Chapter 1 Introduction
1.1 Dengue virusDengue viruses (DVs) are human pathogens and the infections affect up to 100 million individuals per year (McBride and Bielefeldt-Ohmann, 2000).
They belong to the genus Flavivirus within the family of Flaviviridae. Members of the genus are typically transmitted to vertebrates by mosquitoes or ticks and frequently cause significant human morbidity and mortality. In addition to DV, other human pathogens such as Japanese encephalitis virus (JEV), tick borne encephalitis virus, West Nile virus (WNV), yellow fever virus (YFV), and Kunjin virus (KUNV) are classified to this family (Beeck et al., 2004).
DVs are comprised of four distinct serotypes, DV1 through DV4, which are transmitted from human to human by the mosquitoes Adeds aegypti and cause infections mostly in tropical and subtropical regions worldwide. They cause dramatic variations in clinical symptom varying from asymptomatic to dengue fever (DF), or even more severe infection with bleeding and shock, known as the dengue hemorrhagic fever (DHF), and dengue shock syndrome (DSS) (McBride and Bielefeldt-Ohmann, 2000). DF is a self-limited debilitating illness characterized by fever, headache, retro-orbital pain, myalgia, arthralgia, and rash. DHF is marked by increased vascular permeability (plasma leakage), thrombocytopenia, and hemorrhagic manifestations; DSS occurs when fluids leakage into the interstitial spaces results in shock, which without appropriate treatment may lead to death (Clyde et al., 2006). The case-fatality rate of DHF/DSS in most countries is about 5% (CDC, USA, 2005).
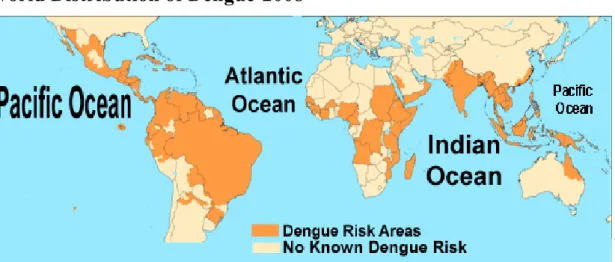
In 2005, dengue viral infection is one of the most important mosquito-borne viral diseases affecting humans. In the 1950s, there were only nine countries reporting the clinical manifestations of dengue. Today the geographic distribution includes more than 100 countries worldwide (Fig. 1.1). Many of these countries had no dengue reported for over 20 years and
several countries have no known history of the diseases. The World Health Organization estimates that more than 2.5 billion people are at risk of dengue infection. First recognized in the 1950s, DHF/DSS has become a leading cause of child mortality in several Asian and South American countries (Guha-Sapir and Schimmer, 2005).
In 2007 the four serotypes of DVs were estimated to cause 50-100 million annual human infections and 22000 deaths worldwide. The number of dengue fever cases per year is increasing steadily, including in the United States where DVs has spread to 36 states since 1985, and the risk of an outbreak is recognized (Zhou et al., 2008; Morens et al., 2008). Nevertheless, there are no known antiviral compounds and no therapeutic treatment against dengue virus. Safe vaccines against the yellow fever virus exist, and progress toward a dengue virus vaccine has being made, but the availability of vaccines is often limited. Therefore, the development of new antiviral drugs and a safe vaccine become imperative (Solomon et al., 2001; Zhou et al., 2008).
1.2 Replication of dengue virus
The intracellular life cycles of the flaviviruses are very similar (Fig. 1.2). Infection with one of the arthropod-borne flaviviruses begins when the vector takes a blood meal and the virus is introduced into the host. The virus attaches to the cell surface, mediated by the E protein, and enters the cell by receptor-mediated endocytosis. Low pH in the endosomal compartment triggers fusion of the viral and host cell membrane mediated by structural reorganization of E, which leads to the release of the nucleocapsid and viral RNA into the cytoplasm. Translation of the input strand takes place; then the virus switches from translation to synthesis of a negative-strand intermediate, which serves as a template for the production of multiple copies of positive-strand viral RNA (vRNA). Successive rounds of translation produce high levels of viral proteins; the structural protein capsid (C), premembrane (prM), and envelope (E) proteins, along with vRNA, are assembled into progeny virions, which are
transported through the Golgi compartment and secreted (Fields et al., 2001; McBride and Bielefeldt-Ohmann, 2000; Clyde et al., 2006; Aruna et al., 2009).
1.3 Genome of dengue virus
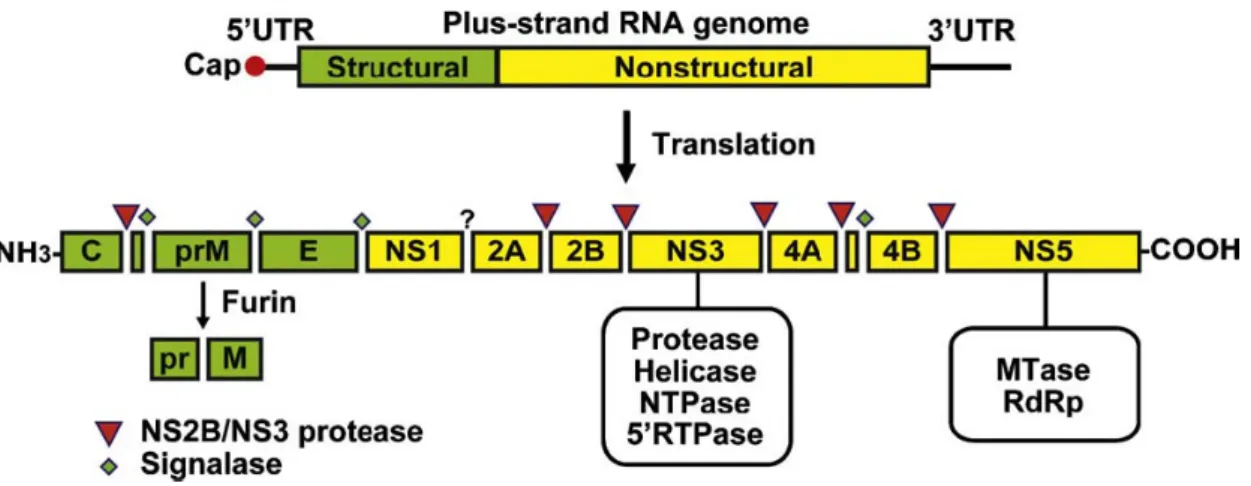
DV possesses a positive sense, single-stranded RNA wrapped in a nucleocapsid protein within an envelope. The whole genome is approximately 10.8 kb in length and contains a single open reading frame. A single polyprotein translated from the viral RNA is cleaved co- and post-translationally by the virus-encoded serine protease, NS2B/NS3, and by host-encoded proteases, including signalase and furin, to produce the three structural proteins, capsid (C), membrane protein (M, which is expressed as prM, the precursor to M), and envelope (E) – that constitute the virus particle and seven nonstructural proteins (NS) that are essential for viral replication encoded by the remainder of the genome. The reported gene order is 5’-C-prM/M-E-NS1-NS2A-NS2B-NS3-NS4A-NS4B-NS5-3’ (Fig. 1.3) (Gubler et al., 1997; Chao et al., 2005; Mukhopadhyay et al., 2005; Sampath et al., 2009).
1.4 Four small non-structural proteins of dengue virus
DV has seven non-structural proteins: NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5 (Fig. 1.4). Except for enzymatic activities contained within NS3 and NS5, the roles of the other proteins in virus replication and pathogenesis are not well defined.
NS2A, 2B, 4A, and 4B are four small non-structural proteins encoded by flavivirus genome. All four proteins are poorly conserved in sequence but exhibit conserved hydrophobicity profiles among flaviviruses, which suggests that they may be membrane-associated (Gublur et al., 1997). The crude membrane fractions of WNV-infected cells contain a set of viral encoding proteins as major constitutes after weak or stringent salt washes. The NS2A, NS2B, and NS4B are membrane or integral membrane proteins that are separated from the membranes after stringent salt washes. In addition, expression in human
A549 cells of the DV nonstructural proteins NS2A, NS4A, or NS4B enhances replication of an IFN-sensitive virus. Moreover, expression of NS4B and, to a lesser extent, of NS2A and NS4A proteins results in down-regulation of IFN-α/β-stimulated gene expression (Munoz-Jordan et al., 2003). This phenomenon is also observed in WNV and KUNV (Liu et
al., 2005).
1.5 Properties of NS2A
NS2A of DV is a small hydrophobic protein of about 24 kDa that migrates anomalously by SDS-polyacrylamide gel electrophoresis (SDS-PAGE). It is about 20 kDa in KUNV (Mackenzie et al., 1998) and 22 kDa in YFV (Lindenbach et al., 1999). Its N-terminus is generated via NS1–2A cleavage by an unknown ER-resident host enzyme (Falgout et al., 1995) and cytosolic cleavage at the NS2A/2B junction by the NS2B-NS3 serine protease. Two forms of NS2A are found in YFV-infected cells. Full-length NS2A (224 amino acids) is the product of cleavage at the NS1/2A and NS2A/2B sites. NS2Aα, a C-terminally truncated form of 190 amino acids, was resulted from partial cleavage by the viral NS2B-NS3 serine protease at the sequence QK↓T within NS2A. Changing lysine to serine at this site (QKT→QST) blocks the production of both NS2Aα and infectious virus, but a mutation in NS2A that blocks virus production can be suppressed by a second mutation on the surface of the NS3 helicase domain (Kummerer et al., 2002). In addition, an upstream mutation in KUNV NS2A has also been shown to block virus assembly (Liu et al., 2003). Therefore, mutations at the YFV and KUNV NS2A provided evidence that NS2A is involved in virus assembly (Lindenbach et al., 2007). In addition, studies in RNA replication with KUNV have shown that NS2A co-localizes with double-strand RNA in discrete cytoplasmic foci and interacts with the 3’ untranslated region of Kunjin viral RNA, as well the proposed replicase components NS3 and NS5 in cell lysates. The result indicates that the flavivirus replication complex includes NS2A (Mackenzie et al., 1998). Thus, NS2A is an attractive candidate for
coordinating the shift between RNA packaging and RNA replication, processes that have been shown to be linked (Khromykh et al., 2001).
Besides its apparent roles in RNA replication and virus assembly, DV2 NS2A has also been shown to act as an interferon (IFN) antagonist by inhibiting IFN signaling (Munoz-Jordan et al., 2003) and specific mutations in KUNV and WNV NS2A have been identified that diminish this inhibitory activity (Liu et al., 2004) and attenuate WNV virulence in mice (Liu et al., 2006). However, the small hydrophobic protein NS2A is poorly characterized, and its specific function for viral replication cycle is still unknown.
