coli >128 >128 >128 >128 >128 >128 >128 >128 >128 >128 >128 >128 >128 >128 >128 S. enterica >128 >128 >128 >128 >128 >128 >128 >128 >128 >128 >128 >128 >128 >128 >128 P. aeruginosa >128 >128 >128 >128 >128 >128 >128 >128 >128 >128 >128 >128 >128 >128 >128
Ampicillin was used as a positive control, and it showed antibacterial activity against S. Aureus, E. Faecalis,
L. monocytogenes, B. cereus, E. coli, S. enterica, and P. aeruginosa with MIC values of 8, 2, 1, 128, 4, 1 and
512 µg/mL, respectively.
The hypoglycaemic activities were determined only for the modified compounds 1–5 and 7–9, the exception being 6 due to insufficient quantities for testing. The hypoglycaemic activities were tested using cell-based screening method, in which glucose uptake of cells treated with the new compound is quantified and compared with that of untreated cells and cells stimulated with insulin [22,23]. It was found that compounds 1–5 but not 7–9 enhanced glucose uptake of treated cells compared with untreated, and that their effects were similar to that of insulin (Figure 4), indicating that compounds 1–
5 possess insulin-like hypoglycaemic activities that can promote the glucose uptake of cells. However, the underlying mechanism is not clear. Whether it involves the activation of the insulin signaling pathway requires further investigation.
Figure 4. Glucose uptake assays for cells treated with compounds (20 μM). A, assays for 1, 2 and 4; B, assay for 5; C, assays for 3, 7, 8 and 9. The total amount of medium glucose consumed by the cells between 0 to 5 h of treatment was calculated, and data expressed as relative glucose uptake versus control (cells with no treatment). Data represent the mean ± standard deviation of triplicate. *p < 0.05 versus control by two-way ANOVA.
Wnts proteins are secreted lipoglycoproteins that function as signaling molecules to regulate embryonic development and tissue homeostatis [24]. Aberrant Wnt signaling can cause an array of human diseases, including schizophrenia, pulmonary fibrosis, rheumatoid arthritis [25,26],
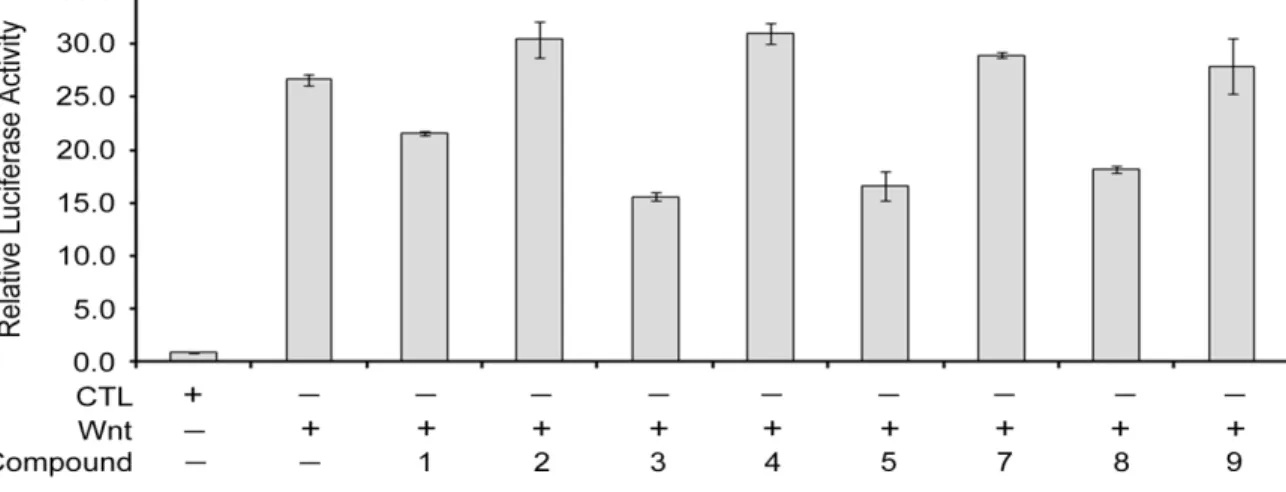
osteoporosis, tetraamelia syndrome, neurodegenerative diseases and various cancers [27–31]. To know more about the biological effects of 1–5 and 7–9, these compounds were tested on the inhibition of canonical Wnt signaling using the TCF/β-catenin-mediated luciferase activity assay [16]. Compound 6 again was not tested due to paucity of the sample. As compared to the assay performed in Wnt-3a conditioned medium without any drug treatment (set as 100%), compounds 1, 3, 5 and 8 at 1 μM of concentration specifically inhibited the Wnt signaling by 20%, 40%, 38% and 32%, respectively, while 2, 4, 7 and 9 were not effective (Figure 5). These data showed that Wnt signaling is sensitive to 1, 3, 5 and 8 and also suggested the potential use of these compounds for the therapy of Wnt-related diseases in the future.
Figure 5. The effect of oleanane triterpene derivatives on -catenin/TCF-mediated luciferase activities in P19 cells. Cells were transfected with Wnt reporter pGL3-OT and normalization vector pTK-Renilla, and treated with control-conditioned medium (CTL), Wnt3a-conditioned medium (Wnt), or different compounds of oleanane triterpene derivatives in Wnt3a-conditioned medium (compounds 1–5 and 7–9) for 20 h, then cell lysates were harvested for dual luciferase activity assays. Each bar is the mean ±S.D. Each experiment was performed in triplicate.
3. Experimental 3.1. General
Optical rotations were determined with a JASCO P2000 digital polarimeter. Ultraviolet (UV) and infrared (IR) spectra were obtained on JASCO V-650 and JASCO FT/IR-4100 spectrophotometers, respectively. The NMR spectra were recorded on a Varian Unity INOVA 600 FT-NMR spectrometer (600 MHz for 1H; 150 MHz for 13C, respectively). Chemical shifts were reported using residual CDCl
3
(δH 7.26 and δC 77.0 ppm) as internal standard. High-resolution ESIMS spectra were obtained on a
LTQ Orbitrap XL (Thermo Fisher Scientific) spectrometer. The crystallographic data were collected on a Bruker D8 Discover SSS X-ray diffractometer equipped with a closed molybdenum tube generator and parabolic Göbel mirror. Silica gel 60 (Merck, 230–400 mesh), LiChroprep RP-18 (Merck, 40–63 μm) and Sephadex LH-20 (Amersham Pharmacia Biotech.) were used for column chromatography (CC). Pre-coated silica gel plates (Merck, Kieselgel 60 F254, 0.25 mm) and pre-coated
RP-18 F254s plates (Merck) were used for analytical thin-layer chromatography (TLC) analyses.
