Liquid-Phase Combinatorial Synthesis of
1,4-Benzodiazepine-2,5-diones as the Candidates of Endothelin
Receptor Antagonism
Ming-Fu Cheng and Jim-Min Fang*
Department of Chemistry, National Taiwan UniVersity, Taipei, Taiwan 106 ReceiVed April 1, 2003
A library of 1,4-benzodiazepine-2,5-dione dicarboxylate derivatives containing aryl substituents at N1- and
N4-positions to mimic the amino acid residues of Try-13, Phe-14, and Asp-18 in endothelin-1 is established
by using the starting materials of R-amino esters, hydroxybenzaldehydes, nitrobenzoyl chlorides, and benzyl bromides in a polyethylene resin-bound liquid-phase synthesis. All of the six synthetic steps are conducted under mild conditions to give the desired products with reasonable yields and purity. The poly(ethylene glycol) support plays as a part of ester linkage that is released at the final step.
Introduction
Endothelins are a group of isopeptides locally produced
in various cell types under different physiological stimuli.1
Human endothelin-1 (ET-1) is a 21 amino acid peptide that exhibits a potent vasoconstrictor activity, conceivably through
its selective interaction with specific receptor subtypes.1
Antagonism on the vasoconstrictor endothelin is a potential approach to the treatment of a variety of human diseases including ischemia, hypertension, congestive heart failure,
pulmonary hypertension, and subarachnoid hemorrhage.1
Among numerous endothelin receptor antagonists,2an indan
derivative SB2096703 (Figure 1) possesses two phenyl
substituents to mimic the amino acid residues of Try-13 and Phe-14 in ET-1. The two carboxylic groups in SB209670 also mimic the Asp-18 residue and the C terminus of ET-1,
which ligates the Zn2+ ion on binding with endothelin
receptor.1
In the process of searching for the nonpeptide endothelin antagonists, we speculated that 1,4-benzodiazepine-2,5-dione derivatives bearing appropriate substituents might serve for this purpose. The benzodiazepine core provides a nearly planar platform as that of the indan ring in SB209670. The
N1- and N4-positions may be implanted with the desired aryl
substituents. For a better binding affinity with the endothelin receptors, carboxylic groups and other substituents may be introduced to various sites of the benzodiazepine scaffold. 1,4-Benzodiazepine-2,5-dione and its analogues represent
an important class of bioactive molecules.4These compounds
show remarkable potency in various biological targets, including antithrombotics, antibiotics, and antitumor
activi-ties.4 Many efforts have been exerted on the synthesis of
this class of bioactive compounds.4,5 The combinatorial or
parallel synthetic approaches are especially noted (Figure 2).5
Using polymer-bound reagents, the combinatorial chemistry
has become an efficient tool to build small molecule libraries
for accelerating the drug discovery process.6
Ellman and co-workers have utilized strategy A to construct a library of 1,4-benzodiazepine-2,5-diones from three components of anthranilic acids, R-amino esters, and
alkylating agents.5a,b Merrifield resin is derivatized by
alkylation with the sodium salt of 4-hydroxy-2,6-dimethoxy-benzaldehyde, and the resin-bound aldehyde (A3) is linked to R-amino esters via reductive amination. Amidation of these resin-bound amino esters with anthranilic acids, followed by cyclization and alkylation, leads to the polymer-bound 1,4-benzodiazepine-2,5-dione derivatives (A1). Treatment of A1
with CF3CO2H/Me2S/H2O releases the solid support to give
1,4-benzodiazepine-2,5-diones without substitution at the N4
-position. This solid-phase synthesis thus successfully pro-vides a library of 2508 members. This study does not mention
the further alkylation at N4-position, which may cause the
general problem of regioselectivity in the N- or O-alkylations
in an amide moiety.7
In the strategy devised by Goff and co-workers,5cR-amino
esters (B4) are first reacted with the resin-bound bromo-acetate reagent B3 and then subjected to amidation with 2-azidobenzoyl chlorides (B2). The azido group is reduced
by Ph3P (Staudinger reaction), and the intermediate is heated
to 130°C to effect an aza-Wittig reaction. Hydrolysis of the
* To whom correspondence should be addressed. E-mail: jmfang@ ccms.ntu.edu.tw.
Figure 1. 1,4-Benzodiazepine-2,5-dione derivatives bearing
car-boxylic groups are designed to mimic the endothelin receptor antagonist SB209670, which contains two aryl substituents flanking the nearly planar core of indan ring.
10.1021/cc030034d CCC: $27.50 © 2004 American Chemical Society Published on Web 10/25/2003
Downloaded by NATIONAL TAIWAN UNIV on August 27, 2009 | http://pubs.acs.org
iminoether intermediate and cleavage of the Rink resin are
achieved concomitantly by treatment with an acid (CF3CO2H/
H2O) to afford the desired 1,4-benzodiazepine-2,5-diones.
The R-amino esters derivatized with Wang resin (C4) are used by Mayer and co-workers to react with 2-nitrobenzoic acid or Fmoc-protected anthranilic acid, giving amides C2
(strategy C).5dThis synthetic strategy has an advantage on
the concurrent release of the resin support during the cyclization step to form 1,4-benzodiazepine-2,5-diones.