1.6 Properties of NS2B
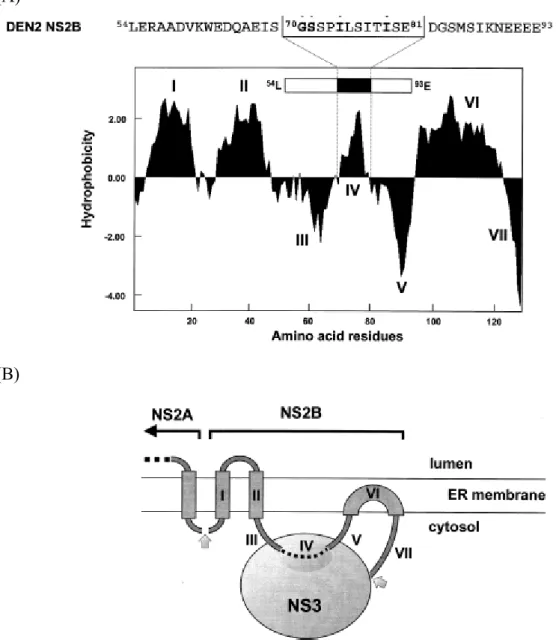
NS2B is a small (≈14 kDa) membrane-associated protein. NS2B forms a stable complex with NS3 and acts as a cofactor for the NS2B-NS3 serine protease. This protease complex catalyzes autocleavage (cis) at the junction between NS2A and NS2B as well as between NS2B and NS3. It also catalyzes the trans cleavage at the junctions between NS3 and NS4A as well as NS4B and NS5 (Clum et al., 1997; Falgout et al., 1991). The initial characterization of the cofactor requirement for the DV NS3 protease revealed that the minimal region required for protease activity was located in a 40-residue central hydrophilic segment of NS2B spanning residues Leu54 to Glu93 (Falgout et al., 1993). Although the hydrophobic regions of NS2B are dispensable for protease activity, they are required for cotranslational membrane insertion of full length NS2B and its efficient activation of the NS3 protease domain (Yusof et al., 2000). Modeling of the putative interactions between the DV2 NS3 protease and its cofactor, NS2B, suggests that a 12 amino acid hydrophobic region within this sequence (70GSSPILSITISE81) may associate directly with NS3 (Fig. 1.5) (Brinkworth et al., 1999).
The alanine substitutions at residues Trp62, Leu75, and Ile79 in the DV NS2B cofactor result in marked effects on autoprocessing at the NS2B/NS3 site (Niyomrattanakit et al.,
2004). Also, mutagenesis experiments with YFV NS2B protein demonstrated that specific residues within this core sequence are critical for protease activation. Deletion of residues 51 to 55, 53 to 55, and 56 to 93 within the conserved central domain of YFV NS2B yielded no detectable processing of an NS2B-NS3pro polyprotein precursor, whereas a four-amino-acid deletion of the sequence 67ISGS70 generated a protease with significantly reduced cleavage efficiency (Chambers et al., 1993). In all, mutation of conserved residues in NS2B can have dramatic effects on autoproteolytic cleavage at the NS2B/NS3 junction and transcleavage activities (Chambers et al., 2005; Niyomrattanakit et al., 2004).
Besides, although the DV NS3 protease exhibits NS2B-independent activity with model substrates for serine proteases such as N-a-benzoyl-arginine-p-nitroanilide, enzymatic cleavage of dibasic peptides is markedly enhanced with the NS2B-NS3 cocomplex, and the presence of the NS2B cofactor was shown to be an absolute requirement for trans cleavage of a cloned polyprotein substrate (Niyomrattanakit et al., 2004). The cofactor, NS2B, is also essential for proper domain motion that contributes to substrate binding (Zuo et al., 2009).
1.7 Properties of NS4A
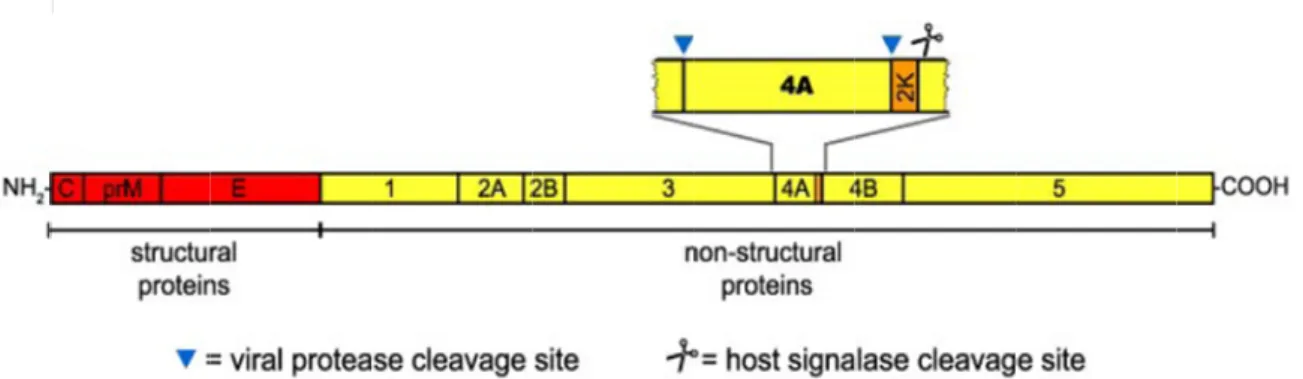
NS4A is a small hydrophobic protein about 16 kDa. The N-terminus of NS4A is generated in the cytoplasm by the viral two-component protease NS2B/NS3, whereas the C-terminal 23 amino acid residues of NS4A seem to act as a signal sequence for the translocation of NS4B into the lumen of the ER (Fig. 1.6). This signal sequence (designated the 2K fragment) is removed from the N terminus of NS4B by the host signalase in the ER lumen. In a previous study, KUNV NS4A-2K precursor can induce intracellular membrane rearrangements, which may form the scaffold for the viral replication complex (Roosendaal et al., 2006).
The observations that KUNV NS4A localizes to the presumed sites of RNA replication and polyprotein processing (Mackenzie et al., 1998) and that an interaction between NS4A and NS1 is required for RNA replication (Lindenbach et al., 1999) suggest that flavivirus
NS4A was involved in some steps of viral RNA amplification.
Recently, it was reported that DV NS4A associates with membranes via 4 internal hydrophobic regions, which are all able to mediate membrane targeting of a cytosolic reporter protein. It was also developed a model for the membrane topology of NS4A in which the N-terminal third of NS4A localizes to the cytoplasm, while the remaining part contains three transmembrane segments, with the C-terminal end localized in the ER lumen (Fig. 1.7). Subcellular localization experiments in DV-infected cells revealed that NS4A resides primarily in ER-derived cytoplasmic that also contain double-strained RNA (dsRNA) and other DV proteins, suggesting that NS4A is a component of membrane-bound viral replication complex. Besides, the individual expression of DV NS4A lacking the 2K fragment resulted in the induction of cytoplasmic membrane alternations resembling virus-induced structures, whereas expression of full-length NS4A does not. Thus, proteolytic removal of the 2K peptide appears to be important for induction of membrane alternations that may harbor the viral replication complex (Miller et al., 2007). However, the small hydrophobic protein NS4A is poorly characterized, and its proper function for viral replication cycle is still unknown.
1.8 Properties of NS4B
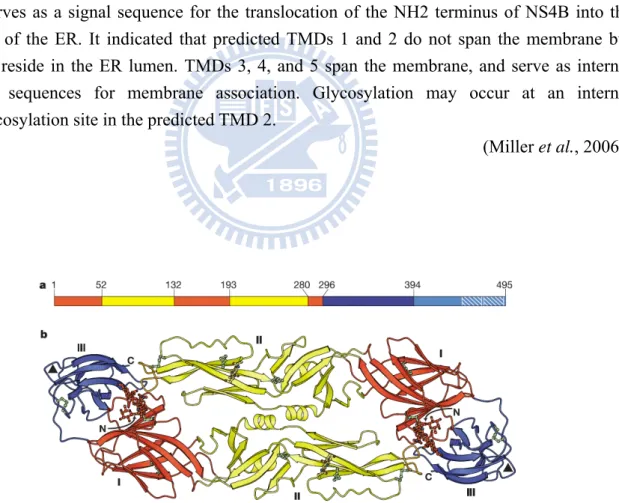
NS4B is the largest of the four small hydrophobic nonstructural proteins and comprises 248 amino acid residues. In DV2-infected cells, NS4B is first produced as a peptide of apparent size of 30 kDa; NS4B is then post-translationally modified, in an unknown way, to produce a polypeptide of apparent size 28 of kDa. The modification of NS4B is found to be cell-dependent and most likely mediated by a cellular enzyme (Preugschat et al., 1991). NS4B proteins of DV serotypes share 78-85 % amino acid sequence identity, whereas those of YFV, WNV and DV share 35 % identity. Hepatitis C virus (HCV) NS4B has only negligible sequence identity to the flavivirus NS4B proteins. Despite this divergence, the topology of NS4B, containing several ER and cytoplasmic domains separated by transmembrane regions
(Fig. 1.8; Miller et al., 2006), is strikingly similar among members of the flaviviridae, suggesting a conserved function of NS4B in the viral life cycle (Lundin et al., 2003).
NS4B of the DV, the WNV, and also the YFV was recently identified as an inhibitor of the IFN-α/β response. Expression of DV NS4B blocks the IFN-α/β-induced signal transduction cascade by interfering with STAT1 (transducer and activator of transcription) phosphorylation. Deletion analyses suggest that the first 125 amino acids of DV 2K-NS4B are sufficient for the inhibition of IFN-α/β signaling, and that proper viral polyprotein processing is required for anti-IFN function (Munoz-Jordan et al., 2005). Likewise NS4A and, to a lesser extent NS2A, appear to block IFN signaling, and the cumulative effect of the three proteins results in robust inhibition of IFN signaling (Munoz-Jordan et al., 2003, 2005).
A number of publications have described mutations in the NS4B protein in attenuated or passage-adapted mosquito-born flaviviruses suggesting this protein plays a vital role in replication and pathogenesis. A single coding mutation (P101L) in DV4 NS4B conferred a small-plaque phenotype in C6/36 cells while at the same time increasing plaque size in Vero cells two-fold and Huh7 cells three-fold. The opposing effects of the NS4B P101L mutation in mosquito and vertebrate systems suggest that the NS4B protein is involved in maintaining the balance between efficient replication in the mosquito vector and the human host (Hanley et al., 2003).
