www.elsevier.comrlocaterpowtec
Crystal growth kinetics of calcite and its comparison with readily
soluble salts
Clifford Y. Tai
), Hsiao-Ping Hsu
Department of Chemical Engineering, National Taiwan UniÕersity, RooseÕelt Road, Taipei 10617, Taiwan
Abstract
The crystal growth kinetics of calcite was investigated in batch crystallizers of stirred tank and fluidized-bed type, which were maintained at a constant pH by an autotitrator. Rhombohedron seed crystals were prepared using different techniques, then the growth experiments were conducted in the metastable region, which was explored as part of this research, to suppress nucleation. The crystal growth rates were evaluated from the consumption rates of sodium carbonate and calcium ion for the stirred-tank and fluidized-bed experiments, respectively. Several operating variables were investigated and the crystal growth rate data of constant pH and ionic strength were analyzed by the two-step crystal growth model. The mass-transfer and surface-reaction coefficients were thus obtained and used to explain the growth behaviors of calcite crystals. Then, the crystal growth kinetics of this sparingly soluble salt was compared with that of readily soluble salts. Although the effects of pH and ionic strength on crystal growth were not reported for the readily soluble systems, these effects were significant to calcite growth. q 2001 Elsevier Science B.V. All rights reserved.
Keywords: Calcite; pH-stat apparatus; Crystal growth kinetics; Stirred tank; Fluidized bed
1. Introduction
Crystal growth from solution of sparingly soluble sys-tem is rarely studied as compared with that of readily soluble system, especially for the growth of large seed crystals. Usually, fine crystals of sparingly soluble salts are generated by spontaneous nucleation via rapid mixing and reaction of two solutions, and they have no chance to grow. Thus, the study on crystal growth of large seed crystal of sparingly soluble salts had been ignored until the pellet reactor was applied to the field of environmental protection. A pellet reactor is a reactive, fluidized-bed, growth-type crystallizer used for water softening, fluoride and phosphate removal, and heavy metal recovery. The water feed and chemical reagents required to cause
deposi-w x
tion are fed to the reactor containing suspended seeds 1,2 . The materials grown on the seed are mostly sparingly soluble salts. For example, the undesired species in water softening of drinking water is calcium ion, which reacts with the added carbonate to form calcium carbonate and
)
Corresponding author. Fax: q886-2-2362-0832.
Ž .
E-mail address: [email protected] C.Y. Tai .
w x
then grow on the seeds 3 . The seeds are later removed from the reactor after they exceed a certain size, say, several hundred micrometers ready for solid–liquid separa-tion.
To design a pellet reactor, the metastable region and the crystal growth rate should be known. The former gives the concentration zone where the reactor is operative, i.e. crystals grow without nucleation, and the latter determines the reactor size. The concept of metastable region for sparingly soluble system is illustrated by Sohnel and Gar-
¨
w x
side 4 . The metastable region is present in a precipitation diagram by plotting p A versus p B, where A and B are the concentrations of ion species and p A represents ylog A. The metastable region of sparingly soluble system is avail-able for only a few systems, perhaps CaCl –Na PO –H O2 3 4 2
w x4 and CaSO P 2H O–H O 5 are the two systems thatw x
4 2 2
can be found, and the latter system is plotted on an ionic product–temperature diagram instead of the precipitation diagram. In the study on crystal growth of sparingly soluble salts using seeded technique, the seed size is
w x
usually less than 10 mm 6–8 because large crystals of sparingly soluble salts are difficult to prepare, and there is no need to collect the growth rate data of large crystals because of a lack in application. The growth rate of a small
w x
crystal is usually lower than that of a large crystal 9 .
0032-5910r01r$ - see front matter q 2001 Elsevier Science B.V. All rights reserved.
Ž .
Therefore, it is not adequate to use the growth rates of small crystals for the design of a pellet reactor.