Strategy D shows the synthetic routes carried out by the
research groups of Hulme5eand Kennedy.5fThe Wang
resin-bound isonitriles (D6) are applied to combine with appropri-ate amines, aldehydes, and anthranilic acids (Ugi four component reaction), giving diamides D2. The terminal amide group (bound to Wang resin) is selectively activated as imidates, which can be substituted by MeONa to give the corresponding methyl esters for a subsequent lactamiza-tion to give 1,4-benzodiazepine-2,5-diones.
All of the above-mentioned methods reside on using solid supports (Merrifield, Rink, and Wang resins) to achieve the
combinatorial syntheses of 1,4-benzodiazepine-2,5-diones. In our present study (strategy E), we considered using the
liquid-phase synthesis6n-v as an alternative method to
construct a library of 1,4-benzodiazepine-2,5-diones. In comparison with solid-phase synthesis, the liquid-phase synthesis is conducted in a homogeneous organic media. Thus, monitoring the progress of poly(ethylene glycol) (PEG)-bound liquid-phase reaction and analysis of the product mixture becomes feasible.
As the target 1,4-benzodiazepine-2,5-dione requires an N4
-aryl substituent with a carboxylic group to mimic the Asp-18 residue of ET-1, we deliberately used the PEG-bound benzaldehydes E3 to couple with R-amino esters E5. By this means, the required carboxylic group can be revealed at the final step along with removal of the PEG support. We demonstrate herein this strategy by a liquid-phase syn-thesis of a small library of 1,4-benzodiazepine-2,5-diones
(10a-p) using PEG5000 monomethyl ether as the support
(Scheme 1).
Figure 2. Strategies for combinatorial syntheses of 1,4-benzodiazepine-2,5-dione libraries. Strategy A, see refs 5a,b; strategy B, see ref 5c;
strategy C, see ref 5d; strategy D, see refs 5e,f; and strategy E, our present study.
Downloaded by NATIONAL TAIWAN UNIV on August 27, 2009 | http://pubs.acs.org
Results and Discussion
The substitution reaction of methyl bromoacetate with m-hydroxybenzaldehyde (1a) or p-hydroxybenzaldehyde (1b)
was realized in the presence of K2CO3. After saponification,
the resulting carboxylic acids were reacted with
MeO-PEG-OH5000 resin to form the PEG-bound esters by using
di-cyclohexylcarbodiimide (DCC) as the activation and dehy-dration agent. The PEG-bound benzaldehydes 2a,b were sub-jected to reductive amination with R-amino esters [aspartic acid dimethyl ester (3a) and valine methyl ester (3b) in this study]. According to the elemental analysis of nitrogen content, the loading of amino esters to the PEG support was nearly quantitative (91-99% of six measurements). The N-(MeO-PEG-bound) amino acid methyl esters 4a-d reacted with 2-nitrobenzoyl chloride (5a) or 4,5-dimethyl-2-nitro-benzoyl chloride (5b) to afford the PEG-bound amides 6a-h. The nitro groups in 6a-d were smoothly reduced by
zinc powder (HOAc, room temperature, 2 h), and the
resulting anilines were treated with CF3CO2H/CF3CO2Na at
room temperature for 13 h to give the PEG-bound 1,4-benzodiazepine-2,5-diones 7a-h. For the more reactive substrates 6e-h, reduction of the nitro group and the subsequent cyclization were conducted in a one pot procedure (zinc powder, HOAc, room temperature, 2 h) to give the PEG-bound benzodiazepines 7e-h. Alkylation of 7a-h with benzyl bromide (8a) or 3-methoxybenzyl bromide (8b) was
achieved in DMF solution by using Cs2CO3as a base, instead
of lithium acetanilide used in strategy A.5a,bOn treatment of
9a-p with sodium methoxide in methanol at room temper-ature for a short period (<5 min), the PEG support was cleaved in a very efficient manner.
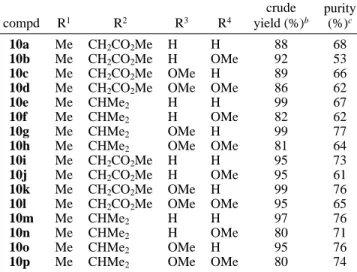
Thus, 16 1,4-benzodiazepine-2,5-dione dimethyl ester derivatives 10a-p were obtained in 80-99% crude yields by a liquid-phase combinatorial synthesis (Table 1). The high-performance liquid chromatography (HPLC) analyses indicated that the purity of 10a-p, prepared from the PEG-bound benzaldehydes 2a,b in six steps, is around 53-76% (corresponding to 88-95% average yield in each synthetic step). This liquid-phase synthesis also showed some notable features. For example, cyclization of 6a-h to 1,4-benzo-diazepine-2,5-diones 7a-h was facilitated by the presence
of the N4-substituents, which enhanced the cis/trans ratio
about these tertiary amides.8All six steps were conducted
under mild conditions (short reaction time at room temper-ature without harsh acid or base) to minimize any possible
racemerization.5bIn this synthetic sequence, the PEG support
played as not only a capture resin but also a part of ester
linkage that was released at the final step.6i As the PEG
support was readily cleaved by treatment with MeONa in MeOH for 10 s, the progress of most reactions (giving compounds 4, 6, 9, and 10) was easily monitored by thin-layer chromatography (TLC) analyses. The final products 10a-p were simply purified by silica gel chromatography Scheme 1. Liquid-Phase Synthesis of
1,4-Benzodiazepine-2,5-dione Dimethyl Ester Derivatives in
This Studya
aReagents and conditions: (i) Methyl bromoacetate, K2CO3, CH3CN,
reflux 4 h, 98%. (ii) 1 N NaOH(aq), THF, room temperature, 2 h, 90%. (iii) Monomethoxy-PEG-OH5000, DCC, DMAP, CH2Cl2, room temperature, 23 h. (iv)L-Aspartic acid dimethyl ester hydrochloride orL-valine methyl ester hydrochloride, NaBH(OAc)3, NaOAc, CH2Cl2, room temperature, 5 h. (v) 2-Nitrobenzoyl chloride or 4,5-dimethoxy-2-nitrobenzoyl chloride, Bu4NI, K2CO3, CH2Cl2, room temperature, 5 h. (vi) Zn, HOAc, room temperature, 2 h. (vii) CF3COOH, CF3COONa, CH3CN, room temperature, 13 h. (viii) Benzyl bromide or 3-methoxybenzyl bromide, Cs2CO3, DMF, room temperature, 2 h. (ix) Na2CO3, MeOH, room temperature, 5 min.