Miller et al. found that NS4B colocalizes with NS3 and double-stranded RNA (dsRNA), an intermediate of viral replication, arguing that NS4B is part of the membrane-bound viral replication complex (Miller et al., 2006). Besides, an interaction between the DV NS4B and NS3 was identified by using a yeast two-hybrid assay and validated in pull-down and immunoprecipitation studies. Furthermore, recombinant NS4B dissociated single-strained RNA (ssRNA) from NS3 and consequently enhanced the overall helicase activity of NS3 in an in vitro unwinding assay. It suggests that NS4B modulates DV replication via its interaction with NS3 (Umareddy et al., 2006). However, the actual role of NS4B in dengue
replication cycle is not clear.
1.9 Properties of E protein
The DV envelope (E) protein is 495 amino acids in length, forms oligomers, and along with the M protein, constitutes most of the accessible virion surface that is covered by the envelope membrane. The DV E protein mediates host cell binding and is essential for infection via a conformation-induced membrane fusion event between the host cell and the virion. In addition, it is also the primary antigen that induces protective immunity and the major antigen for virus neutralization (Monath et al., 1996).
The X-ray crystallographic structures of ectodomains of TBEV E and DV2 E proteins have been determined (Modis et al, 2003, 2004). Each monomer of the crystallized dimeric E protein contains three domains (Fig. 1.9): domain I is a centrally located β barrel, domain II contains a dimerization region and fusion peptide, and domain III harbors the receptor-binding activity.
The glycosylated E protein mediates both receptor binding and fusion activities (Anderson
et al., 1992; Guirakhoo et al., 1991). Infectious entry occurs within the endosome, following
uptake of intact virus particles through receptor-mediated endocytosis. Protonation of histidines in the low-pH environment of the endosome is the likely trigger for the conformational change that leads to the reversible dissociation of virion surface E dimmers into monomers, followed by an irreversible transition to homotrimers (Modis et al, 2004; Kampmann et al., 2006). A hydrophobic stretch of amino acids, referred to as the “fusion peptide”, is exposed during this conformational change and interacts with the target membrane, initiating the fusion process and ultimately the merging of the two bilayers (Heinz
et al., 2001; Yennamalli et al., 2009).
Furthermore, crystallization of a DV2 E protein fragment in the presence of the detergent β-N-octylglucoside (β-OG) has led to structures with and without occupation of the hinge
pocket by β-OG. Occupation of the pocket by β-OG requires an altered conformation of the kl loop toward an “open” position, and the angle that domain II makes with domain I-domain III also differs between two crystal structures with and without β-OG. The β-OG pocket (Fig. 1.10) may therefore represent an ideal target for structure-based design of potential antiviral agents because ligands that bind there could alter the conformational equilibrium associated with the hinge angle and inhibit virus entry (Modis et al., 2003, 2004).
1.10 The β-OG pocket of E as a target
Flavivirus E proteins have been shown to play a pivotal role in virus assembly, morphogenesis, and infection of host cells. A crystal structure of the soluble ectodomain of E from DV2 reveals a hydrophobic pocket, the β-OG pocket, lined by residues that influence the pH threshold for fusion. The β-OG pocket, which accepts a hydrophobic ligand, opens and closes through a conformational shift in a β-hairpin at the interface between domain I and domain II. Therefore, the opening up of the β-OG pocket just at the locus of a likely hinge suggests that compounds inserted at this position might hinder further conformational change and hence inhibit the fusion transition (Modis et al., 2003). It opens up a new avenue for identifying antiviral agents against early steps of dengue virus infection.
Recently, this pocket has been targeted using virtual screening, and tetracycline derivatives and the other small molecules with antiviral inhibitory activity have been identified (Yang et al., 2007; Zhou et al., 2008; Wang et al., 2009). First, Yang et al. has identified the tetracyclic ring structure compounds, tetracycline derivatives with the help of a molecular docking platform using the detergent ligand (β-OG) binding site on the DV2 E protein as target and performing virtual screening with comprehensive medical chemistry database (Yang et al., 2007). Among the tetracycline derivatives, rolitetracycline and doxycycline displayed significant inhibitory effects on the propagation of DV2 PL046 strain in cell cultures. Next, Zhou et al. used a hierarchical four-stage computational HTS to identify
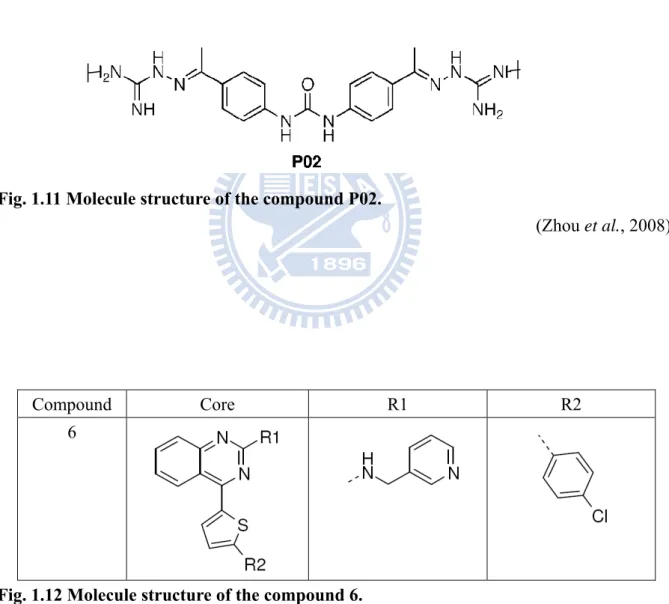
small-molecule compounds that bind to the β-OG pocket of the E protein of DV. Among the 23 top-ranked compounds, which were tested for antiviral activity in biological assays, P02 (Fig. 1.11) was demonstrated both to bind E protein and to have antiviral activity. Moreover, P02 was shown by STD NMR to compete with β-OG for binding to E protein, which unambiguously demonstrates association of P02 with E protein and supports a binding site that overlaps the β-OG pocket targeted by computational screening (Zhou et al., 2008). This year, Wang et al. found a small compound, labeled compound 6 (Fig. 1.12), which was identified as one of the inhibitors with an average EC50 of 119 nanomolar against DV2 in a human cell line. Mechanism of action studies demonstrate that compound 6 acts at early stage during DV infection. It arrests DV in vesicles co-localizing with endocytosed dextran, and inhibits NS3 expression (Wang et al., 2009).
In addition to DV, arrays of 23 compounds were selected for antiviral testing against YFV replication. Bioassay results identified a class of thiazole compounds with antiviral potency in cell-based assays. Modification of these lead compounds led to a series of analogues with improved antiviral activity and decreased cytotoxicity (Li et al., 2008).
Furthermore, the pre-fusion and post-fusion structures of DV E protein were analyzed to identify potential novel sites that could bind small molecules, which could interfere with the conformational transitions that mediate the fusion process. Seven structurally diverse molecules were selected to test experimentally for inhibition of DV propagation. The best compound showed an IC50 in the micromolar range against DV2 (Yennamalli et al., 2009).
1.11 The overview of the experimental design
(I) Functional expression of nonstructural proteins NS2A, NS2B, NS4A, and NS4B of dengue virus type 2 PL046 strain.
Since the functions of these four small nonstructural proteins regarding to dengue viral replication have not been well-studied, it was decided to construct expression clones of
individual genes for protein production, functional study, subunit vaccine development, and, for the construction of DNA vaccines in the future.
Here, I started with constructing the expression clones of these four nonstructural proteins from DV2 PL046 strain with the enhanced green fluorescence protein gene (EGFP) as tags. Then, I expressed the four nonstructural proteins in mammalian cells, BHK-21, to select for the stable cell lines. Finally, I have initiated the functional study with plaque assay.
(II) Biological assay of inhibitor candidates to dengue virus type 2 and dengue virus type 3.
The incidence of dengue fever epidemics has increased dramatically over the last few decades. However, no commercial vaccine or antiviral therapy is available. Therefore, a need for the search of safe and effective antiviral drugs becomes imperative. Entry of DV into a host cell is mediated by its major envelope protein, E. The crystal structure of the ectodomain of DV2 revealed a hydrophobic pocket, the β-OG pocket, which was proposed as a suitable target site for small-molecule inhibitors of the fusion process. Previously, research in the lab has identified certain tetracyclic ring structure compounds, tetracycline derivatives as inhibitor candidates, targeting the detergent ligand (β-OG) binding site on the DV2 E protein from comprehensive medical chemistry database.
In this study, I attempted to assess (1) whether other tetracycline derivatives and other ringed compounds have inhibitory effect on DV and (2) whether the sequence variations of E protein between DV2 and DV3 affect the specificity of the candidate compounds. Therefore, I performed plaque formation assay using the compounds with different concentrations against DV2 and DV3 separately.
Chapter 2 Materials and Methods
2.1 Materials
2.1.1 Virus
Dengue virus type 2 PL046 strain (Taiwan local strain) Dengue virus type 3 H87 strain
2.1.2 Cell lines
BHK-21 (baby hamster kidney cell) C6/36 (Aedes albopictus larva cell)
2.1.3 Bacterial strains
Escherichia coli DH5α strain: for general cloning (Invitrogen)
2.1.4 Plasmids
Plasmid Description Reference
pcDNA3.0 T7 promoter and CMV promoter. (selection marker: Ampicillin)
(Appendix 1) Invitrogen pNS2A-HAHis pNS2B-HAHis pNS4A-HAHis pcDNA3-D24B-HAHis
BamHI-XbaI fragment containing
2A,2B, 4A, and 4B with C-terminal HA and His tag in pcDNA3
(Appendix 2) 徐婕琳,2003, 楊馥嘉,2006 交大碩士論文; Yang laboratory collection
pNS2A-EGFP pNS2B-EGFP pNS4A-EGFP pcDNA3-D24B-EGFP
BamHI-XbaI fragment containing
2A,2B, 4A, and 4B with C-terminal EGFP tag in pcDNA3
This study
pEGFP-N2
pEGFP-N2 encodes a red-shifted variant of wild-type GFP which has been optimized for brighter fluorescence and
higher expression in mammalian cells.
