國 立 交 通 大 學
材 料 科 學 與 工 程 學 系
碩 士 論 文
氧化現象對碳化矽/氧化鋁/氧化鋯複合材料
機械性質的影響
Effect of Oxidation Phenomenon on the Mechanical Properties
of SiC/Al
2O
3/ZrO
2Composites
研 究 生:游力建
指導教授:林健正 博士
氧化現象對碳化矽/氧化鋁/氧化鋯複合材料
機械性質的影響
Effect of Oxidation Phenomenon on the Mechanical Properties
of SiC/Al2O3/ZrO2 Composites
研 究 生:游力建 Student : Li-Gen Yu 指導教授:林健正 教授 Advisor:Chien-Cheng Lin 國 立 交 通 大 學 材料科學與工程學系 碩 士 論 文 A ThesisSubmitted to Department of Material Science and Engineering College of Engineering
National Chiao Tung University in partial Fulfillment of the Requirements
for the Degree of Master in Material Science and Engineering
June 2004
Hsinchu, Taiwan, Republic of China
氧化現象對碳化矽/氧化鋁/氧化鋯複合材料
機械性質的影響
研究生:游力建 指導教授:林健正 國立交通大學材料科學與工程研究所摘要
本研究以熱壓法製備碳化矽體積比為 30% 的複合材料,基地為 不同體積比的氧化鋯與氧化矽,以 AZxx 表之,其中 xx 代表氧化鋯 在基地中的體積百分比;複合材料則以 AZxx/SiC 符號表示。測試試 片以SENB(Single Edge Notched Beam)方法製備,並分別於 1350℃下 進行不同氧化時間處理,最後以四點抗折儀器量測機械性質。並輔以 X 光繞射儀(X-ray)、掃描式電子顯微鏡(SEM/EDS)、穿透式電子顯微 鏡(TEM/EDS)觀察分析斷裂面的顯微結構。實驗結果顯示氧化試片的 weight gain 隨氧化鋯含量的增加而增加,這是由於氧化鋯的添加促使 氧在基地相的擴散速率增加所致。試片氧化後的斷裂面型態也因氧化 鋯含量不同而相異,AZ0/SiC 試片斷裂面型態氧化前後相近,斷裂面 平整且氧化現象不明顯,破壞模式為穿晶破壞;而 AZ10/SiC 和 AZ20/SiC 氧化後斷裂面相似,出現因氧化而生成的氧化層及孔洞, 對試片的破壞強度及韌性是負面效應;而 AZ50/SiC 的斷裂面則滿佈因氧化而生成的孔洞及裂縫。破壞強度與韌性的測試結果可分為兩部 分,未氧化試片的氧化鋯含量越多對試片破壞強度與韌性的提高幫助 越大,可歸因於碳化矽與氧化鋯強化相的幫助;而氧化後結果則因試 片氧化現象不同而相異,AZ0/SiC 與 AZ10/SiC 試片氧化程度輕微, 試片破壞強度與韌性受氧化影響較小,短時間氧化出現破壞強度與韌 性提高;AZ20/SiC 與 AZ50/SiC 試片氧化程度嚴重,試片破壞強度與 韌性受氧化影響大,氧化後試片的破壞強度與韌性都比氧化前差。
Phenomenon of Oxideation on the Mechanical Properties of
SiC/Al2O3/ZrO2 Composites
Student:Li-Gen-Yu Advisor:Chien-Cheng-Lin Department of Material Science and Engineering
Nation Chiao Tung University
Abstract
The composites contented 30 vol% SiC was fabricated by hot pressing. It with different volume percent of zirconia and alumina in matrix, expressed as AZxx (xx meaned the percentage of zirconia in matrix). The composites represented as AZxx/SiC. The specimens are made of SENB (Single Edge Notched Beam) method, and then measured with four-point fracture test. The microstructures of the fracture surface of various composites after exposured at a temperature of 1350℃, were studied using X-ray diffraction, SEM, TEM, and energy-dispersive spectroscopy ( EDS ). The result showed the oxidation weight gain increase with the raise of zirconia content. Because of the raise of zircon content caused the increase of diffusion rate. The fracture surface morphology differ to ZrO2 content. The fracture surface morphology of
AZ0/SiC is similar and planar, without obvious oxidation. The fracture mode is transgrainular. The fracture surface of AZ10/SiC and AZ20/SiC are similar, with the oxidation layer and pore born of the oxidation. It give the negative effect to the fracture strength and toughness of specimen. The fracture surface of AZ50/SiC is with pore and crack of oxidation. The result of fracture strength and toughness can be divided into two parts. The fracture strength and toughness are better with the more ZrO2. It is the helpness of SiC and ZrO2 particle. The result of
oxidation depend on the phenomenon of oxidation. The degree of oxidation of AZ0/SiC and AZ10/SiC is light, the effect of oxidation on fracture strength and toughness is small. After short oxidation time, the fracture strength and toughness rise. The degree of oxidation of AZ50/SiC is heavy. The effect of oxidation on fracture strength and toughness is giant. The fracture strength and toughness of specimen after oxidation is weaker than that before oxidation.