Table 1. Yields and Purities of the
1,4-Benzodiazepine-2,5-dione Dimethyl Ester Derivatives Prepared in This Study (Scheme 1)a
compd R1 R2 R3 R4 crude yield (%)b purity (%)c 10a Me CH2CO2Me H H 88 68 10b Me CH2CO2Me H OMe 92 53 10c Me CH2CO2Me OMe H 89 66 10d Me CH2CO2Me OMe OMe 86 62 10e Me CHMe2 H H 99 67 10f Me CHMe2 H OMe 82 62 10g Me CHMe2 OMe H 99 77
10h Me CHMe2 OMe OMe 81 64
10i Me CH2CO2Me H H 95 73 10j Me CH2CO2Me H OMe 95 61 10k Me CH2CO2Me OMe H 99 76 10l Me CH2CO2Me OMe OMe 95 65 10m Me CHMe2 H H 97 76 10n Me CHMe2 H OMe 80 71
10o Me CHMe2 OMe H 95 76
10p Me CHMe2 OMe OMe 80 74
aCompounds 10a-h Belong to the meta-series, whereas com-pounds 10i-p Belong to para-series.bThe crude yield of 10a-p in six step liquid-phase reactions (steps iv-ix).cThe purity of
10a-p after cleavage of the PEG support with Na2CO3.
Downloaded by NATIONAL TAIWAN UNIV on August 27, 2009 | http://pubs.acs.org
and fully characterized by physical and spectroscopic methods.
The interaction of ET-1 with endothelin receptor is coupled
to the increase of intracellular Ca2+concentration ([Ca2+]
i), a consequence of multistep biological events initiated by
G-protein.9According to our preliminary examination, some
dicarboxylic acid analogues derived from the dimethyl esters (10a-p) showed significant inhibition against the ET-1
induced [Ca2+]
iincrease in the transfected Chinese hamster
ovary cells. We are currently engaged in the biological evaluation of various 1,4-benzodiazepine-2,5-diones as the endothelin receptor antagonists.
Experimental Section
Melting points are uncorrected. 1H NMR spectra were
recorded at 300 or 400 MHz;13C NMR spectra were recorded
at 75 or 100 MHz. CDCl3(δΗ) 7.24 and δC) 77.0 (central
line of triplet)) was used as an internal standard in1H and
13C NMR spectra, unless otherwise stated. Mass spectra were
recorded at an ionizing voltage of 70 or 20 eV. HPLC (Hewlett-Packard 1100) analysis was performed on a vp250/
10 Nucleosil 100-7 column (25 cm× 1 cm i.d.) with UV
detection atλ ) 254 nm using the eluents of EtOAc/hexane
(3:2 or 4:1) at a flow rate of 1 mL/min. Merck silica gel 60F sheets were used for analytical TLC. Merck silica gel
60F glass plates (20 cm× 20 cm with 2 mm thickness) were
used for preparative TLC. Column chromatography was performed on silica gel (70-230 mesh) using gradients of EtOAc/hexane as eluents. Tetrahydrofuran (THF) was
dis-tilled from sodium benzophenone ketyl under N2.
Mono-methoxy-PEG-OH was dried by azeotropical removal of water with refluxing acetonitrile.
Methyl (3-Formylphenoxy)acetate and Methyl (4-Formylphenoxy)acetate. A mixture of 3-hydroxybenzalde-hyde (or 4-hydroxybenzalde3-hydroxybenzalde-hyde, 10.0 g, 81.9 mmol) and
K2CO3 (34.0 g, 245.7 mmol) in CH3CN was stirred under
reflux. After 1 h, methyl bromoacetate was added, and the mixture was allowed to stir under reflux for another 3 h.
After the reaction was finished, CH3CN was removed, and
the residue was extracted with EtOAc (40 mL × 3). The
organic layer was dried over MgSO4and evaporated to give
methyl (3-formylphenoxy)acetate [or methyl (4-formyl-phenoxy)acetate] (15.6 g, 80.3 mmol, 98%) as a yellow oil without purification.
