Synthesis and Properties of Triptycene-Diaminostilbene Hybrid Systems
Jye-Shane Yanga* ( ) and Jyu-Lun Yanb( )a
Department of Chemistry, National Taiwan University, Taipei 10617, Taiwan, R.O.C. b
Department of Chemistry, National Central University, Chungli 32054, Taiwan, R.O.C.
The synthesis, electronic spectra, and thermal and electrochemical properties of two triptycene-capped trans-4,4¢-diaminostilbenes (1Me and 1Ph) are reported in order to evaluate their applicability as blue light-emitting materials in organic light-emitting diodes (OLEDs). For comparison, the fluorescence properties of the planar analogs 2Me and 2Ph are also investigated. High fluorescence quantum yields (0.72-0.78) are observed for 1Me and 1Ph in THF, presumably due to a large barrier for the nonradiative double-bond torsion reaction in the singlet excited state. In the spin-cast thin films, the fluorescence quan-tum efficiencies become lower (0.15-0.26) and the fluorescence spectra are red-shifted. Compared to 1Me and 1Ph, 2Me and 2Ph display even larger fluorescence shifts and fluorescence quenching, which can be attributed to their more planar structures that favor intermolecularp-stacking in the solid state. The glass transition temperatures for 1Me and 1Ph are 113 and 120oC, respectively, and the decomposition temper-atures 341 and 375oC, respectively. Cyclic voltammograms of 1Me and 1Ph show two reversible redox waves, corresponding to two monoelectronic oxidation processes of the amino groups forming radical cat-ion and dicatcat-ion species.
Keywords: Triptyene; Diaminostilbene; OLED; Cyclic voltammogram.
INTRODUCTION
The fluorescence quantum efficiency (Ff) of trans-stilbenes strongly depends on the position and nature of substituents.1-3While most substituted trans-stilbenes re-semble the parent trans-stilbene with weak fluorescence at room temperature, there are four categories of stilbene de-rivatives that have fluorescence as the dominant mode of excited-state decay (i.e.,Ff> 0.5). One category consists of 4,4¢-disubstituted trans-stilbenes with strong electron-donating amino and alkoxy groups.4-7This is manifested by the fact that 4,4¢-diaminostilbene and its derivatives have long been employed as optical brighteners.8A second cate-gory is formed by trans-stilbenes with extended conjuga-tion by aromaticp-substituents such as 4-phenylstilbene (4-styrylbiphenyl),94-styrylstilbene (1,4-bis(styryl)ben-zene),104,4¢-diphenylstilbene,11and 2-strylnaphthalene.12 Oligo(p-phenylenevinylene)s can be considered as ex-tended forms of this category. The third category refers to 3-amino substituted trans-stilbenes as a result of the unique “meta-amino effect”.13The fourth category was recently established for N-aryl substituted trans-4-aminostilbenes,
where the “amino-conjugation effect” could be considered as “pseudo-p-substituents” belonging to the second cate-gory.14Since the major nonradiative decay that competes with fluorescence for trans-stilbenes is the activated trans ® cis photoisomerization reaction, an increase in the dou-ble bond torsional barrier accounts for the fluorescence en-hancement in these stilbene derivatives.1,13,14
Many highly fluorescent stilbene fluorophores have been investigated as the light-emitting materials in organic light-emitting diodes (OLEDs).15-20However,B-stacking of the planarB-conjugated backbones in the solid state of-ten result in poor device performance due to molecular crystallization, excimer formation, emission band broaden-ing, and/or fluorescence quenching. To reduce these unde-sired intermolecular interactions, bulky substituents are generally introduced.18,21For example, Chen and co-work-ers introduced ortho substituents to the diphenylamino group and sterically crowded tetraphenylphenyl group to replace the non-amino substituted phenyl ring in trans-4-(N,N-diphenylamino)stilbene.18 The resulting stilbene fluorophores in the solid state display amorphous film mor-phology and solution-like fluorescence properties, which Dedicated to the memory of the late Professor Ho Tong-Ing.
have great potentials for fabricating nondoping OLEDs. To search for new stilbene-based blue fluorophores, we have investigated the fluorescence properties of two triptycene-capped trans-4,4¢-diaminostilbenes (1Me, and 1Ph) in solutions and in the solid state. The fluorescence quantum yields for trans-4,4¢-diaminostilbene and its N,N,N¢,N¢-tetramethyl derivative in ethereal solvents are 0.76 and 0.45, respectively.4,22 Introduction of N-aryl groups to diaminostilbene might further enhance the fluo-rescence quantum efficiency. The presence of nonplanar rigid triptycene groups in 1Me and 1Ph might prevent p-stacking of the stilbene moieties and thus retain high flu-orescence quantum efficiency in the solid state. We report herein the synthesis, fluorescence, thermal, and electro-chemical properties of 1Me and 1Ph. For comparison, the fluorescence quantum yields of the non-triptycene-incor-porated analogs 2Me and 2Ph are also reported. The results show that diaminostilbenes 1Me and 1Ph possess higher fluorescence quantum yields than the corresponding 2Me and 2Ph in both solutions and thin solid films.