致謝
經過兩年的努力後終於完成這份論文,其間面臨許多難題與困 難,慶幸在同學、朋友、師長的鼓勵及協助下讓我能夠克服並順利畢 業。 在此,要特別感謝一些在我的研究上協助我的師長、同學們;首 先,要感謝的是林健正老師,感謝林老師在專業的知識上給我許多的 指導,且給我獨立思考的精神,並督促我們以嚴謹的態度來面對研究 上的事物。另外,還要感謝慈莉學姊,在研究的方向上給我許多的指 導,讓我的研究得以順利的進行。此外,特別要感謝昆霖、宏達、家 祥學長,在研究分析上給我許多的建議與幫助,他們樹立了實驗室良 好的研究典範,讓我們得到的許多研究上的啟發。也感謝家政、明昌 同學,他們在實驗上以及生活上給我許多關懷及建議,這情誼令人難 忘。還有要感謝研究室的偉舜、桓德、文紹學弟,協助我在實驗上的 研究,且給實驗室帶來許多歡笑,讓我愉快的渡過在研究室的時光。 此外,感謝可愛的怡雯姊及諸位璞玉姊姊,謝謝妳們的支持與鼓勵, 很高興能和妳們相知相識。 最後,特別感謝我的家人,在我這兩年的研究生活中給我最大的 支持,讓我得以順利地完成這兩年的學業。 謹以此篇論文獻給曾經給予我幫助的家人、師長、朋友們;謝謝。目錄
中文摘要………...………..i 英文摘要………..……….iii 致謝……….………...v 目錄………..…vi 表目錄………..….ix 圖目錄……….…..x 第一章 前言………..………...1 第二章 文獻回顧……….4 2.1 分散原理…………..………..…………4 2.2 溶劑的選擇………...……….……4 2.3 調整 PH 值……….5 2.4 SiC 強化氧化物基地的複合材料之氧化行為………..…….5 2.5 ZrO2的添加對複合材料之影響機制………..…...7 2.6 氧化矽的相變化行為………...9 2.7 Al2O3和SiO2的二元相圖………..…10 2.8 韌化機制簡介……….………....……13 第三章 實驗步驟………...……..15 3.1 粉體之特性……….…153.2 分散實驗……….……….…...…15 3.3 熱壓試片的製備…..………..…...16 3.3.1 SiC/ZrO2/Al2O3複合材料粉末的製備…….…….16 3.3.2 SiC/ZrO2/Al2O3複合材料熱壓試片………….….16 3.4 試片加工.………….………..….……….…..…….17 3.5 氧化試驗……….…………..……..17 3.5.1 氧化試驗前準備………..…17 3.5.2 氧化試驗……….….18 3.6 單位面積之重量變化(△W/A)分析……….…….…..18 3.7 四點抗折測試………...…………19 3.8 X-RAY 繞射分析………...………20 3.9 掃描式電子顯微鏡(SEM/EDS)………..20 3.10 掃描穿透式電子顯微鏡觀察(STEM/EDS)………..….21 第四章實驗結果………...22 4.1 氧化重量增加分析…...………22 4.2 X 光繞射分析……….…...………...24 4.2.1 複合材料氧化表面之 X 光繞射分析………..24 4.2.2 複合材料破壞斷面之 X 光繞射分析………..26 4.3 複合材料斷裂面微觀結構分析..….………..29