Clontech
pNS2B-EGFP(pro) pNS4A-EGFP(pro) pcDNA3-D24B-EGFP(pro)
BamHI-XbaI fragment containing
2A,2B, 4A, and 4B with C-terminal EGFP tag in pcDNA3 (EGFP tag
without ATG)
This study
2.1.5 Primers
Primer Primer sequence (5’ to 3’)
Position in Dengue genome* 2AE-F ATGGGACATGGGCAGATTGACAACTTCTCA 3478-3504 2BE-F GAGCTGGCCACTAAATGAGGCTAT 4131-4154 4AE-F CCCTGACCCTGAACCTAATCACA 6377-6399 4BE-F TCCATGAACGAGATGGGTTTCCTGGA 6826-6845 4BE-FP GAACGAGATGGGTTTCCTGGAA 6826-6846
*According to the sequence of Dengue virus type 2, PL046 strain (GenBank: AJ968413.1) Boldface: the genes on the dengue virus genome; underline: start codon or restriction site.
start codon
Primer Primer sequence (5’ to 3’) Position in EGFP genome* 2AE-R TGTACAGCTCGTCCATGCCGAGAGTGATC 1369-1397 EGFP-F CCGCTCGAGATGGTGAGCAAGGG 683-696 EGFP-R GCTCTAGATTACTTGTACAGCTCGTCCATG 1381-1402 EGFP-F2 CCGCTCGAGGTGAGCAAGGGCG 686-698 EGFP-R2 GCTCTAGATTACTTGTACAGCTCGTCCATGCCG 1378-1402
*According to the sequence of cloning vector pEGFP-N2 with enhanced green fluorescent protein gene (GenBank: U57608.1). Boldface: the genes on the dengue virus genome; underline: start codon or restriction site.
2.1.6 Chemicals, enzymes and reagents
Chemical Source Catalog
number
Application
1 kb DNA ladder SibEnzyme SEM11C001 DNA
electrophoresis
2-propanol Sigma I 9516 RNA extraction
9-amino-1,2,3,4-tetrahydr oacridine hydrochloride
hydrate
Sigma A79922 Plaque assay
Acetic acid Fluka 33209 Western Blot
Acryl/Bis 37.5:1 solution AMRESCO 0245 Western Blot
Agarose VEGONIA 9201-05 DNA/RNA
electrophoresis XbaI
XbaI
XhoI XhoI
Ampicillin Applichem A0839 Bacterial culture
APS Bio-Rad 161-0700 Western Blot
B-mercaptoethanol MERCK 1.1543.0100 Reducing reagent
Berberine Sigma B3251 Plaque assay
Chloroform Riedel-de Haën
32211 Genomic DNA
extraction
Chlortetracycline Sigma C-4881 Plaque assay
Coomassie Brilliant Blue R-250
J.T.Baker F792-01 Protein-staining
Crystal Violet Sigma C-3886 Cell staining
DEPC Sigma D 5758 RNase inactivation
for RT-PCR
DMSO Sigma D-8418 Freeze cells
DNA Polymerase I, Large (Klenow) Fragment
BioLabs M0210S Klenow fill in
Doxycycline Sigam D-9891 Plaque assay
ECL substrate PIERCE 34079 Western Blot
EDTA AMRESCO 0105 TE buffer
EtBr Sigma E-7637 DNA/RNA staining
Fetal Bovine Serum Biological industries
04-001-1A Cell culture
Formaldehyde Riedel-de Haën
33220 Cell fixation
G418 (GENETICIN) GIBCO 11811-031 Cell culture
plaque assay
LB agar Alpha
Biosciences
L12-111 Bacterial medium
LB broth Scharlau 02-385 Bacterial medium
Lipofectamine 2000 Invitrogen 11668-019 Transfection
MEM GIBCO 41500-034 Cell culture
medium
Methanol Mallinckrodt 3016-08 Western Blot
Methylcellulose Sigma M 0512 Plaque assay
NaCl AMRESCO 0241 Buffer
NaHCO3 Sigma S-5761 Cell culture
NaOH Riedel-de Haën 30620 Buffer Nitrocellulose Transfer Membrane Schleicher & Schuell 10401396 Western Blot
Nonfat powdered milk New Zealand Milk Brands
Ltd
EMB 53048-D Western Blot
Oxytetracycline Sigma O-5750 Plaque assay
Phenol saturated solution Amresco 0945 Genomic DNA
extraction
PMSF Fluka 78830 Protein inhibitor
Protein marker Fermentas SM0671 SDS-PAGE
Restriction enzyme TakaRa, BioLabs
--- Plasmid construction
Rolitetracycline Sigma R-2253 Plaque assay SDS Riedel-de
Haën
62862 Western Blot
T4 DNA ligase Fermentas 1812 Plasmid
construction
TEMED Sigma T-9281 Western Blot
Tetracycline Sigma T-3258 Plaque assay
Tris (Base) AMRESCO 0826 Western Blot
Trypsin GIBCO 12605-010 Cell culture
Tween-20 Sigma P-1379 Western Blot
Urea Fluka SK-2644U Denature reagent
X-ray film Midwest
Scientific
LA7111 Western Blot
2.1.7 Antibodies
Antibody source Catalog number
Calnexin (H-70) (from rabbit)
Santa Cruz SC-11397
Alexa flour 594 goat anti-rabbit IgG
Invitrogen A11037
Goat Polyclonal anti-GFP, conjugated HRP
2.1.8 Kits
Kit Source Catalog
number
Application
ExcelPureTM Plasmid Miniprep Purification Kit
Premier N-PM050 Plasmid extraction
QIAGEN Plasmid Midi Kit QIAGEN 12143 Plasmid extraction
PCR Clean-up/Gel Extraction Kit
Premier N-DCE050 DNA purification
RNeasy Mini Kit QIAGEN 74104 RNA extraction
QIAshredderTM QIAGEN 79654 RNA extraction
SuperScript One-Step RT-PCR with Platinum taq
Invitrogen 10928-042 RT-PCR
2.1.9 Buffers
● 0.25% Coomassive blue stain solution
2.5 g Coomassive brilliant blue,50% methanol,10% acetic acid added ddH2O to 1000 ml ● 0.5% crystal violet solution (500 ml)
2.5 g crystal violet,25 ml 37% formaldehyde,250 ml EtOH,4.25 g NaCl ● 10X SDS-PAGE running buffer
0.25 M Tris base,1.92 M Glycin,1% SDS ● 10X transfer buffer
39 mM Glycin,48 mM Tris base,10% SDS, 20% methanol ● 1X PBS (pH 7.4)
137 mM NaCl,10 mM Na2HPO4,2.7 mM KCl,1.8 mM KH2PO4 ● 2X SDS-PAGE loading buffer
0.5% bromphenol blue,0.5 MTris-HCl (pH 6.8),10% SDS,100% glycerol ● 5% Blocking buffer
2.5 g nonfat powdered milk dissolved in 50 ml 1X TBS buffer ● 50X TAE buffer
48.4 g Tris base,0.5 M EDTA (pH 8.0) 20 ml,11.42 ml acetic acid added ddH2O to 200 ml
● Cell lysis buffer (RIPA buffer)
0.1% SDS,1% Triton X-100,1% NP-40,10 mM Tris-HCl (pH 7.4),1 mM MgCl2,1 mM PMSF
● Digestion buffer
100 mM NaCl,10 mM Tris-Cl pH8,25 mM EDTA pH8,0.5% SDS,0.1 mg/ml proteinase K
● TBS buffer (Tris-buffered saline) 10 mM Tris (pH 8.0),150 mM NaCl ● TBST buffer
10 mM Tris (pH 8.0),150 mM NaCl,0.05% Tween 20
2.1.10 Media
● LB (Luria-Bertani) / Ampicillin agar
1% tryptone,0.5% yeast extract,1% NaCl,1.5% agar,50 μg/ml Ampicillin ● LB (Luria-Bertani) / Kanamycin agar
1% tryptone,0.5% yeast extract,1% NaCl,1.5% agar,50 μg/ml Kanamycin ● LB broth
2.1.11 Equipments
● -20℃ low temperature refrigerator (WHITE-WESTINGHOUSE) ● -80℃ low temperature refrigerator 925/926 (FIRSTEK SCIENTIFIC) ● 4℃ refrigerator KS-101MS (MINI KINGCON)
● Auto dry box DX100 (Taiwan Dry Tech Co. Ltd) ● Bench top orbital shaker S101 (FiRSTEK) ● Centrifuge model 5100 (Kubota Corporation) ● CO2 incubator 5420-0NEW (NAPCO)
● Confocal (OLYMPUS, FV 500)
● Digital scale PB153-S (METTLER TOLEDO) ● Fluorescence microscope
● GeneQuant pro DNA/RNA calculator (AMERSHAM PHARMACIA BIOTECH) ● Gene CyclerTM (BIO-RAD)
● Hemacytomete (MARIEMFELD)
● Image system GEL DOC 2000 (BIO-RAD)
● Microcentrifuge MICRO 240A (DENVILLE SCIENTIFIC INC.) ● Mini-Protein electrophoresis cell 3 (BIO-RAD)
● Mini Trans-Blot Electrophoretic Transfer Cell (BIO-RAD) ● Orbital shaker IKA-VIBRAX-VXR
● pH meter F 360 (BACKMAN)
● SHORTER MINI Horizontal Gel electrophoresis Apparatus MJ-105 (MEDCLUB) ● Thermal Cycler PTC-100RT (MJ RESEARCH INC.)
● Vertical acrylamide electrophoresis unit (BIO-RAD) ● VORTEX-GENIE2 G560 (SCIENTIFIC INDUSTRICS) ● Water bath B206-T1 (FIRSTEK SCIENTIFIC)
2.2 Methods
2.2.1 Transformation of E. coli
2.2.1.1 Preparation of competent cells (for chemical method)
A single colony of E. coli was inoculated in 5 ml of LB broth and grew overnight at 37℃ with vigorous shaking (~180 rpm). 2 ml of the overnight culture was transferred into 100 ml LB broth (containing 5% glucose and 2 mM MgCl2) and was then incubated at 37℃ with shaking until the OD600 is between 0.4 and 0.7. The cultures were stored on ice for 20 minutes. The cells were recovered by centrifugation at 1620×g for 10 minutes and then resuspended in 50 ml ice-cold 0.1 M CaCl2. The cells were let stand on ice for 30 minutes and were pelleted by centrifugation at 720×g for 10 minutes at 4℃. The pellet was resuspended in 10 ml ice-cold 0.1 M CaCl2. The cells were incubated at 4℃ for 18 hours and were then recovered by centrifugation at 720×g for 10 minutes. The pellet was resuspended in 10 ml ice-cold 0.05 M CaCl2 (containing 15 % glycerol). The cells were dispensed as 100 μl per eppendorf tube and then were stored at -80℃.