RESULTS AND DISCUSSION
As shown in Scheme I, diaminostilbenes 1Me and 1Ph were prepared through Pd-catalyzed amination reac-tion23between trans-4,4¢-dibromostilbene and the amino-triptycene 3 followed by N-methylation of the intermediate 4 with methyl iodide under basic conditions and by N-phen-ylation with bromobenzene under Pd-catalyzed amination
conditions, respectively. The precursor trans-4,4¢-dimostilbene was in turn prepared with 4-bromobenzyl bro-mide and 4-bromobenzaldehyde according to the standard Horner-Wadsworth-Emmons procedures for olefin synthe-sis,14,24and the other precursor 3 was prepared through a five-step route starting from anthracene and benzoquinone (Scheme II).
The first two synthetic steps in Scheme II for the syn-thesis of the triptycene hydroquinone 6 was conducted by following the same procedures reported by Barlett et al.25 O-Alkylation of 6 was performed under a standard SN2 re-action condition.26Nitration27of the resulting compound 7 followed by reduction of the nitro group by tin(II) chlo-ride28afforded compound 3.
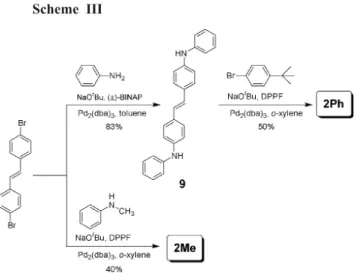
Compounds 2Me and 2Ph were also synthesized via Pd-catalyzed amination reactions starting with trans-4,4¢-dibromostilbene and commercially available anilines and 1-bromo-4-(tert-butyl)benzene (Scheme III).
The absorption and fluorescence spectra of 1Me and
N N R R R = CH3 R = 4-(tBu)Ph C4H9O OC4H9 N OC4H9 C4H9O NR R R = CH3 1Me R = Ph 1Ph 2Me 2Ph Scheme I + O O Toluene HBr, HOAc 99 % 60 % O O OH OH K2CO3, KI Acetone C4H9Br 81 % HNO3, HOAc OC4H9 OC4H9 OC4H9 OC4H9 NO2SnCl22H2O THF/ethanol 83% OC4H9 OC4H9 NH2 3 5 6 8 7 reflux 99 % Scheme II
1Ph in THF are shown in Fig. 1. Whereas 1Me displays a single intense long wavelength absorption band, there is a second absorption band at a shorter wavelength (317 nm) for 1Ph. According to our previous studies on aminostil-benes,14,29the long wavelength band can be attributed to electronic transitions delocalized in the triphenylamine moieties. Similar spectral features are also found in the ab-sorption spectra of 2Me and 2Ph (spectra not shown). De-spite the difference in the N-triptycenyl and the N-phenyl groups, the absorption and fluorescence maxima of 1Me and 1Ph resemble those of 2Me and 2Ph, respectively (Ta-ble 1). However, the fluorescence quantum yield for 1Ph is significantly higher than that for 2Ph in THF (Table 1).
The high fluorescence quantum yields observed for 1Me, 1Ph, 2Me, and 2Ph in THF are consistent with our expectations based on the strongly fluorescent parent com-pound, trans-4,4¢-diaminostilbene.4-6
However, the “amino conjugation effect” observed for trans-4-aminostilbene14 appears to be unimportant for the diaminostilbene system, since these N-phenyl substituted diaminostilbenes have nearly the same fluorescence quantum yields as the parent
compound. It should be noted that N-phenyl substitution of aminostilbenes could lead to two opposite effects on the fluorescence quantum yield. The first effect is to raise the singlet barrier for trans® cis photoisomerization and thus to enhance the fluorescence (a positive effect).14The sec-ond effect is to induce the intersystem crossing from S1to T1so that the fluorescence quantum yield is reduced (a neg-ative effect).31Accordingly, the similarity in the fluores-cence quantum yield for 1Me, 1Ph, and 2Me in comparison to the parent trans-4,4¢-diaminostilbene might result from a balance of the above two opposite effects. However, in the case of 2Ph, the negative effect is stronger than the positive one, which accounts for the lower fluorescence quantum yield observed for 2Ph vs 2Me. The smaller difference in fluorescence quantum yield between 1Me and 1Ph might be associated with the bulkier triptycene group, which re-duces both the positive and the negative effect.
