行政院國家科學委員會補助專題研究計畫
■成果報告
□期中進度報告
核醣核酸二級結構預測與分群分析
計畫類別:■ 個別型計畫 □ 整合型計畫
計畫編號:NSC
97-2221-E-009-131-
執行期間: 97 年 8 月 1 日至 98 年 7 月 31 日
計畫主持人:胡毓志
共同主持人:
計畫參與人員: 鄭家胤,
成果報告類型(依經費核定清單規定繳交):■精簡報告 □完整報告
本成果報告包括以下應繳交之附件:
□赴國外出差或研習心得報告一份
□赴大陸地區出差或研習心得報告一份
■出席國際學術會議心得報告及發表之論文各一份
□國際合作研究計畫國外研究報告書一份
處理方式:除產學合作研究計畫、提升產業技術及人才培育研究計畫、
列管計畫及下列情形者外,得立即公開查詢
□涉及專利或其他智慧財產權,□一年□二年後可公開查詢
執行單位:交通大學資訊工程系
中 華 民 國 98 年 9 月 1 日
行政院國家科學委員會專題研究計畫成果報告
國科會專題研究計畫成果報告撰寫格式說明
Preparation of NSC Project Reports
計畫編號:NSC 97-2221-E-009 -131-
執行期限:97 年 8 月 1 日至 98 年 7 月 31 日
主持人:胡毓志 交通大學資訊工程系
計畫參與人員:
鄭家胤,陳柏志,黃全榮
交通大學資訊工程系
一、中文摘要 核醣核酸在後轉譯調控上扮演重要的 角色,然而與去氧核醣核酸不同的是,去 氧核醣核酸的 motif 大多可在序列間發掘 其保留區,而核醣核酸的 motif 則必須在 結構間找尋。目前已有部分分析系統工具 可以在一群功能性相同的核醣核酸中尋找 可能的共通結構元,但是其大多僅能從功 能性相同的核醣核酸中搜尋,本計畫提出 並完成新的核醣核酸共同結構元預測及分 群的系統,且已利用多個已知的核醣核酸 家族做系統測試。 關鍵詞: 核醣核酸,二級結構,分群 AbstractRNA plays a crucial role in post-transcriptional regulation. Unlike DNA binding proteins, which recognize motifs composed of conserved sequences, RNA protein binding sites are more conserved in structures than in sequences. Though some current approaches can now identify common structure motifs from a set of RNAs, they typically assume the given set forms a single family, which is not necessarily correct. We proposed and developed a new adaptive method that conducts structure prediction as well as clustering simultaneously. We demonstrated its performance on several real RNA families
.
Keywords: RNA, secondary structures, clustering
Introduction
Like proteins, RNA functions generally depend on their structures. Although structural genomics, the systematic study of all macro-molecular structures in a genome, is currently focused more on proteins, thousands of genes produce transcripts exerting their functions without ever producing protein products [1]. It can be easily argued that the comprehensive understanding of the biology of a cell requires the knowledge of identity of
all functional RNAs (both non-coding and protein-coding) and their molecular structures. Since it is often difficult to acquire the 3D spectrum data of RNA molecules for structure determination, versatile and reliable computational methods that can predict RNA structures are highly desirable.
Many computational methods for the prediction of RNA secondary structures have been developed. According to the search strategies used, they can be roughly classified into the following categories: (1) thermodynamics, (2) comparative sequence analysis, (3) stochastic context-free grammars, (4) heuristics and (5) hybrid. By applying dynamic programming, thermodynamic methods are aimed to find the optimal secondary structure for single RNA sequences with the global minimum free energy [2-4]. If homologous sequences are available, from the alignment of these sequences, comparative approaches look for covariance evidence between base pairs to identify consensus structures [5-8]. Some researchers applied stochastic context-free grammars (SCFG) to build a probabilistic model for consensus structures in a family of related RNAs, which is considered the SCFG-based analogue of profile HMMs [9,10]. The tree structure of this SCFG-based analogue allows for a convenient graphical representation that intuitively and compactly reflects the structure of the RNA family being modeled [11]. Due to the fact that the time and space complexity of most methods are still too high to be practical, some approaches adapt useful heuristics to alleviate the problem [12-15]. Although these methods are not guaranteed to fine the optimal structure, they are able to produce the approximately best solutions. There also exist some hybrid approaches that combine the virtue of various strategies such as thermodynamic stability, sequence covariance, phylogenetic analysis, etc. [16-18]. Besides the search strategy that describes how to find the solutions, RNA secondary structure
prediction methods can also be classified by
what to find. Some methods focus on finding
the optimal structure for an entire single RNA sequence [2,3,12,13,20]; others, the consensus structure elements shared by a family of related RNA sequences [19,21,22].
Many functional RNAs have evolutionarily conserved secondary structures in order to fulfill their roles in a cell. For protein-coding RNAs, some of the functions can be presented by functional motifs. For example, several best-understood structurally conserved RNA motifs are found in viral RNAs, such as the TAR and RRE structures in HIV and the IRES regions in Picornaviridae [23]. Apparently, structural information is very useful in characterizing a class of functional RNAs. Based on characteristic structures, we can likely identify novel functional RNAs or partition given RNAs into biologically meaningful families. Several systems have been developed to find consensus structural elements within a family of functionally related RNAs [9,15,19]; however, there is little work on clustering of unaligned RNAs based on characteristic secondary structures. Given a set of unaligned RNA sequences without prior knowledge of the number or identity of families in the set, our goal is to automate both clustering and secondary structure prediction simultaneously. In this paper, we propose an adaptive approximation approach combined with a genetic programming-based structure prediction method to identify from unaligned RNAs reasonable clusters associated with characteristic secondary structure elements. To demonstrate its performance, we tested it on several real datasets.
