(MS ID: MBE-12-0129)
Assessing determinants of exonic evolutionary rates in mammals
Feng-Chi Chen1,2,3,*, Ben-Yang Liao1,*, Chia-Lin Pan1, Hsuan-Yu Lin1, Andrew Ying-Fei Chang1
1 Division of Biostatistics and Bioinformatics, Institute of Population Health Sciences, National Health Research Institutes, Zhunan, Miaoli County, 350 Taiwan, Republic of China.
2 Department of Life Science, National Chiao-Tung University, Hsinchu, 300 Taiwan, Republic of China.
3 Department of Dentistry, China Medical University, Taichung, 404 Taiwan, Republic of China.
*Correspondence should be addressed to:
Ben-Yang Liao Feng-Chi Chen
Division of Biostatistics & Bioinformatics Division of Biostatistics & Bioinformatics
Institute of Population Health Sciences National Health Research Institutes 35, Keyan Road, Zhunan Town, Miaoli County 350, Taiwan, R.O.C. Phone: 886-37-246-166 ext 36118 Institute of Population Health Sciences
National Health Research Institutes 35, Keyan Road, Zhunan Town, Miaoli County 350, Taiwan, R.O.C. Phone: 886-37-246-166 ext 36111
Fax: 886-37-586-467 Fax: 886-37-586-467
Keywords: exon evolution, alternative splicing, structural-functional constraint, gene
expression, gene compactness
Abstract
From studies investigating the differences in evolutionary rates between genes, gene compactness and gene expression level have been identified as important
determinants of gene-level protein evolutionary rate, as represented by
nonsynonymous-to-synonymous substitution rate (dN/dS) ratio. However, the causes of exon-level variances in dN/dS are less understood. Here we use principal component regression to examine to what extent thirteen exon features explain the variance in dN, dS, and the dN/dS ratio of human-rhesus macaque or human-mouse orthologous exons. The exon features were grouped into six functional categories: expression features, mRNA splicing features, structural-functional features, compactness features, exon duplicability, and other features, including G+C content and exon length. Although expression features are important for determining dN and dN/dS between exons of different genes, structural-functional features and splicing features explained more of the variance for exons of the same genes. Furthermore, we show that compactness features can explain only a relatively small percentage of variance in exon-level dN or dN/dS in either between-gene or within-gene comparison. By contrast, dS yielded inconsistent results in the human-mouse comparison and the human-rhesus macaque comparison. This inconsistency may suggest rapid evolutionary changes of the mutation landscape in mammals. Our results suggest that between-gene and within-gene variation in dN/dS (and dN) are driven by different evolutionary forces, and that the role of mRNA splicing in causing the variation in evolutionary rates of coding sequences may be underappreciated.
Introduction
The evolutionary rates of different protein-coding genes in a genome can vary by several orders of magnitude (Li 1997). This variation has been extensively studied and is typically explained by differences in mutation rate and selection intensity among genes. In the past few years, data generated by whole-genome sequencing and functional genomic assays have provided biologists an unprecedented opportunity to address this issue systematically. As a result, several biological factors associated with and potentially underlying evolutionary rate variations of protein-coding genes have been identified. These factors include gene essentiality (Hirsh and Fraser 2001; Jordan et al. 2002; Zhang and He 2005; Liao, Scott, and Zhang 2006), gene expression level (Pal, Papp, and Hurst 2001a; Akashi 2003; Subramanian and Kumar 2004;
Drummond, Raval, and Wilke 2006), tissue specificity of gene expression (Hastings 1996; Duret and Mouchiroud 2000; Subramanian and Kumar 2004; Winter, Goodstadt, and Ponting 2004; Zhang and Li 2004), presence of a duplicate paralog (Nembaware et al. 2002; Castillo-Davis and Hartl 2003; Yang, Gu, and Li 2003), properties in the protein interaction network (Fraser et al. 2002; Fraser 2005; Hahn and Kern 2005; Kim, Korbel, and Gerstein 2007), tendency to form misinteracting protein complex (Yang et al. 2012), local recombination rate (Pal, Papp, and Hurst 2001b), pleiotropy (He and Zhang 2006), amino acid composition (Xia, Franzosa, and Gerstein 2009), structural features of protein folding (Bloom et al. 2006; Zhou, Drummond, and Wilke 2008; Franzosa and Xia 2009), G+C content (Xia, Franzosa, and Gerstein 2009), gene compactness (Liao, Scott, and Zhang 2006), and subcellular localization (Liao, Weng, and Zhang 2010).