High-performance liquid chromatography (HPLC) was carried out using a Hitachi L-2130 pump equipped with a Hitachi L-2420 UV-vis detector at 220 nm and a semi-preparative reversed-phase column (Merck, Hibar Purospher RP-18e, 5 μm, 250 × 10 mm).
3.2. Plant Material
Leaves and twigs of Fatsia polycarpa (7.1 kg) were collected at Hohuan Mountain (2,105 m elevation), Taiwan, in November 2009, and identified by one of the authors (C.-H. C.). A voucher specimen (FPL) was deposited in the Research Center for Biodiversity, China Medical University, Taiwan. 3.3. Acetylation
Fatsicarpain D (2.0 mg) was dissolved in pyridine (0.5 mL) and allowed to react overnight at room temperature with acetic anhydride (one drop). Then, the reaction was quenched by the addition of 1.0 mL of H2O, followed by extraction with EtOAc (3 × 1.0 mL). The EtOAc extracts were combined and
evaporated. The resulting residue was subjected to a short silica gel column using n-hexane–EtOAc (3:1) to yield an acetylated product 1 (1.9 mg). Additionally, fatsicarpain C (3.0 mg), 11-en-28,13β-olide (3.0 mg), fatsicarpain F (3.0 mg) and 3α-hydroxyolean-11,13(18)-dien-28-oic acid (3.0 mg) were subjected to acetylation, and chromatographic purification of the product according to the same procedure mentioned under acetylation of fatsicarpain D, to give 2 (2.9 mg), 3 (2.8 mg), 4 (2.7 mg) and 5 (2.9 mg), respectively.
3α-Acetoxyolean-9,12-dien-28-oic acid (1). Yield 91%. white amorphous powder; [α]25
D +75 (c = 0.1,
CHCl3); UV (MeOH) λmax (log ε) 212 (4.40), 274 (3.85) nm; IR (KBr) νmax 3,458, 2,941, 1,733,
1,700, 1,455, 1,374, 1,245, 758 cm−1; 1H-NMR (CDCl 3, 600 MHz) δH 1.59 (1H, m, H-1α), 1.76 (1H, m, H-1β), 1.73 (1H, m, H-2α), 1.94 (1H, m, H-2β), 4.64 (1H, br t, J = 3.0 Hz, H-3), 1.31 (1H, m, H-5), 1.50 (2H, m, H-6), 1.72 (1H, m, H-7α), 1.32 (1H, m, H-7β),5.60 (1H, d, J = 6.0 Hz, H-11), 5.58 (1H, d, J = 6.0 Hz, H-12), 1.16 (1H, m, H-15α), 1.83 (1H, m, H-15β), 1.99 (1H, m, H-16α), 1.68 (1H, m, H-16β), 2.98 (1H, dd, J = 13.8, 4.2 Hz, H-18), 1.62 (1H, m, H-19α), 1.20 (1H, m, H-19β), 1.23 (1H, m, H-21α), 1.36 (1H, m, H-21β), 1.79 (1H, m, H-22α), 1.66 (1H, m, H-22β),0.89 (3H, s, H-23), 0.91 (1H, s, H-24), 1.19 (3H, s, H-25), 0.99 (3H, s, H-26), 1.08 (3H, s, H-27), 0.95 (3H, s, H-29), 0.90 (3H, s, H-30), 2.04 (3H, s, 3-OAc); 13C-NMR (CDCl 3, 150 MHz) δC32.2 (t, C-1), 23.3 (t, C-2), 77.8 (d, C-3), 36.8 (s, C-4), 46.0 (d, C-5), 18.0 (t, C-6), 31.8 (t, C-7), 42.4 (s, C-8), 154.8 (s, C-9), 38.7 (s, C-10), 115.2 (d, C-11),120.7 (d, C-12), 144.7 (s, C-13),40.8 (s, C-14),26.9 (t, C-15), 23.6 (t, C-16), 45.9 (s, C-17), 39.4 (d, C-18), 45.9 (t, C-19),30.6 (s, C-20), 33.7 (t, C-21), 32.1 (t, C-22), 27.8 (q, C-23), 21.9 (q, C-24),24.8 (q, C-25), 20.4 (q, C-26), 20.1 (q, C-27), 182.8 (s, C-28),23.5 (q, C-29), 32.9 (q, C-30), 170.8 (s, 3-OAc), 21.3 (q, 3-OAc); ESIMS m/z 519 [M+Na]+; HRESIMS m/z 519.3445 [M+Na]+
(Calcd for C32H48O4Na, 519.3445).
3α-Acetoxy-24-formylolean-11,13(18)-dien-28-oic acid (2). Yield 85%. white amorphous powder; [α]25
νmax 3,454, 2,941, 2,861, 1,734, 1,698, 1,450, 1,373, 1,242, 759 cm−1; 1H-NMR (CDCl 3, 600 MHz) δH 1.33 (1H, m, H-1α), 1.51 (1H, m, H-1β), 1.78 (1H, m, H-2α), 1.93 (1H, m, H-2β), 4.87 (1H, br s, H-3), 2.01 (1H, m, H-5), 1.50 (2H, m, H-6), 1.33 (1H, m, H-7α), 1.25 (1H, m, H-7β), 2.20 (1H, br s, H-9), 5.64 (1H, d, J = 10.2 Hz, H-11), 6.47 (1H, dd, J = 10.2, 3.0 Hz, H-12), 1.10 (1H, m, H-15α), 1.70 (1H, m, H-15β), 2.01 (1H, m, H-16α), 1.71 (1H, m, H-16β), 2.54 (1H, d, J = 14.4 Hz, H-19α), 1.69 (1H, d, J = 14.4 Hz, H-19β), 1.32 (1H, m, H-21α), 1.39 (1H, m, H-21β), 2.28 (1H, m, H-22α), 1.40 (1H, m, H-22β), 1.08 (3H, s, H-23), 9.41 (1H, s, H-24), 0.97 (3H, s, H-25), 0.82 (3H, s, H-26), 1.06 (3H, s, H-27), 0.81 (3H, s, H-29), 0.96 (3H, s, H-30), 2.06 (3H, s, 3-OAc); 13C-NMR (CDCl 3, 150 MHz) δC 31.8 (t, C-1), 22.2 (t, C-2), 74.6 (d, C-3), 51.1 (s, C-4), 43.9 (d, C-5), 20.3 (t, C-6), 31.9 (t, C-7), 41.2 (s, C-8), 53.9 (d, C-9), 36.1 (s, C-10), 126.2 (d, C-11), 125.5 (d, C-12), 136.8 (s, C-13), 42.2 (s, C-14), 24.8 (t, C-15), 32.6 (t, C-16), 48.0 (s, C-17), 131.5 (s, C-18), 40.5 (t, C-19), 32.4 (s, C-20), 36.8 (t, C-21), 35.4 (t, C-22), 14.0 (q, C-23), 205.9 (d, C-24), 17.8 (q, C-25), 16.6 (q, C-26), 19.9 (q, C-27), 180.9 (s, C-28), 24.0 (q, C-29), 32.2 (q, C-30), 170.0 (s, 3-OAc), 21.2 (q, 3-OAc); ESIMS m/z 533 [M + Na]+; HRESIMS m/z 533.3207 [M + Na]+ (Calcd for C
32H46O5Na, 533.3237).