System
In order to find a reasonable partition for a given set of unaligned RNAs without knowing beforehand how many clusters actually existing in this set, we assume that each cluster is likely a functional family that contains characteristic structure elements. Based on this assumption, our new method is focused on finding significant consensus structure elements that can be used to characterize the families of RNAs. Since the number of clusters and its size are not known in advance, we adapt a generate-and-test strategy that iteratively adjusts the hypothesized cluster size until some significant consensus structure elements can
be found associated with this cluster. After a cluster is obtained, all its members are then removed from the given set of RNAs. We can repeat the same separate-and-conquer strategy to identify other clusters until the set of RNAs is emptied.
Consensus structure element prediction can be considered a supervised learning problem which involves both positive and negative examples [15]. Positive examples are a given set of RNA sequences; negative examples are some number of sequences randomly generated based on the observed frequencies of sequence alphabet in positive examples. The objective here is to learn the structure elements that can be used to distinguish the given functionally related sequences from the random sequences.
We modify GPRM [22], an RNA consensus secondary structure prediction tool, to find significant structure elements from a dataset that may contain multiple variable-sized clusters of unaligned sequences. GPRM has been tested on several real RNA families, including pseudoknots, and shown its effectiveness in predicting conserved structure elements in a given RNA family. To describe the characteristic structure elements for a cluster, we adapt the same representation that is expressive enough to even represent pseudoknots. We also apply the same genetic operators to optimize candidate structure elements during evolutionary process. What is different from the previous work is the fitness function.
The fitness function is used to measure the quality of individuals (i.e. candidate structure elements) in a population. The higher the fitness of an individual, the better its chances of survival to the next generation. In the previous work, the input dataset was assumed to be a single class of functionally related RNA sequences. We were interested in those structure elements that can reflect the characteristics conserved in a family, e.g. the RNA protein binding sites. Derived from the F-score, the fitness function was aimed to balance the importance of two measures, recall (i.e. sensitivity) and precision (i.e. positive predictive value) [15]. It assigns higher values to those structural motifs commonly shared by the given family of RNAs, and rarely contained in random sequences. For a given set of RNA sequences that form a single family only, the fitness function used in [15,22] can effectively guide
the evolutionary process in genetic programming. Nevertheless, when the input dataset contains multiple functional classes, the recall measure may dominate the calculation of F-score if the fitness function treats the entire dataset as a single class. This will mislead the system to find over-general elements shared by most sequences. To alleviate the bias, we defined a new measure of recall. By taking cluster size into account, we can better constrain the search space and allow conserved clusters to emerge more likely instead of being buried in bigger but much less coherent clusters.
The GP (Genetic Programming)-based structure prediction method can find the fittest secondary structure elements according to a given range of the cluster size, while the significance of the cluster found along with its characteristic structure elements is highly dependent on the range we choose. With proper adjustment of cluster size through the generate-and-test procedure combined with the GP-based prediction method, we can identify a meaningful cluster and the associated characteristic structure elements.
The adaptive adjustment of cluster size in the generate-and-test procedure is controlled by the consensus structure specificity. It is defined as the Laplace prior precision. The Laplace prior approach has also been applied to inductive leaning to evaluate the significance of inductive rules [24]. We incorporate the Laplace prior into the calculation of precision with the aim to avoid well conserved but too small clusters. Note that the Laplace prior precision is only used to determine the significance of a cluster found, unlike the F-score, which is used to direct the optimization process to find the best structure elements under the constraints of the cluster size. By the comparison of the Laplace prior precision with a pre-specified threshold, we can adjust the range of cluster size accordingly, and then re-run the GP-based method to predict a new structure element and derive the new cluster it characterizes.
Once a significant cluster is found, we separate all its members out of the given dataset of RNA sequences. We then apply the same procedure to those that still remain in the dataset until the entire set is emptied. This separate-and-conquer strategy is effective even when no prior knowledge of the identities of the clusters is given. It can automatically partition the given dataset into
meaningful clusters, and also identify their characteristic structure elements.
Experimental Results
Two types of quality were considered to evaluate the performance of our method. One is to measure the agreement between the predicted clusters and the actual cluster identities; the other, to quantify the agreement between the predicted structure elements and the actual structure assignment. We applied the adjusted Rand index [28] and the Matthews correlation coefficient [29] to measure the qualities.
Our algorithm is designed to automatically partition a given set of unaligned RNA sequences into meaningful clusters, each associated with characteristic conserved secondary structure elements. The number of real clusters and the distribution of cluster size may affect the prediction of partitions and characteristic structure elements. To measure their effect on the performance, we tested our method on different datasets with various number and size of clusters. We used three families, including 16S RNA, IRE (Iron Response Element) and viral 3’UTR, to prepare the test datasets. They have been used in previous experiments and published in literature [15,19]. The sequence data and the correct structure elements can be accessed at public databases [26,27]. The 16S RNA dataset contains 34 archaea 16S ribosomal sequences originally derived from a set of 311 sequences extracted from the SSU rRNA database. The archaea set of 311 sequences was further reduced to 34, filtering out the sequences that miss base assignments or are greater than 90% identical. The IRE dataset was constructed by Gorodkin et al. [19] from 14 sequences from the UTR database. They modified the IREs and their UTRs to make the search more difficult. By iteratively shuffling the sequences and randomly adding one nucleotide to the IRE conserved region, they built a set of 56 IRE-like sequences from the 14 IRE UTRs. The third data set includes 18 viral 3'UTRs each of which contains a pseudoknot. Seven of the RNA sequences are the soil-borne rye mosaic viruses; the others are the soil-borne wheat mosaic viruses.
With the three real families of RNA sequences, we first tested our method on each possible pair of the families, i.e. 16S RNA/IRE, 16S RNA/viral 3’UTR, and IRE/viral 3’UTR. We then applied our method
to the union of all the three families. In each run of the experiment, no information regarding the number of families or the family size was given to the algorithm beforehand. One purpose of this experiment is to analyze the effect incurred by the number of clusters in a dataset. Furthermore, as the real conserved structure elements differ in various families, we can also observe how the interleaving of distinct structure elements within a single dataset may affect the prediction process. The results are presented in Table 1, and some partial predicted secondary structures are shown in Figure 1.