2.2.1.2 Transformation
Stored competent cells were thawed on ice. 0.1~1 μg of plasmid DNA was mixed with 50 μl competent cells and was then set on ice for 30 minutes. The mixture was incubated in a preheated 42℃ circulation water bath for 1 minute and then on ice for 1 minute. Then 500 μl of LB broth was added to the cells. The culture was incubated at 37℃ with shaking (180 rpm) for 1 hour. 100 μl of the culture was plated on LB agar plate with 50 μg/ml ampicillin or kanamycin. The plate was set at room temperature until the liquid had been absorbed. The plate was inverted and was then incubated at 37℃ for 12~18 hours.
2.2.2 Plasmid DNA extraction
Plasmid DNA in E. coli was extracted with ExcelPureTM Plasmid Miniprep Purification
Kit (Premier, Cat. N-PM050). The procedure is as following:
A single colony of E. coli was inoculated in 5 ml of LB broth (with antibiotics) and grew overnight at 37℃ with vigorous shaking (180 rpm). The cells were recovered by centrifugation at 1037×g for 12 minutes and then resuspended in 200 μl Solution I buffer (Premier, Inc.; San Diego, USA). The E. coli solution was transferred to an eppendorf. 200 μl of Solution II buffer (Premier, Inc.; San Diego, USA) was added and mixed gently. 200 μl of Solution III buffer (Premier, Inc.; San Diego, USA) was added to the mixture and mixed gently again. Cells were spun at 16100×g for 5 minutes at room temperature (RT). The supernatant was transferred to Mini-MTM Column. The solution was centrifuged at 16100×g for 1 minute, and the filtrate in the collection tube was discarded. 700 μl of Washing solution (Premier, Inc.; San Diego, USA) was added in and the column was spun for 1 minute. This step was repeated once again. After the filtrate was discarded, the column was centrifuged at 16100×g for 3 minutes to remove residual ethanol. Mini-MTM Column was transferred to a new eppendorf and incubated at 60℃ for 5 minutes to evaporate the ethanol. Finally, DNA was eluted by 30-50 μl Elution Solution (Premier, Inc.; San Diego, USA) and centrifuged at 16100×g for 1 minute. Plasmid DNA was stored at -20℃.
2.2.3 Restriction enzyme digestion
0.5~1 μg of DNA was dissolved in appropriate volume of water and was digested with
restriction enzyme (following the commercial protocol). Generally, 1 μg DNA was digested with 1 unit of restriction enzyme in a 10 μl reaction at 37℃ for 1 hour or longer. The reaction was stopped by heat inactivation (65℃ for 10 minutes in general).
2.2.4 Cell culture
BHK-21 cells were grown in Minimum Essential Medium (MEM; Gibco) supplemented with 5% fetal bovine serum (FBS) and 0.22% NaHCO3. Cells were incubated in tissue culture incubator at 37℃ with 5% CO2. C6/36 cells were grown at 28℃ in MEM medium (Gibco) supplemented with 0.22% NaHCO3 and 10% FBS.
2.2.5 Transfection of mammalian cell
Cells were transfected with different expression plasmid with LipofectamineTM 2000
(Invitrogen, Cat. 11668-019) and appropriate amount of plasmids according to the manufacturer’s instructions. For example, in a 35 mm culture dish, 4 μg DNA was diluted in 250 μl serum-free medium and mixed gently. 10 μl LipofectamineTM 2000 was gently mixed with 250 μl serum-free medium and incubated for 5 minutes at RT. The diluted DNA was combined with diluted LipofectamineTM 2000 and incubated for 20 minutes at RT. The mixture was added to 90-95% confluent cells and incubated at 37℃ in CO2 incubator for 4-6 hours. The medium was replaced with fresh 5% FBS-MEM medium. Cells were incubated at 37℃ in CO2 incubator for 24 hours prior to the following assay.
2.2.6 Immunofluorescence and confocal microscopy.
BHK-21 cells were grown on coverslips, 24 hours after transfection, fixed in 4% paraformaldehyde for 30 minutes, followed by washing 2 times for 5 minutes in PBS, and permeabilized with 0.5% Triton X-100 for 10 minutes at room temperature. After the cells were washed as described above, the cells were blocked with 1% bovin serum albumin (BSA) at RT for 1 hour. Then cells were incubated with polyclonal anti-calnexin antibody (1:100) to stain the ER for 1 hour, and then washed 3 times for 5 minutes. Goat anti-rabbit IgG secondary antibody (1:1000) incubation was performed for 1 hour in the dark, and the cells were then washed 3 times for 5 minutes each. Subsequently, coverslips were mounted on
glass slides and the cells were analyzed by confocal laser scanning microscopy(OLYMPUS, FV 500).
2.2.7 Selection of stable transfected cells
24 hours after transfection, transfected BHK-21 cells were passaged down by trypsin. A
culture of 1/500 cells were seeded in 6 cm dishes with MEM medium containing 5% FBS and 800 μg/ml G418 for ten days. Then the single colonies were selected to 24 well dishes with MEM medium containing 5% FBS and 800 μg/ml G418 and the medium was changed every four days until the cells were confluent. After that, the cells were passaged to 6 cm dishes for amplification with 500 μg/ml G418. The cell colonies were selected for other experiments.
2.2.8 Preparation of proteins from mammalian cells
The transfectants, 24 hours after transfection, or the stable transfected cells were lysed
with 100~200 μl RIPA buffer (containing 1 mM PMSF) at RT. The lysates were sedimented by centrifugation at 12000×g for 5 minutes at 4℃. The supernatant and pellet were respectively mixed with 1 volumn 2X SDS-PAGE loading dye (containing 8 M urea, 200 mM DTT, and 200 mM β-mercaptoethanol) and analyzed by Western blotting.
2.2.9 Western blot analysis
Mammalian cells expressed proteins were separated by SDS-PAGE (12% gel) using a
Mini-Protein electrophoresis cell 3 (Bio-Rad) and electrophoretically transferred to Nitrocellulose membrane (Schleicher&Schuell) using Mini Trans-Blot Electrophoretic Transfer Cell (Bio-Rad) at 0.09A for 37 minutes. The membrane was blocked for 12-14 hours at 4℃ with 5% nonfat powdered milk (New Zealand Milk Brands Ltd) in TBS buffer. Then, the membrane was incubated with Goat Polyclonal anti-GFP, conjugated HRP for 1 hour at RT and washed in PBST 3 times for 5 minutes. Finally, the blots were developed by ECL and
then exposed to an X-ray film.
2.2.10 Plaque formation of stable cell lines
MEM without FBS was mixed with virions of DV2 PL046 strain in the amount of
200-250 PFU/well. The mixtures were mixed gently and added onto the stable transfected BHK-21 cells in 35mm dishes and then incubated at 37℃ with 5% CO2 for 1 hour. MEM containing 5% FBS and 1.1% methylcellulose were added to the well and incubated at 37℃ with 5% CO2 for 5-7 days. The medium was discarded before the cells were fixed with 3.7% formaldehyde. After 30 minutes, the solution was removed and the cells were stained with 0.5% crystal violet in 3.7% formaldehyde. The plated were washed with water before the plaque numbers were scored.
2.2.11 Statistical analysis
Analysis of variance (ANOVA) of the data was evaluated by the Statistics Package for Social Science (SPSS 10.0). Scheffe test was employed to determine the statistical significance of the differences between the means (P<0.05).
2.2.12 Preparation of genomic DNA from mammalian tissue
The stable transfected cells of BHK-21 were trypsinized, and collected cells from the flask. The cells were then centrifuged at 500×g for 5 minutes and the supernatant was discarded. The cell pellet was resuspended with 1 to 10 ml ice-cold PBS then centrifuged for 5 minutes at 500×g and the supernatant was discarded. This step was then repeated once. Then, 0.3 ml digestion buffer was added to resuspend cells and the samples were incubated with shaking at 50℃ for 12-18 hours in tightly capped tubes. Then samples were extracted by adding an equal volume of phenol/chloroform/isoamyl alcohol and then centrifuged for 10 minutes at 1080×g at 4℃. The aqueous (top) layer were transferred to a new tube and 1/2 volume of 7.5 M
ammonium acetate and 2 (original) volume of 100% ethanol were added. The DNA should immediately form a stringy precipitate and recovered by centrifugation at 16100×g 4℃ for 10 minutes. The pellet was rinsed with 70% ethanol and centrifuged at 16100×g for 10 minutes to decant ethanol and air dry. Finally, DNA was resuspended with autoclaved water until dissolved. It can be shaken gently at room temperature or at 65℃ for several hours to facilitate solubilization. The extracted genomic DNA was stored at -20℃.
2.2.13 RNA extraction
RNA in mammalian cells was extracted with RNeasy Mini Kit (Qiagen). The procedure is as following:
After the media were removed, the stable transfected cells were lysed directly in the cell culture dish (10 cm diameter) by adding 350μl Buffer RLT (QIAGEN) to the cell culture dish and then pipetting the lysate well into a microcentrifuge tube to ensure that no cell clumps are visible. To homogenize the lysate, it was pipetted directly into a QIAshredder spin column (QIAGEN) placed in a 2 ml collection tube (QIAGEN). Then, the whole set up was centrifuged for 2 minutes at 16100×g. 350μl (1 volume) 70% ethanol was added to the homogenized lysate, and it was mixed well by pipetting. The samples were transferred, including any precipitate that may be formed, to an RNeasy spin column (QIAGEN) placed in a 2 ml collection tube (QIAGEN) and centrifuged for 30 seconds at ≧8000×g. Then the flow through was discarded. 350μl buffer RW1 (QIAGEN) was added to the RNeasy spin column, and the whole set up was centrifuged for 30 seconds at ≧8000×g to wash the spin column membrane and then the flow through was discarded. 80μl DNase I incubation mix (10μl DNase I stock and 70μl buffer RDD, QIAGEN) was added directly to the RNeasy spin column membrane, and the whole set up was placed on the benchtop (20-30℃) for 15 minutes. 350μl buffer RW1 was added to the RNeasy spin column and the whole set up was centrifuged for 30 seconds at ≧8000×g to wash the spin column membrane. 500μl buffer
RPE was added to the RNeasy spin column, which was centrifuged for 30 seconds at ≧ 8000×g to wash the spin column. 500μl buffer RPE was added to the RNeasy spin column, which was centrifuged for 2 minutes at ≧ 8000×g to wash the spin column. The RNeasy spin column was then transferred to a new 2 ml collection to centrifuge at 16100×g for 1 minute. Finally, the RNeasy spin column was placed in a new 1.5 ml collection tube and 30-50 μ l RNase-free water was added directly to the spin column membrane for centrifugation 1 minute at ≧ 8000×g to elute the RNA. The extracted RNA was stored at -80℃.