All of the abovementioned studies focused on sequence evolution of protein coding genes as a whole. However, evolutionary rates also differ among regions of the
same protein. For example, structurally ordered protein regions evolve more slowly than intrinsically disordered regions (IDRs) (Brown et al. 2002; Brown, Johnson, and Daughdrill 2010; Chen, Pan, and Lin 2011), and protein regions encoded by
alternatively spliced exons (ASEs) evolve more rapidly than those encoded by
constitutively spliced exons (CSEs) (Chen et al. 2006; Chen et al. 2007; Ramensky et al. 2008; Chen, Pan, and Lin 2011). Thus, exon features have a profound effect on within-gene variation in evolutionary rate. Because exon-intron structure is an important characteristic of multicellular eukaryotic genes that causes complexity (Sorek, Shamir, and Ast 2004; Xing and Lee 2007) and diversity (Xing and Lee 2005; Chen et al. 2006; Chen and Chuang 2007; Keren, Lev-Maor, and Ast 2010; Chen, Pan, and Lin 2011) of proteomes, a systematic analysis to delineate the individual and integrative contributions of exon features to within-gene evolutionary rate variation is necessary to understand the molecular evolution of complex organisms.
To address this issue, we analyzed the effects of exon features (described below) on the variation of exonic evolutionary rates in mammals. We calculated the
nonsynonymous substitution rate (dN), synonymous substitution rate (dS), and the dN/dS ratio for exons in human-mouse and human-rhesus macaque one-to-one orthologous genes. To account for the inter-correlations between evolutionary rate determinants, principal component regression (PCR) was used to analyze the relative contributions of exon features on the variances of dN, dS, and dN/dS ratio. PCR
outperformed multivariate regression and partial correlation in delineating the relationships among multiple inter-correlated factors when the data were noisy (Drummond, Raval, and Wilke 2006). In this study, thirteen exon features that may affect evolutionary rates were analyzed (table 1): weighted exon frequency (WEF, see Materials and Methods), ASE/CSE exon type, exonic expression level, coefficient of
variation in exonic expression levels across multiple tissues, exonic expression breadth, percent of IDR, percent of annotated protein domain(s), proportion of amino acids predicted to be solvent-accessible, the lengths of 5’ and 3’ flanking introns, exon duplicability (Materials and Methods), exon length, and G+C content. We
demonstrate that the features related to the splicing and structural-functional constraints of exons are the most important in causing within-protein variation in evolutionary rates in mammals.
Materials and Methods
Source data and calculation of evolutionary rates
The human-mouse and human-rhesus macaque one-to-one orthologous genes and the corresponding transcript and peptide sequences were retrieved from Ensembl v59 through the BioMart interface (http://www.biomart.org) (Guberman et al. 2011). To ensure data quality, we retained only full-length transcripts (with start and stop codons) that have known protein products. To avoid unequal weighting between genes, we selected the longest transcript from each human gene as the representative. We then aligned the human peptide sequence against the orthologous mouse/macaque peptide sequences (i.e. peptides derived from one-to-one orthologous gene pairs) using MUSCLE (Edgar 2004). The longest alignable mouse/macaque peptide
orthologue was retained. The peptide sequence alignments were then back-translated to nucleotide sequences. The boundaries of “orthologous exons” were defined according to Ensembl human exon annotations. All gaps in the transcript alignments were discarded, so our approach did not consider lineage-specific gains/losses of exons. We calculated the dN, dS, and dN/dS of each pair of orthologous exons using the codeml module of PAML 4 (Yang 2007). To ensure accurate estimates of evolutionary
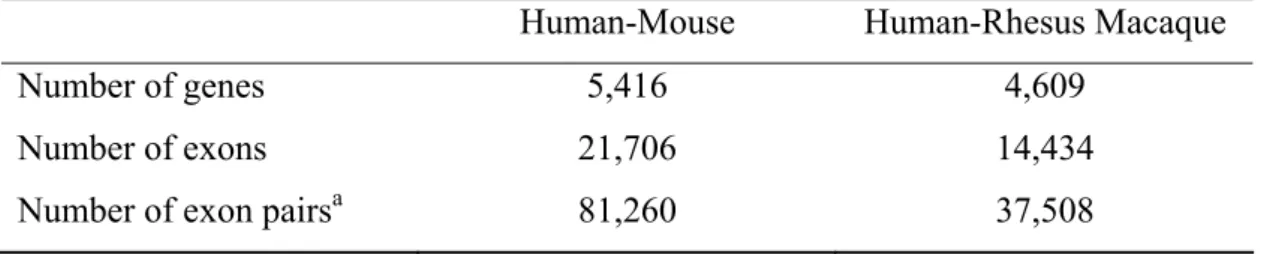
rates, only exons longer than 81 bp were included (Nekrutenko, Makova, and Li 2002; Chen, Pan, and Lin 2011). For the human-mouse comparison, our final dataset
included 5,416 human-mouse orthologous gene pairs, comprised of 21,706
orthologous exon pairs (table 2). For the human-rhesus macaque comparison, our final dataset included 4,609 orthologous genes, which were comprised of 14,434
orthologous exon pairs (table 2). Compared to the number of human-mouse
orthologous genes, there were fewer human-rhesus macaque orthologous genes in our analysis because the macaque draft genome had not been completely sequenced.