Discussion
In this project, we proposed a new approach that can perform structure prediction and clustering simultaneously for RNA analysis. The predicted results provide biologists with reasonable hypotheses and suggest further biological verifications. The performance of the new strategy has been demonstrated on several real RNA functional families. The system can be extended in the following directions. First, in case domain knowledge is available, we expect the results can be better improved by incorporating the
background knowledge into the optimization process to effectively constrain the search space. Second, the discovery of important clusters in data usually goes through a repeated process cycle of finding clusters, interpreting results and augmenting data. No current unsupervised clustering system can produce maximally useful results if operated alone [11]. We plan to design a human-machine interface, so that biologists can easily monitor the system status and adapt the system parameter settings. Third, the algorithm itself is highly modular and most of the modules are independent of each other. This property may lead to a parallel-processing version of the system to significantly reduce its computational time.
References
1. The Genome Sequencing Consortium (2001) “Gene content of the human genome”, Nature, 409, p860-921.
2. Zuker, M and Stiegler, P. (1981) ”Optimal computer folding of larger RNA sequences using therdynamics and auxiliary information”, Nucleic Acids Res., 9, p133-148.
3. Zuker, M. (1989) “On finding all suboptimal foldings of an RNA molecule”, Science, 244, p48-52.
4. Hofacker, I., Fontana, W., Stadler, P., Bonhoeffer, L., Tacker, M. And Schuster, P. (1994) “Fast folding and comparison of RNA secondary structures”, Monatsh. Chem., 125, p167-188.
5. Chiu, D. and Kolodziejczak, T. (1991) “Inferring consensus structure from nucleic acid sequences”, Comput. Appl. Biosci., 7, p347-352.
6. Gutell, R., Power, A., Hertz, G., Putz, E. and Stormo, G. (1992) “Identifying constraints on the higher-order structure of RNA: continued development and application of comparative sequence analysis methods”, Nucleic Acids Res., 20, p5785-5795.
7. Gulko, B. and Haussler, D. (1996) “Using multiple alignments and phylogenetic trees to detect RNA secondary structure”, Proc. Pac. Symp. Biocompt., p350-367.
prediction”, Proc. Int. Conf. Intell. Syst. Mol. Biol., p10-17.
9. Eddy, S. and Durbin, R. (1994) “RNA sequence analysis using covariance models”, Nucleic
Acids Res., 22, p2079-2088.
10. Sakakibara, Y., Brown, M., Hughey, R., Mian, I., Sjolander, K., Underwood, R. and Haussler, D. (1994) “Stochastic context-free grammars for tRNA modeling”, Nucleic Acids Res., 22, p5112-5120.
11. Durbin, R., Eddy, S., Krogh, A. and Mitchison, G. (1998) “Biological sequence analysis”, Cambridge University Press.
12. Abraham, J., van denBerg, M., van Batenburg, F. and Pleij, C. (1990) ”Prediction of RNA secondary structure, including pseudoknotting, by computer simulation”, Nucleic Acids Res., 18, p3035-3044.
13. Gultyaev, A., van Batenburg, F. and Pleij, C. (1995) “The computer simulation of RNA folding pathways using a genetic algorithm”, J. Mol. Biol., 250, p37-51.
14. van Batenburg F., Gultyaev, A. and Pleij, C. (1995) “An APL-programmed genetic algorithm for the prediction of RNA secondary structure”, J. Theor. Biol., 174, p269-280.
15. Hu, Y. (2002) "Prediction of consensus structural motifs in a family of coregulated RNA sequences", Nucleic Acids Res., 30, p3886-3893.
16. Luck, R., Graf, S. and Steger, G. (1999) “ConStruct: a tool for thermodynamic controlled prediction of conserved secondary structure”, Nucleic Acids Res., 27, p4208-4217.
17. Hofacker, I., Fekete, M. and Stadler, P. (2002) “Secondary structure prediction for aligned RNA sequences”, J. Mol. Biol., 319, p1059-1066.
18. Juan, V. and Wilson, C. (1999) “RNA secondary structure prediction based on free energy and phylogenetic analysis”, J. Mol. Biol., 289, p935-947.
19. Gorodkin, J., Stricklin, S. L. and Stormo, G. D. (2001) Discovering common stem-loop motifs in unaligned RNA sequences. Nucleic Acids Res., 29, 2135-2144.
20. Rivas, E. and Eddy, S. (1999) “A dynamic programming algorithm for RNA structure prediction including pseudoknots”, J. Mol. Biol., 285, p2053-2068.
21. Mathews, D. and Turner, D. (2002) “Dynalign: an algorithm for finding the secondary structure common to two RNA sequences”, J. Mol. Biol., 317, p191-203.
22. Hu, Y. (2003) "GPRM: a genetic programming approach to finding common RNA secondary structure elements", Nucleic Acids Res., 31, p3446-3449.
23. Hofacker, I., Priwitzer, B. and Stadler, P. (2004) “Prediction of locally stable RNA secondary structures for enome-wide surveys”, Bioinformatics, 20, p186-190.
24. Clark, P and Boswell, R. (1991) “Rule Induction with CN2: some recent improvements”, in Proceedings of the Fifth European Conference on Machine Learning, p151-163.
25. Cheeseman, P. and Stutz. J. (1996) “Bayesian Classification (AUTOCLASS): Theory and Results”, in Advances in Knowledge Discovery and Data Mining, p153-180, AAAI.
26. Batenburg, F.H.D. van, Gultyaev, A.P. and Pleij, C.W.A. (2001) “PseudoBase: structural information on RNA pseudoknots”, Nucleic Acids Res., 28, 1, 201-204.