The seeded technique for studying the crystal growth rate of readily soluble system looks straightforward as compared with that of sparingly soluble system. It seems that there are many problems to encounter in studying the latter system, including at least the following: the prepara-tion of large seed crystals, the determinaprepara-tion of supersatu-ration, the identification of metastable region, and the control of pH and supersaturation of solution. The large seed crystals of sparingly soluble salts seem difficult to prepare; however, this problem can be solved by using the gel growth method, which is a well-known technique to
w x
grow high-quality single crystals at a low cost 10 . The expression of supersaturation is different between the read-ily and sparingly soluble systems. The definition of
rela-w x
tive supersaturation proposed by Nielsen and Toft 11 is adopted by many researchers. The concentrations of species, which are required to calculate ionic activity and thus supersaturation, were determined by using a computer program based on the algorithm of successive
approxima-w x
tion of the ionic strength 12 . To study crystal growth, the solution supersaturation should be controlled within the metastable region; otherwise, nucleation would occur at higher supersaturation and mess up the operation. Al-though the metastable region of CaCO is not reported in3
w x
the literature, experimental techniques are available 4 and experiments can be conducted to locate the region. During a crystal growth process, the solution pH will change, especially for the system that the species will form com-plex with hydrogen ion such as CaCO –H O system, and3 2
the supersaturation will shift either to a low value where crystal growth rate is negligible or to a high value where nucleation occurs. Therefore, the control of pH is essential
w x
and can be achieved by using an autotitrator 7,8 . In our laboratory, the growth kinetics of calcite crystals was studied in batch, pH-stat crystallizers of a stirred tank and fluidized-bed type. This report summarizes the experi-mental study regarding crystal growth rates of calcite in laboratory-scale equipments. After the seed crystals were prepared and the metastable region was located, the crystal growth data were evaluated from the consumption rate of titrant or calcium ion using an autotitrator. The factors that affect the crystal growth rates were investigated. Then the growth rate data were analyzed by the two-step growth model, thus the mass-transfer and surface-reaction coeffi-cients were determined. Finally, the crystal growth kinetics of sparingly soluble systems was compared with that of readily soluble systems in the two types of crystallizer. This kind of comparison is worthwhile from the academic point of view. In the past, experiments of the two systems were conducted by different research groups, probably due to different experimental techniques involved.
The two-step growth model used in the analysis of crystal growth rate data takes account of the mass-transfer resistance and surface-reaction resistance in series and
neglects all other resistances in a crystal growth process, which may be expressed mathematically by the following equations:
G s Kd
Ž
s y si.
sKdoLaŽ
s y si.
Bulk transportŽ .
1G s K srsK Lbsr Surface reaction 2
Ž .
r i ro i
where G is the linear crystal growth rate, s the overall supersaturation, si the interfacial supersaturation, L the crystal size, r the surface-reaction order, and Kdo and
K , the mass-transfer and surface-reaction coefficients,ro
respectively.
2. Determination of relative supersaturation
The relative supersaturation of a readily soluble salt is usually expressed by the difference between solution con-centration and saturation concon-centration divided by satura-tion concentrasatura-tion. This simplified expression of driving force is difficult to apply to sparingly soluble system because the species of crystallizing compound are usually not in equal concentration. In the literature, different ex-pressions have been used. The relative supersaturation
w x
proposed by Nielsen and Toft 11 is adopted here:
s s K1r2y
K1r2 rK1r2
Ž .
3Ž
IP sp.
spwhere KIP is the ionic product, defined as K sIP aCa2qaCO2y, and Ksp is the solubility product of CaCO .3
3
The activity of species, aCa2q or aCO2y, is the product of 3
activity coefficient and ionic concentration of the respec-tive species. The concentrations of ionic species are com-puted from the measured pH, total calcium and carbonate concentration by successive approximation for the ionic
w x
strength 12,13 . The computer program contains the mass action and mass balance equations and the modified De-bye–Huckle equation for calculating the activity coeffi-
¨
w x
cient 14 . In the operation of crystal growth experiment, supersaturation should be controlled in a suitable range; a low supersaturation gives a growth rate that is insignifi-cant, and a high supersaturation causes nucleation that messes up the operation. The supersaturation is signifi-cantly affected by the solution pH because the concentra-tion of CO2y is a function of pH at a fixed concentration
3
of total carbonate. As judged from the distribution of the total carbonate ions, existing as CO2y, HCOy
and H CO ,
3 3 2 3
the suitable operation range of CaCO system is roughly3
between pH 8.5 and 10.5.