3α-Acetoxyolean-11-en-28,13β-olide (3). Yield 82%. white amorphous powder; [α]25
D +52 (c = 0.1, CHCl3); IR (KBr) vmax 2,941, 1,750, 1,736, 1,456, 1,376, 1,247, 1,158, 756 cm−1; 1H-NMR (CDCl3, 600 MHz) δH 1.17 (1H, m, H-1α), 1.62 (1H, m, H-1β), 1.63 (1H, m, H-2α), 1.95 (1H, m, H-2β), 4.65 (1H, t, J = 2.4 Hz, H-3), 1.24 (1H, m, H-5), 1.50 (2H, m, H-6), 1.45 (1H, m, H-7α), 1.26 (1H, m, H-7β), 2.01 (1H, br s, H-9), 6.05 (1H, d, J = 10.2 Hz, H-11), 5.42 (1H, dd, J = 10.2, 3.0 Hz, H-12), 1.21 (1H, m, H-15α), 1.73 (1H, m, H-15β), 2.13 (1H, td, J = 13.2, 6.6 Hz, H-16α), 1.36 (1H, m, H-16β), 2.08 (1H, m, H-18), 1.82 (1H, t, J = 13.2 Hz, H-19α), 1.36 (1H, m, H-19β), 1.32 (1H, m, H-21α), 1.36 (1H, m, H-21β), 1.65 (1H, m, H-22α), 1.69 (1H, m, H-22β), 0.85 (3H, s, H-23), 0.90 (3H, s, H-24), 0.93 (3H, s, H-25), 1.07 (3H, s, H-26), 1.12 (3H, s, H-27), 0.88 (3H, s, H-29), 0.97 (3H, s, H-30), 2.08 (3H, s, 3-OAc); 13C-NMR (CDCl 3, 150 MHz) δC 33.5 (t, C-1), 22.6 (t, C-2), 78.1 (d, C-3), 36.7 (s, C-4), 49.6 (d, C-5), 17.4 (t, C-6), 30.9 (t, C-7), 41.4 (s, C-8), 52.9 (d, C-9), 36.4 (s, C-10), 135.8 (d, C-11), 126.8 (d, C-12), 89.8 (s, C-13), 41.7 (s, C-14), 25.3 (t, C-15), 21.3 (t, C-16), 44.0 (s, C-17), 50.5 (d, C-18), 37.3 (t, C-19), 31.4 (s, C-20), 34.3 (t, C-21), 27.1 (t, C-22), 27.6 (q, C-23), 21.2 (q, C-24), 17.7 (q, C-25), 19.0 (q, C-26), 18.5 (q, C-27), 180.1 (s, C-28), 23.5 (q, C-29), 33.2 (q, C-30), 170.7 (s, 3-OAc), 21.4 (q, 3-OAc); ESIMS m/z 519 [M + Na]+; HRESIMS m/z 519.3422 [M + Na]+
(Calcd for C32H48O4Na, 519.3445).
3α-Acetoxy-11α-methoxyolean-12-en-28-oic acid (4). Yield 83%. white amorphous powder; [α]25 D −76 (c = 0.1, CHCl3); IR (KBr) vmax 2,941, 1,737, 1,697, 1,450, 1,373, 1,243, 1,110, 756 cm−1; 1H-NMR (CDCl3, 600 MHz) δH 1.49 (1H, m, H-1α), 1.60 (1H, m, H-1β), 1.61 (1H, m, H-2α), 2.06 (1H, m, H-2β), 4.62 (1H, br s, H-3), 1.24 (1H, m, H-5), 1.46 (2H, m, H-6), 1.50 (1H, m, H-7α), 1.22 (1H, m, H-7β), 1.79 (1H, d, J = 8.4 Hz, H-9), 3.74 (1H, dd, J = 8.4, 3.6 Hz, H-11), 5.53 (1H, d, J = 3.6 Hz, H-12), 1.18 (1H, m, H-15α), 2.01 (1H, m, H-15β), 2.03 (1H, m, H-16α), 1.63 (1H, m, H-16β), 2.87 (1H, dd, J = 13.2, 3.6 Hz, H-18), 1.61 (1H, m, H-19α), 1.19 (1H, m, H-19β), 1.24 (1H, m, H-21α), 1.37 (1H, m, 21β), 1.80 (1H, m, 22α), 1.62 (1H, m, 22β), 0.85 (3H, s, 23), 0.89 (3H, s, H-24), 1.03 (3H, s, H-25), 0.78 (3H, s, H-26), 1.26 (3H, s, H-27), 0.96 (3H, s, H-29), 0.93 (3H, s, H-30), 2.07 (3H, s, 3-OAc); 13C-NMR (CDCl 3, 150 MHz) δC 34.3 (t, C-1), 29.4 (t, C-2), 78.4 (d, C-3), 36.5 (s,
4), 49.9 (d, 5), 18.1 (t, 6), 32.8 (t, 7), 42.8 (s, 8), 53.0 (d, 9), 38.2 (s, 10), 76.0 (d, C-11), 121.6 (d, C-12), 148.3 (s, C-13), 41.9 (s, C-14), 27.8 (t, C-15), 22.8 (t, C-16), 46.0 (s, C-17), 40.7 (d, C-18), 45.6 (t, C-19), 30.7 (s, C-20), 33.7 (t, C-21), 32.2 (t, C-22), 28.0 (q, C-23), 21.9 (q, C-24), 16.8 (q, C-25), 18.7 (q, C-26), 25.5 (q, C-27), 180.4 (s, C-28), 23.5 (q, C-29), 33.0 (q, C-30), 170.9 (s, 3-OAc), 21.4 (q, 3-OAc); ESIMS m/z 551 [M + Na]+; HRESIMS m/z 551.3686 [M + Na]+ (Calcd for
C33H52O5Na, 551.3707).