(3-Formylphenoxy)acetic Acid and (4-Formylphenoxy)-acetic Acid. To a solution of (3-formylphenoxy)acetate [or methyl (4-formylphenoxy)acetate] (14.9 g, 76.8 mmol) in
THF (75 mL) was added 1 N NaOH(aq)(45 mL) at 0°C for
30 min. The reaction mixture was allowed to stir at room temperature for 1.5 h. After the reaction was completed, 1
N NH4Cl(aq) (65 mL) was added, and the mixture was
extracted with EtOAc (40 mL× 5). The organic layer was
dried over Na2SO4and purification by recrystallization from
EtOAc to give (3-formylphenoxy)acetic acid [or (4-formyl-phenoxy)acetic acid] (12.4 g, 69.1 mmol) as colorless solids. Methoxy-PEG (3-Formylphenoxy)acetate (2a) and Meth-oxy-PEG (4-Formylphenoxy)acetate (2b). A solution of
methoxy-PEG-OH5000 (15.0 g, 3.0 mmol) was treated with
DCC (1.9 g, 9.0 mmol), DMAP (367 mg, 3.0 mmol), and
(3-formylphenoxy)acetic acid [or (4-formylphenoxy)acetic
acid, 1.6 g, 9.0 mmol] in CH3CN (75 mL) at room
temperature for 23 h. The mixture was filtered through Celite, and the filtrate was concentrated to about 15 mL. The residue
was cooled in an ice bath and triturated with Et2O (105 mL)
to give precipitates, which were filtered and washed
suc-cessively with Et2O/2-propanol (1:1, 60 mL). White powders
of PEG-bound ester 2a (or 2b) were obtained by drying under reduced pressure.
N-(PEG-Bound) Amino Acid Methyl Esters (4a-d). A
mixture ofL-aspartic acid dimethyl ester hydrochloride (3a,
553.3 mg, 2.8 mmol) [orL-valine methyl ester hydrochloride
(3b, 469.4 mg, 2.8 mmol)] and PEG-bound ester 2a (or 2b)
(7.0 g, 1.4 mmol) in CH2Cl2 (40 mL) was treated with
sodium triacetoxyborohydride (918 mg, 4.2 mmol) and
NaOAc (230 mg, 2.8 mmol) at 0°C. The suspension was
allowed to warm to room temperature and stirred for 5 h. The progress of reaction was monitored by TLC analysis (EtOAc/hexane, 3:2) of aliquots, which were treated with NaOMe/MeOH for 10 s before analysis. After the reaction was completed, brine (30 mL) was added, and the mixture
was extracted with CH2Cl2(40 mL× 3). The organic layer
was dried over MgSO4and concentrated to about 7 mL. The
residue was triturated with Et2O (50 mL) to give precipitates.
The mixture was cooled in an ice bath; the precipitates were
filtered and washed successively with Et2O/2-propanol (1:
1, 30 mL) to give 4a-d. Elemental analysis (six measure-ments) showed a nitrogen content of 0.24-0.27%, equivalent to a loading of 0.182 mmol/g on average (91-99%).
Linkage of N-(PEG-Bound) Amino Acid Methyl Esters 4a-d with 2-Nitrobenzoyl Chloride or 4,5-Dimethyl-2-nitrobenzoyl Chloride, Giving Amides 6a-h. To a solution of N-(PEG-bound) amino acid methyl ester 4a (or 4c,d) (3.0
g, 0.6 mmol) in CH2Cl2 (20 mL) was added K2CO3 (312
mg, 2.4 mmol) and Bu4NI (111 mg, 0.3 mmol).
2-Nitro-benzoyl chloride (5a, 0.17 mL, 1.2 mmol) [or 4,5-dimethyl-2-nitrobenzoyl chloride (5b, 295 mg, 1.2 mmol)] was then added dropwise into the mixture. The mixture was stirred at room temperature, and the progress of reaction was moni-tored by TLC analysis (EtOAc/hexane, 3:2) of aliquots, which were treated with NaOMe/MeOH for 10 s before analysis. After 5 h, the mixture was filtered through Celite, and the filtrate was concentrated to about 5 mL. The residue
was cooled in an ice bath and triturated with Et2O (30 mL)
to give precipitates, which were filtered and washed
suc-cessively with Et2O/2-propanol (1:1, 25 mL). Yellow
pow-ders of PEG-bound amide 6a (or 6b-h) were obtained by drying under reduced pressure.
PEG-Bound Benzodiazepines 7a-h. PEG-bound amide 6a (or 6b-d) (3.0 g, 0.6 mmol) was dissolved in HOAc (20 mL) and treated with zinc powder (392 mg, 6 mmol) at room temperature for 2 h. The suspension was filtered through Celite, and the filtrate was evaporated under reduced pressure to give a thick mass of PEG-bound anilines without further purification.
To a solution of the PEG-bound anilines (3.0 g, 0.6 mmol)
in CH3CN (60 mL) was added trifluoroacetic acid (1.5 mL)
and its sodium salt (600 mg) at room temperature. The
mixture was stirred for 13 h, after which CH3CN was
Downloaded by NATIONAL TAIWAN UNIV on August 27, 2009 | http://pubs.acs.org
removed under reduced pressure. After addition of brine (25
mL), the mixture was extracted with CH2Cl2(25 mL× 3).