3. Experimental
3.1. Identification of metastable region
To suppress nucleation in a crystal growth experiment, supersaturation should be kept in the metastable region,
which is the area between supersolubility and solubility
w x
curves in a precipitation diagram 4 . The experimental procedures to identify the metastable region of CaCl –2
w x
Na CO –H O system are described elsewhere 15 . Briefly2 3 2
speaking, the supersolubility curve of CaCO is the bound-3 ary where nucleation occurs at a specified induction period for different species concentrations, i.e. CaCl and Na CO2 2 3 solution of known concentrations are mixed and stirred for a period of time at which the solution becomes turbid. The metastable region of calcium carbonate is shown in Fig. 1 as p aCa2q versus p aCO2y, where aCa2q and aCO2y are the
3 3
activities of Ca2q and CO2y, respectively. The activities 3
are calculated from the concentrations by the same com-puter program for estimating supersaturation. The solubil-ity curve is plotted in Fig. 1 using the solubilsolubil-ity product of
y9 w x
CaCO , K s 4.872 = 103 sp 16 . The two curves are almost straight and parallel in the concentration range studied, and they are independent of pH values between 8.5 and 10.5. The metastable zone width so determined is based on primary nucleation. In the experiments, the oper-ation range was kept within one-half of the width to suppress the primary and secondary nucleation.
3.2. Preparation of seed crystals
Seed crystals used in the experiment were from two sources. The large sizes greater than 50 mm were prepared
by the gel growth technique or by curing natural calcite, and smaller sizes were obtained by curing the product from Nacalai Tseque. The size of large crystals was deter-mined by sieving, i.e. the mean size of two close-cut sieves, and the size of smaller crystals was converted from the surface area measured by a BET surface area analyzer
ŽMicroneritics, 2100 D , taking the crystals as rhombohe-.
dron identified by SEM photographs. The identification of calcite was accomplished by using an IR spectra and X-ray powder diffraction pattern.
3.3. Crystallization apparatus
The crystallization system that contains a pH-stat flu-idized-bed crystallizer, a storage tank, and a pH-control system is shown in Fig. 2. In the study of crystal growth in a stirred tank, the fluidized bed is removed and the storage tank is used as the crystallizer. The key component of the
Ž
set-up is an autotitrator or pH-stat apparatus Kyoto
Elec-.
tronic, AT 200 that maintains the pH constant during an experiment. The experimental procedures are described in
w x
detail by Tai et al. 13,15 . In the reports, the procedures for estimating the linear crystal growth rates are also given. Briefly speaking, in the stirred-tank experiment, CaCl is in excess and the control of pH is done by adding2 Na CO solution, thus the crystal growth rate is estimated2 3
Fig. 1. Metastable region of calcium carbonate: p aCa2q vs. p aCO2y. — Solubility curve; – – supersolubility curve; ^ pH s 8.5, e pH s 9.5, 3
Ž . Ž . Ž . Ž . Ž .
Fig. 2. A pH-stat crystallization system. 1 pH and temperature indicator, 2 reagent bottle, 3 pumping system of reagent, 4 reagent delivering line, 5
Ž . Ž . Ž . Ž . Ž . Ž . Ž . Ž .
burette, 6 thermometer, 7 water bath, 8 storage tank, 9 temperature controller, 10 motor, 11 glass electrode, 12 reference electrode, 13
Ž . Ž . Ž . Ž . Ž . Ž .
thermocompensator, 14 axial-flow impeller, 15 magnetic motor, 16 flowmeter, 17 fluidized-bed crystallizer, 18 recycle valve, 19 distributor.
by the consumption rate of the titrant; in the fluidized-bed experiment, the pH is controlled by adding NaOH solution and the crystal growth rate is evaluated by the consump-tion rate of calcium ion.