Fig. 1. Absorption and fluorescence spectra of (a) 1Me and (b) 1Ph in THF and in spin-cast films. Scheme III
Table 1. Spectral, thermal, and electrochemical data of diaminostilbemes
lmaxfl, nm (Ffl) Tg Tm Td E1/2
Compd. lmaxabs, nma,b
Solutiona Solidc (°C)d (°C)d (°C)e (V)f 1Me 376 423 (0.78) 450 (0.26) 113 249 341 0.45, 0.63 1Ph 387 (317) 434 (0.72) 441 (0.15) 120 268 375 0.62, 0.73 2Me 372 424 (0.73) 462 (0.03) 2Ph 385 (303) 434 (0.45) 483 (0.05) a
in THF.bThe second absorption band in parentheses.cSolid films prepared by spin casting the chloroform solutions of the substrate.dGlass transition temperature taken by DSC.eDecomposition temperature measured by TGA at 5% weight loss.fHalf-wave redox potentials vs SCE by using the redox of ferrocene/ferrocenium (E1/2= 0.39 V vs SCE)
30
Significant reduction in fluorescence quantum yields is observed for 1Me, 1Ph, 2Me, and 2Ph on going from THF to the thin solid films, particularly for the latter two cases (Table 1). The films of optical densities in the range of 0.1 and 0.2 at the absorption maxima were prepared by spin casting with 5 × 10-3M chloroform solutions. Along with the reduction inFflis the dramatic red shift of the fluo-rescence spectra, where the amount of spectral shifts is also larger for 2Me and 2Ph (Dlmaxfl= 38-49 nm) than for 1Me and 1Ph (Dlmaxfl= 7-27 nm). These observations indicate that substantial intermolecular interactions are present in 2Me and 2Ph due to their planar molecular structures that allow intermolecularp-stacking interactions. Intermolecu-larp-stacking interactions might also occur in the case of 1Me but not in 1Ph, because the spectral shifts in 1Ph are rather small. Since the presence of a small amount of exter-nal fluorescence quenchers generally dictates the observed fluorescence quantum yield for systems in the solid state due to intermolecular exciton migration,32the larger fluo-rescence quenching effects observed for 2Me and 2Ph vs 1Me and 1Ph might reflect more population of the non-fluorescent energy traps and more efficient exciton migra-tion to nonfluorescent energy traps in the former films. The nonfluorescent energy traps could bep-stacked excimers or minor impurities.32It should also be noted that the effi-ciency of intermolecular exciton migration increases with decreasing the intermolecular distances, which accounts for the large reduction in fluorescence quantum yields on going from solutions to the solid state.33According to our recent studies on the fluorescence behavior of pentipty-cene-derived oligo(p-phenyleneethynylene)s in solutions and spin-cast films, the red shifts of fluorescence and the changes in the relative intensities of vibrational bands for 1Ph in films vs solutions could be attributed to the reab-sorption effect.34
The thermal stabilities of 1Me and 1Ph have been in-vestigated. As shown in Table 1, 1Ph has a higher glass transition temperature (Tg), melting temperature (Tm), and decomposition temperature (Td) than 1Me. Improved ther-mal stability for 1Ph vs 1Me is consistent with the previous observations for other N-phenyl vs N-alkyl substituted aro-matic amines.35
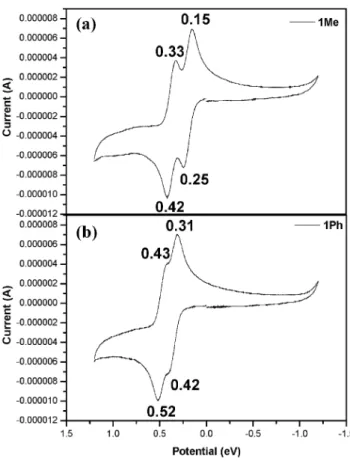
Fig. 2 shows the cyclic voltammograms recorded for 1Me and 1Ph in CH2Cl2(1.0 × 10-3M) in the presence of Bu4NPF6(0.1 M) supporting electrolyte. Both 1Me and 1Ph display two reversible redox events in the regions of 0.2-0.5 V vs Ag/AgNO3quasi-reference electrode. The
half-wave potentials (E1/2) of 1Me and 1Ph vs SCE cali-brated with the redox potential of ferrocene/ferrocenium under the same conditions (0.14 V) are shown in Table 1.
The two redox events displayed by 1Me and 1Ph are the first and second monoelectronic processes for the ami-no groups (eq. 1):
(1)
It is interesting to note that the redox potentials for 1Me are lower than those for 1Ph (Table 1) and that the splitting of
Fig. 2. Cyclic voltammograms of (a) 1Me and (b) 1Ph. Working electrode: Pt. Reference electrode: Ag/AgNO3. Scan rate = 100 mV/s. Supporting electrolyte: Bu4NPF6.
the two redox waves is larger for 1Me (DE1/2= 0.18 V) than that for 1Ph (DE1/2= 0.11 V). Two reversible redox events are also observed for N,N,N¢,N¢-tetraethyl-1,4-phenylene-diamine and its derivatives, which display a even larger splitting of the redox waves (DE1/2~ 0.5 V).36These obser-vations indicate that the degree of electronic delocalization of the radical cation is lower when the length of the p-spacer between the two amino group is larger. In addition, the radical cation appears to be more localized in the tri-phenylamino vs N-methylditri-phenylamino group.