To control for differences in exon features of different genes, we calculated the differences in evolutionary rates (dN, dS, and dN/dS) and the differences in exon
features for all possible two-exon combinations of the same transcript. Using PCR, we examined how much of the variance in exon-level dN, dS, or dN/dS was explained by exon features. Our dataset for this within-gene analysis included 81,260
human-mouse exon pairs (combinations) and 37,508 human-rhesus macaque exon pairs (table 2).
Measuring exon features
ASE and CSE classification (Shabalina et al. 2010) and WEF calculation were done using in-house PERL scripts. WEF is defined as the length-weighted average of the frequency of an exon (supplementary fig. S1). Here, the frequency of an exon measures its relative importance and is calculated as the percent of transcript isoforms of a gene that include this specific exon. For example, CSEs have an exon frequency of 100% because they always occur in different isoforms. CSEs are considered to be indispensable for the biological functions of their transcript/protein. We assume that an exon’s importance is reflected in how frequently it appears in different transcript
isoforms. In the case of partially overlapping exons (supplementary fig. S1), the exon boundaries may be ambiguous, and the frequencies of these exons are hard to define. For such cases, WEF gives a reasonable quantitative measure of the frequency an exon is used in splicing events.
Intrinsically disordered regions were predicted by using Disopred (Ward et al. 2004). Pfam protein domain information was retrieved from the Ensembl Database (http://www.ensembl.org) and the percent of annotated protein domain(s) of each exon was calculated. Solvent-accessible amino acid residues were predicted by using the ACCpro module of the SCRATCH package (Cheng et al. 2005) with a 30% exposure threshold.
5’ and 3’ intron length, exon length, and G+C content of the exons were
calculated using in-house PERL scripts on the genomic sequences downloaded from BioMart. The first and last coding exons of each transcript were excluded because they contain only 3’ intron and 5’ intron, respectively. Exon duplicability was
evaluated by BLAST-aligning each exon to the entire transcriptome. A potential exon duplicate was defined as a BLAST hit that is ≥90% alignable and ≥90% identical to the query exon. The total number of BLAST hits matching these criteria was defined as the duplicability of an exon.
The expression features of the exons were derived from two published human RNA-seq datasets (GSE12946 and GSE13652) (Pan et al. 2008; Wang et al. 2008) archived in Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/). These datasets cover eleven human tissues (adipose, brain, breast, cerebral cortex, colon, heart, liver, lung, lymph node, skeletal muscle, and testes). The 32-mer RNA-seq sequences were mapped to the human genome (hg 19) using SeqMap (Jiang and
2010), the exon-specific transcriptional abundance was defined as the total number of RNA-seq reads uniquely mapped onto the exon divided by the number of unique n-mers per exon, where n=32. The exon-specific transcriptional abundances were averaged over all of the analyzed tissues to represent the expression level of an exon. To measure the expression breadths of exons, the exon-specific transcriptional
abundances were sorted for each tissue, and the top 50% of the exons were defined as expressed in a certain tissue. The expression breadth of an exon was then calculated as the proportion of tissues in which this exon was expressed (transcriptional abundance >0). The coefficient of variation was calculated by dividing the standard deviation of exon-specific transcriptional abundances by the mean of exon-specific transcriptional abundances across the eleven tissues for each exon. The PCR analyses were
conducted in R (http://www.r-project.org/) using modified scripts from (Drummond, Raval, and Wilke 2006).
Results
Charicteristics of exons
To evaluate the determinants of exon-level evolutionary rates, we had to control for gene-level differences. For example, expression level may differ by a much larger extent between genes than between exons of the same genes. Therefore, the results from PCR analyses based on pooled exon comparisons may to some extent reflect the gene-level differences. To address this issue, we calculated the between-exon
differences in human-mouse evolutionary rates (dN/dS, dN, and dS) and the thirteen exon features for exons of the same transcript. We performed PCR analyses separately for dN/dS, dN, and dS against the exon features.
into six biologically meaningful categories (table 1): (1) mRNA splicing features: WEF, and ASE/CSE exon type (ASE=0; CSE=1); (2) exon-level RNA expression features: expression level, coefficient of variation in expression level, and expression breadth; (3) structural-functional features: percent of IDR, percent of Pfam protein domain(s), and proportion of amino acid residues predicted to be solvent-accessible; (4) gene compactness features: the lengths of 5’ and 3’ flanking introns; (5) exon duplicability; and (6) other features: exon length and G+C content (Supplementary Table S1).