4. Crystal growth kinetics in a stirred tank
The working equation for estimating the parameters of
Ž .
the two-step growth model is derived by combing Eqs. 1
Ž . and 2 . 1rr G G q ss
Ž .
4 až
b/
KdoL K LroWhen experimental data of all crystal sizes were used in a
Ž .
regression analysis, they did not fit well with Eq. 4 . Therefore, growth rate data of small crystals for 2, 5, and 8
mm, and large crystals between 58 and 230 mm were
analyzed separately. The results are shown below:
G s 1.999 = 10y9 Ly0 .00256 s y s 5
Ž
i.
Ž .
G s 1.345 = 10y1 1 L0 .627s2 .20 6Ž .
ifor small crystals, where G is in mrs, L in mm, and s and s are dimensionless. In addition,i
G s 5.887 = 10y1 0L0 .434 s y s 7
Ž
i.
Ž .
G s 1.292 = 10y1 1 L0 .604s2 .02 8Ž .
ifor large crystals.
It is concluded that the mass-transfer rate is independent of crystal size for small crystals but increases with
increas-ing size for large crystals. This is because the exponent of
Ž .
L is a very small number in Eq. 5 and a significant
Ž .
positive figure in Eq. 7 . On the other hand, the surface-reaction rate is similar for the whole size range by
compar-Ž . Ž .
ing Eqs. 6 and 8 and increases with an increase in crystal size.
Once the parameters in the two-step growth model are available, a judgement on the importance of each step can
w x
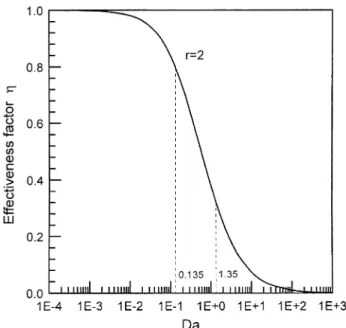
be made using the concept of effectiveness factor 17 . The
Fig. 3. Operating range of Damkohler number and corresponding effec-¨
Fig. 4. Operating range of Damkohler number and corresponding effec-¨
tiveness factor for potassium alum system.
surface-reaction effectiveness factor h for an r th-order rate is:
r
h s 1 y hDa
Ž
.
Ž .
9where Da is expressed by:
Da s K Lby asry 1r
K
Ž
10.
ro do
When h approaches 1, the crystal growth process is sur-face-reaction controlled; on the other hand, the growth process is mass-transfer controlled when h approaches
Ž .
zero. Eq. 9 is plotted in Fig. 3, which shows that the mass-transfer resistance and surface-reaction resistance are both significant for large crystals, and the mass-transfer resistance becomes less important at lower supersaturation for smaller crystals. The result of large crystals is consis-tent with that of one of the readily soluble salts, potassium alum, as shown in Fig. 4 for particle size being between
w x
100 and 700 mm 19 . On the other hand, the result of small crystals approaches the conclusion made by Garside
w x
and Jancic 18 that the growth process of potassium alum,
ˇ ´
a readily soluble salt, is controlled by the surface reaction for micron-sized particles.