27. Hu, Y. (2002) “The NCTU BioInfo Archive”
28. Matthews, B.W. (1975) Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochem. Biophys. Acta, 405, 442-451.
29. Rand, W. M. (1971) Objective criteria for the evaluation of clustering methods. Journal of
(a)
IRE+viral 3’UTR Recall Precision Matthews
IRE 0.97 0.99 0.97
viral 3’UTR 0.71 0.95 0.79
(b)
16S RNA+viral 3’UTR Recall Precision Matthews
16S RNA 0.97 0.95 0.83
viral 3’UTR 0.77 0.98 0.77
(c)
IRE+16S RNA Recall Precision Matthews
IRE 0.73 0.99 0.85
16S RNA 0.81 0.73 0.67
Table 1. Summary of the experimental results. Table (a), (b) and (c) present the result for the dataset containing IRE and viral 3’UTR, 16S RNA and viral 3’UTR, IRE and 16S RNA, respectively. ***** IRE ***** > seq_D15071.1 41 45 47 51 58 62 63 67 t g c g g u c c u g g c c a g u g a g c u g g g c c g c predicted: . ( ( ( ( ( . ( ( ( ( ( . . . ) ) ) ) ) ) ) ) ) ) published: . ( ( ( ( ( . ( ( ( ( ( . . . ) ) ) ) ) ) ) ) ) ) ***** 16S RNA ***** > U51469 13 20 23 31 37 46 52 61 g u u u c a u u g a a g u u u g c u u u u a g u g a g g u g a c g u c u a a u u g g c g u u a u c g 62 67 75 78 85 a a c u u g u g g u a a g c g a c a a g g g a a a a predicted: . ( ( ( ( ( ( ( ( . . ( ( ( ( ( ( ( ( ( . . . ( ( ( ( ( ( ( ( ( ( . . . ) ) ) ) ) ) ) ) ) ) . . . ) ) ) ) ) ) ) ) ) . . ) ) ) ) ) ) ) ) . . published: . ( ( ( ( ( ( ( ( . . ( ( ( ( ( ( ( ( ( ( ( ( ( . . ( ( ( ( ( ( ( ( ( . . . ) ) ) ) ) ) ) ) ) . . ) ) ) ) ) ) ) ) ) ) ) ) ) . . ) ) ) ) ) ) ) ) . . ***** viral 3’UTR ***** > PKB183 14 16 18 24 25 27 32 38 a c g u c g u g c a g u a c g g u a a a c u g c a c a u predicted: . ( ( ( . [ [ [ [ [ [ [ ) ) ) . . . . ] ] ] ] ] ] ] . . published: . ( ( ( . [ [ [ [ [ [ [ ) ) ) . . . . ] ] ] ] ] ] ] . .
Figure 1. A partial result of the predicted RNA motifs. The numbers above the sequences are the indices of the nucleotides. The predicted and the published motifs are both shown for reference.
行政院國家科學委員會補助國內專家學者出席國際學術會議報告
98 年 9 月 1 日報告人姓
名
胡 毓 志
服務
機構
及職稱
交通大學資訊工程系
副教授
時間
會議
地點
07/13/2009-07/16/2009
Las Vegas, USA
本會核定
補助文號
NSC 97-2221-E-009-131-
會議
名稱
(中文) 生物資訊暨計算生物學國際研討會
(英文)2009 International Conference on Bioinformatics and
Computational Biology
發表
論文
題目
1.(中文)
利用蛋白質結構字元及一維模組搜尋法分析金屬結合蛋白的 結構模組(英文)
Discovery of Structural Motifs in Metalloproteins Using Protein Structural Alphabets and 1D Motif-finding Methods報告內容應包括下列各項: 一、參加會議經過
於 07/12 辦理註冊報到,07/13 參加 Opening Remarks by Dr. Arabnia from University of Georgia,於 07/13-07/16 期間,參加與會學者之論文發表,同時,07/13 發表論文。並 與多位國外學者討論相關研究議題。會議論文不乏有關基因表現分析,蛋白質結構分 析,調控訊號檢視,生物網路分析等等,對於我國內生物資訊的發展,將提供非常多的 助益與新的發展方向。 二、與會心得 本次參加人數及國家眾多,其研究領域更包括計算機科學、醫學、生物學等之應用,藉 由討論及論文發表,獲得寶貴經驗,對於未來研究提供了新的方向。其中更結識他國友 人,經由研討,可明白其他國家的發展經驗。 目前系統生物學已經成為國際重要的研究課題,可預見的是,在不久的將來,大量的基 因表現資料以及蛋白質結構將如同 DNA 及蛋白質序列般,不斷地被產生及發表,如何 能從這些不同類型的生化資料中發掘有用的訊息將是重要課題。藉由這次與會學習的經 驗,我們可以得知國外研究之重點,作為我國在生物科技的發展依據。 三、考察參觀活動(無是項活動者省略) 四、建議 由於生物科技是目前國內新興研究發展之重要產業,懇請國科會及相關單位,能多支持 與獎勵國內學者多參與此類國際研討會,除了增加我國在國際相關領域的能見度,同 時,提供相互學習之機會,這是直接提昇我國在生技發展地位的最有效做法。 五、攜回資料名稱及內容
The Proceedings of Biocomp2009 六、其他
Discovery of Structural Motifs in Metalloproteins Using Protein Structural
Alphabets and 1D Motif-finding Methods
Shih-Yen Ku13,Chih-Ying Wei4 and Yuh-Jyh Hu12
Department of Computer Science1, Institute of Biomedical Engineering2, National Chiao Tung
University, Molecular and Computational Biology Program, Department of Biological Sciences3,
University of Southern California, Department of Management Information Systems4, National Chengchi University
Abstract
Though the increasing number of available 3D proteins structures has made possible a wide variety of computational protein structure research, yet the success is still hindered by the high 3D computational complexity. Based on 3D information, several 1D protein structural alphabets have been developed, which not only can describe the global folding structure of a protein as a 1D sequence, but can also characterize local structures in proteins. In this paper, we introduce an approach that combines standard 1D motif detection method with structural alphabets to discover metal-binding sites in metalloproteins. We tested our method on different metal-binding proteins. The results show that our combinatorial strategy can efficiently and successfully identify the structural preferences in metal-binding sites.