Structural-functional features and splicing features are the two most important determinants of within-gene dN/dS variations
As shown in fig. 1, the primary principal component for exonic dN/dS is composed mainly of variation in structural-functional features (component 3 in the left panel in fig. 1A), and the secondary component (component 2) consists mainly of variation in splicing features. Although variation in expression features (component 1) is a well-known determinant of dN/dS at the protein-level (Pal, Papp, and Lercher 2006), it ranked sixth in explaining exon-level dN/dS variation. A similar trend was observed for dN (fig. 1B; left panel). Meanwhile, other features dominated the third and the fourth most important components for exonic dN/dS. For dS, the two most important components were composed mainly of splicing features and
structural-functional features (fig. 1C; left panel).
We then calculated the total contribution of each feature category to the variance in dN/dS. Some feature categories dominated multiple components, so we summed the percentages of variance explained by components that were dominated by the same feature category. A component was dominated by a feature category if the feature
category accounted for more than 50% of this component. If none of the feature categories exceeded the 50% threshold, the component was designated as a “mixed” component. For dN/dS, structural-functional features, splicing features, and expression features, accounted for approximately 3.4%, 2.8%, and 0.5% of the variance
explained, respectively (fig. 1A; right panel). For dN, the three features accounted for 3.5%, 2.6%, and 0.5% of the variance explained, respectively (fig. 1B; right panel). For dS, the three features accounted for 1.7%, 1.5%, and 0.2% of the variance explained, respectively. Unexpectedly, other features accounted for a considerable percent of variance explained for dN/dS (~2.2%), dN (~2.3%), and dS (0.6%) (fig. 1A, 1B, and 1C; right panel). Although compactness features were previously suggested to be a dominant factor affecting gene-level dN/dS (Liao, Scott, and Zhang 2006), they accounted for a relatively small percent (<0.1%) of exonic dN/dS, dN, and dS. Similarly, although gene duplication has a strong effect on evolutionary rates, exon duplicability explained <0.1% of the variance of exon-level evolutionary rates (fig. 1, right panel). (For detailed human-mouse PCR results broken down by each exon feature see supplementary tables S2-S4.)
To evaluate these results across smaller genetic distances, we repeated the analyses for the human-rhesus macaque comparison. We obtained similar results for dN/dS and dN, with splicing features, structural-functional features, and expression features account for ~1.5%, 0.7%, and 0.3% of dN / dS variance, respectively (fig. 2A). For dN, these features accounted for 1.4%, 0.7%, and 0.2%, respectively (fig. 2B). Notably, the percent of variance in evolutionary rates explained by the exon features were generally larger in the human-mouse comparison than in the human-rhesus macaque comparison, possibility due to the relatively low sequence quality of the rhesus macaque genome and the small genetic distance between human and rhesus
macaque. The principal components for variation in dS were different between the two datasets. In the human-rhesus macaque comparison, the importance of splicing and structural-functional features was significantly reduced, whereas the importance of other features increased (fig. 2C). Therefore, the primary determinant of exon-level dS in mammals remains inconclusive. The detailed human-rhesus macaque PCR results (broken down to thirteen exon features) are given in supplementary tables S5~S7.
The effects of gene-level characteristics on the evolutionary rates of exons
Unlike previous findings based on analyses of full-length mammalian proteins (Liao, Scott, and Zhang 2006; Drummond and Wilke 2008), expression features and compactness features accounted for only a small percent of variance in exon-level dN/dS and dN. This is possibly due to that these two feature categories, especially expression features, differed to a greater extent between genes than between exons of the same genes. Therefore, the effects of these two features were less significant in the intragenic analyses. To examine this possibility, we randomly selected 81,260
human-mouse and 37,508 human-rhesus macaque exon pairs from different genes without replacement and conducted a between-gene PCR analysis. We then summed the contributions of each of the feature categories as described above. We repeated this analysis on randomly selected exon sets for 500 times and generated boxplots of percent variance explained for each feature category (figs. 3 and 4). In the
human-mouse comparison (fig. 3), expression features are more important in affecting the variances in dN/dS and dN than splicing features and structural-functional features (fig. 3A and 3B). For the variance in exon-level dS, splicing features were the most important, followed by structural-functional features and expression features (fig. 3C).
and dN (fig. 4A and 4B). For dS, splicing features remained relatively important. However, the contributions of structural-functional features and mixed features varied to a large extent. This is because in many cases, structural-functional features
accounted for either slightly less or slightly more than 50% of a component. In the former case, the component was designated as dominated by structural-functional features, whereas in the latter case, it was considered a mixed component. These variations in component designation caused the large variations in fig. 4C.