The organic layer was dried over MgSO4and concentrated
to about 5 mL. The residue was cooled in an ice bath and
triturated with Et2O (30 mL) to give precipitates. The
precipitates were filtered and washed successively with Et2O/
2-propanol (1:1, 30 mL) to give light yellow solids of the PEG-bound benzodiazepines 7a (or 7b-d). For the more reactive substrates 6e-h, reduction of the nitro group and the subsequent cyclization were conducted in a one pot procedure (zinc powder, HOAc, room temperature, 2 h) to give the PEG-bound benzodiazepines 7e-h.
Alkylation of PEG-Bound Benzodiazepines 7a-h, Giv-ing Benzodiazepines 9a-p. To a mixture of PEG-bound
benzodiazepines 7a-h (1.5 g, 0.3 mmol) and Cs2CO3(1.5
g, 4.5 mmol) in DMF (15 mL) was added benzyl bromide (8a, 0.7 mL, 6.0 mmol) [or 3-methoxybenzyl bromide (8b, 0.9 mL, 6.0 mmol)] at room temperature. The progress of reaction was monitored by TLC analysis (EtOAc/hexane, 3:2) of aliquots, which were treated with NaOMe/MeOH for 10 s before analysis. The reaction was completed in 2 h, and DMF was then removed under reduced pressure. After addition of brine (10 mL), the mixture was extracted with
CH2Cl2 (10 mL × 3). The organic layer was dried over
MgSO4and concentrated to about 5 mL. The residue was
cooled in an ice bath and triturated with Et2O (15 mL) to
give precipitates, which were filtered and washed
succes-sively with Et2O/2-propanol (1:1, 20 mL). Light yellow
powders of the PEG-bound benzodiazepines 9a-p were obtained by drying under reduced pressure.
1,4-Benzodiazepine-2,5-dione Dimethyl Ester Deriva-tives 10a-p. The PEG-bound benzodiazepine (9a-p, 1.5 g, 0.3 mmol) was dissolved in MeOH (5 mL) and treated
with Na2CO3(31.7 mg, 0.3 mmol) at room temperature. The
progress of reaction was monitored by TLC analysis (EtOAc/ hexane ) 3:2) of aliquots, which were treated with NaOMe/ MeOH for 10 s before analysis. After the reaction was completed (stirring for 5 min), the mixture was cooled in an
ice bath and triturated with Et2O (5 mL) to give precipitates
of MeO-PEG resin. The mixture was filtered through Celite, and the filtrate was concentrated under reduced pressure to give a crude product of 1,4-benzodiazepine-2,5-diones (10a-p). The purity of these crude products was determined by HPLC analyses. Purification of the crude product was carried out by preparative TLC by elution with gradients of EtOAc/ hexane (2:3 to 3:2).
1-Benzyl-3-(methoxycarbonyl)methyl-4-[3-(methoxy-carbonyl)methoxybenzyl]-1,4-benzodiazepine-2,5-dione (10a). Colorless solid, mp ) 52.0-54.0°C; [R]22
D) +61.8 (CHCl3, c ) 0.59). IR (KBr): 2958, 1740, 1648 cm-1.1H NMR (CDCl3, 400 MHz): δ 7.92 (1H, d, J ) 7.6 Hz), 7.45-7.41 (1H, m), 7.32-7.19 (6H, m), 7.13 (2H, d, J ) 7.2 Hz), 6.90 (1H, d, J ) 7.2 Hz), 6.86 (1H, s), 6.77 (1H, d, J ) 8.0 Hz), 5.22 (1H, d, J ) 16.0 Hz), 5.13 (1H, d, J ) 16.0 Hz), 4.91 (1H, d, J ) 16.0 Hz), 4.80 (1H, dd, J ) 10.4 Hz, 4.4 Hz), 4.60 (2H, s), 4.31 (1H, d, J ) 16.0 Hz), 3.78 (3H, s), 3.56 (3H, s), 3.26 (1H, dd, J ) 16.8 Hz, 10.4 Hz), 2.64 (1H, dd, J ) 16.8 Hz, 4.4 Hz).13C NMR (CDCl 3, 100 MHz): δ 170.5 (C), 169.3 (C), 168.8 (C), 168.6 (C), 158.2 (C), 140.1 (C), 139.5 (C), 136.6 (C), 132.5 (CH), 130.9 (CH), 129.9 (CH), 129.3 (C), 128.8 (2× CH), 127.3 (CH), 126.5 (2 × CH), 126.2 (CH), 121.4 (CH), 120.5 (CH), 113.7 (CH), 113.3 (CH), 65.3 (CH2), 53.0 (CH), 52.3 (CH3), 52.1 (CH3), 51.7 (CH2), 47.0 (CH2), 31.9 (CH2). HRMS calcd for C29H28N2O7 (M + H+), 517.1975; found, 517.1978.
Acknowledgment. We thank the National Science Coun-cil for financial support.