2.2.14 Semi-quantitative RT-PCR-SuperscriptTM One-Step RT-PCR
The SuperScript™ One-Step RT-PCR with PlatinumR Taq System is designed for the
convenient, sensitive, and reproducible detection and analysis of RNA molecules by RT-PCR. Components for both cDNA synthesis and PCR are combined in a single tube, using gene-specific primers and target RNAs from either total RNA or mRNA. Reverse transcription automatically follows PCR cycling without additional steps (Invitrogen).
The extracted RNA was performed with SuperScripTM One-Step RT-PCR, the procedure is as following:
The template RNA and components, including 2X reaction mix, sense primers, anti-sense primers, RT/platinum Taq mix, and autoclaved distilled water were prepared as depicted in table 1 (Invitrogen) and added to the microcentrifuge tubes on the ice. The mixture was gently mixed to make sure that all the components are at the bottom of the amplification tube. The mixture was then centrifuged briefly if needed. The thermal cycle was performed according to the program in table 2 (Invitrogen Cat. 10928-042).
2.2.15 Amplification of Dengue virus
MEM medium with 10% FBS. Infection of C6/36 cells with DV2 PL046 strain or DV3 H87 strain was carried out at multiplicity of infection 0.1 (M.O.I.) in 10% FBS MEM medium at 37℃ in 5% CO2 for 2 hours and then cultured in 28℃ in the absence of CO2. After incubating at 28℃ for 4-5 days, cell culture supernatant was collected after clarification of cell debris, and stored at -80℃.
2.2.16 Plaque formation assay for the inhibitory effects of compounds in DV2 and DV3 propagation
Mammalian BHK-21 host cells were cultured at 37℃ with 5% CO2 in MEM medium (Gibco) supplemented with 0.22% sodium bicarbonate and 5% fetal bovine serum (FBS) (Gibco). BHK-21 cells were plated at a density of 3 × 105 cells per well in 6-well plates and incubated at 37℃ with 5% CO2 for 24 hours. Different dilutions of the compounds were added to the 6-well plates incubated at 37℃ with 5% CO2 for 1 hour, following by 0.5 ml of medium containing 200 PFUs of the DV2 PL046 strain or DV3 H87 strain per well for 1 hour incubated at 37℃ with 5% CO2. Then a 1:1 mixture of 10% MEM medium: 2.2% methylcellulose was added to the cultures. The cultures were then incubated at 37℃ with 5% CO2 for 6~7 days. The medium was aspirated prior to fixation of the cells with 3.7% formaldehyde for 30 minutes. Then, the fixing solution was removed and the cells were stained with 1% crystal violet in 3.7% formaldehyde. Finally, the plates were washed with water prior to scoring of plaques.
Chapter 3 Functional expression of nonstructural proteins NS2A, NS2B,
NS4A, and NS4B of dengue virus type 2 PL046 strain
(I) Results
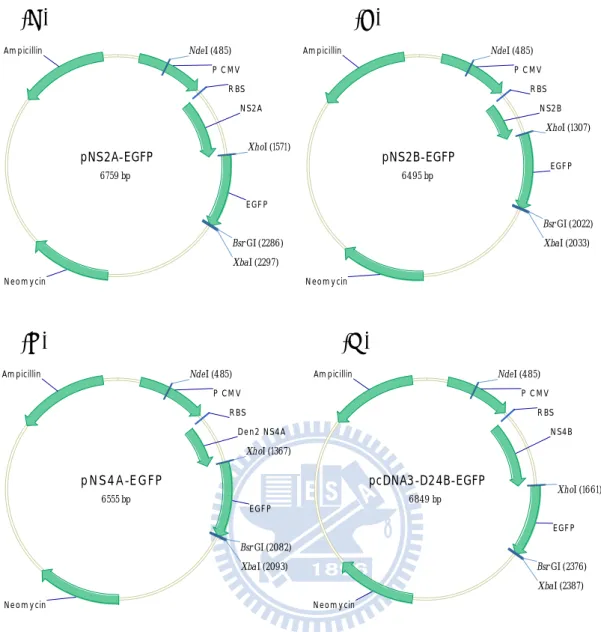
3.1 Construction of pNS2A-EGFP, pNS2B-EGFP, pNS4A-EGFP, and pcDNA3-D24B-EGFP expression plasmids
pEGFP-N2 (Clontech) encodes a red-shifted variant of wild-type GFP (Prasher et al.,
1992; Chalfie et al., 1994; Inouye et al., 1994) which has been optimized for brighter fluorescence and higher expression in mammalian cells ( Fig. 3.1). The EGFP fragment was obtained by polymerase chain reaction (PCR) amplification with primers EGFP-F and EGFP-R on pEGFP-N2 as the template. The contents of the PCR reaction mixture are listed in table 3 and the condition of the PCR reaction was performed with the program described in table 4. The PCR products were gel-purified after being digested with restriction enzyme XhoI at the 5’-end and XbaI at the 3’-end. pNS2A-HAHis, pNS2B-HAHis, pNS4A-HAHis (徐婕琳, 2003, 交大碩士論文), and pcDNA-D24B-HAHis (楊馥嘉, 2006, 交大碩士論文) were also treated with the same restriction enzyme digestion to remove the HA-His tag and then to be gel purified. The XhoI-XbaI fragment containing EGFP was ligated to the XhoI-XbaI vector fragments of pNS2A-HAHis, pNS2B-HAHis, pNS4A-HAHis, and pcDNA3-D24B-HAHis (pcDNA3 based). Thus the four nonstructural genes in pcDNA3 carried the C-terminal EGFP tag and the new constructs were named pNS2A-EGFP, pNS2B-EGFP, pNS4A-EGFP, and pcDNA3-D24B-EGFP (Fig. 3.2 A, B, C, and D), respectively. These constructs were assessed by BsrGI and NdeI digestions. pNS2A-EGFP was digested into 1.8 kb and 5 kb fragments ( Fig. 3.3). pNS2B-EGFP was digested into 1.5 kb and 5 kb fragments ( Fig. 3.3). pNS4A-EGFP was digested into 1.6 kb and 5 kb fragments ( Fig. 3.3). pcDNA3-D24B-EGFP was digested into 1.9 kb and 5 kb fragments ( Fig. 3.3).
3.2 Expression and subcellular localization of pNS2A-EGFP, pNS2B-EGFP, pNS4A-EGFP, and pcDNA3-D24B-EGFP in mammalian cells, BHK-21 3.2.1 Confocal fluorescence microscope analysis
pNS2A-EGFP, pNS2B-EGFP, pNS4A-EGFP, pcDNA3-D24B-EGFP, and the control
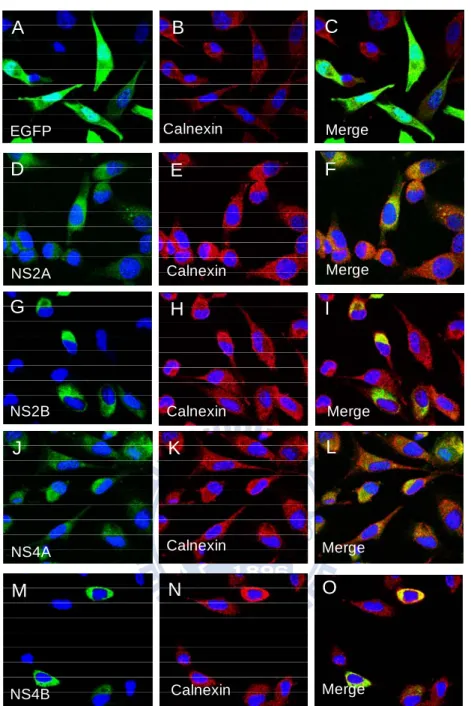
pEGFP-N2 were transfected into BHK-21 to investigate the expression of the recombinant proteins by IF assay (immunofluorescence assay) and confocal fluorescence microscopy. Those four constructs all exhibited the green fluorescence through the C-terminal EGFP tag (enhanced green fluorescence protein) as shown in Fig. 3.4 (D, G, J, and M), which confirmed the expression of NS2A-EGFP, NS2B-EGFP, NS4A-EGFP, and NS4B-EGFP.
Additionally, a colocalization study with the cellular marker protein calnexin, a marker for the reticular subdomain of rough ER was performed. And the green fluorescence of the four proteins displayed at the same location as the red fluorescence, from calnexin, in cytoplasm (Fig. 3.4 E, H, K, and N). While the EGFP displayed the green fluorescence around whole cells (Fig. 3.4 A.). The merged were shown in Fig. 3.4 (C, F, I, L, and O).
3.2.2 Assay of the transient expression of pNS2A-EGFP, pNS2B-EGFP,
pNS4A-EGFP, and pcDNA3-D24B-EGFP in BHK-21 by Western blot analysis
Expression of four nonstructural protein constructs, pNS2A-EGFP, pNS2B-EGFP, pNS4A-EGFP, and pcDNA3-D24B-EGFP were driven by CMV immediate-early gene promoter on vector pcDNA3 in BHK-21 cells. BHK-21 cells transfected with these four plasmids separately were harvested 24 hours posttransfection and were lysed by RIPA buffer. The cell lysates were analyzed by SDS-PAGE with Coomassie blue staining and Western blot with antibody against the C-terminal EGFP tag (Fig. 3.5). The predicted molecular weights of NS2A-EGFP, NS2B-EGFP, NS4A-EGFP, and NS4B-EGFP are 50.9, 41.2, 43.7, 54 kDa, respectively. Compared with cells transfected with vector only, there was no extra band on the Coomassie blue stained gel (Fig. 3.5 A). Next, the blots were examined with Western blot
analysis. For cells transfected with pNS2A-EGFP, the dominant band was between 34 and 26 kDa which was the same as EGFP positive control (Fig. 3.5 B b). There was also a band at about 43 kDa, which was a little smaller than the predicted 50.9 kDa by Western blot (Fig. 3.5 B a).For cells transfected with pNS2B-EGFP, there were two bands at about 43 kDa (Fig. 3.5 B c). For cells transfected with pNS4A-EGFP, a band was detected at about 43 kDa, which was as expected (Fig. 3.5 B d ) but the dominant band was between 34 and 26 kDa which was the same as EGFP control (Fig. 3.5 B e). As for NS4B, there was a band at about 55 kDa, as expected (Fig. 3.5 B f).