Discussion
In this study, we analyzed the contributions of thirteen exon features (table 1) to exonic evolutionary rates using both within- and between-gene comparisons. The thirteen features of exons were classified into six major components based on the principal component analysis: splicing features, expression features,
structural-functional features, gene compactness features, duplicability, and other features composed of G+C content and exon length. Although other features contributed to dN, dS, and dN / dS at an appreciable level (figs. 1-4), a biological interpretation of this component is currently lacking and requires further exploration. We cannot exclude the possibility that our datasets contain noises that cannot be easily eliminated using PCR analyses. Alternatively, some important exon features might not have been included, leaving a considerable proportion of variances in evolutionary rates unexplained.
The within-gene analyses (figs. 1 and 2) controlled for between-gene differences in exon features and indicated that structural-functional features and splicing features are the two most important determinants of exon-level dN/dS and dN. By contrast, between-gene analyses (figs. 3 and 4) indicated that expression features have larger
effects on exon-level dN/dS and dN variance than structural-functional and splicing features. Taken together, our results suggest that the differences in gene-level
biological features (especially expression features) may set the coarse background of protein evolution at the gene level, upon which exon features fine-tune the
within-gene variations in protein evolutionary rates. Because between-gene variation in expression features has strong effects on dN/dS and dN, controlling for the
expression features revealed the significant effects of exon-level features, such as splicing features and structural-functional features.
Gene compactness was identified as more important than expression level in affecting dN/dS at the gene level (Liao, Scott, and Zhang 2006). However, at the exon level, gene compactness only has minor contributions to dN/dS (figs.1~4). By contrast, expression features were an important contributor to exonic dN/dS variations in the between-gene analysis (figs. 3 and 4). The increased importance of gene expression and decreased importance of gene compactness on exon-level dN/dS variance may reflect differences in the source data between the gene-level and exon-level analyses. Notably, the gene-level study incorporated microarray expression data, whereas the present exon-level study incorporated RNA-seq expression data (Pan et al. 2008). For the microarray data, probes were not located within all exons of a gene. As a result, microarrays do not precisely measure mRNA abundance, especially for alternatively spliced genes. Furthermore, the expression signals measured by hybridization methods are affected by probe-target affinity, which can vary for probes within the same transcript (Liao and Zhang 2006). Therefore, sequencing-based methods, such as RNA-seq, may have better accuracy and resolution than array-based methods in measuring exon-level expression properties. Another potential reason for the decreased effect of gene compactness on dN/dS might be the result of including
splicing features (which have been overlooked in previous studies) in the present analyses.
It is unexpected that splicing features and structural-functional features are more important than expression features and compactness features in affecting within-gene exon-level dN/dS differences (figs. 1 and 2). Many unicellular organisms, such as yeast, contain few introns and rarely implement alternative splicing. By contrast, complex multicellular organisms implement alternative splicing as a mechanism for gene regulation. Thus, lineage-specific properties, such as splicing features, can be an important determinant in affecting within-gene variation in protein evolutionary rate.
In a previous PCR study that examined gene-level evolutionary rates in yeast, the percent of dN/dS variation accounted for by the most influential factors (expression level, codon bias, and protein abundance) were as large as ~40% (Drummond, Raval, and Wilke 2006). By contrast, the contributions to exon-level evolutionary rate
variation of the most influential categories are smaller (~3%, fig. 1). There are several possible reasons for this difference in explainable variance. First, the significantly reduced effective population sizes in multicellular organisms have led to a decrease in the efficiency of selection, thereby weakening the correlation between dN/dS and the examined exon features. Second, in multicellular organisms, tissue differentiation results in genes that are expressed in multiple tissues and subject to complex
regulation and selection pressures. In mammals, natural selection may have targeted not only individual biological factors, such as exon features, but also factors
associated with spatial-temporal interactions (Gu and Su 2007). Thus, the relatively small percent of explainable dN/dS variance may reflect our limited knowledge of the targets of selection in complex organisms. Consistent with this notion, previous studies showed that the percent of variance in dN/dS explained by a single biological
factor is smaller in mammals than in yeast (Liao, Scott, and Zhang 2006; Liao, Weng, and Zhang 2010). Third, although we filtered out short exons (Materials and Methods), the average length of the analyzed exons are shorter than the lengths of genes.
Therefore, the estimates of exonic evolutionary rates and other exon features may be less accurate and subject to large variation. In other words, exon-level data may be noisier than gene-level data. In addition, the within-gene differences in biological features and evolutionary rates can be fairly small. The signal-to-noise ratio in the exon-level analysis is thus limited, which may have reduced the explaining power of the exon features. Regardless of the amount of variance explained, the human-mouse and human-rhesus macaque comparisons yielded consistent results for how exon features explain variation in dN/dS and dN.