Supporting Information Available. Physical and spec-tral data of compounds 10b-p. This material is available free of charge via the Internet at http://pubs.acs.org. References and Notes
(1) For recent reviews of endothelin-1, endothelin receptors, and biological activities, see (a) Ohlstein, E. H.; Ruffolo, R. R., Jr. In Endothelin Receptors; Ruffolo, R. R., Jr., Ed.; CRC: Boca Raton, FL, 1995; 1-14. (b) Ohlstein, E. H.; Elliott, J. D.; Feuerstein, G. Z.; Ruffolo, R. R., Jr. Med. Res. ReV. 1996, 16, 365-390. (c) Mateo, A. O.; De Artinano, A. A. Pharmacol. Res. 1997, 36, 339-351. (d) Webb, M. L.; Krystek, S. R., Jr. In Endothelin Receptor Signaling Mech-anisms; Pollock, D. M., Highsmith, R. F., Eds.; Springer: Berlin, 1998; pp 67-88. (e) Masaki, T.; Ninomiya, H.; Sakamoto, A.; Okamoto, Y. Mol. Cell. Biochem. 1999, 190, 153-156. (f) Hlavacek, J.; Marcova, R. Collect. Czech. Chem. Commun. 1999, 64, 1211-1252. (g) Goldie, R. G. Clin. Exp. Pharmacol. Physiol. 1999, 26, 145-148. (h) Schiffrin, E. L. Am. J. Hypertension 2001, 14, 83S-89S. (i) Ergul, A. Pharmacotherapy 2002, 22, 54-65. (j) Green-berg, B. H. CongestiVe Heart Failure 2002, 8, 257-261. (2) For recent reviews for the synthetic antagonists of endothelin
receptors, see (a) Doherty, A. M. In Chemical Structures Approaches to Rational Drug Design; Weiner, D. B., Williams, W. V., Eds.; CRC: Boca Raton, FL, 1995; pp 85-123. (b) Elliott, J. D.; Lago, M. A.; Peishoff, C. E. In Endothelin Receptors; Ruffolo, R. R., Jr., Ed.; CRC: Boca Raton, FL, 1995; pp 79-107. (c) Walsh, T. F. Annu. Rep. Med. Chem. 1995, 30, 91-100. (d) Filep, J. G. Drugs Today
1995, 31, 155-171. (e) Doherty, A. M. Drug DiscoVery
Today 1996, 1, 60-70. (f) Cheng, X.-M.; Ahn, K.; Haleen, S. J. Annu. Rep. Med. Chem. 1997, 32, 61-70. (g) Webb, M. L.; Meek, T. D. Med. Res. ReV. 1997, 17, 17. (h) Tasker, A. S.; Pollock, D. M. In Endothelin Receptor Signaling Mechanisms; Pollock, D. M., Highsmith, R. F., Eds.; Springer: Berlin, 1998; pp 3-15. (i) Elliott, J. D.; Ohlstein, E. H.; Peishoff, C. E.; Ellens, H. M.; Lago, M. A. Pharm. Biotechnol. 1998, 11, 113-129. (j) Benigni, A.; Remuzzi, G. Lancet 1999, 353, 133-138. (k) Dao, H. H.; Moreau, P. Exp. Opin. InVest. Drugs 1999, 8, 1807-1821. (l) Goddard, J.; Webb, D. J. Drugs R&D 1999, 2, 1-12. (m) Roux, S.; Breu, V.; Ertel, S. I.; Clozel, M. J. Mol. Med. 1999, 77, 364-376. (n) Subbiah, V.; Katwa, L. Drugs 2000, 3, 190-197. (o) Boss, C.; Bolli, M.; Weller, T. Curr. Med. Chem. 2002, 9, 349-383. (p) Dasgupta, F.; Mukherjee, A. K.; Gangadhar, N. Curr. Med. Chem. 2002, 9, 549-575.
(3) For SB209670 and the related endothelin receptor antago-nists, see (a) Elliott, J. D.; Lago, M. A.; Cousins, R. D.; Gao, A.; Leber, J. D.; Erhard, K. F.; Nambi, P.; Elshourbagy, N. A.; Kumar, C.; Lee, J. A.; Bean, J. W.; DeBrosse, C. W.; Eggleston, D. S.; Brooks, D. P.; Feuerstein, G.; Ruffolo, R. R.; Weinstock, J.; Gleason, J. G.; Peishoff, C. E.; Ohlstein, E. H. J. Med. Chem. 1994, 37, 1553-1557. (b) Clark, W. M.; Tickner-Eldridge, A. M.; Huang, G. K.; Pridgen, L. N.; Olsen, M. A.; Mills, R. J.; Lantos, I.; Baine, N. H. J. Am. Chem. Soc. 1998, 120, 4550-4551. (c) Clark, W. M. Curr. Opin. Drug DiscoVery DeV. 1999, 2, 565-577. (d) Song, Z. J.; Zhao, M.; Desmond, R.; Devine, P.; Tschaen, D. M.;
Downloaded by NATIONAL TAIWAN UNIV on August 27, 2009 | http://pubs.acs.org
Tillyer, R.; Frey, L.; Heid, R.; Xu, F.; Foster, B.; Li, J.; Reamer, R.; Volante, R.; Grabowski, E. J. J.; Dolling, U. H.; Reider, P. J.; Okada, S.; Kato, Y.; Mano, E. J. Org. Chem. 1999, 64, 9658-9667. (e) Zhang, J.; Didierlaurent, S.; Fortin, M.; Lefrancois, D.; Uridat, E.; Vevert, J. P. Bioorg. Med. Chem. Lett. 2000, 10, 2575-2578. (f) Morimoto, H.; Fukushima, C.; Yamauchi, R.; Hosino, T.; Kikkawa, K.; Yasuda, K.; Yamada, K. Bioorg. Med. Chem. 2001, 9, 255-268. (g) Haylor, J. L.; Morcos, S. K.; Nephrology, D. Transplantation 2001, 16, 1336-1337. (h) Niiyama, K.; Mase, T.; Takahashi, H.; Naya, A.; Katsuki, K.; Nagase, T.; Ito, S.; Hayama, T.; Hisaka, A.; Ozaki, S.; Ihara, M.; Yano, M.; Fukuroda, T.; Noguchi, K.; Nishikibe, M.; Ishikawa, K. Bioorg. Med. Chem. 2002, 10, 2461-2470.