In summary, we have synthesized two triptycene-diaminostilbene systems (1Me and 1Ph), and their photo-physical, thermal, and electrochemical properties are in-vestigated. When compared with the planar model com-pounds 2Me and 2Ph, 1Me and 1Ph display higher fluores-cence quantum yields in both solutions and thin solid films. In addition, the thin-film fluorescence is located in the blue region. Compounds 1Me and 1Ph also possess good ther-mal and electrochemical stability. Nonetheless, the large decreases in the fluorescence intensity and the significant red shifts in the fluorescence spectra for 1Me and 1Ph on going from dilute solutions to thin films indicate the pres-ence of significant intermolecularp-stacking interactions, which can create non-emissive excimer traps, and dipole-dipole interactions, which can facilitate intermolecular exciton migration to the traps, in the solid state. Further structural modification by bulkier substituents might im-prove the light-emitting properties of diaminostilbene sys-tems in the solid state.
EXPERIMENTAL SECTION Methods
Electronic spectra were recorded at room temperature (23± 1 °C). UV-visible spectra were measured on a Cary 300 double beam spectrophotometer. Fluorescence spectra were recorded on a PTI QuantaMaster C-60 spectrometer and corrected for the response of the detector. The optical density (OD) of all solutions and thin films was about 0.1 at the wavelength of excitation. A N2-bubbled solution of anthracence (Ffl= 0.27 in hexane)37was used as a standard for the fluorescence quantum yield determination of com-pounds 1Me, 1Ph, 2Me and 2Ph under N2-bubbled THF solutions with solvent refractive index correction. An error of ±10% is estimated for the fluorescence quantum yields. Fluorescence quantum yields for 1Me and 1Ph in spin-cast
films (OD ~ 0.05-0.15) were determined relative to equi-absorbing films of 9,10-diphenylanthracene dispersed in poly(methyl methacrylate) (PMMA) (Ffl= 0.83).38 Elec-trochemical data of compound 1Me and 1Ph were deter-mined by cyclic voltammetry (CV) in 0.1 M Bu4NPF6/CH2Cl2 solutions on a platinum button electrode with a platinum coil auxiliary electrode and an isolated Ag/AgNO3 refer-ence electrode using an CHI 611A Electrochemical Ana-lyzer. The data were corrected by the CV of ferrocene un-der the same conditions. DSC thermographs were carried out on a Mettler DSC 822 and calibrated by a pure indium sample. All phase transitions are determined by a scan rate of 5.0°C/min. Thermogravimetric analysis (TGA) was performed with a Perkin-Elmer TGA-7 themal analysis system using dry nitrogen as a carrier gas at a flow rate of 100 mL/min. The TGA experiments were conducted from room temperature to 900°C with a linear heating rate of 10 °C/min.
Materials
Solvents for organic synthesis were reagent grade or HPLC grade, but all were HPLC grade for spectra and quantum yield measurements. Compounds 5,25 6,25and trans-4,4¢-dibromostilbene39
are known compounds, and our1H NMR spectra conform to the literature values. Compound 1Me
Under an atmosphere of argon, sodium hydride (0.05 g, 1.2 mmol) was added to a solution of 4 (0.2 g, 0.2 mmol) in 3 mL DMF. Methyl iodide (0.2 mL, 2.4 mmol) was added dropwise to the solution at 0°C. The mixture was warmed to room temperature and stirred for 2 h. The solu-tion was poured into ice water and the insoluble residue was filtered off, providing the yellow solid of 1Me with a yield of 99%: mp: 230-233 °C, 1H-NMR (500 MHz, CDCl3): 0.91 (t, J = 7.3 Hz, 6H), 1.01 (t, J = 7.3 Hz, 6H), 1.45 (t, J = 7.4 Hz, 4H), 1.51-1.55 (m, 4H), 1.67 (t, J = 7.1 Hz, 4H), 1.77 (t, J = 7.0 Hz, 4H), 3.16 (bs, 6H), 3.71 (t, J = 6.3 Hz, 4H), 3.82 (t, J = 6.1 Hz, 4H), 5.82 (s, 2H), 5.85 (s, 2H), 6.38 (m, 2H), 6.63 (m, 4H), 6.79 (m, 2H), 7.00 (m, 8H), 7.24 (m, 4H), 7.40 (m, 8H) ppm;13C-NMR (125 MHz, CDCl3): 13.9, 14.0, 19.4, 19.5, 31.5, 32.5, 39.2, 47.3, 48.6, 68.8, 74.2, 109.8, 113.8, 123.7, 123.8, 124.8, 125.0, 125.1, 126.7, 127.5, 132.3, 137.8, 140.9, 144.9, 145.6, 145.8, 147.9, 150.1 ppm; IR (KBr): 1694, 2956, 3021 cm-1; FAB-HRMS Cacld for C72H74N2O4: 1030.5649, found 1030.5702.