By analyzing the effects of thirteen exon features on exon-level evolutionary rate, we demonstrate the predominant roles of splicing features and structural-functional features in determining dN/dS and dN of mammalian exons. Our results clearly
demonstrate that gene-level and exon-level variations in dN/dS and dN are affected by different biological properties of DNA. Our findings thus may shed new lights on the sources of evolutionary rate variations within mammalian genes.
Acknowledgement
This study was supported by the intramural funding of National Health Research Institutes, Taiwan, to both F.-C.Chen and B.-Y. Liao and research grant (NSC
99-2311-B-400-003-MY2) from the National Science Council, Taiwan, to B.-Y. Liao. We thank Dr. Pei-Shen Lin for statistical advice. We also thank two anonymous reviewers for constructive comments.
Figure legends Figure 1
The principal components that affect human-mouse within-gene exonic evolutionary rate variations (left panel) and the percent of variance explained by each category of exon features (right panel) for (A) dN/dS ratio; (B) dN; and (C) dS. Note that only the six most important components are shown.
Figure 2
The principal components that affect human-macaque within-gene exonic
evolutionary rate variations (left panel) and the percent of variance explained by each category of exon features (right panel) for (A) dN/dS ratio; (B) dN; and (C) dS. Note that only the six most important components are shown.
Figure 3.
The distributions of percent variance in human-mouse evolutionary rates explained by different categories of exon features as generated by 500 random sets of exon pairs from different genes: (A) dN/dS ratio; (B) dN; and (C) dS. Upper quartile, median, and lower quartile values are indicated in each box. Bars outside the box indicate 1.5-fold interquartile range from the upper and lower quartile.
Figure 4.
The distributions of percent variance in human-macaque evolutionary rates explained by different categories of exon features as generated by 500 random sets of exon pairs from different genes: (A) dN/dS ratio; (B) dN; and (C) dS. Upper quartile, median, and lower quartile values are indicated in each box. Bars outside the box indicate 1.5-fold
interquartile range from the upper and lower quartile.
Supplementary figure S1 The calculation of weighted exon frequency (WEF). The
WEF of an exon is the length-weighted average of the frequency of occurrence of this exon in all of the alternatively spliced transcripts of the gene of interest.
Table 1. Exon features included in PCR.
Exon features (1) Splicing features Weighted exon frequency
ASE/CSE exon type (2) Expression features Average expression level
Coefficient of variation of expression level Expression breadth (3) Structural-functional features Proportion of intrinsically disordered region
Proportion of Pfam domain Proportion of solvent accessible region
(4) Compactness features Length of 5’ flanking intron Length of 3’ flanking intron (5) Exon duplicability Exon duplicability
(6) Other features Exon length
Table 2. The human-mouse and human-rhesus macaque orthologous genes and exons for within-gene and between-gene analyses.
Human-Mouse Human-Rhesus Macaque
Number of genes 5,416 4,609
Number of exons 21,706 14,434
Number of exon pairsa 81,260 37,508
References
Akashi H. 2003. Translational selection and yeast proteome evolution. Genetics
164:1291-1303.
Bloom JD, Drummond DA, Arnold FH, Wilke CO. 2006. Structural determinants of the rate of protein evolution in yeast. Mol Biol Evol 23:1751-1761.
Brown CJ, Johnson AK, Daughdrill GW. 2010. Comparing models of evolution for ordered and disordered proteins. Mol Biol Evol 27:609-621.
Brown CJ, Takayama S, Campen AM, Vise P, Marshall TW, Oldfield CJ, Williams CJ, Dunker AK. 2002. Evolutionary rate heterogeneity in proteins with long disordered regions. J Mol Evol 55:104-110.
Castillo-Davis CI, Hartl DL. 2003. Conservation, relocation and duplication in genome evolution. Trends Genet 19:593-597.
Chen FC, Chaw SM, Tzeng YH, Wang SS, Chuang TJ. 2007. Opposite evolutionary effects between different alternative splicing patterns. Mol Biol Evol
24:1443-1446.
Chen FC, Chuang TJ. 2007. Different alternative splicing patterns are subject to opposite selection pressure for protein reading frame preservation. BMC Evolutionary Biology 7:179.
Chen FC, Pan CL, Lin HY. 2011. Independent effects of alternative splicing and structural constraint on the evolution of mammalian coding exons. Mol Biol Evol 29:187-193.
Chen FC, Wang SS, Chen CJ, Li WH, Chuang TJ. 2006. Alternatively and
constitutively spliced exons are subject to different evolutionary forces. Mol Biol Evol 23:675-682.