3.3 Selection of stable transfected cells of pNS2A-EGFP, pNS2B-EGFP, pNS4A-EGFP, pcDNA3-D24B-EGFP, and pEGFP-N2
3.3.1 Fluorescence microscopy and Western blot analysis of the selected stable cell
lines
pNS2A-EGFP, pNS2B-EGFP, pNS4A-EGFP, pcDNA3-D24B-EGFP, and pEGFP-N2 were transfected into BHK-21 for stable clone selection by G418. The selected colonies were analyzed by Western blot with anti-GFP-HRP antibody against the C-terminal EGFP tag and were observed by fluorescence microscope.
There were fluorescence signals in the selected cells including EGFP, NS2A-EGFP, NS2B-EGFP, NS4A-EGFP, and NS4B-EGFP (Fig. 3.6 B, 3.7 B, 3.8 B, 3.9 B, and 3.10 B). The predicted molecular weights of EGFP, NS2A-EGFP, NS2B-EGFP, NS4A-EGFP, and NS4B-EGFP are 27, 50.9, 41.2, 43.7, and 54 kDa, respectively. For all the selected colonies, only the pellets and the supernatants of EGFP had the expected bands at 27 kDa (Fig. 3.6 C and D), and the NS2A-EGFP had the bands at about 43 kDa (Fig. 3.7 C) in the pellet and the supernatant, which was a little smaller than the expected. The others all had the unexpected bands at 27 kDa just as the molecular weight of EGFP (Fig. 3.8 C, 3.9 C, and 3.10 C).
3.3.2 PCR analysis on the genomic DNA of the selected stable cell lines of NS2A-EGFP, NS2B-EGFP, NS4A-EGFP, and NS4B-EGFP
Because of the bands in Western blot of NS2A-EGFP, NS2B-EGFP, NS4A-EGFP, and NS4B-EGFP were smaller than expected, I decided to check the constructs by PCR amplification of the genomic DNA extracted from the stable transfected cell lines.
The PCR amplification of the genomic DNA of the stable cell line NS2A-EGFP was performed with primers 2AE-F and 2AE-R to obtain a band of 1.4 kb, which was consistent with the expected 1377 bp (indicated by a in Fig. 3.11 A). The negative control was the stable transfected cells of pcDNA3 and the positive control was the pNS2A-EGFP plasmid, indicated by b in Fig. 3.11 A. The PCR amplification of the genomic DNA of the stable cell line, NS2B-EGFP, was performed with primers 2BE-F and 2AE-R to obtain a band of 1.1 kb, which is consistent with the expected 1111 bp (indicated by c in Fig. 3.11 B). The negative control was the stable transfected cells of pcDNA3 and the positive control was the pNS2B-EGFP plasmid, indicated by d in Fig. 3.11 B. For NS4A-EGFP genomic DNA, the PCR primers were 4AE-F and 2AE-R, and a fragment of 1169 bp was obtained, which was as expected (indicated by e in Fig. 3.11 C) when compared with the negative control (stable transfected cells of pcDNA3) and the positive control (pNS4A-EGFP plasmid, indicated f in Fig. 3.11 C). As for NS4B-EGFP, the PCR amplification was with primers 4BE-F and 2AE-R. But no band was detected when compared with the negative control (stable transfected cells of pcDNA3) and the positive control (pcDNA3-D24B-EGFP plasmid, indicated by g in Fig. 3.11 D).
3.3.3 RNA expression of the stable cell lines of 2A-EGFP, 2B-EGFP, 4A-EGFP, and 4B-EGFP
The total RNAs were extracted from selected stable cell lines of NS2A-EGFP, NS2B-EGFP, NS4A-EGFP, NS4B-EGFP, and pcDNA3 by RNeasy Mini Kit. The extracted
RNAs were used for performing superscript one-step RT-PCR with individual primers. For NS2A-EGFP, the primers were 2AE-F and 2AE-R and a fragment of 1.4 kb was obtained, which was as the expected 1377 bp (indicated by a in Fig. 3.12) when compared to the negative control (stable transfected cells pcDNA3, Fig. 3.12 lane 4). For NS2B-EGFP, the primers were 2BE-F and 2AE-R and a fragment of 1.1 kb was obtained, which was as the expected 1111 bp (indicated by b in Fig. 3.12) when compared to the negative control (stable transfected cells pcDNA3, Fig. 3.12 lane 5). As for NS4A-EGFP, the primers were 4AE-F and 2AE-R and a fragment of 1.2 kb was obtained, which was as the expected 1169 bp (indicated by c in Fig. 3.12) when compared to the negative control (stable transfected cells pcDNA3, Fig. 3.12 lane 6). But for NS4B-EGFP with primers 4BE-FP and 2AE-R, no band was detected (Fig. 3.12 lane 7). In conclusion, the expressions of mRNAs of NS2A-EGFP, NS2B-EGFP, and NS4A-EGFP in the stable lines were detected but not NS4B-EGFP.
3.4 Construction of pNS2B-EGFP(pro), pNS4A-EGFP(pro), and pcDNA3-D24B-EGFP(pro) expression plasmids
The EGFP’ fragment without the start codon ATG was obtained by polymerase chain reaction (PCR) amplification with primers EGFP-F2 and EGFP-R2 on pEGFP-N2 as the template. The PCR products were digested with restriction enzymes XhoI at the 5’-end and
XbaI at the 3’-end and followed by gel purification. pNS2B-EGFP, pNS4A-EGFP, and
pcDNA-D24B-EGFP were also treated with the same restriction enzyme digestions to remove the EGFP tag with the start codon, and gel purified. The XhoI-XbaI fragment containing EGFP’ was ligated to the larger XhoI-XbaI fragments of pNS2B-EGFP, pNS4A-EGFP, and pcDNA3-D24B-EGFP (Fig. 3.13). Thus I replaced the original EGFP tag with the EGFP’ tag, which did not contain the start codon. They were named pNS2B-EGFP(pro), pNS4A-EGFP(pro), and pcDNA3-D24B-EGFP(pro) (Fig. 3.14 A, B, C).
1.5 kb (1534 bp) and 5 kb (4958 bp) (Fig. 3.14 D). pNS4A-EGFP(pro) was assessed by BsrGI and NdeI. There were fragments of 1.6 kb (1594 bp) and 5 kb (4958 bp) (Fig. 3.14 D). pcDNA3-D24B-EGFP(pro) was assessed by BsrGI and NdeI. There were fragments of 1.9 kb (1888 bp) and 5 kb (4958 bp) (Fig. 3.14 D). Besides, pNS2B-EGFP(pro), pNS4A-EGFP(pro), and pcDNA3-D24B-EGFP(pro) were sequenced to confirm the deletion of the EGFP start codon, ATG, in Fig. 3.15.
3.5 Expression of pNS2B-EGFP(pro), pNS4A-EGFP(pro), and pcDNA3-D24B-EGFP(pro) in mammalian cells, BHK-21
BHK-21 cells transfected with pNS2B-EGFP(pro), pNS4A-EGFP(pro), and pcDNA3-D24B-EGFP(pro) separately were harvested 24 hours posttransfection and were lysed by RIPA buffer. The cell lysates were analyzed by SDS-PAGE with Coomassie blue staining and Western blot with antibody against the C-terminal EGFP tag (Fig. 3.16 A, B). The predicted molecular weights of NS2B-EGFP(pro), NS4A-EGFP(pro), and NS4B-EGFP(pro) are 41.1, 43.6, and 53.9 kDa, respectively. Compared with cells transfected with vector only, there was no extra band on the Coomassie blue stained gel in the lanes (Fig. 3.16 A). With Western analysis, for cells transfected with pNS2B-EGFP(pro), there was a dominant band at about 43 kDa (Fig. 3.16 B, a). For cells transfected with pNS4A-EGFP(pro), a band was detected at about 43 kDa, which was as expected (Fig. 3.16 B, b) but the dominant band was between 34 and 26 kDa, which was the same as the EGFP control (Fig. 3.16 B, c). As for NS4B, there was a band at about 55 kDa, which was as expected (Fig. 3.16 B, d).
3.6 Selection of stable transfected cells of pNS2B-EGFP(pro), pNS4A-EGFP(pro), and pcDNA3-D24B-EGFP(pro).
pNS2B-EGFP(pro), pNS4A-EGFP(pro), and pcDNA3-D24B-EGFP(pro) were transfected into BHK-21 for stable clone selection by G418. The selected cells were analyzed by Western
blot analysis with anti-GFP-HRP antibody against the C-terminal EGFP tag and were observed by fluorescence microscope. There was fluorescence signal in the selected cells NS4A-EGFP(pro) (Fig. 3.17 F), but the fluorescence signals of NS2B-EGFP(pro) and NS4B-EGFP(pro) were not prominent (Fig. 8. D, H), compared to the BHK-21 cells (Fig. 3.17 J).
The predicted molecular weights of NS2B-EGFP(pro), NS4A-EGFP(pro), and NS4B-EGFP(pro) are 41.1, 41.2, and 53.9 kDa, respectively. For stable cell line NS2B-EGFP(pro), a band was detected below 43 kDa by Western blot analysis, which was as expected (Fig. 3.17 A b). For stable cell line NS4B-EGFP(pro), a band was detected at about 55 kDa, which was as expected (Fig. 3.17 B. c). But NS2B-EGFP(pro) and NS4A-EGFP(pro) also had a band at the same position (Fig. 3.17 A d), at about 55 kDa. For NS4A-EGFP(pro), no band at the predicted position was detected instead, but there was the unexpected band at about 27 kDa, the same as the molecular weight of EGFP (Fig. 3.17 A a).
3.7 Plaque formation on stable cell lines
The selected stable cell lines NS2A-EGFP, NS2B-EGFP(pro), NS4B-EGFP(pro), EGFP, and pcDNA3 were infected with DV2 virus PL046 strain. Seven days post-infection, the cells were fixed and stained with crystal violet. The number of plaques of BHK-21 was defined as 100% and the relative percentage of plaque numbers of the stable cell lines were recorded in Table 5 and Fig. 3.18. The data were analyzed by ANOVA and Scheffe test. For NS2A-EGFP, there was a 44% reduction (down to 81.46% from that of control EGFP, 126.32%) with significance (P<0.05). Whereas, the differences of the percentage of plaque numbers between NS2B-EGFP(pro), NS4B-EGFP(pro) and controls, pcDNA3, and EGFP, were not significant.