The parameters in the two-step growth model of the
Ž .
sparingly soluble salt CaCO3 are compared with those of
w x
readily soluble salts as shown in Table 1 13 . The varia-tion in parameters can be understood because various types of crystals are included in the table, i.e. readily soluble
Ž .
inorganic potassium sulfate, potassium alum , readily
sol-Ž .
uble organic succinic acid , and sparingly soluble
inor-Ž .
ganic calcite . The kinetic behaviors of large inorganic crystals of readily soluble and sparingly soluble salts are similar, i.e. mass-transfer and surface-reaction coefficients increase with increasing crystal size. However, the organic
Ž .
crystal succinic acid behaves differently in the crystal size effect on mass-transfer coefficient, which decreases with an increase in crystal size. There is no explanation for this type of growth kinetics at the present time. For small crystals, no comparison can be made because the parame-ters of the two-step model are not available for the readily soluble crystals.
The findings on the mass-transfer rates of various crys-tal sizes are consistent with the growth behaviors inferred
w x
by Mullin 9 : large particles have higher terminal velocity and, in cases where diffusion plays a significant role in the growth process, the larger the crystals, the higher the growth rate; for crystals smaller than about 10 mm, their sizes are smaller than that of turbulent eddies and they may be growing in a virtually stagnant medium even in a well-agitated system, thus the mass-transfer coefficient is independent of size. As to the surface-reaction rate, the
w x
size effect has been postulated by Garside and Davey 22 . The larger the crystals, the more energetically will they collide in agitated suspensions and the greater is the potential for surface damage, or the number of dislocations in a crystal increases with size due to mechanical stresses
w x9 . Both of these effects favor faster surface-reaction
kinetics and lead to higher growth rates with increasing crystal size.
5. Crystal growth kinetics in a fluidized bed
Depending on the superficial velocity applied in an operation, two types of fluidized bed are classified, i.e. the dense bed and the lean bed. The former is conducted at a
Table 1
Parameters of the two-step growth model for calcite and other substances grown in an agitated tank: mass-transfer coefficient, K s Kd doLa; surface-integration coefficient, K s K Lr ro b
Author Systems Agitation Seed size a b r
Ž . Ž .
speed rpm mm
Ž . w x
Garside et al. 1974 19 Potassium sulfate 500–800 570–1180 0.130 0.700 2.40
Ž . w x
Tai and Yu 1989 20 Potassium alum 950 100–700 0.581 0.583 2.00
Ž . w x
Qiu and Rasmuson 1990 21 Succinic acid 355–800 400–710 y0.170 1.600 3.00
Ž . w x
Tai et al. 1993 13 Calcite 800 58–230 0.434 0.604 2.02
superficial velocity lower than the particle terminal veloc-ity to give the bed a clear solution–suspension boundary; in the latter, the superficial velocity is approaching the terminal velocity and the particles may move freely in the bed with no boundary. In the operation of a pellet reactor, a dense bed is more advantageous as far as seed loading is concerned. Therefore, factors that affect the calcite growth rate in a dense bed were investigated, including superficial velocity, particle size, supersaturation, pH, and ionic strength.
When keeping the ionic strength and pH constant, the crystal growth rate increases with an increase in crystal size ranging between 460 and 920 mm; on the other hand, the growth rates of 460-mm seed are rather constant at various superficial velocities of 1.42, 2.36, 3.54 and 4.73
w x
mrs 15 , in spite of a great change in bed voidage and bed expansion. The mechanisms of crystal growth related to crystal size and superficial velocity will be clarified after the growth rate data are analyzed by the two-step growth model.
According to different sources, the surface-reaction
or-w x
der of calcite growth is approximately 2 6,8,13 . Taking
Ž . Ž .
r s 2, Eqs. 2 and 3 are combined to give:
s 1 1
'
s G q ,
Ž
11.