Cheng J, Randall AZ, Sweredoski MJ, Baldi P. 2005. SCRATCH: a protein structure and structural feature prediction server. Nucleic Acids Res 33:W72-76.
Drummond DA, Raval A, Wilke CO. 2006. A single determinant dominates the rate of yeast protein evolution. Mol Biol Evol 23:327-337.
Drummond DA, Wilke CO. 2008. Mistranslation-induced protein misfolding as a dominant constraint on coding-sequence evolution. Cell 134:341-352.
Duret L, Mouchiroud D. 2000. Determinants of substitution rates in mammalian genes: expression pattern affects selection intensity but not mutation rate. Mol Biol Evol 17:68-74.
Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792-1797.
Franzosa EA, Xia Y. 2009. Structural determinants of protein evolution are context-sensitive at the residue level. Mol Biol Evol 26:2387-2395. Fraser HB. 2005. Modularity and evolutionary constraint on proteins. Nat Genet
37:351-352.
Fraser HB, Hirsh AE, Steinmetz LM, Scharfe C, Feldman MW. 2002. Evolutionary rate in the protein interaction network. Science 296:750-752.
Gu X, Su Z. 2007. Tissue-driven hypothesis of genomic evolution and
sequence-expression correlations. Proc Natl Acad Sci U S A 104:2779-2784. Guberman JM, Ai J, Arnaiz O et al. (60 co-authors) 2011. BioMart Central Portal: an
open database network for the biological community. Database (Oxford)
2011:bar041.
Hahn MW, Kern AD. 2005. Comparative genomics of centrality and essentiality in three eukaryotic protein-interaction networks. Mol Biol Evol 22:803-806. Hastings KE. 1996. Strong evolutionary conservation of broadly expressed protein
Mol Evol 42:631-640.
He X, Zhang J. 2006. Toward a molecular understanding of pleiotropy. Genetics
173:1885-1891.
Hirsh AE, Fraser HB. 2001. Protein dispensability and rate of evolution. Nature
411:1046-1049.
Jiang H, Wong WH. 2008. SeqMap: mapping massive amount of oligonucleotides to the genome. Bioinformatics 24:2395-2396.
Jordan IK, Rogozin IB, Wolf YI, Koonin EV. 2002. Essential genes are more
evolutionarily conserved than are nonessential genes in bacteria. Genome Res
12:962-968.
Keren H, Lev-Maor G, Ast G. 2010. Alternative splicing and evolution: diversification, exon definition and function. Nat Rev Genet 11:345-355.
Kim PM, Korbel JO, Gerstein MB. 2007. Positive selection at the protein network periphery: evaluation in terms of structural constraints and cellular context. Proc Natl Acad Sci U S A 104:20274-20279.
Li W-H. 1997. Molecular Evolution. Sinauer.
Liao BY, Scott NM, Zhang J. 2006. Impacts of gene essentiality, expression pattern, and gene compactness on the evolutionary rate of mammalian proteins. Mol Biol Evol 23:2072-2080.
Liao BY, Weng MP, Zhang J. 2010. Impact of extracellularity on the evolutionary rate of mammalian proteins. Genome Biol Evol 2:39-43.
Liao BY, Zhang J. 2006. Low rates of expression profile divergence in highly expressed genes and tissue-specific genes during mammalian evolution. Mol Biol Evol 23:1119-1128.
Nekrutenko A, Makova KD, Li WH. 2002. The K(A)/K(S) ratio test for assessing the protein-coding potential of genomic regions: an empirical and simulation study. Genome Res 12:198-202.
Nembaware V, Crum K, Kelso J, Seoighe C. 2002. Impact of the presence of paralogs on sequence divergence in a set of mouse-human orthologs. Genome Res
12:1370-1376.
Pal C, Papp B, Hurst LD. 2001a. Highly expressed genes in yeast evolve slowly. Genetics 158:927-931.
Pal C, Papp B, Hurst LD. 2001b. Does the recombination rate affect the efficiency of purifying selection? The yeast genome provides a partial answer. Mol Biol Evol 18:2323-2326.
Pal C, Papp B, Lercher MJ. 2006. An integrated view of protein evolution. Nat Rev Genet 7:337-348.
Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. 2008. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput
sequencing. Nat Genet 40:1413-1415.
Qian W, Liao BY, Chang AY, Zhang J. 2010. Maintenance of duplicate genes and their functional redundancy by reduced expression. Trends Genet 26:425-430. Ramensky VE, Nurtdinov RN, Neverov AD, Mironov AA, Gelfand MS. 2008.
Positive selection in alternatively spliced exons of human genes. Am J Hum Genet 83:94-98.