4.4 四點抗折測試分析…….……...……….……...…..31 4.4.1 破壞強度分析………...……….….31 4.4.2 破壞韌性分析……….36 4.4.3 韌化機制分析……….37 第五章 結論……….…...41 第六章 參考文獻………...42
表目錄
Table 1 試片成分、熱壓條件及 XRD 相….……….……...…..50 Table 2 AZ10/SiC 試片於 1350℃氧化不同時間之 m-ZrO2 比例變
化...51 Table 3 AZ20/SiC 試片於 1350℃氧化不同時間之 m-ZrO2 比例變
化...52 Table 4 AZ50/SiC 試片於 1350℃氧化不同時間之 m-ZrO2 比例變
化...53 Table 5 試片材料性質表...54
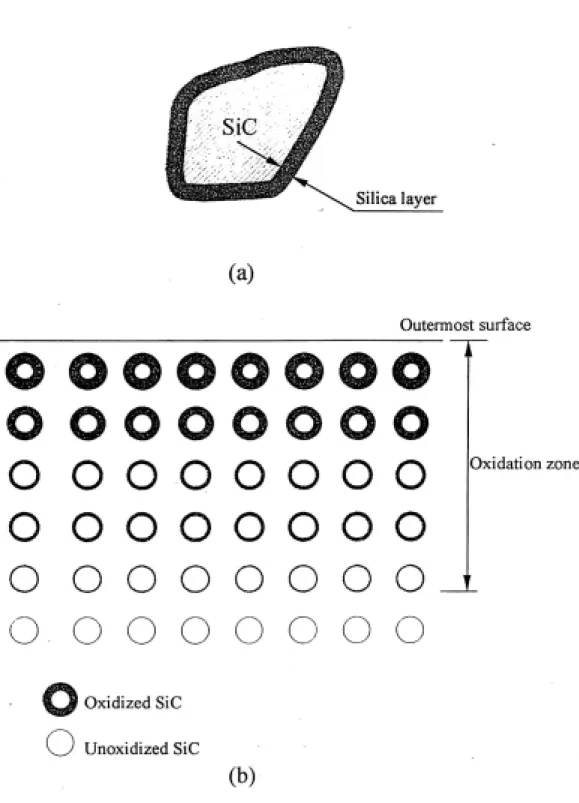
圖目錄 Fig. 2-1 (a)silica 層示意圖……….………....……..55 (b)SiC 複合材料氧化後的氧化深度示意圖………….……55 Fig. 2-2 Mode I 於不同氧化時間的氧化型態圖示………...……….56 Fig. 2-3 Mode I 於不同氧化時間的氧化型態圖示…..………..…….56 Fig. 2-4 碳化矽試片表層之截面微觀結構圖示 (a) 未負載………57 (b) 負載後………57 Fig. 3-1 實驗步驟流程圖………..……...………...58
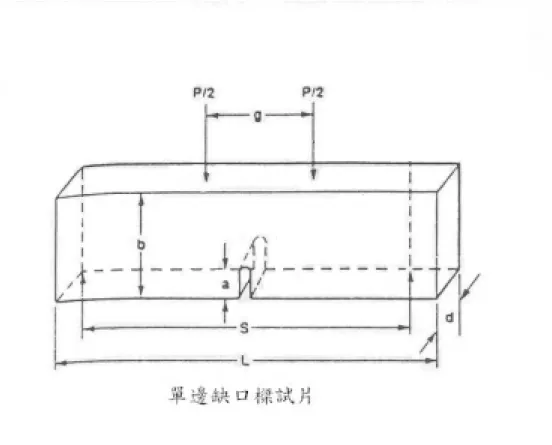
Fig. 3-2 SENB(single edge notched beam)試片……….….59
Fig. 4-1 於 1350℃氧化溫度不同氧化鋯含量之氧化時間對氧化重量 增加量平方關係圖………..60 Fig. 4-2 AZ0/SiC 於 1350℃不同氧化時間之試片氧化表面 X 光繞射光 譜圖………..61 Fig. 4-3 AZ10/SiC 於 1350℃不同氧化時間之試片氧化表面 X 光繞射 光譜 圖……….………...……….61 Fig. 4-4 AZ20/SiC 於 1350℃不同氧化時間之試片氧化表面 X 光繞射 光譜 圖………...………...………..62 Fig. 4-5 AZ50/SiC 於 1350℃不同氧化時間之試片氧化表面 X 光繞射 光譜 圖……….……….…....……….62 Fig. 4-6 AZ10/SiC 於 1350℃不同氧化時間之試片斷裂面 X 光繞射光 譜 圖……….……….……63 Fig. 4-7 AZ20/SiC 於 1350℃不同氧化時間之試片斷裂面 X 光繞射光 譜圖...63 Fig. 4-8 AZ50/SiC 於 1350℃不同氧化時間之試片斷裂面 X 光繞射光
譜圖...64 Fig. 4-9 AZ0/SiC 於 1350℃氧化溫度之(a)未氧化 (b)氧化 9hr (c)氧化
50hr (d)氧化 200hr 之斷裂面 SEM 微觀結構………65 Fig. 4-10 AZ10/SiC 於 1350℃氧化溫度之(a)未氧化 (b)氧化 9hr (c)
氧化50hr (d)氧化 200hr 之斷裂面 SEM 微觀結構……...66 Fig. 4-11 AZ20/SiC 於 1350℃氧化溫度之(a)未氧化 (b)氧化 9hr (c)