Keyword: metalloproteins, protein structural alphabets, structural motifs
1. Introduction
As the rapid growth of protein structural information, biologists require accurate
classification to understand and rationalize the variety in proteins [1]. To ensure the classification can be more easily constructed and better comprehensible, it is desired we provide only essential characteristic structural descriptions of protein functional parts. With such a classification, we can assign a novel protein to known categories, and thus predict its structures and functions. The task of extracting characteristic structural features for classification becomes more challenging for small proteins, where the characteristic statistics are marginal owing to short protein chains, or for proteins that only share low sequence similarity, e.g. some metal binding proteins. In this paper, we evaluate the feasibility of using structural alphabets and 1D motif detection methods to discover the structural motifs in Mg2+-binding and zinc-binding proteins.
Metalloproteins requires metal cofactors in cellular biochemistry, which play important roles in both intra- and extracellular catalytic activities and structural stabilization [2-4], as metal binding increases thermal and conformational stability of small domains. Among many, here we focus our study on zinc-binding and Mg-binding proteins.
The C2H2 zinc finger is one of the best-studied metal binding domains. It was first observed as a repeated zinc-binding motif with DNA-binding properties in the Xenopus transcription factor IIIA, and the term `zinc finger' is now largely used to denote any compact domain stabilized by a zinc ion [5-7]. Previous studies of zinc fingers include automatic neural network-based numerical taxonomy methods that identify evolutionary relationships among proteins [8-9] or the analysis of sequence and structure similarity using BLAST-based sequence alignment method in combination with DaLiLite followed by visual inspection [10][11].
Although these methods have made significant advances in structural classification, they still left plenty of space for improvement in classification accuracy and efficiency.
Like zinc, magnesium is also a versatile and important metal cofactor. It helps stabilize a variety of protein structures, e.g., the interface of the ribonucleotide reductase subunits [12]. It is also used to stabilize nucleic acids by alleviating electrostatic repulsion between negatively charged phosphates [13]. A few relatively short sequence motifs have been discovered for Mg2+ proteins with close sequence homology. Examples include the NADFDGD motif, found in different RNA polymerases, DNA Pol I and HIV reverse transcriptase, and the YXDD or LXDD motifs in reverse transcriptase and telomerase, where the residues in bold are the Mg2+ ligands [2]. Nevertheless, these Mg2+ sequence motifs are sometimes too short to be statistically specific to Mg2+-binding sites, and may easily escape detection. On the other hand, the Mg2+-binding sites share sufficient structural similarity that can characterize Mg2+-proteins.
As the conserved local structural features can be identified in various ways and described in different representations, e.g., the relationships between local sequences and structures [14-16], we took a simpler approach and applied the widely used 1D motif detection algorithms to protein structural alphabet sequences. Representing conserved local structural features by 1D structural alphabets instead of 3D co-ordinates is more efficient in comparison and more economical in storage. The 1D-based approaches can also serve as a pre-processor to filter out remotely related or irrelevant proteins before we apply other more accurate but more computationally intensive structure analysis tool. To demonstrate its applicability, we applied the 1D-based approach to discovering structural motifs in Zn-binding and Mg-binding proteins, and compared the motifs found against those reported in literature.
2. Materials and Methods
We first transformed protein sequences into a 1D representation, and later identified significant motifs from the 1D alphabet sequences that could characterize the local structural features. There are several protein structural alphabets available [17-19]. As these alphabets were derived from different design philosophies, their sizes can vary from a dozen to nearly a hundred. They address different structural characteristics and have various applications. Therefore, in different domains, we can apply an appropriate structural alphabet to transform amino acid sequences or protein 3D structures into 1D structural alphabet sequences as required. Currently, we use the alphabet designed for SA-FAST [20]. It contains 18 letters, five of which represent the helix structure, eight for the sheet, and the rest for the coil.
Given a set of functionally or structurally related proteins, after the conversion into 1D structural alphabet sequences, we can apply a sequence motif detection algorithm to discover significant motifs. There has been significant amount of research on motif discovery with
different objective functions, motif representations and search strategies [21-23]. In our study, we used MEME [24] to detect structural motifs, which adopts an expectation maximization
approach to find motifs represented as weight matrices. Unlike IUPAC-IUB codes, motifs described in weight matrices are more flexible because a weight matrix can show each alphabet letter preference in every motif position. Besides, a weight matrix can be easily transformed to IUPAC-IUB codes or regular expressions when necessary, but not vice versa.
We call the motifs found by MEME in the structural alphabet sequences simple motifs. When the local properties in protein structures are too complicated, e.g. multiple binding sites or sub-domains, to capture in a simple motif, we combine several simple motifs into a compound
motifs to regular expressions first, and then combine them to a compound motif. A compound motif example looks like the following.
M1[20,50]M2[0,6]M3, where M1, M2 and M3 are simple motifs, and the numbers in the brackets
denote the range of residue separation between motifs.
M1= SP[PS][SN]N[NE]EE,
M2= [WE][NE]EEACWGQS,
M3= TTTTTTTTTLK[TG][SH]WNMR[DQ],
where letters in brackets denote the possible structural alphabet letters in the respective motif position.
We illustrate the system flow in Figure 1. We proposed for structural motif discovery a general framework in which the structural alphabet and the motif finding algorithm can be replaced with others when needed in different applications.
Protein Set
Wet Lab
Protein 3D Structure Database (e.g. PDB)
Protein Structures
Structural Alphabet Conversion
Structural Alphabet Sequences
Motif Finding Algorithms (e.g. MEME)
Simple Structural Motifs
Compound Structural Motifs
Fig 1. System flow of structural motif discovery
3. Experimental Results
The domains from C2H2-like fingers consist of a β-hairpin followed by an α-helix that forms a left-handed ββα-unit, where two zinc ligands are contributed by a zinc knuckle at the end of the β-hairpin and the other two ligands come from the C-terminal end of the α-helix [25,26]. The C2H2 zinc finger motif (classic zinc finger) was first discovered in the Xenopus laevis
transcription factor IIIA, and has since been found in many transcription factors and in other DNA-binding proteins.