Compound 1Ph
A mixture of 4 (0.20 g, 0.20 mmol), NaOBut(0.13 g, 1.35 mmol), DPPF (0.01g, 0.015 mmol), and Pd2(dba)3 (0.01 g, 0.01 mmol) in 1 mL of anhydrous toluene under ar-gon was heated at 90°C. Bromobenzene (0.20 mL, 1.44 mmol) was then added to the solution and stirred at 90°C for 24 h. The solution was cooled and then 40~50 mL of THF was added. The insoluble residue was filtered off and the filtrate was concentrated under reduced pressure to af-ford the crude product. Further purification was performed by column chromatography using CH2Cl2/hexane (1:1) as the eluent to provide the light yellow solid of 1Ph with a yield of 90%: mp: 267-269°C,1H-NMR (500 MHz, CDCl3): 0.88 (t, J = 7.4 Hz, 6H), 0.98 (t, J = 7.4 Hz, 6H), 1.32-1.33 (m, 4H), 1.48-1.52 (m, 4H), 1.56-1.59 (m, 4H), 1.71-1.74 (m, 4H), 3.69-3.77 (m, 8H), 5.74 (s, 2H), 5.83 (s, 2H), 6.30 (s, 2H), 6.88-7.00 (m, 20H), 7.14 (m, 4H), 7.23-7.24 (m, 4H), 7.34-7.40 (m, 8H) ppm;13C-NMR (125 MHz, CDCl3): 14.0 (2C), 19.4 (2C), 31.4, 32.2, 47.2, 48.5, 68.8, 74.1, 110.6, 121.7, 122.0, 122.3, 123.7, 123.9, 124.9, 125.1, 126.3, 126.8, 128.8, 131.2, 132.1, 136.6, 141.0, 144.8, 145.6, 145.8, 146.6, 147.2, 150.3 ppm; IR (KBr): 1603, 2958, 3025 cm-1; FAB-HRMS Cacld for C82H78N2O4: 1154.5962, found 1154.5994.
Compound 2Me
The procedures resemble those of compound 1Ph but replacing toluene with o-xylene (yield of 40%): mp: 183-185°C,1H-NMR (200 MHz, CDCl3): 3.34 (s, 6H), 6.94-7.10 (m, 12H), 7.24-7.51 (m, 8H) ppm; 13C-NMR (50 MHz, CDCl3): 40.2, 119.6, 121.3, 121.9, 126.0, 127.1, 129.2, 130.5, 148.0, 148.7 ppm; IR (KBr): 1600, 2886, 3052 cm-1. Compound 2Ph
The procedures resemble those of compound 1Ph but replacing toluene with o-xylene (yield of 50%): mp: 110-112°C,1H-NMR (200 MHz, CDCl3): 1.29 (s, 18H), 6.85-7.12 (m, 16H), 7.20-7.37 (m, 12H) ppm;13C-NMR (50 MHz, CDCl3): 31.4, 34.3, 122.6, 123.4, 124.1, 124.2, 126.1, 126.4, 127.0, 129.1, 131.6, 144.7, 146.0, 147.1, 147.7 ppm; IR (KBr): 1593, 2966, 3032 cm-1. Compound 3
Compound 8 (0.9 g, 2.0 mmol) was dissolved in 20 mL THF and then a mixture of SnCl2×2H2O (2.8 g, 12.0 mmol) and 20 mL ethanol was added dropwise to the
solu-tion. The mixture was stirred at 70°C for 24 h. The solvent was removed under reduced pressure, and to the residue was added CH2Cl2and then washed with brine. The organic layer was dried over anhydrous MgSO4, and the filtrate was concentrated under reduced pressure. Column chromatog-raphy with ethyl acetate/hexane (1:4) as eluent afforded the white solid with a yield of 81%: mp: 132-134°C,1H-NMR (500 MHz, CDCl3): 1.02 (t, J = 7.4 Hz, 3H), 1.06 (t, J = 7.4 Hz, 3H), 1.54-1.57 (m, 2H), 1.59-1.63 (m, 2H), 1.77-1.80 (m, 2H), 1.85-1.88 (m, 2H), 3.59 (bs, 2H), 3.86 (q, J = 6.9 Hz, 4H), 5.65 (s, 1H), 5.72 (s, 1H), 5.95 (s, 1H), 6.93-6.97 (m, 4H), 7.34 (d, J = 6.3 Hz, 4H) ppm;13C-NMR (125 MHz, CDCl3): 14.0, 14.1, 19.5, 19.6, 31.6, 32.6, 46.8, 48.6, 68.8, 74.0, 68.8, 74.0, 97.9, 123.5 (2C), 124.6, 124.7, 125.1, 136.3, 137.5, 139.4, 145.4, 146.4, 150.4 ppm; IR (KBr): 1615, 2959, 3359 cm-1; FAB-HRMS Cacld for C28H31NO2: 413.2355, found 413.2354.