氧化50hr (d)氧化 200hr 之斷裂面 SEM 微觀結構……...67 Fig. 4-12 AZ0/SiC 於 1350℃氧化溫度之(a)未氧化 (b)氧化 9hr (c)氧
化 50hr (d)氧化 200hr 之斷裂面 SEM 微觀結構…………68 Fig. 4-13 (a)不同氧化鋯含量試片於 1350℃之破壞強度對氧化鋯含 量關係圖……..………69
(b)不同氧化誥含量試片於 1350℃之氧化前後的破壞強度 變化量對氧化鋯含量關係圖...69 Fig. 4-14 AZ0/SiC 經 1350℃分別氧化(a)9hr (b)50hr(c)200hr 之氧化
表面BEI 微觀結構……….70 Fig. 4-15 (a)不同氧化鋯含量試片於 1350℃之破壞韌性對氧化鋯含 量關係圖...71 (b)不同氧化鋯含量試片於 1350℃之氧化前後的破壞韌
Fig. 4-16 AZ10/SiC 於 1350℃/50hr 之試片斷裂面 SEM 微觀結構..72
Fig. 4-17 AZ50/SiC 經 1350℃分別氧化(a)9hr(b)50hr(c)200hr 之斷裂面 SEM 微觀結構……….………73
Fig. 4-18 AZ0/SiC 於 1350℃/50hr 之試片 TEM 圖示………..74
(a) 裂縫之 TEM 圖示………..……….…….74
(b) 碳化矽之 EDS 分析光譜……….74
Fig. 4-19 AZ0/SiC 於 1350℃/50hr 之試片斷裂面 SEM 微觀結構…..75
Fig. 4-20 AZ10/SiC 於 1350℃/50hr 之試片斷裂面 TEM 圖示………76
(a) 裂縫之 TEM 圖示………76
(b) 氧化鋯之 EDS 分析光譜………76
Fig. 4-21 AZ10/SiC 於 1350℃/50hr 之試片斷裂面 SEM 微觀結構…77 Fig. 4-22 AZ10/SiC 於 1350℃/50hr 之試片斷裂面 SEM 微觀結構…78 Fig. 4-23 AZ20/SiC 於 1350℃/50hr 之試片 BEI 微觀結構…………..79 Fig. 4-24 AZ20/SiC 於 1350℃/50hr 之試片斷裂面 SEM 微觀結構…80
第一章 前言
陶瓷材料具有許多優異的物理以及化學特性,如高硬度、高強 度、耐磨耗、化學安定性佳等優點,在耐高溫方面亦是其特別受矚目 的特性。自 1960 年起,陶瓷材料即被使用在耐火材料和建材上,然 而因為其本質上的低韌性及強度分散等脆性材料之特徵,而使陶瓷材 料在運用上受到很大的限制。因此自 1965 年起,科學家投入許多心 力研究陶瓷材料的強度以及破壞韌性等研究,積極發展出陶瓷材料來 改善這些缺陷。陶瓷基複合材料即是在陶瓷基材中添加強化相,通常 為非氧化物相SiC、Si3N4,可為顆粒狀、片狀、鬚晶、長纖維或是短 纖維等,使得裂痕在成長傳遞時的能量能夠被大量吸收,藉此使材料 的韌性大量提昇。 陶瓷在機械性質方面在這二十幾年間因顆粒(particle)和纖維 (fiber)的增強作用,已有了大大地改進。特別是碳化矽鬚晶強化氧 化鋁,在作為切割工具或高溫的結構材料上有其很大的優點。這些複 合材料具有高強度、斷裂韌性且兼具在高溫下有好的熱衝擊性和整體 潛變阻抗。