This classic C2H2 zinc finger typically contains a repeated ~30 amino sequences. To demonstrate that our approach is capable of detecting the structural ββα-unit, we first transformed the 156 zinc finger proteins in SCOP C2H2 zinc finger family into structural alphabet sequences, and then applied standard motif-finding algorithms to these sequences to identify common motifs that can characterize the ββα-unit.
We used 8 as the motif width and ran MEME to find motifs. A motif found was considered as corresponding to a sub-domain correctly if more than half of the residues in the sub-domain were included in the motif. If any simple motif or compound motif correctly corresponded to a
sub-domain, we claimed this sub-domain was recovered successfully (i.e. a hit). In Table 1, we present the simple motif or compound motif found to characterize the sub-domains, and its coverage. The results suggest that using standard motif-finding algorithms, e.g. MEME,
combined with an appropriate structural alphabet was able to recover the structural sub-domains in C2H2 zinc finger proteins. We show some C2H2 zinc finger proteins with structural motifs highlighted in color in Figure 2.
Unlike Zn2+ binding sites, Mg2+ binding sites have less sequence similarity, but sufficient structural similarity, which make an appropriate test case to verify our motif-finding method’s capability of discovering structural motifs with low sequence homology. Previously, Dudev and Lim applied a similar idea to identify the structural motifs in Mg2+-binding proteins [13]. In the study, they successfully discovered four motifs corresponding to the Mg2+ binding sites in 16 out of 70 Mg2+-binding proteins. For comparison, we used the same 70 proteins in our experiments. Instead of the structural alphabet in PBE [27], we used the alphabet designed for SA-FAST [20] to represent the 70 Mg2+-binding proteins. Furthermore, we used a widely-used motif-finding system, MEME, rather than a method solely based on motif occurrence frequency [28], to identify common motifs (simple motifs) first, and then combined those significant simple motifs (i.e. with low E-value) into compound motifs.
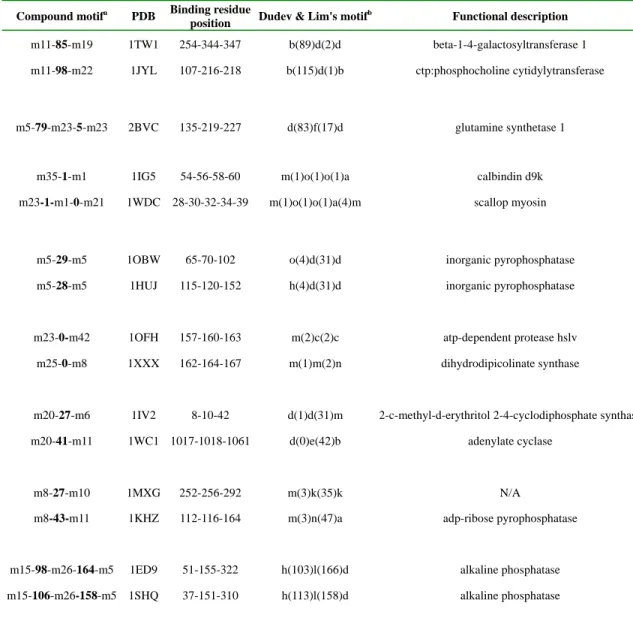
We show in Table 2 some of the compound motifs that cover the Mg2+-binding residues. Each of the simple motifs has an E-value lower than 1.9e-015 as presented in Table 3. Based on the compound motifs generated, we noticed that Mg2+-specific structural compound motifs are not commonly shared among the 70 Mg2+-binding proteins. This observation is similar to that by Dudev and Lim [13], who found only four first-shell structural motifs shared by more than three Mg2+-binding proteins, with a total of 16 proteins containing these motifs. Unlike Dudev and Lim, who defined a structural motif based on its occurrence frequency, we constructed
compound motifs, more flexible and expressive than Dudev and Lim’s, from significant simple motifs found by MEME. By constraining the number of simple motifs, e.g. setting a reasonable
E-value threshold, we can reduce the search space for compound motif candidates, and still avoid
the risk of overlooking less frequent but significant structural motifs.
4. Conclusion
In this paper, we introduced a general framework for structural motif discovery, and applied it to two types of metalloproteins, Mg2+-binding proteins and C2H2 zinc finger proteins. Two major components in our framework are the structural alphabet used to describe protein
our experiments, we used the alphabet designed for SA-FAST, and a widely used motif detection algorithm, MEME. These components can be flexibly replaced with others when necessary to increase the applicability in different domains. The experimental results showed that using structural alphabets combined with standard motif-finding algorithms could successfully identify biologically meaningful sub-domains in proteins.
With the positive results, we plan to carry out the future work as what follows. First, many structural alphabets and quite a few motif detection algorithms have been developed based on different design philosophies and application domains. We intend to incorporate other structural alphabets and motif-finding algorithm into our system. We expect to discover more kinds of motifs in a wider variety of protein structures. Second, we plan to build a structural alphabet motif database. Given the effectiveness and the economy in characterizing and storing structural properties of proteins, a structural alphabet motif database can complement most protein
sequence and 3D structure databases. Third, we will design a protein function predictor using structural motifs as important features. Based on the motifs, the functions of novel proteins can be predicted by classifying them to protein groups with known functions. Finally, as there have been many protein structure or function prediction systems available, we also plan to evaluate the feasibility of using structural alphabet-based methods as a preprocessor. Compared with most prediction strategies typically based on 3D information, alphabet-based methods have much lower computational complexity. They can help other predictors constrain the search space efficiently by filtering out irrelevant predictions.
d1a1ia2 d1jk1a1 d1a1ga1 d1a1ja1
d1p47a1 d1f2ig1 d1llmc1 d3znf
Fig 2. Examples of C2H2 zinc finger protein structures. The simple motifs that map to the β-hairpin and the α-helix are highlighted in color,
where M1=[GN][HE][NE]AC[AW]RQ, M2=[FH]CWNA[RC]QK and M3= TTTTTT[PL][KPL]. The compound motif mapping to the ββα-unit is [FH]CWNA[RC]QK (0-2) [GN][HE][NE]AC[AW]RQ (0-5) TTTTTT[PL][KPL].