'
G Kd(
Krthus the mass-transfer and surface-reaction coefficients can be evaluated by plotting sr6G versus 6G. The obtained
Kd and K are listed in Table 2. Despite the variation inr
particle size and superficial velocity, the values of K ared
rather constant. The largest deviation from the average value of K , 1.32 = 10y2
, is about 15%. The
surface-reac-d
tion coefficient Kr increases with an increase in crystal size when we compare the first three runs in Table 2, and
K is independent of superficial velocity as shown by ther
last three runs. The dependence of K on crystal size isr
w x
similar to the readily soluble systems 23,24 and has been explained in the Section 4. The independence of Kr on superficial velocity is easily understood. Because surface reaction is an interfacial phenomenon, it should not be disturbed by liquid flow under a mild agitation, such as in
Table 2
Mass-transfer and surface-reaction coefficients of calcite crystal estimated
Ž . 3
by using Eq. 11 at pH s9.5 and I s 0.0025 kmolrm Run no. Particle Superficial Kd Kr
y10 y9 Ž . Ž . size velocity 10 mrs 10 mrs Žmm. Žmrs. A-4-1-O20 920 3.54 1.45 13.78 A-4-2-O13 650 3.54 1.42 2.25 A-2-2-M14 460 3.54 1.33 0.30 A-1-4-M08, 16 460 2.36 1.11 0.31 A-2-4-M12, 13 460 1.42 1.29 0.34
Fig. 5. logG vs. log s showing effect of ionic strength on the growth rate of CaCO3 crystal at Ls 460 mm; pH s9.5; T s 258C. Symbols and
Ž 3. )
corresponding ionic strength kmolrm : [ 0.0340; v 0.0185; 0.0105;
B0.0025.
a fluidized bed. To explain the constant K , an argumentd based on the relative velocity between solution and particle is presented here. As the superficial velocity is reduced, the bed voidage and thus the cross-sectional area for the passage of solution becomes smaller. As a result, the relative velocity, which determines the mass-transfer coef-ficient, does not vary much for different crystal sizes and superficial velocities. Besides, we can arrive at the con-stant K using a correlation proposed by Kunii and Leven-d
w x w x
spiel 25 . The argument is presented somewhere else 15 . The variation of Kd with particle size or superficial velocity is actually less than 15%. This result is similar to
w x
that reported by Budz et al. 23 in Table 3 of their paper for the K–alum system, in which the highest deviation from the average value is about 10%. The constant
mass-w x
transfer coefficient is also reported by Tournie et al. 26 for crystal dissolution process, after they analyzed exten-sive dissolution data of both lean and dense fluidized bed in the literature.
Once K and K are obtained, the effects of crystal sized r
and superficial velocity on the kinetic behaviors of CaCO3
growth can be explained in terms of the two-step growth model. The surface-reaction step is responsible for the size-dependent growth because K increases with increas-r
ing crystal size and Kd is independent of crystal size. On the other hand, the crystal growth rates of the same size are constant at various superficial velocities because Kd
and K are independent of superficial velocity.r
The effects of supersaturation and ionic strength on calcite growth rate are shown in Fig. 5. The ionic strength varying from 0.0025 to 0.0340 kmolrm3 was adjusted by
adding NaCl solution. For all levels of ionic strength, the growth rates of calcite increase with an increase in super-saturation. At the same supersaturation, the growth rates increase with ionic strength from 0.0025 to 0.0185 kmolrm3; however, a further increase in ionic strength is
no more effective. The slope of logG y log s curve changes at the lowest ionic strength, indicating a change in the weighting of various resistances to crystal growth. The kinetic behavior of PbF crystal reported by Stubicar et al.2
w27 is similar to that of CaCO as far as the effect of ionicx
3
strength is concerned.
Fig. 6 shows the effects of pH at two levels, 8.5 and 9.5, on the growth rate of calcite. Similar to the effects of ionic strength, different slopes of logG–log s plot are obtained at different pH, and the influence of pH becomes
w x
insignificant at higher supersaturation. Stubicar et al. 27 reported that the crystal growth rate of a-fluoride in-creased with pH when the solution pH was lower than the isoelectric point at pH 5.6. The results obtained in this experiment of calcite is consistent with a-lead fluoride because the isoelectric point of calcite is between 9 and 10
w28 . Hohl et al. 29 also demonstrated that the CaHPO Px w x
4
2H O and hydroxyapatite crystals grew more slowly at pH2 values below, than they do above, the isoelectric point. Besides, the pH effect for CaF growth is reported by Tai2
w x
et al. 30 as shown in Fig. 7. The pH effect implies that the charge on the crystal face is important in the building
w x
of growth units into the lattice 4 . The effects of ionic strength and pH are seldom reported for readily soluble salts; however, they are significant to the sparingly soluble salts. This is probably because the ionic strength is already very high in a supersaturated solution of readily soluble
Fig. 6. logG vs. log s showing crystal growth rate of CaCO at two pH3
values: I s 0.018 kmolrm3; Ls 460 mm; T s 258C; us 2.36=10y2
mrs; v pH s8.5, I pH s9.5.