Compound 4
The procedures resemble those of compound 1Ph but replacing DPPF with (±)-BINAP (yield of 92%): mp: 235-237°C,1H-NMR (500 MHz, CDCl3): 1.00-1.03 (m, 12H), 1.52-1.59 (m, 8H), 1.77-1.80 (m, 4H), 1.82-1.85 (m, 4H), 3.84-3.88 (m, 8H), 5.71 (s, 2H), 5.79 (s, 2H), 5.99 (bs, 2H), 6.54 (s, 2H), 6.88 (s, 2H), 6.97-7.00 (m, 12H), 7.34-7.35 (m, 4H), 7.36-7.39 (m, 8H) ppm; 13C-NMR (125 MHz, CDCl3): 14.0 (2C), 19.5, 19.6, 31.6 (2C), 32.5, 47.0, 48.7, 69.0, 74.7, 99.3, 117.6, 123.6 (2C), 124.9, 125.2, 125.8, 126.7, 127.3, 130.5, 133.6, 138.3, 139.5, 142.3, 145.3, 146.0, 150.1 ppm; IR (KBr): 1603, 2959, 3065, 1408 cm-1; FAB-HRMS Cacld for C70H70N2O4: 1002.5336, found 1002.5373.
Compound 7
A mixture of 6, 1-bromobutane (3.5 g, 25.0 mmol) and potassium carbonate (4.2 g, 30.0 mmol) in 50 mL of ac-etone was refluxed at 70°C for 48 h. The solvent was re-moved under reduced pressure, and the residue was dis-solved in CH2Cl2and washed with brine. The organic layer was dried over anhydrous MgSO4, and the filtrate was con-centrated under reduced pressure. Column chromatogra-phy with CH2Cl2/hexane (1:4) as eluent afforded a white solid with a yield of 81%.1H-NMR (500 MHz, CDCl3): 1.02 (t, J= 7.4 Hz, 6H), 1.54-1.59 (m, 4H), 1.79-1.81 (m, 4H), 3.92 (t, J= 6.4 Hz, 4H), 5.86 (s, 2H), 6.47 (s, 2H), 6.95-6.97 (m, 4H), 7.36-7.38 (m, 4H) ppm;13C-NMR (125 MHz, CDCl3): 14.0, 19.4, 31.6, 47.5, 69.4, 110.7, 123.7,
124.9, 135.7, 145.8, 148.5 ppm. Compound 8
Compound 7 (1.0 g, 2.5 mmol) was dissolved in 10 mL acetic acid and stirred at 0°C for 90 minutes. A mixture of 10 mL acetic acid and 2 mL nitric acid was added drop-wise to the solution at 0°C. The mixture was warmed to room temperature and stirred for 48 h. To the solution was added CH2Cl2and then washed with cold water. The or-ganic layer was dried over anhydrous MgSO4, and the fil-trate was concenfil-trated under reduced pressure. Column chromatography with CH2Cl2as eluent afforded the yellow solid with a yield of 99%: mp: 162-163°C,1H-NMR (500 MHz, CDCl3): 1.04 (t, J = 7.4 Hz, 3H), 1.07 (t, J = 7.5 Hz, 3H), 1.55-1.59 (m, 2H), 1.61-1.64 (m, 2H), 1.84-1.87 (m, 2H), 1.91-1.95 (m, 2H), 3.99 (td, J = 6.4 and 2.0 Hz, 4H), 5.83 (s, 1H), 5.92 (s, 1H), 7.00-7.04 (m, 4H), 7.11 (s, 1H), 7.38-7.43 (m, 4H) ppm; 13C-NMR (125 MHz, CDCl3): 13.9, 14.0, 19.4, 31.2, 32.3, 47.5, 48.4, 69.0, 76.3, 106.5, 123.9, 124.2, 125.6 (2C), 140.6, 141.2, 142.3, 142.8, 144.3, 144.5, 149.4 ppm; IR (KBr): 1527, 2960 cm-1; FAB-HRMS Cacld for C28H29NO4: 443.2097, found 443.2094; Anal. Calcd for C28H29NO4: C, 75.82, H, 6.59, N, 3.16. Found: C, 75.93, H, 6.59, N, 3.16.