(II) Discussion
3.8 Construction of pNS2A-EGFP, pNS2B-EGFP, pNS4A-EGFP, and pcDNA3-D24B-EGFP expression plasmids
Previously, researchers in the laboratory have cloned the four nonstructural genes of DV2 PL046 strain into the expression vector pcDNA3 along with C-terminal HA and His tags (徐 婕 琳, 2003, 交 大 碩 士 論 文 ; 楊 馥 嘉 , 2006, 交 大 碩 士 論 文 ). They were named pNS2A-HAHis, pNS2B-HAHis, pNS4A-HAHis, and pcDNA3-D24B-HAHis (Appendix 2). Therefore, it was convenient to investigate the expression and to select the stable cell lines of these four nonstructural genes by the C-terminal EGFP tag. I replaced the HA-His tags with the EGFP tag and obtained the expression clones of the four nonstructural genes on the pcDNA3 backbone, named pNS2A-EGFP, pNS2B-EGFP, pNS4A-EGFP, and pcDNA3-D24B-EGFP. These four constructs were assessed by restriction digestions (Fig. 3.3) and confirmed by sequencing.
3.9 Expression of pNS2A-EGFP, pNS2B-EGFP, pNS4A-EGFP, and pcDNA3-D24B-EGFP in mammalian cells, BHK-21
3.9.1 Confocal fluorescence microscopy analysis
The EGFP gene has been optimized for brighter fluorescence and higher expression in mammalian cells (excitation maximum= 488 nm; emission maximum=507 nm). To confirm the expression of pNS2A-EGFP, pNS2B-EGFP, pNS4A-EGFP, and pcDNA3-D24B-EGFP, these four expression plasmids and pEGFP-N2 were transfected into BHK-21. All have the green fluorescence expression through the C-terminal EGFP tag in BHK-21 (Fig. 3.4 A, D, G, J, and M). Additionally, the four nonstructural proteins were colocalized against the ER, using calnexin as the marker. 2A-EGFP, 2B-EGFP, 4A-EGFP, and 4B-EGFP proteins co-localized with the ER markers in the cytoplasm of BHK-21, compared to EGFP proteins displayed the
whole cells (Fig. 3.4 A). It suggested that the constructs of the four nonstructural genes could express in mammalian cells.
3.9.2 Transient expression of pNS2A-EGFP, pNS2B-EGFP,
pNS4A-EGFP, and pcDNA3-D24B-EGFP in BHK-21
The predicted molecular weights of NS2A-EGFP, NS2B-EGFP, NS4A-EGFP, and NS4B-EGFP are 50.9, 41.2, 43.7, and 54.0 kDa, respectively. The nonstructural proteins NS2B and NS4B could be detected both in the supernatants of the cells with the expected size by Western blot (Fig. 3.5 B c, f). For NS4A, even though there was a band at about 43 kDa, the dominant band was between 34 and 26 kDa (Fig. 3.5 B c). For NS2A, the dominant band was between 34 and 26 kDa, and there was a band at about 43 kDa which was smaller than expected. This was perhaps due to the protein charge affecting mobilityor a partial cleavage at the N-terminal of NS2A. All in all, the transient expression of nonstructural proteins NS2B and NS4B in mammalian cells were confirmed by Western blot clearly, but NS2A and NS4A were not. Nevertheless, I proceeded to select the stable cell lines expressing these four proteins.
3.10 Selection of stable transfected cells of pNS2A-EGFP, pNS2B-EGFP, pNS4A-EGFP, pcDNA3-D24B-EGFP, and pEGFP-N2
3.10.1 Western blot analysis of the selected stable cell lines
pNS2A-EGFP, pNS2B-EGFP, pNS4A-EGFP, pcDNA3-D24B-EGFP, and pEGFP-N2 were transfected into BHK-21 for stable clone selection by G418. The selected cells were analyzed by Western blot analysis with anti-GFP-HRP antibody against the C-terminal EGFP tag and were observed by fluorescence microscope (Fig. 3.6. b; 3.7. b; 3.8. b; 3.9. b; 3.10. b). The predicted molecular weights of NS2A-EGFP, NS2B-EGFP, NS4A-EGFP, NS4B-EGFP, and EGFP are 50.9, 41.2, 43.7, 54.0, and 27 kDa, respectively. The EGFP were detected in the
supernatants and pellets of the cells with the expected size by Western blot (Fig. 3.6 c, d). The 2A-EGFP was detected at about 43 kDa in the supernatants and pellets which was smaller than the expected 50.9 kDa (Fig 3.7. c). It was similar with the transient expression of NS2A-EGFP (Fig. 3.5 B. a). The smaller size may be due to the protein charge affecting mobilityor a partial cleavage at the N-terminal of NS2A. For NS2B-EGFP, NS4A-EGFP, and NS4B-EGFP, no expected bands were detected but a band at about 27 kDa, which was the same as the molecular weight of EGFP (Fig. 3.8. c, 3.9. c, 3.10. c).
3.10.2 Materials and RNA expression confirmation of the selected stable cell lines of NS2A-EGFP, NS2B-EGFP, NS4A-EGFP, and NS4B-EGFP
Because of the incorrect bands in Western blot of NS2A-EGFP (smaller than expected), NS2B-EGFP, NS4A-EGFP, and NS4B-EGFP, first I tried to examine the materials by PCR amplification of the genomic DNA extracted from the stable cell lines. The PCR amplification of the genomic DNA extracted from the stable cell lines of NS2A-EGFP, NS2B-EGFP, NS4A-EGFP, and NS4B-EGFP were performed with forward primers 2AE-F, 2BE-F, 4AE-F, and 4BE-F and reverse primers 2AE-R located at the C-terminal of EGFP tag. For NS2A-EGFP, NS2B-EGFP, and NS4A-EGFP, expected bands were obtained by PCR amplification, but no band was detected with NS4B-EGFP. It showed that the DNA in the selected stable cell lines of NS2A-EGFP, NS2B-EGFP, and BS4A-EGFP were correct while NS4B-EGFP was not.
Second, I isolated the total RNA of the selected stable cell lines NS2A-EGFP, NS2B-EGFP, NS4A-EGFP, and NS4B-EGFP by using RNeasy Mini Kit. The extracted RNAs were subjected to superscript one-step RT-PCR (Invitrogen) with individual primers to amplify the full length RNA of NS2A-EGFP, NS2B-EGFP, NS4A-EGFP, and NS4B-EGFP, separately. According to the results in Fig. 3.12, RNAs of NS2A-EGFP, NS2B-EGFP, and NS4A-EGFP were detected in the stable lines but not NS4B-EGFP. Therefore, genomic DNA
and RNA expression of NS2A-EGFP, NS2B-EGFP, and NS4A-EGFP were verified by PCR and RT-PCR except for NS4B-EGFP. Thus, I presumed that the transient and long-term expressions of NS2A-EGFP were achieved but the smaller size in Western blot perhaps due to the protein charge. Nevertheless, the selection of stable cell lines of NS2B-EGFP and NS4A-EGFP could not be achieved.
There were two possible reasons: (1) The C-terminal EGFP tag of NS2B-EGFP and NS4A-EGFP clones had its own start codon, ATG. Overexpression of NS2B and NS4A proteins is toxic to cells, therefore, the translation starts at the ATG of the EGFP sequence. (2) The sequence of NS2B and NS4A may contain internal ribosome entry sites (IRESs). Many pathogenic viruses, for example HCV and piconavirus, use an alternative, cap-independent mechanism that substitutes RNA structure for the cap and proteins involved in translation initiation. The RNA structure driving this process is called internal ribosome entry sites (IRESs) (Jeffrey et al., 2008; Baird et al., 2006).
Therefore, I decided to solve the problem by deleting the ATG start codon at the N-terminal of the EGFP tag of pNS2B-EGFP, pNS4A-EGFP, and pcDNA3-D24B-EGFP. Thus, I re-cloned 2B-EGFP, 4A-EGFP, and 4B-EGFP constructs as described in the next section.
3.11 Construction of pNS2B-EGFP(pro), pNS4A-EGFP(pro), and pcDNA3-D24B-EGFP(pro) expression plasmids
The possible reason why the stable cell line selection of NS2B-EGFP, NS4A-EGFP, and NS4B-EGFP could not be carried out maybe because cells selectively expressed the C-terminal EGFP gene, which had its own start codon. Thus I obtained the EGFP fragment without the start codon by PCR amplification. This fragment is named EGFP’. The EGFP fragments of pNS2B-EGFP, pNS4A-EGFP, and pcDNA3-D24B-EGFP were then replaced by the EGFP’ fragments. The constructs were named pNS2B-EGFP(pro), pNS4A-EGFP(pro),
and pcDNA3-D24B-EGFP(pro) (Fig. 3.14 A, B, C). All constructs were assessed by BsrGI and NdeI to expected results (Fig. 3.14 D). The constructs were further confirmed by sequencing to ensure that the ATG at the EGFP tag of pNS2B-EGFP(pro), pNS4A-EGFP(pro), and pcDNA3-D24B-EGFP(pro) were actually deleted (Fig. 3.15).
3.12 Expression of pNS2B-EGFP(pro), pNS4A-EGFP(pro), and pcDNA3-D24B-EGFP(pro) in mammalian cells, BHK-21
The predicted molecular weights of NS2B-EGFP(pro), NS4A-EGFP(pro), and NS4B-EGFP(pro) are 41.1, 43.6, and 53.9 kDa, respectively. The nonstructural proteins NS2B and NS4B could be detected both in the supernatants of the cells with the expected size by Western blot (Fig. 3.16 B a, d). For NS4A, the dominant band was between 34 and 26 kDa (Fig. 3.16 B c), even though the expected band was detected at about 43 kDa (Fig. 3.16 B b).
All in all, the transient expression of nonstructural proteins NS2B-EGFP(pro) and NS4B-EGFP(pro) in mammalian cells were confirmed by Western blot but NS4A-EGFP(pro) was not. The sequence of NS4A may contain internal ribosome entry sites (IRESs) or a partial cleavage at the end of NS4A (Preugschat et al., 1991). Nevertheless, these three re-cloned constructs were proceeded to the stable cell line selection.
3.13 Selection of stable transfected cells of pNS2B-EGFP(pro), pNS4A-EGFP(pro), and pcDNA3-D24B-EGFP(pro)
pNS2B-EGFP(pro), pNS4A-EGFP(pro), and pcDNA3-D24B-EGFP(pro) were transfected into BHK-21 for stable clone selection by G418. First the selected cells were observed by fluorescence microscope. The fluorescence signal of NS4A-EGFP(pro) was observed (Fig. 3.17 F), but NS2B-EGFP(pro) and NS4B-EGFP(pro) were not observed obviously (Fig. 3.17 D, H), compared to the BHK-21 cells (Fig. 3.17 J). It was a possibility that the fluorescence signals were too weak to be observed.Besides, I further confirmed the expression by Western