3α-Acetoxy olean-11,13(18)-dien-28-oic acid (5). Yield 85%. white amorphous powder; [α]25 D −51
(c = 0.1, CHCl3); UV (MeOH) λmax (log ε) 232 (4.10), 245 (3.68), 264 (3.65) nm; IR (KBr) vmax3,464,
2,941, 1,731, 1,696, 1,456, 1,376, 1,247, 756 cm−1; 1H-NMR (CDCl 3, 600 MHz) δH 1.25 (1H, m, H-1α), 1.64 (1H, m, H-1β),1.64 (1H, m, H-2α), 1.96 (1H, m, H-2β), 4.66 (1H, br s, H-3), 1.30 (1H, m, H-5), 1.50 (2H, m, H-6), 1.38 (1H, m, H-7α), 1.34 (1H, m, H-7β),2.05 (1H, br s, H-9), 5.65 (1H, d, J = 10.2 Hz, H-11), 6.43 (1H, dd, J = 10.2, 2.4 Hz, H-12), 1.09 (1H, m, H-15α), 1.72 (1H, m, H-15β), 2.00 (1H, m, H-16α), 1.72 (1H, m, H-16β), 2.54 (1H, d, J = 14.4 Hz, H-19α), 1.67 (1H, d, J = 14.4 Hz, H-19β), 1.29 (1H, m, H-21α), 1.40 (1H, m, H-21β), 2.27 (1H, m, H-22α), 1.40 (1H, m, H-22β), 0.89 (3H, s, H-23), 0.85 (3H, s, H-24), 0.93 (3H, s, H-25), 0.80 (3H, s, H-26), 1.02 (3H, s, H-27), 0.81 (3H, s, H-29), 0.95 (3H, s, H-30), 2.09 (3H, s, 3-OAc); 13C-NMR (CDCl 3, 150 MHz) δC 33.3 (t, C-1), 22.6 (t, C-2), 78.3 (d, C-3), 36.7 (s, C-4), 49.7 (d, C-5), 18.0 (t, C-6), 32.2 (t, C-7), 40.8 (s, C-8), 54.1 (d, C-9), 36.7 (s, C-10), 127.0 (d, C-11), 125.2 (d, C-12), 137.0 (s, C-13), 42.0 (s, C-14), 24.8 (t, C-15), 32.6 (t, C-16), 48.0 (s, C-17), 130.9 (s, C-18), 40.4 (t, C-19), 32.5 (s, C-20), 36.8 (t, C-21), 35.5 (t, C-22), 27.7 (q, C-23), 21.3 (q, C-24), 17.8 (q, C-25), 16.4 (q, C-26), 19.9 (q, C-27), 181.9 (s, C-28), 24.0 (q, C-29), 32.2 (q, C-30), 170.9 (s, 3-OAc), 21.4 (q, 3-OAc); ESIMS m/z 519 [M + Na]+;
HRESIMS m/z 519.3422 [M+Na]+ (Calcd for C
32H48O4Na, 519.3445).
3.4. Acylation
To a solution of fatsicarpain A (5.0 mg) in CH2Cl2 (1.0 mL) EDC·HCl (1.0 mg) was added. The
solution was stirred for 3 h at 50 °C. After the completion of the reaction the solvent was evaporated under reduced pressure to give a crude product which was subjected to a short silica gel column eluting with n-hexane–EtOAc (1:1) to yield 6 (3.2 mg).
N-(3-(Dimethylamino)propyl)-N-(ethylcarbamoyl)-3α,23-dihydroxyolean-11,13(18)-dien-28-amide (6).Yield 48%. white amorphous powder; [α]25
D –13 (c = 0.1, CHCl3); UV (MeOH) λmax (log ε) 236
(4.11), 247 (3.88), 262 (3.74) nm; IR (KBr) vmax 3,414, 2,933, 1,696, 1,686, 1,651, 1,463, 1,363, 1,256, 1,225, 1,051, 756 cm−1; 1H-NMR (CDCl 3, 600 MHz) δH 1.39 (1H, m, H-1α), 1.64 (1H, m, H-1β), 1.53 (1H, m, H-2α), 2.02 (1H, m, H-2β), 3.72 (1H, br s, H-3), 1.74 (1H, m, H-5), 1.45 (2H, m, H-6), 1.42 (2H, m, H-7), 2.14 (1H, br s, H-9), 5.65 (1H, d, J = 10.2 Hz, H-11), 6.43 (1H, d, J = 10.2, 3.0 Hz, H-12), 1.07 (1H, m, H-15α), 1.65 (1H, m, H-15β), 2.63 (1H, m, H-16α), 1.64 (1H, m, H-16β), 2.53 (1H, d, J = 14.4 Hz, H-19α), 1.47 (1H, d, J = 14.4 Hz, H-19β),1.28 (1H, m, H-21α), 1.42 (1H, m, H-21β), 2.41 (1H, m, H-22α), 1.26 (1H, m, H-22β), 3.55 (1H, d, J = 11.4 Hz, H-23a), 3.42 (1H, d, J = 11.4 Hz, H-23b),0.70 (3H, s, H-24), 0.95 (3H, s, H-25), 0.89 (3H, s, H-26), 1.00 (3H, s, H-27), 0.83 (3H, s, H-29), 0.92 (3H, s, H-30), 4.08 (1H, m, H-1'a), 3.38 (1H, m, H-1'b), 2.02 (1H, m, H-2'a), 1.75 (1H, m, H-2'b), 2.64 (1H, m, H-3'a), 2.33 (1H, m, H-3'b), 2.30 (3H, s, H-4'), 2.30 (3H, s, H-5'),
3.28 (1H, m, H-6'a), 3.22 (1H, m, H-6'b), 1.16 (3H, t, J = 6.6 Hz, H-7');13C-NMR (CDCl 3, 150 MHz) δC 32.4 (t, C-1), 26.2 (t, C-2), 76.7 (d, C-3), 40.4 (s, C-4), 42.6 (d, C-5), 18.0 (t, C-6), 32.0 (t, C-7), 40.8 (s, C-8), 54.1 (d, C-9), 36.7 (s, C-10), 126.8 (d, C-11), 125.5 (d, C-12), 135.9 (s, C-13), 41.5 (s, C-14), 24.2 (t, C-15), 30.4 (t, C-16), 52.0 (s, C-17), 134.1 (s, C-18), 40.7 (t, C-19), 33.0 (s, C-20),37.7 (t, C-21), 29.7 (t, C-22), 71.3 (t, C-23), 17.4 (q, C-24), 18.0 (q, C-25), 16.7 (q, C-26), 19.5 (q, C-27), 178.2 (s, C-28), 24.7 (q, C-29), 31.9 (q, C-30), 45.5 (t, C-1'), 24.7 (t, C-2'), 55.2 (t, C-3'), 44.0 (q, C-4'), 44.0 (q, C-5'), 35.6 (t, C-6'), 14.6 (q, C-7');ESIMS m/z 626 [M + H]+; HRESIMS m/z 626.4901 [M + H]+ (Calcd for C 38H64O4N3, 626.4891).