Compound 9
The procedures resemble those of compound 1Ph. (yield: 83%): mp: 199-200°C,1H-NMR (200 MHz, DMSO-d6): 6.80-6.87 (t, J = 7.2 Hz, 2H), 6.96 (s, 2H), 7.05-7.13 (m, 8H), 7.21-7.29 (m, 4H), 7.43 (d, J = 8.6 Hz, 4H), 8.29 (s, 2H) ppm;13C-NMR (50 MHz, DMSO-d6): 116.7, 117.2, 120.0, 125.3, 127.3, 129.3, 129.4, 142.7, 143.3 ppm; IR (KBr): 1600, 3025, 3397 cm-1; Anal. Calcd for C26H22N2: C, 86.15, H, 6.12, N, 7.73. Found: C, 85.80, H, 6.15, N, 7.67.
ACKNOWLEDGMENT
Financial support for this research was provided by the National Science Council of Taiwan, ROC. We greatly appreciate Professor C. K. Lai (NCU) for allowing use of his DSC apparatus.
Received July 28, 2006.
REFERENCES
1. Görner, H.; Kuhn, H. J. Adv. Photochem. 1995, 19, 1-117. 2. Waldeck, D. H. Chem. Rev. 1991, 91, 415-436.
3. (a) Saltiel, J.; Waller, A. S.; Sears, D. F., Jr.; Hoburg, E. A.; Zeglinski, D. M.; Waldeck, D. H. J. Phys. Chem. 1994, 98, 10689-10698. (b) Saltiel, J.; Waller, A. S.; Sears, D. F., Jr.; Garrett, C. Z. J. Phys. Chem. 1993, 97, 2516-2522. (c) Saltiel, J.; Sun, Y.-P. Photochromism, Molecules and
Sys-tems; Dürr, H.; Bouas-Laurent, H., Eds.; Elsevier:
Amster-dam, 1990; pp 64-164. (d) Saltiel, J.; Charlton, J. L.
Rear-rangements in Ground and Excited States; de Mayo, P., Ed.;
Academic Press: New York, 1980; Vol. 3, pp 25-89. 4. Lewis, F. D.; Weigel, W.; Zuo, X. J. Phys. Chem. A. 2001,
105, 4691-4696.
5. Papper, V.; Pines, D.; Likhtenshtein, G.; Pines, E. J. Photochem. Photobio. A. Chem. 1997, 111, 87-96.
6. Smit, K. J.; Ghiggino, K. P. Chem. Phys. Lett. 1985, 122, 369-374.
7. Zeglinski, D. M.; Waldeck, D. H. J. Phys. Chem. 1988, 92, 692-701.
8. Meier, H. Angew. Chem. Int. Ed. Engl. 1992, 31, 1399-1420, and references cited therein.
9. Bokeriya, É. N.; Viktorova, V. S.; Karegishvili, L. I.; Kovyrzina, K. A.; Kushakevich, Y. P.; Radaikina, L. A. J.
Org. Chem. USSR (Engl. Transl.), 1979, 15, 1944-1949.
10. Nakatsuji, S.; Matsuda, K.; Uesugi, Y.; Nakashima, K. Akiyama, S.; Katzer, G.; Fabian, W. J. Chem. Soc. Perkin
Trans. 2 1991, 861-867.
11. Tan, X.; Gustafson, T. L. J. Phys. Chem. A 2000, 104, 4469-4474.
12. Saltiel, J.; Sears, D. F., Jr.; Choi, J.-O.; Sun, Y.-P.; Eaker, D. W. J. Phys. Chem. 1994, 98, 35-46 and references cited therein.
13. (a) Lewis, F. D.; Yang, J.-S. J. Am. Chem. Soc. 1997, 119, 3834-3835 (b) Lewis, F. D.; Kalgutkar, R. S.; Yang, J.-S. J.
Am. Chem. Soc. 1999, 121, 12045-12053. (c) Lewis, F. D.;
Weigel, W. J. Phys. Chem. A. 2000, 104, 8146-8153. 14. (a) Yang, J.-S.; Chiou, S.-Y.; Liau, K.-L. J. Am. Chem. Soc.
2002, 124, 2518-2527. (b) Yang, J.-S.; Liau, K.-L.; Wang, C.-M.; Hwang, C.-Y. J. Am. Chem. Soc. 2004, 126, 12325-12335.
15. (a) Adachi, C.; Tsutsui, T.; Saito, S. Appl. Phys. Lett. 1990,
56, 799-801. (b) Adachi, C.; Tsutsui, T.; Saito, S. Appl. Phys. Lett. 1990, 56, 531-533. (c) Adachi, C.; Tsutsui, T.; Saito, S. Appl. Phys. Lett. 1989, 55, 1489-1491. (d) Kitamura, T.;
Yokoyama, M. J. Appl. Phys. 1991, 69, 821-826.