(4) For recent articles on the syntheses and bioactivities of 1,4-benzodiazepine-2,5-diones, see (a) McDowell, R. S.; Gadek, T. R.; Barker, P. L.; Burdick, D. J.; Chan, K. S.; Quan, C. L.; Skelton, N.; Strumble, M.; Thorsett, E. D.; Tischler, M.; Tom, J. Y. K.; Webb, T. R.; Burnier, J. J. Am. Chem. Soc.
1994, 116, 5069-5076. (b) McDowell, R. S.; Blackburn,
B. K.; Gadek, T. R.; McGee, L. R.; Rawson, T.; Reynolds, M.; Robarge, K. D.; Somers, T. C.; Thorsett, E. D.; Tischler, M.; Webb, R. R.; Venuti, M. C. J. Am. Chem. Soc. 1994, 116, 5077-5083. (c) Webb, R. R.; Barker, P. R.; Baier, M.; Reynolds, M. E.; Robarge, K. D.; Blackburn, B. K.; Tichler, M. H.; Weese, K. J. Tetrahedron Lett. 1994, 35, 2113-2116. (d) Armstrong, R.; Combs, A.; Tempest, P.; Brown, D.; Keating, T. Acc. Chem. Res. 1996, 29, 123-131. (e) Ellman, J. A. Acc. Chem. Res. 1996, 29, 9, 132-143. (f) Moroder, L.; Lutz, J.; Grams, F.; Rudolph-Bo¨hner, S.; O¨ sapay, G.; Goodman, M.; Kolbeck, W. Biopolymers 1996, 38, 295-300. (g) Hulme, C.; Cherrier, M.-P. Tetrahedron Lett. 1999, 40, 5295-5299. (h) Cutler, S. J.; Cutler, H. G.; Hamdy, M. K. Proc. Plant Growth Regul. Soc. Am. 27th 2000, 33-41. (i) Osman, A. N.; El-Gendy, A. A.; Omar, R. H.; Wagdy, L.; Omar, A. H. Indian J. Chem., B 2002, 41B, 871-874. (j) Kamal, A.; Nallan, C. L.; Gujjar, R.; Poddutoori, R.; Olepu, S. U.S. Patent 6362331 B1, 2002.
(5) For solid-phase synthesis of 1,4-benzodiazepine-2,5-diones, see (a) Boojamra, C. G.; Burow, K. M.; Ellman, J. A. J. Org. Chem. 1995, 60, 5742-5743. (b) Boojamra, C. G.; Burow, K. M.; Thompson, L. A.; Ellman, J. A. J. Org. Chem.
1997, 62, 1240-1256. (c) Goff, D. A.; Zuckermann, R. N.
J. Org. Chem. 1995, 60, 5744-5745. (d) Mayer, J. P.; Zhang, J.; Bjergarde, K.; Lenz, D. M.; Gaudino, J. J. Tetrahedron Lett. 1996, 37, 8081-8084. (e) Hulme, C.; Peng, J.; Morton, G.; Salvino, J. M.; Herpin, T.; Labaudiniere, R. Tetrahedron Lett. 1998, 39, 7227-7230. (f) Kennedy, A. L.; Fryer, A. M.; Josey, J. A. Org. Lett. 2002, 4, 1167-1170.