3.5. Anhydride Coupling Esterification
To fatsicarpain A (2.0 mg) in CH2Cl2 (1.0 mL) at room temperature were successively added
DMAP (1.0 mg), DMAP·HCl (0.1 mg) and EDC·HCl (1.0 mg) and the mixture was allowed to react overnight. The reaction was quenched by water, followed by extraction with EtOAc (3 × 1.5 mL). The EtOAc extract was successively washed with 5% aqueous HCl, saturated aqueous NaHCO3, and brine.
The organic layer was dried over anhydrous MgSO4 and evaporated to give a crude product, which
was subjected to a short silica gel column eluting with n-hexane–EtOAc (3:1) to yield 7 (1.7 mg). Similarly, 3α-hydroxyolean-11,13(18)-dien-28-oic acid (2.0 mg) and 3α-acetoxyolean-11,13(18)-dien-28-oic acid (5) (2.0 mg) in CH2Cl2 (1.0 mL) were submitted to anhydride coupling
esterification with DMAP, DMAP·HCl and EDC·HCl at room temperature overnight to yield 8 (1.8 mg) and 9 (1.8 mg), respectively.
3α,23-Dihydroxyolean-11,13(18)-dien-28-oic anhydride (7). Yield 42%. white amorphous powder; [α]25
D–24 (c 0.2, CHCl3); UV (MeOH) λmax (log ε) 237 (4.16), 248 (3.90), 264 (3.73) nm; IR (KBr) vmax
3,446, 2,937, 1,793, 1,772, 1,734, 1,457, 1,374, 1,243, 997cm−1; 1H-NMR (CDCl
3, 600 MHz) δH 1.41
(2H, m, H-1α and H-1'α), 1.69 (2H, m, H-1β and H-1'β), 1.53 (2H, m, H-2α and H-2'α), 2.01 (2H, m, H-2β and H-2'β), 3.73 (2H, br s, H-3 and H-3'), 1.76 (2H, br d, J = 10.8 Hz, H-5 and H-5'), 1.47 (4H, m, H-6 and H-6'), 1.43 (4H, m, H-7 and H-7'), 2.13 (2H, br s, H-9 and H-9'), 5.69 (2H, d, J = 10.2 Hz, H-11 and H-11'), 6.44 (2H, dd, J = 10.2, 2.4 Hz, H-12 and H-12'), 1.09 (2H, m, H-15α and H-15'α), 1.64 (2H, m, H-15β and H-15'β), 2.05 (2H, m, H-16α and H-16'α), 1.71 (2H, m, H-16β and H-16'β), 2.54 (2H, d, J = 14.4 Hz, H-19α and H-19'α), 1.80 (2H, dd, J = 14.4, 1.8 Hz, H-19β and H-19'β), 1.39 (2H, m, H-21α and H-21'α), 1.33 (2H, m, H-21β and H-21'β), 2.21 (2H, m, H-22α and H-22'α), 1.47 (2H, m, H-22β and H-22'β), 3.55 (2H, d, J = 11.4 Hz, H-23a and H-23'a), 3.43 (2H, d, J = 11.4 Hz, H-23b and H-23'b), 0.70 (6H, s, H-24 and H-24'), 0.94 (6H, s, H-25 and H-25'), 0.79 (6H, s, H-26 and H-26'), 1.00 (6H, s, H-27 and H-27'), 0.81 (6H, s, H-29 and H-29'), 0.96 (6H, s, H-30 and H-30'); 13C- NMR (CDCl
3, 150 MHz) δC 32.5 (t, C-1 and C-1'), 26.3 (t, C-2 and C-2'), 77.0 (d, C-3 and
C-3'), 40.4 (s, C-4 and C-4'), 42.6 (d, C-5 and C-5'), 18.0 (t, C-6 and C-6'), 31.9 (t, C-7 and C-7'), 40.8 (s, C-8 and C-8'), 54.2 (d, C-9 and C-9'), 36.7 (s, C-10 and C-10'), 127.4 (d, C-11 and C-11'), 125.1 (d, C-12 and C-12'), 137.6 (s, C-13 and C-13'), 42.2 (s, C-14 and C-14'), 24.9 (t, C-15 and C-15'), 31.7 (t, C-16 and C-16'), 49.8 (s, C-17 and C-17'), 130.2 (s, C-18 and C-18'), 40.0 (t, C-19 and C-19'), 32.4 (s, C-20 and C-20'), 36.8 (t, C-21 and C-21'), 35.4 (t, C-22 and C-22'), 71.3 (t, C-23 and C-23'), 17.4 (q, C-24 and C-24'), 18.0 (q, C-25 and C-25'), 16.6 (q, C-26 and C-26'), 19.8 (q, C-27 and C-27'), 172.7 (s,
C-28 and C-28'), 24.2 (q, C-29 and C-29'), 32.1 (q, C-30 and C-30'); ESIMS m/z 945 [M+Na]+;
HRESIMS m/z 945.6595 [M + Na]+ (calcd for C
60H90O7Na, 945.6579).