Fig. 7. pH effect on the crystal growth of CaF , using 388-mm fluoride2
crystal seed. I s 0.11 kmolrm3; us 2.4=10y2 mrs; I pH s10, e
pH s9, v pH s8.
systems, and the pH does not change during the solid formation.
6. Concluding remarks
The pH-stat apparatus is suitable for studying crystal growth kinetics of sparingly soluble salts. Growth experi-ments should be conducted in the metastable region that lies between two parallel straight lines, i.e. solubility curve and supersolubility curve, when the precipitation diagram is constructed by plotting p aCa2q versus p aCO2y. Factors
3
that affect the calcite growth rates are identified, including supersaturation, superficial velocity, particle size, pH, and ionic strength. Then the two-step crystal growth model is employed to analyze the calcite growth data of constant pH and ionic strength and much useful kinetic information is revealed. The obtained Kd and K , mass-transfer andr surface-reaction coefficients, can be used to explain the crystal growth behaviors. In a stirred tank operated at fixed agitation rate, both coefficients are size-dependent except
Ž .
for the mass-transfer of small crystals - 10 mm , and the surface reaction is responsible for the size-dependent growth of small crystals. In a fluidized bed operated in a dense mode, the mass-transfer coefficient is independent of crystal size and superficial velocity and the surface-reac-tion coefficient is size-dependent but not influenced by superficial velocity; thus the superficial velocity has little effect on the growth rates of calcite of the same size and the surface reaction is responsible for the size effect. The crystal growth behaviors of this sparingly soluble salt are similar to that of readily soluble salts. For the sparingly soluble systems, two more variables, i.e. pH and ionic
strength, appear important. This finding together with the similar results previously reported imply that the charge on crystal face is important in the building of growth units into the crystal lattice.
Notations
a Exponent of crystal size in Eq. 1Ž .
aCa2q Activity of calcium ion kmolrmŽ 2.
a 2y
CO3 Ž
2.
Activity of carbonate ion kmolrm
b Exponent of crystal size in Eq. 2Ž .
Da Damkohler number
¨
G Linear crystal growth rate mrsŽ .
I Ionic strength kmolrmŽ 3.
Kd Mass-transfer coefficient mrsŽ .
Kdo Size-independent mass-transfer coefficient mrsŽ .
KIP Ž
3.2
Ionic product kmolrm
Kr Surface-reaction coefficient mrsŽ .
Kro Size-independent mass-transfer coefficient mrsŽ .
Ksp Ž
3.2
Solubility product kmolrm
L Crystal size in m or mm as specified in the textŽ .
r Surface-reaction order
T Temperature 8CŽ .
h Effectiveness factor of crystal growth
s Bulk relative supersaturation
si Interfacial relative supersaturation
Acknowledgements
The authors gratefully acknowledge the financial sup-port of the National Science Council of the Republic of China through the years in the area of crystal growth.
References
w x1 J.C. van Dijk, D.A. Wilms, J. Water SRT-Aqua. 40 1991 263–280.Ž . w x2 M.M. Seckler, O.S.L. Bruinsma, G.M. van Rosmalen, J.C. van Dijk,
Ž .