Table 1. Summary of compound motifs mapping to C2H2 zinc finger ββα-unit that consists of β-hairpin and α-helix. SCOP 1.73 (C2H2 zinc finger)
g.37.1.1 Structural (sub-)domain
Compound motif
Hita Coverageb
β-hairpin [FH]CWNA[RC]QK(0-2) [GN][HE][NE]AC[AW]RQ 131 83.9 % α-helix [GN][HE][NE]AC[AW]RQ(0-5)TTTTTT[PL][KPL] 142 91.0% ββα-unit [FH]CWNA[RC]QK(0-2) [GN][HE][NE]AC[AW]RQ
(0-5) TTTTTT[PL][KPL]
124 79.5%
Total --- 156 100%
aWe called it a hit for a structural (sub-)domain when more than half of the (sub-)domain residues were contained in a motif. We presented the
count of hits of different (sub-)domains.
bCoverage was defined as the ratio of the count of hits to the number of zinc finger proteins, e.g., if No.=156 and Hits=131, then
Coverage=131/156=83.9%.
Table 2. Structural motifs found in Mg2+-binding proteins.
Compound motifa PDB Binding residue
position Dudev & Lim's motif
b Functional description
m11-85-m19 1TW1 254-344-347 b(89)d(2)d beta-1-4-galactosyltransferase 1 m11-98-m22 1JYL 107-216-218 b(115)d(1)b ctp:phosphocholine cytidylytransferase
m5-79-m23-5-m23 2BVC 135-219-227 d(83)f(17)d glutamine synthetase 1
m35-1-m1 1IG5 54-56-58-60 m(1)o(1)o(1)a calbindin d9k m23-1-m1-0-m21 1WDC 28-30-32-34-39 m(1)o(1)o(1)a(4)m scallop myosin
m5-29-m5 1OBW 65-70-102 o(4)d(31)d inorganic pyrophosphatase
m5-28-m5 1HUJ 115-120-152 h(4)d(31)d inorganic pyrophosphatase
m23-0-m42 1OFH 157-160-163 m(2)c(2)c atp-dependent protease hslv m25-0-m8 1XXX 162-164-167 m(1)m(2)n dihydrodipicolinate synthase
m20-27-m6 1IV2 8-10-42 d(1)d(31)m 2-c-methyl-d-erythritol 2-4-cyclodiphosphate synthase m20-41-m11 1WC1 1017-1018-1061 d(0)e(42)b adenylate cyclase
m8-27-m10 1MXG 252-256-292 m(3)k(35)k N/A
m8-43-m11 1KHZ 112-116-164 m(3)n(47)a adp-ribose pyrophosphatase
m15-98-m26-164-m5 1ED9 51-155-322 h(103)l(166)d alkaline phosphatase m15-106-m26-158-m5 1SHQ 37-151-310 h(113)l(158)d alkaline phosphatase
m5-20-m20 1ZPD 440-467-469 k(26)h(1)a pyruvate decarboxylase m5-23-m39 1POX 447-474-476 k(29)h(1)a pyruvate oxidase
m5-27-m4 1UMD 175-204-206 k(28)h(1)a 2-oxo acid dehydrogenase alpha subunit m5-25-m39 2C3M 963-991-993 k(27h(1)a pyruvate-ferredoxin oxidoreductase
m16-155-m18 1NUY 1118-1121-1280 f(1)h(158)m fructose-1-6-bisphosphatase m16-146-m18 1KA1 142-145-294 f(1)h(148)m halotolerance protein hal2 m16-123-m18 2BJI 1090-1093-1220 f(1)h(126)m inositol-1(or 4)-monophosphatase
m24-155-m12 1O08 1008-1010-1170 f(1)h(159)b beta-phosphoglucomutase m24-106-m12 1U7P 11-13-123 f(1)h(109)b magnesium-dependent phosphatase-1 m24-186-m12 2C4N 9-11-201 f(1)h(189)b nagd
m24-118-m12 2B82 44-46-167 f(1)h(119)b class b acid phosphatase
a
Compound motifs are composed of significant simple motifs, e.g. m24-161-m12 is a compound motif composed of simple motif m24 and m12, where 161 is the number of residues in between. The significance of a simple motif is determined by its E-value. The E-value of all simple motifs in table is less than or equal to1.9e-015 (the smaller E-value, the more significant).
bDudev & Lim considered a motif significant if the number of its occurrence is greater than or equal to 3. Significance motifs are marked in bold.
Table 3. Summary of simple motifs used in compound motifs. Simple Motif
(regular expression) Motif Index E-value
LKGHN m1 5.2e-954 M[DA]DHN m4 9.0e-547 EEARQ m5 4.8e-531 TLKGH m6 6.6e-477 ACWNE m8 8.6e-422 MADWN m10 1.7e-303 [LM]KGHN m11 1.2e-287 ACARQ m12 7.7e-309 EMAD[HQ] m15 3.9e-231 EEACW m16 8.5e-204 [FA]RQ[TP]T m18 1.5e-176 TLKGM m19 1.6e-163 EEEE[MAE] m20 3.0e-136 ARQTT m21 2.8e-095
TTLKG m22 1.8e-098 TTPP[PS] m23 5.7e-088 ACW[NF]Q m24 1.0e-075 SF[RP]CN m25 7.6e-078 SFRQT m26 6.9e-055 TT[ST]PP m35 3.3e-022 MM[DA]DH m39 1.9e-015 AR[CQW]PP m42 7.1e-020
5. References
[1]Berman,H.M., Battistuz,T., Bhat,T.N., Bluhm,W.F., Bourne,P.E., Burkhardt,K., Feng,Z., Gilliland,G.L., Iype,L., Jain,S. et al. “The protein data bank”, Acta Crystallogr. D Biol.