3α-Hydroxyolean-11,13(18)-dien-28-oic anhydride (8). Yield 45%. white amorphous powder; [α]25 D
–84 (c 0.2, CHCl3); UV (MeOH) λmax (log ε) 236 (4.08), 248 (3.94), 264 (3.70) nm; IR (KBr) vmax
3,437, 2,937, 1,800, 1,770, 1,736, 1,457, 1,376, 1,243, 997 cm−1; 1H-NMR (CDCl
3, 600 MHz) δH1.38
(2H, m, H-1α and H-1'α), 1.63 (2H, m, H-1β and H-1'β), 1.60 (2H, m, H-2α and H-2'α), 2.02 (2H, m, H-2β and H-2'β), 3.44 (2H, br s, H-3 and H-3'), 1.35 (2H, m, H-5 and H-5'), 1.48 (4H, m, H-6 and H-6'), 1.33 (4H, m, H-7 and H-7'), 2.07 (2H, br s, H-9 and H-9'), 5.68 (2H, d, J = 10.8 Hz, H-11 and H-11'), 6.42 (2H, dd, J = 10.8, 3.0 Hz, H-12 and H-12'), 1.09 (2H, m, H-15α and H-15'α), 1.61 (2H, m, H-15β and H-15'β), 2.05 (2H, m, H-16α and H-16'α), 1.70 (2H, m, H-16β and H-16'β), 2.53 (2H, d, J = 14.4 Hz, H-19α and H-19'α), 1.78 (2H, d, J = 14.4 Hz, H-19β and H-19'β), 1.31 (2H, m, H-21α and H-21'α), 1.39 (2H, m, H-21β and H-21'β), 2.22 (2H, m, H-22α and H-22'α), 1.45 (2H, m, H-22β and H-22'β), 0.95 (6H, s, H-23 and H-23'), 0.84 (6H, s, H-24 and H-24'), 0.91 (6H, s, H-25 and H-25'), 0.77 (6H, s, H-26 and H-26'), 0.97 (6H, s, H-27 and H-27'), 0.80 (6H, s, H-29 and H-29'), 0.95 (6H, s, H-30 and H-30'); 13C-NMR (CDCl
3, 150 MHz) δC 32.7 (t, C-1 and C-1'), 25.2 (t, C-2 and C-2'), 76.2
(d, C-3 and C-3'), 37.5 (s, C-4 and C-4'), 48.5 (d, C-5 and C-5'), 18.2 (t, C-6 and C-6'), 32.2 (t, C-7 and 7'), 40.9 (s, 8 and 8'), 54.1 (d, 9 and 9'), 36.8 (s, 10 and 10'), 127.7 (d, 11 and C-11'), 125.0 (d, C-12 and C-12'), 137.6 (s, C-13 and C-13'), 42.2 (s, C-14 and C-14'), 24.9 (t, C-15 and C-15'), 31.7 (t, C-16 and C-16'), 49.8 (s, C-17 and C-17'), 130.1 (s, C-18 and C-18'), 40.0 (t, C-19 and C-19'), 32.5 (s, C-20 and C-20'), 36.8 (t, C-21 and C-21'), 35.4 (t, C-22 and C-22'), 28.1 (q, C-23 and C-23'), 21.7 (q, C-24 and C-24'), 17.8 (q, C-25 and C-25'), 16.6 (q, C-26 and C-26'), 19.9 (q, C-27 and C-27'), 172.8 (s, C-28 and C-28'), 24.2 (q, C-29 and C-29'), 32.1 (q, C-30 and C-30'); ESIMS m/z913 [M+Na]+; HRESIMS m/z 913.6683 [M + Na]+ (calcd for C
60H90O5Na, 913.6680).
3α-Acetoxyolean-11,13(18)-dien-28-oic anhydride (9). Yield 47%. colorless needles; [α]25
D–67 (c 0.2,
CHCl3); UV (MeOH) λmax (log ε) 236 (4.18), 248 (3.98), 265 (3.75) nm; IR (KBr) vmax 2,935, 1,797,
1,771, 1,733, 1,456, 1,373, 1,241, 998 cm−1; 1H-NMR (CDCl
3, 600 MHz) δH 1.26 (2H, m, 1α and
H-1'α), 1.65 (2H, m, H-1β and H-1'β), 1.66 (2H, m, H-2α and H-2'α), 1.96 (2H, m, H-2β and H-2'β), 4.66 (2H, br s, H-3 and H-3'), 1.29 (2H, m, H-5 and H-5'), 1.49 (4H, m, H-6 and H-6'), 1.35 (4H, m, H-7 and H-7'), 2.05 (2H, br s, H-9 and H-9'), 5.67 (2H, d, J = 10.2 Hz, H-11 and H-11'), 6.43 (2H, dd, J = 10.2, 2.4 Hz, H-12 and H-12'), 1.11 (2H, m, H-15α and H-15'α), 1.64 (2H, m, H-15β and H-15'β), 2.07 (2H, m, H-16α and H-16'α), 1.72 (2H, m, H-16β and H-16'β), 2.55 (2H, d, J = 15.6 Hz, H-19α and H-19'α), 1.79 (2H, d, J = 15.6 Hz, H-19β and H-19'β), 1.34 (2H, m, H-21α and H-21'α), 1.39 (2H, m, 21β and 21'β), 2.22 (2H, m, 22α and 22'α), 1.47 (2H, br d, J = 13.8 Hz, 22β and H-22'β), 0.85 (6H, s, H-23 and H-23'), 0.89 (6H, s, H-24 and H-24'), 0.93 (6H, s, H-25 and H-25'), 0.78 (6H, s, H-26 and H-26'), 1.01 (6H, s, H-27 and H-27'), 0.81 (6H, s, H-29 and H-29'), 0.96 (6H, s, H-30 and
H-30'), 2.09 (3H, s, 3-OAc); 13C NMR (CDCl
3, 150 MHz) δC33.3 (t, 1 and 1'), 22.7 (t, 2 and
C-2'), 78.3 (d, C-3 and C-3'), 36.7 (s, C-4 and C-4'), 49.8 (d, C-5 and C-5'), 18.0 (t, C-6 and C-6'), 32.1 (t, C-7 and C-7'), 40.9 (s, C-8 and C-8'), 54.1 (d, C-9 and C-9'), 36.7 (s, C-10 and C-10'), 127.4 (d, C-11 and C-11'), 125.1 (d, C-12 and C-12'), 137.6 (s, C-13 and C-13'), 42.2 (s, C-14 and C-14'), 24.9
(t, C-15 and C-15'), 31.7 (t, C-16 and C-16'), 49.8 (s, C-17 and C-17'), 130.2 (s, C-18 and C-18'), 40.0 (t, C-19 and C-19'), 32.5 (s, C-20 and C-20'), 36.8 (t, C-21 and C-21'), 35.4 (t, C-22 and C-22'), 27.7 (q, C-23 and C-23'), 21.3 (q, C-24 and C-24'), 17.8 (q, C-25 and C-25'), 16.6 (q, C-26 and C-26'), 19.8 (q, C-27 and C-27'), 172.7 (s, C-28 and C-28'), 24.2 (q, C-29 and C-29'), 32.1 (q, C-30 and C-30'), 170.8 (s, 3-OAc), 21.4 (q, 3-OAc); ESIMS m/z 997 [M + Na]+; HRESIMS m/z 997.6893 [M + Na]+
(calcd for C64H94O7Na, 997.6892). Crystal data: C64H94O7·H2O (formula weight 993.41), approximate crystal size, 0.64 × 0.56 × 0.30 mm, monoclinic, space group, P21, T = 110(2) K, a = 14.1986(4) Å,
b = 13.3529(4) Å, c = 15.5987(5) Å, = 90°, β = 104.518(3)°, γ = 90°, V = 2862.96(15) Å3, D
c = 1.152 Mg/m3, Z = 2, F(000) = 1088,
(MoK) = 0.074 mm1.A total of 14049 reflections were collected in the
range 2.93° < < 29.39°, with 9750 independent reflections [R(int) = 0.0216], completeness to max was
99.8%; psi-scan absorption correction applied;full-matrix least-squares refinement on F2,the number
of data/restraints/parameters were 9750/1/681; goodness-of-fit on F2 = 1.046; final R indices [I >
2σ(I)], R1 = 0.0419, wR2 = 0.0808; R indices (all data), R1 = 0.0611, wR2 = 0.0838, absolute structure
parameter 0.7(8), largest difference peak and hole, 0.423 and −0.206 e/Å3.