[1] 鋁基複合材料具有高度的耐磨耗性、硬度、強度、抗化學腐蝕性 等優點,氧化鋁陶瓷具有耐熱性、絕熱性、高硬度和耐磨耗等種種特 性,自古以來常被廣泛運用到不同的領域。由於他在高溫下的安定性,常被應用作為爐材、耐熱管、保護管、坩堝等高溫材料。然而因 為其脆性而使得其不適合作為熱衝擊或是結構方面需要高度韌性地 運用。[2] 為了克服鋁基及其他具高度利用性陶瓷材料的機械性質,可加入 一些氧化物去改變其韌性,例如把 ZrO2顆粒加入陶瓷基材,這種韌 化主要是在裂痕生長過程,或是燒結冷卻的過程中,氧化鋯顆粒發生 正方晶相至單斜晶相的的麻田散鐵相型變化,可明顯的提高陶瓷基複 合材料的破壞韌性,此即所謂的相變化韌化機構。此外,也可加入 SiC 鬚晶等強化相提昇其韌性。由於 SiC 鬚晶在 Al2O3基質中氧化會 減少撓曲強度,ZrO2顆粒被添加到SiC-Whisker 強化地氧化基材中, 明顯的改善其在室溫下的破裂韌性。[2] 因此我們應用前述的韌化方式,以Al2O3以及部分安定化氧化鋯
為基地,在其中添加SiC 為強化相,製作出一個 SiC/ZrO2/Al2O3的複
合材料。因此一複合材料主要應用於高溫結構方面,所以需要考慮其 高溫抗氧化性,而我們在其中所添加之非氧化物強化相SiC,在高溫 的環境中會產生氧化作用,而導致複合材料的機械性質下降。因此, 對於複合材料的高溫氧化行為之研究就有其不容忽視的重要性。而另 外一方面添加氧化鋯對陶瓷材料雖然有韌化作用,但也因氧在其中擴 散速率較快,而影響其氧化速率。所以氧化鋯的含量對於高溫氧化的
影響及其後續機械性質方面的影響值得深入探討。 本實驗主要目的在於探討Al2O3/SiC/ZrO2的機械性質以及韌化機 制;因此,將以四點抗折儀器的測試以及 X-射線繞射儀鑑定,來分 析此複合材料在不同氧化鋯含量下經過氧化處理後的機械性質變 化,並輔以 SEM、TEM 對其抗折測試後的斷面作微觀分析,藉以分析 不同氧化鋯含量試片的韌化機制為何。
第 2 章文獻回顧
本章將首先對製作Al2O3/SiC/ZrO2複合材料流程中所需要運用的 原理做一簡單介紹;接著,對SiC、ZrO2的氧化行為以及其氧化速率 的計算方式做簡單回顧。最後,再對機械性質測試中發生的可能韌化 機制做文獻回顧。 2.1 分散原理 陶瓷製程成形前的粉末處理分為乾式混合和濕式混合,許多的研 究[3-7]指出濕式混合可以得到良好的分散性,Sacks 等人[7]更提出濕式 混合較乾式混合於燒結前的生胚有較小的開放性空孔和較高的相對 密度,可降低燒結時的收縮和減少燒結後缺陷的產生。在濕式混合裡 良 好 的 分 散 性 , 可 減 少 混 合 過 程 中 相 同 的 粉 末 凝 聚 成 粉 團 ( agglomerate ),對部份氧化釔安定的氧化鋯而言,若能減少燒結前凝 團的產生,即使利用一般的常壓燒結密度也可達 99.5%[8]。 另外,粉體的分散必須考慮、溶劑的選擇、粉體的表面電位、電 解質、分散劑和表面活性劑等因素的影響。 2.2 溶劑的選擇 溶劑的選擇必須是介電常數值高,可增加擴散電偶層的厚度,而得到較佳的分散效果;且溶劑需有好的潤濕性和解離性,使粉體能夠 和溶劑潤濕,並充分解離於溶劑中。此外,如果溶劑的沸點過高,則 會造成混合後不易乾燥的問題。文獻[6,9,10]中顯示醇類的溶劑對碳化矽 有好的分散效果,且 Parish 等人[11]亦指出醇類對於金屬氧化物有好 的分散效果,而甲醇和乙醇二種溶劑符合介電常數高、沸點低之優 點,因此本實驗將以乙醇做為溶劑的選擇。 