Shabalina SA, Spiridonov AN, Spiridonov NA, Koonin EV. 2010. Connections between alternative transcription and alternative splicing in mammals. Genome Biol Evol 2:791-799.
Subramanian S, Kumar S. 2004. Gene expression intensity shapes evolutionary rates of the proteins encoded by the vertebrate genome. Genetics 168:373-381. Sultan M, Schulz MH, Richard H et al. 2008. A global view of gene activity and
alternative splicing by deep sequencing of the human transcriptome. Science
321:956-960.
Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB. 2008. Alternative isoform regulation in human tissue transcriptomes. Nature 456:470-476.
Ward JJ, McGuffin LJ, Bryson K, Buxton BF, Jones DT. 2004. The DISOPRED server for the prediction of protein disorder. Bioinformatics 20:2138-2139. Winter EE, Goodstadt L, Ponting CP. 2004. Elevated rates of protein secretion,
evolution, and disease among tissue-specific genes. Genome Res 14:54-61. Xia Y, Franzosa EA, Gerstein MB. 2009. Integrated assessment of genomic correlates
of protein evolutionary rate. PLoS Comput Biol 5:e1000413.
Xing Y, Lee C. 2007. Relating alternative splicing to proteome complexity and genome evolution. Adv Exp Med Biol 623:36-49.
Xing Y, Lee C. 2005. Evidence of functional selection pressure for alternative splicing events that accelerate evolution of protein subsequences. Proc Natl Acad Sci U S A 102:13526-13531.
Yang J, Gu Z, Li WH. 2003. Rate of protein evolution versus fitness effect of gene deletion. Mol Biol Evol 20:772-774.
Yang JR, Liao BY, Zhuang SM, Zhang J. 2012. Protein misinteraction avoidance causes highly expressed proteins to evolve slowly. Proc Natl Acad Sci U S A. Yang Z. 2007. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol
24:1586-1591.
Zhang J, He X. 2005. Significant impact of protein dispensability on the instantaneous rate of protein evolution. Mol Biol Evol 22:1147-1155.
Zhang L, Li WH. 2004. Mammalian housekeeping genes evolve more slowly than tissue-specific genes. Mol Biol Evol 21:236-239.
Zhou T, Drummond DA, Wilke CO. 2008. Contact density affects protein evolutionary rate from bacteria to animals. J Mol Evol 66:395-404.
% Variance explained A. B. C. 0 0.5 1 1.5 2 2.5 3 comp 3 comp 2 comp 9 comp 4 comp 8 comp 1 expression features splicing features structural-fucn onal features compactness features duplicability other features 0 0.5 1 1.5 2 2.5 3 comp 3 comp 2 comp 9 comp 4 comp 8 comp 1 expression features splicing features structural-fucn onal features compactness features duplicability other features 0 0.2 0.4 0.6 0.8 1 1.2 1.4 1.6 comp 2 comp 8 comp 9 comp 11 comp 13 comp 12 expression features splicing features structural-fucn onal features compactness features duplicability other features 0 0.5 1 1.5 2 2.5 3 3.5 4 0 0.5 1 1.5 2 2.5 3 3.5 4 0 0.2 0.4 0.6 0.8 1 1.2 1.4 1.6 1.8
dN/dS
dN
dS
% Variance explained A. B. C. 0 0.2 0.4 0.6 0.8 1 1.2 1.4 comp 3 comp 4 comp 2 comp 9 comp 8 comp 1 expression features splicing features structural-fucn onal features compactness features duplicability other features 0 0.2 0.4 0.6 0.8 1 1.2 1.4 comp 3 comp 2 comp 4 comp 9 comp 8 comp 1 expression features splicing features structural-fucn onal features compactness features duplicability other features 0 0.2 0.4 0.6 0.8 1 1.2 comp 4 comp 9 comp 2 comp 7 comp 1 comp 10 expression features splicing features structural-fucn onal features compactness features duplicability other features 0 0.2 0.4 0.6 0.8 1 1.2 1.4 1.6 0 0.2 0.4 0.6 0.8 1 1.2 1.4 1.6 0 0.5 1 1.5 2 2.5
dN/dS
dN
dS
0.0 0.5 1.0 1.5 2.0 2.5 0.0 0.5 1.0 1.5 2.0 2.5 0.0 0.5 1.0 1.5 2.0 2.5 expressionsplicing mixed other expressionsplicing mixed other expressionsplicing mixed other A. B. C. % Variance explained
dN/dS
dN
dS
0.0 0.5 1.0 1.5 2.0 2.5 0.0 0.5 1.0 1.5 2.0 2.5 0 1 2 3 4 mixed
other other othermixed
A. B. C.
%
Variance explained