3.6. Cytotoxicity, Anti-hepatitis B Virus (HBV) Assay and Antibacterial Activity
The experimental details of these assays were carried out according to a previously described procedure [2].
3.7. Glucose Uptake Assay
FL83B cells were purchased from the American Type Culture Collection (Rockville, MD, USA) and cultured in F12K medium as described previously [22]. Cells were seeded in 12-well plates (4 × 105 cells/well), cultured overnight, washed with phosphate-buffered saline (PBS, pH 7.4), and
subjected to glucose uptake assays in triplicate as described previously with modifications [22,23]. Briefly, each compound was dissolved in DMSO (dimethyl sulfoxide). Cells were incubated in 450 μL of Eagle’s minimum essential medium (MEM) containing 100 nM of insulin, or 20 μM of the indicated compound. In the control, cells were incubated in MEM containing DMSO in a concentration equivalent to that contained in the other groups. At 0, 1, 2, 3, 4, and 5 h, 30 μL of the culture medium was withdrawn and centrifuged at 500 g for 5 min. Five microliters of the resulting supernatant was mixed with 250 μL of a glucose assay kit (Glucose GOD FS, DiaSys Diagnostic Systems, Holzheim, Germany) in a 96-well plate and incubated at 37 °C for 10 min. Absorbance at 500 nm was then determined using a microplate reader (Molecular Devices, Sunnyvale, CA, USA). A standard curve was established simultaneously using solutions of glucose in concentrations between 1–10 mM. The data of glucose uptake assays were analysed against the control by two-way analysis of variance (ANOVA), with cell treatment and time (the time points at which medium glucose concentration were measured) as the two parameters. Significance was considered when the p value between groups of cells and the p value of interaction between the two parameters were both < 0.05. 3.8. Dual Luciferase Activity Assay
P19 cells (1 × 105 cells/well) were seeded into 24-well dish for overnight. On the next day, cells
were transfected with the Wnt reporter construct pGL3-OT (Dr. Bert Vogelstein, the Johns Hopkins University; Addgene No. 16558) and the normalization construct pTK-Renilla, treated with control conditioned medium, Wnt-3a conditioned medium or 1 μM of the compounds (1–5 and 7–9) in Wnt-3a conditioned medium for 16 hours, then the cell lysate was collected and assayed for dual luciferase activities by using Dual Luciferase Reporter Asssay system (Promega). Each assay was done in triplicate. The data of firefly luciferase activity was normalized by that of Renilla luciferase activity. Control conditioned medium and Wnt-3a conditioned medium were prepared as described previously [16]. 4. Conclusions
After varying their structures at the C-3 and C-28 positions, compounds 1–6 revealed greater cytotoxic and antibacterial potential than their parent compounds. Compounds 1–5 obviously enhanced glucose uptake of treated cells as compared with untreated cells, and their effects were similar to that of insulin, indicating that compounds 1–5 possessed insulin-like hypoglycaemic activities. Compounds 1, 3, 5 and 8 at 1 μM of concentration specifically inhibited the Wnt signaling by 20%, 40%, 38% and 32%, respectively. According to our studies, the oleanane-type derivatives endowed with various biological activities and F. polycarpa identified as a potential plant source for the discovery of promising new drugs. Advanced bioactivity assays and chemical modifications for these compounds will be carried out if sufficient material can be recollected from the plant.
Acknowledgments
This work was financially supported by research grants from the Committee on Chinese Medicine and Pharmacy, Department of Health, Executive Yuan (CCMP99-RD-208), and the National Science Council Taiwan (Grant No. NSC102-2113-M-390-001, NSC101-2811-B-039-013 and NSC101-2621-B-039-002) awarded to S.-Y. C. and C.-H. C.
References and Notes
1. Hill, R.A.; Connolly, J.D. Triterpenoids. Nat. Prod. Rep. 2013, 30, 1028–1065.
2. Cheng, S.Y.; Wang, C.M.; Hsu, Y.M.; Huang, T.J.; Chou, S.C.; Lin, E.H.; Chou, C.H. Oleanane-type triterpenoids from the leaves and twigs of Fatsia polycarpa. J. Nat. Prod. 2011, 74, 1744–1750. 3. Sun, H.X.; Zheng, Q.F.; Tu, J. Induction of apoptosis in HeLa cells by
3β-hydroxy-12-oleanen-27-oic acid from the rhizomes of Astilbe chinensis. Bioorg. Med. Chem. 2006, 14, 1189–1198. 4. Rojas, R.; Caviedes, L.; Aponte, J.C.; Vaisberg, A.J.; Lewis, W.H.; Lamas, G.; Sarasara, C.;
Gilman, R.H.; Hammond, G.B. Aegicerin, the first oleanane triterpene with wide-ranging antimycobacterial activity, isolated from Clavija procera. J. Nat. Prod. 2006, 69, 845–846.
5. Chang, C.I.; Kuo, C.C.; Chang, J.Y.; Kuo, Y.H. Three new oleanane-type triterpenes from Ludwigia octovalvis with cytotoxic activity against two human cancer cell lines. J. Nat. Prod. 2004, 67, 91–93.
6. Zeng, N.; Shen, Y.; Li, L.Z.; Jiao, W.-H.; Gao, P.Y.; Song, S.J.; Chen, W.S.; Lin, H.W. Anti-inflammatory triterpenes from the leaves of Rosa laevigata. J. Nat. Prod. 2011, 74, 732–738.