(6) For recent reviews of combinatorial and parallel syntheses of organic molecules, see (a) Thompson, L. A.; Ellman, J. A. Chem. ReV. 1996, 96, 555-600. (b) Gordon, E. M.; Gallop, M. A.; Patel, D. V. Acc. Chem. Res. 1996, 29, 144-154. (c) Balkenhohl, F.; von dem Bussche-Huennefeld, C.; Lansky, A.; Zechel, C. Angew. Chem., Int. Ed. Engl. 1996, 35, 2289-2337. (d) Wilson, S. R., Czarnik, A. W., Eds.; Combinatorial Chemistry, Synthesis and Application; Wiley: New York, 1997. (e) Obrecht, D.; Villalgordo, J. M. In Solid-Supported Combinatorial and Parallel Synthesis of Small-Molecular-Weight Compound Libraries; Baldwin, J. E., Williams, R. M., Eds.; Elsevier Science: Oxford, U.K.,
1998. (f) Kerwin, J. F. In Combinatorial Chemistry and Molecular DiVersity in Drug DiscoVery; Gordon, E. M., Kerwin, J. F., Eds.; Wiley-Liss: New York, 1998; p 475. (g) Booth, R. J.; Hodges, J. C. Acc. Chem. Res. 1999, 32, 18-26. (h) Franzen, R. G. J. Comb. Chem. 2000, 2, 195-214. (i) Kirschning, A.; Monenschein, H.; Wittenberg, R. Chem. Eur. J. 2000, 6, 4445-4450. (j) Bhattacharyya, S. Curr. Med. Chem. 2001, 8, 1383-1404. (k) Bra¨se, S.; Gil, C.; Knepper, K. Bioorg. Med. Chem. 2002, 10, 2415-2437. (l) Ganesan, A. Drug DiscoVery Today 2002, 7, 47-55. (m) Boyle, N. A.; Janda, K. D. Curr. Opin. Chem. Biol. 2002, 6, 339-346. (n) Sun, C.-M. Comb. Chem. High Throughput Screening 1999, 2, 299-318. (o) Wentworth, P. Trends Biotechnol. 1999, 17, 448-452. (p) Boger, D. L.; Goldberg, J. In Combinatorial Chemistry; Fenniri, H., Ed.; Oxford University Press: Oxford, 2000; pp 303-326. (q) Sugawara, T.; Cork, D. G. In Combinatorial Chemistry; Fenniri, H., Ed.; Oxford University Press: Oxford, 2000; pp 373-400. (r) Bhattacharyya, S. Indian J. Chem., B 2001, 40B, 878-890. (s) Weinbrenner, S. Methods Principles Med. Chem.
2000, 9, 22-46. (t) Krepinsky, J. J.; Douglas, S. P. In Solid
Support Oligosaccharide Synthesis and Combinatorial Car-bohydrate Libraries; Seeberger, P. H., Ed.; Wiley: New York, 2001; pp 175-199. (u) Kirschning, A.; Monenschein, H.; Wittenberg, R. Angew. Chem., Int. Ed. 2001, 40, 650-679. (v) Hird, N.; Itoh, K. Yuki Gosei Kagaku Kyokaishi
2002, 60, 508-509.
(7) Blass, B. E.; Burt, T. M.; Liu, S.; Portlock, D. E.; Swing, E. M. Tetrahedron Lett. 2000, 41, 2063-2066.
(8) (a) Dale, J.; Titlestad, K. J. Chem. Soc., Chem. Commun.
1969, 656-659. (b) Titlestad, K. Acta Chem. Scand., B 1977,
31, 641-661. (c) Pastuszak, J.; Gardner, J. H.; Singh, J.; Rich, D. H. J. Org. Chem. 1982, 47, 2982-2987. (d) Jacquier, R.; Lazaro, R.; Raniriseheno, H.; Viallefont, P. Int. J. Pept. Protein Res. 1987, 30, 22-32. (e) Steiner, J. R.; Barnes, C. L. Int. J. Pept. Protein Res. 1988, 31, 212-219. (f) Cavelier-Frontin, F.; Pe`pe, G.; Verducci, J.; Siri, D.; Jacquier, R. J. Am. Chem. Soc. 1992, 114, 8885-8890. (9) (a) Highsmith, R. F.; Blackburn, K.; Schmidt, D. J. Annu.
ReV. Physiol. 1992, 54, 257-77. (b) Sakurai, T.; Yanagisawa, M.; Masaki, T. Trends Pharmacol. Sci. 1992, 13, 10-38. (c) Masaki, T.; Yanagisawa, M.; Goto, K. Med. Res. ReV.
1992, 12, 391-421. (d) Lin, W. W.; Kiang, J. G.; Chuang,
D. M. J. Neurosci. 1992, 12, 1077-1085. (e) Kohno, M. Clin. Calcium 1994, 4, 1510-1514. (f) Pollock, D. M.; Keith, T. L.; Highsmith, R. F. FASEB J. 1995, 9, 1196-1204. (g) Wenzel, R. R.; Luscher, T. F. In Endothelium CardioVascular Disease; Luescher, T. F., Ed.; Springer: Berlin, 1995; pp 129-147. (h) Schiffrin, E. L.; Touyz, R. M. J. CardioVasc. Pharmacol. 1998, 32 (Suppl. 3), S2-S13. (i) Decker, E. R.; Brock, T. A. In Endothelin Receptor Signaling Mechanisms; Pollock, D. M., Highsmith, R. F., Eds.; Springer: Berlin, 1998; pp 131-146. (j) Ruschitzka, F. T.; Noll, G.; Luscher, T. F. J. Clin. Basic Cardiol. 1999, 2, 175-180. (k) Neylon, C. B. Clin. Exp. Pharmacol. Physiol. 1999, 26, 149-153. (l) Triggle, C. R. In Potassium Channels in CardioVascular Biology; Archer, S. L., Rusch, N. J., Eds.; Kluwer Academic/ Plenum Publishers: New York, 2001; pp 667-689.
CC030034D
Downloaded by NATIONAL TAIWAN UNIV on August 27, 2009 | http://pubs.acs.org