16. Ko, C.-W.; Tao, Y.-T.; Danel, A.; Krzemiñska, L.; Tomasik, P. Chem. Mater. 2001, 13, 2441-2446.
17. Li, C.-L.; Shieh, S.-J.; Lin, S.-C.; Liu, R.-S. Org. Lett. 2003,
5, 1131-1134.
18. Chen, C.-T.; Chiang, C.-L.; Lin, Y.-C.; Chan, L.-H.; Huang, C.-H.; Tsai, Z.-W.; Chen, C.-T. Org. Lett. 2003, 5,
1261-1264 and references cited therein.
19. Lincker, F.; Bourgun, P.; Masson, P.; Didier, P.; Guidoni, L.; Bigot, J.-Y.; Nicoud, J.-F.; Donnio, B.; Guillon, D. Org. Lett. 2005, 7, 1505-1508.
20. Tang, R.; Tan, Z.; Li, Y.; Xi, F. Chem. Mater. 2006, 18, 1053-1061.
21. (a) Yang, J.-S.; Swager, T. M. J. Am. Chem. Soc. 1998, 120, 5321-5322. (b) Yang, J.-S.; Swager, T. M. J. Am. Chem. Soc. 1998, 120, 11864-11873.
22. Sakurovs, R.; Ghiggino, K. P. Aust. J. Chem. 1981, 34, 1367-1372.
23. (a) Wolfe, J. P.; Wagaw, S.; Marcoux, J.-F.; Buchwald, S. L.
Acc. Chem. Res. 1998, 31, 805-818. (b) Hartwig, J. F. Angew. Chem. Int. Ed. 1998, 37, 2046-2067.
24. Wadsworth, W. S., Jr. Org. React. 1977, 25, 73-253. 25. Barlett, P. D.; Ryan, M. M.; Cohen, S. G. J. Am. Chem. Soc.
1942, 64, 2649-2653.
26. Yang, J.-S.; Lee, C.-C.; Yau, S.-L.; Chang, C.-C.; Lee, C.-C.; Leu, J.-M. J. Org. Chem. 2000, 65, 871-877.
27. Sankararaman, S.; Haney, W. A.; Kochi, J. K. J. Am. Chem.
Soc. 1987, 109, 5235-5249.
28. Bellamy, F. D.; Ou, K. Tetrahedron Lett. 1984, 25, 839-842. 29. (a) Yang, J.-S.; Wang, C.-M.; Hwang, C.-Y.; Liau, K.-L.;
Chiou, S.-Y. Photochem. Photobiol. Sci. 2003, 2, 1225-1231. (b) Yang, J.-S.; Liau, K.-L.; Hwang, C.-Y.; Wang, C.-M. J. Phys. Chem. A 2006, 110, 8003-8010.
30. Zanello, P.; de Biani, F. F.; Glidewell, C.; Koenig, J.; Marsh, S. J. Polyhedron 1998, 17, 1795-1801.
31. Yang, J.-S.; Liau, K.-L.; Tu, C.-W.; Hwang, C.-Y. J. Phys.
Chem. A 2005, 109, 6450-6456.
32. Donley, C. L.; Zaumseil, J.; Andreasen, J. W.; Nielsen, M. M.; Sirringhaus, H.; Friend, R. H.; Kim, J.-S. J. Am. Chem.
Soc. 2005, 127, 12890-12899.
33. Kim, J.; Levitsky, I. A.; McQuade, D. T.; Swager, T. M. J.
Am. Chem. Soc. 2002, 124, 7710-7718.
34. Yang, J.-S.; Yan, J.-L.; Hwang, C.-Y.; Chiou, S.-Y.; Liau, K.-L.; Tsai, H.-H. G.; Lee, G.-H.; Peng, S.-M. J. Am. Chem.
Soc. 2006, 128, 14109-14119.
35. (a) Bedworth, P. V.; Cai, Y.; Jen, A.; Marder, S. R. J. Org.
Chem. 1996, 61, 2242-2246. (b) Verbiest, T.; Burland, D. M.;
Jurich, M. C.; Lee, V. Y.; Miller, R. D.; Volksen, W. Science 1995, 268, 1604-1606. (c) Gilmour, S.; Montgomery, R. A.; Marder, S. R.; Cheng, L.-T.; Jen, A. K.-Y.; Cai, Y.; Perry, J. W.; Dalton, L. R. Chem. Mater. 1994, 6, 1603-1604. (d) Marder, S. R.; Perry, J. W. Science 1994, 263, 1706-1707. 36. Zhou, Q.; Swager, T. M. J. Org. Chem. 1995, 60, 7096-7100. 37. Dawson, W. R.; Windsor, M. W. J. Phys. Chem. 1968, 72,
3251-3260.
38. Osaheni, J. A.; Jenekhe, S. A. J. Am. Chem. Soc. 1995, 117, 7389-7398.
39. Janben, C. E.; Kraus, N. Eur. J. Org. Chem. 2005, 2322-2329.