F. Delgorge, in: A. Mersmann Ed. , Industrial Crystallization, vol. 90, Garmisch-Parten Kirchen, FRG, 1990, pp. 134–148.
w x3 P. Dirken, E. Baars, A. Graveland, F.C. Woensdregt, in: A. Mers-Ž .
mann Ed. , Industrial Crystallization, vol. 90, Garmisch-Parten Kirchen, FRG, 1990, pp. 95–100.
w x4 O. Sohnel, J. Garside, Precipitation—Basic Principles and Industrial¨
Applications. Butterworth-Heinemann, Boston, 1992, p. 149.
w x5 D.M. Killer, R.E. Massey, O.E. Hileman, Can. J. Chem. 56 1978Ž .
831–838.
w x6 G.H. Nancollas, M.M. Reddy, J. Colloid Interface Sci. 37 1971Ž .
824–830.
w x7 E.K. Giannimaras, P.G. Koutsoukos, J. Colloid Interface Sci. 116 Ž1987 423–430..
w x8 J. Christoffersen, M.R. Christoffersen, J. Cryst. Growth 100 1990Ž .
203–211.
w x9 J.W. Mullin, Crystallization. 3rd edn., Butterworth-Heinemann,
Ox-ford, 1993, pp. 237–238.
w10 H.K. Henisch, Crystals in Gels and Liesegang Rings. 2nd edn.,x
Cambridge Univ. Press, Cambridge, 1988, p. 48.
w11 A.E. Nielsen, J.M. Toft, J. Cryst. Growth 67 1984 278–288.x Ž . w12 G.H. Nancollas, Interactions in Electrolyte Solutions. Elsevier, Ams-x
terdam, 1966, p. 85.
w13 C.Y. Tai, P.-C. Chen, S.-M. Shih, AIChE J. 39 1993 1472–1482.x Ž . w14 J.N. Butler, Ionic Equilibrium. Addison-Wesley, Reading, MA, 1964,x
p. 432.
w15 C.Y. Tai, W.-C Chien, C.Y. Chen, AIChE J. 45 1999 1605–1614.x Ž . w16 O. Sohnel, J. Garside, Precipitation—Basic Principles and Industrialx ¨
Applications. Butterworth-Heinemann, Boston, 1992, p. 296.
w17 J. Garside, Chem. Eng. Sci. 26 1971 1425–1431.x Ž . w18 J. Garside, S.J. Jancic, AIChE J. 22 1976 887–894.x ˇ ´ Ž .
w19 J. Garside, J.W. Mullin, S.W. Das, Ind. Eng. Chem. Fundam. 13x Ž1974 299–305..
w20 C.Y. Tai, K.H. Yu, J. Cryst. Growth 96 1989 849–855.x Ž . w21 Y. Qiu, C. Rasmuson, AIChE J. 36 1990 665–676.x Ž .
w22 J. Garside, R.J. Davey, Chem. Eng. Commun. 4 1980 393–424.x Ž . w23 J. Budz, P.H. Karpinski, Z. Nuruc, AIChE J. 30 1984 710–717.x Ž . w24 C.Y. Tai, C.-Y. Chen, J.-F. Wu, Chem. Eng. Commun. 56 1987x Ž .
329–340.
w25 D. Kunii, O. Levenspiel, Fluidization Engineering. Wiley, Newx
York, 1969, p. 167.
w26 P. Tournie, C. Larguerie, J.P. Couderc, Chem. Eng. Sci. 34 1979x Ž .
1247–1255.
w27 N. Stubicar, B. Markoric, A. Tonejc, M. Stubicar, J. Cryst. Growthx
Ž .
130 1993 300–304.
w28 J.S. Reed, Introduction to the Principles of Ceramic Processing.x
Wiley, Singapore, 1989, p. 134.
w29 H. Hohl, P.G. Koutsoukos, G.H. Nancollas, J. Cryst. Growth 57x Ž1982 325–335..
w30 C.Y. Tai, T.M. Tsao, P.-C. Chen, M.S. Lee, Industrial Crystalliza-x