7. Wang, F.; Hua, H.; Pei, Y.; Chen, D.; Jing, Y. Triterpenoids from the resin of Styrax tonkinensis and their antiproliferative and differentiation effects in human leukemia HL-60 cells. J. Nat. Prod. 2006, 69, 807–810.
8. Djoumessi, A.V.B.; Sandjo, L.P.; Liermann, J.C.; Schollmeyer, D.; Kuete, V.; Rincheval, V.; Berhanu, A.M.; Yeboah, S.O.; Wafo, P.; Ngadjui, B.T.; Opatz, T. Donellanic acids A–C: new cyclopropanic oleanane derivatives from Donella ubanguiensis (Sapotaceae). Tetrahedron 2012, 68, 4621–4627.
9. Ho, J.C.; Chen, C.M.; Row, L.C. Oleanane-type triterpenes from the flowers, pith, leaves, and fruit of Tetrapanax papyriferus. Phytochemistry 2007, 68, 631–635.
10. Cáceres-Castillo, D.; Mena-Rejón, G.J.; Cedillo-Rivera, R.; Quijano, L. 21β-Hydroxy-oleanane-type triterpenes from Hippocratea excelsa. Phytochemistry 2008, 69, 1057–1064.
11. Nishino, H.; Nishino, A.; Takayasu, J.; Hasegawa, T.; Iwashima, A.; Hirabayashi, K.; Iwata, S.; Shibata, S. Inhibition of the tumor-promoting action of 12-O-tetradecanoylphorbol-13-acetate by some oleanane-type triterpenoid compounds. Cancer Res. 1988, 48, 5210–5215.
12. Liu, J. Pharmacology of oleanolic acid and ursolic acid. J. Ethnopharmacol. 1995, 49, 57–68. 13. Kashiwada, Y.; Hashimoto, F.; Cosentino, L.M.; Chen, C.H.; Garrett, P.E.; Lee, K.H. Betulinic
acid and dihydrobetulinic acid derivatives as potent anti-HIV agents. J. Med. Chem. 1996, 39, 1016–1017.
14. Qian, K.; Kuo, R.Y.; Chen, C.H.; Huang, L.; Morris-Natschke, S.L.; Lee, K.H. Anti-AIDS agents 81. Design, synthesis, and structure−activity relationship study of betulinic acid and moronic acid derivatives as potent HIV maturation inhibitors. J. Med. Chem. 2010, 53, 3133–3141.
15. Crystallographic data for 9 has been deposited with the Cambridge Crystallographic Data Centre (deposition number CCDC 837341). Copies of the data can be obtained, free of charge, on application to the Director, CCDC, 12 Union Road, Cambridge CB21EZ, UK (fax: +44-1223-336033 or e-mail: [email protected]).
16. Chen, H.J.; Lin, C.M.; Lin, C.S.; Perez-Olle, R.; Leung, C.L.; Liem, R.K. The role of microtubule actin cross-linking factor 1 (MACF1) in the Wnt signaling pathway. Genes Dev. 2006, 20, 1933–1945.
17. Volonterio, A.; Ramirez de Arellano, C.; Zanda, M. Synthesis of 1,3,5-trisubstituted hydantoins by regiospecific domino condensation/aza-Michael/O→N acyl migration of carbodiimides with activated α,β-unsaturated carboxylic acids. J. Org. Chem. 2005, 70, 2161–2170.
18. Ganem, D.; Prince, A.M. Hepatitis B virus infection – natural history and clinical consequences. N. Engl. J. Med. 2004, 350, 1118–1129.
19. Egan, B.J.; Marzio, L.; O'Connor, H.; O'Morain, C. Treatment of Helicobacter pylori infection.
Helicobacter 2008, 13, 35–40.
20. Chang, J.M.; Chen, T.H. Bacterial foodborne outbreaks in central Taiwan, 1991-2000. J. Food Drug Anal. 2003, 11, 53–59.
21. Tanachatchairtana, T.; Bremner, J.B.; Chokchaisiri, R.; Suksamrarn, A. Antimycobacterial activity of cinnamate-based esters of the triterpenes betulinic, oleanolic and ursolic acids. Chem. Pharm Bull. 2008, 56, 194–198.
22. Cheng, H.L.; Huang, H.K.; Chang, C.I.; Tsai, C.P.; Chou, C.H. A cell-based screening identifies compounds from the stem of Momordica charantia that overcome insulin resistance and activate AMP-activated protein kinase. J. Agric. Food Chem. 2008, 56, 6835–6843.
23. Chang, C.I.; Tseng, H.I.; Liao, Y.W.; Yen, C.H.; Chen, T.M.; Lin, C.C.; Cheng, H.L. In vivo and in vitro studies to identify the hypoglycaemic constituents of Momordica charantia wild variant WB24. Food Chem. 2011, 125, 521–528.
24. MacDonald, B.T.; Tamai, K.; He, X. Wnt/β-catenin signaling: components, mechanisms, and diseases. Developmental cell 2009, 17, 9–26.
25. Sen, M.; Lauterbach, K.; El-Gabalawy, H.; Firestein, G.S.; Corr, M.; Carson, D.A. Expression and function of wingless and frizzled homologs in rheumatoid arthritis. Proc. Natl. Acad. Sci. U S A 2000, 97, 2791–2796.
26. Sen, M.; Chamorro, M.; Reifert, J.; Corr, M.; Carson, D.A. Blockade of Wnt-5A/Frizzled 5 signaling inhibits rheumatoid synoviocyte activation. Arthritis Rheum. 2001, 44, 772–781.
27. Moon, R.T.; Kohn, A.D.; De Ferrari, G.V.; Kaykas, A. WNT and β-catenin signalling: diseases and therapies. Nat. Rev. Genet. 2004, 5, 691–701.
28. Clevers, H. Wnt/β-catenin signaling in development and disease. Cell 2006, 127, 469–480.
29. De Ferrari, G.V.; Moon, R.T. The ups and downs of Wnt signaling in prevalent neurological disorders. Oncogene 2006, 25, 7545–7553.
30. Johnson, M.L.; Rajamannan, N. Diseases of Wnt signaling. Rev. Endocr. Metab. Disord. 2006, 7, 41–49.
31. Luo, J.; Chen, J.; Deng, Z.L.; Luo, X.; Song, W.X.; Sharff, K.A.; Tang, N.; Haydon, R.C.; Luu, H.H.; He, T.C. Wnt signaling and human diseases: what are the therapeutic implications? Lab. Invest. 2007, 87, 97–103.
Sample Availability: Samples of the compounds may not be available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).