Crystallogr, 2002, 58, 899-907.
[2] Cowan, J.A. “Metal activation of enzymes in nucleic acid biochemistry”, Chem Rev 1998, 98, 1067-1087.
[3] Cowan, J.A. “Biological chemistry of magnesium”, New York, VCH, 1995.
[4] Bohm, S., Frishman, D. and Mewes, H.W. “Variations of the C2H2 zinc finger motif in the yeast genome and classification of yeast zinc finger proteins”, Nucleic Acids Research, 1997, 25, 2464-2469.
[5] Laity, J.H., Lee,B.M. and Wright,P.E. “Zinc finger proteins: new insights into structural and functional diversity”, Curr. Opin. Struct. Biol., 2001, 11, 39-46.
[6] Iuchi, S. “Three classes of C2H2 zinc finger proteins”, Cell. Mol. Life Sci., 2001, 58, 625-635. [7] Klug, A. and Schwabe, J.W. “Protein motifs 5. Zinc fingers”, FASEB J., 1995, 9, 597-604. [8] Dietmann,S. and Holm,L. “Identification of homology in protein structure classification”,
Nature Struct. Biol., 2001, 8, 953-957.
[9] Dietmann, S., Park, J., Notredame, C., Heger, A., Lappe, M. and Holm, L. “A fully automatic evolutionary classification of protein folds: Dali Domain Dictionary version 3”, Nucleic Acids
Research, 2001, 29, 55-57.
[10] Holm, L. and Park, J. “DaliLite workbench for protein structure comparison”,
Bioinformatics, 2000, 16, 566-567.
[11] Krishna, S.S., Majumdar, I. and Frishin, N.V. “Structural classification of zinc fingers”,
Nucleic Acids Research, 2003, 31, 523-550.
[12] Nordlund P., Sjoberg B.M., Eklund H. “Three-dimensional structure of the free radical protein of ribonucleotide reductase”, Nature 1990, 345-593.
[13] Dudev, M. and Lim, C. “Discovering structural motifs using a structural alphabet: Applications to magnesium-binding sites”, BMC Bioinformtics, 2007, 8, 106.
[14] R. Unger and J.L. Sussman “The importance of short structural motifs in protein structure analysis”, J. Comput. Aided Mol. Des., 1993, 457-472.
[15] K.F. Han and D. Baker “Recurring local sequence motifs in proteins”, J. Mol. Biol., 1995, 176-187.
[16] K.T. Simons, C. Kooperberg, E. Huang and D. Baker “Assembly of protein tertiary
structures from fragments with similar local sequences using simulated annealing and Bayesian scoring functions”, J Mol Biol., 1997, 209–225.
[17] de Brevern, A.G. “New assessment of a structural alphabet”, In Sillico Biology, 2005, 5, 26. [18] Schuchhardt, J., Schneider, G., Reichelt, J., Schomburg, D. and Wrede, P. “Local structural motifs of protein backbones are classified by self-organizing neural networks”, Protein
Engineering, 1996, 9, 833-842.
[19] Unger, R., Harel, D., Wherland, S. and Sussman, J.L. “A 3D building blocks approach to analyzing and predicting structure of proteins”, Proteins, 1989, 5, 355-373.
[20] Ku, S. and Hu, Y. “Protein structure search and local structure characerization”, BMC
Bioinformtics, 2008, 9, 349.
[21] Lawrence, C., Altschul, S., Boguski, M., Liu, J., Neuwald, A. and Wootton, J. “Detecting subtle sequence signals: a Gibbs sampling strategy for multiple alignment”, Science, 1993, 262, 208-214.
[22] Hertz, G., Hartzell III, G. and Stormo, G. “Identification of consensus patterns in unaligned DNA sequences known to be functionally related”, Computer Applications in Biosciences, 1990, 6, 81-92.
[23] van Helden, J., Andre, B. and Collado-Vides, J. “Extracting regulatory sites from the
upstream region of yeast genes by computational analysis of oligonucleotide frequencies”, J. Mol.
Bio, 1998, 281, 827-842.
[24] Bailey, T. and Elkan, C. “Unsupervised learning of multiple motifs in biopolymers using rxpectation maximization”, Machine Learning, 1995, 21, 51-80.
[25] Grishin,N.V. “Treble clef finger - a functionally diverse zincbinding structural motif”,
Nucleic Acids Res., 2001, 29, 1703-1714.
[26] Wang,B., Jones,D.N., Kaine,B.P. and Weiss,M.A. “Highresolution structure of an archaeal zinc ribbon defines a general architectural motif in eukaryotic RNA polymerases”, Structure, 1998, 6, 555-569.
[27] Tyagi M, Sharma P, Swamy CS, Cadet F, Srinivasan N, de Brevern AG, Offman B. “Protein block expert (PBE): a web-based protein structure analysis server using a structural alphabet”,
Nucleic Acids Res., 2006, 34, W119-W123.
[28] Jonassen I, Eidhammer I, Conklin D, Taylor WR “Structure motif discovery and mining the PDB”, Bioinformatics, 2001, 18, 362-367.


![Fig 2. Examples of C2H2 zinc finger protein structures. The simple motifs that map to the β-hairpin and the α-helix are highlighted in color, where M 1 =[GN][HE][NE]AC[AW]RQ, M 2 =[FH]CWNA[RC]QK and M 3 = TTTTTT[PL][KPL]](https://thumb-ap.123doks.com/thumbv2/9libinfo/8533632.187431/15.918.148.772.548.892/examples-finger-protein-structures-simple-hairpin-highlighted-tttttt.webp)