2.3 調整 PH 值 當兩粉體互相靠近時,會因粉體帶相同的電荷,產生靜電排斥力 而無法接近對方,調整溶液的PH 值可改變粉體表面的電荷量,而產 生較大的排斥力,使分散效果提高;Homemy 等人[4]研究Al2O3和SiC(W) 複合材料的分散行為中,利用NH4OH 調整 pH 大於 10 時,可以使粉 體間不會產生凝固現象,並於燒結後得到良好的機械性質;Porter 等 人[11]於去離子水中調整 pH 值為 11,可使 Al2O3和 SiC(W)得到良好的 分散性。 2.4 SiC 強化氧化物基地的複合材料之氧化行為 近期有許多關於SiC 鬚晶或顆粒強化氧化物基地的複合材料之氧 化行為研究[31-39,44-52],kriven 等人[31]研究在 Al2O3或 mullite 基地中加
入20vol%SiC 鬚晶,暴露於 1200℃的氧化環境時,Al2O3/SiC 複合材
料的機械性質急速下降,而 mullite/SiC 的強度則無明顯的變化,表示 SiC 的氧化速率在 Al2O3基地中較快的原因。而 Porter 和 Chokshi[44]
在 Al2O3基地中加入 18vol%SiC 鬚晶,於 1500~1700℃中研究其氧化
行為,發現氧化產物SiO2和基地產生mullite 。Borom 等人[38,39]亦於
Al2O3或mullite 基地中加入 SiC 鬚晶或顆粒或 MoSi2顆粒,他們發現
在1200 至 1500℃時,氧化層厚度的成長遵循拋物線速率定律,且氧 化行為不僅受氧化產物 SiO2 的影響,並且受到氧化產物和基地之間
的反應影響,導致最後成分的改變。Luthra 和 Park [37]研究 Al2O3/SiC(P)
和 mullite/SiC(P)複合材料的氧化行為中,其氧化行為遵循拋物線速率
定 律 , 且 於 氧 化 區 域 和 未 氧 化 區 域 間 發 現 氧 化 產 物 mullite 、 aluminosilicate 和部份氧化的 SiC。
有 關 SiC 強 化 ZrO2 基 地 複 合 材 料 的 氧 化 行 為 研 究 中 ,
Backhaus-Ricoult [45]對 SiC/ZrO2/Al2O3複合材料在空氣中氧化,發現
其氧化後的截面有三個以顏色區分的區域,灰色氧化最外層的區域為 aluminosilicate;中間的白色區域有 ZrO2、Al2O3、反應產物mullite、
和大的孔洞;內層黑色的區域包含zircon、carbon 和部份“dissolved" 的SiCw。
2.5 ZrO2的添加對複合材料之影響機制
氧化鋯所具有的韌化機構,主要歸因於其具有三種同素異構體。 單斜晶 Monoclinic、正方晶 Tetragonal、立方晶 Cubic。在高於 1700 ℃時,正方晶是穩定態,到了2370℃變成了立方晶(2680℃為熔點); 由此可之,低溫時氧化鋯呈現對稱性較低的相,在降溫過程時其晶格 體積會膨脹,氧化鋯的韌化方式便是利用加入其它氧化物使得正方晶 相能存在於室溫下。當氧化鋯本體受到外來能量(應力)時,氧化鋯 由正方晶變成單斜晶相,使得晶格產生約 9.7%地膨脹而再裂縫尖端 產生一縮應力,或是有所謂的 Microcracking 微裂痕的現象出現,以 增加其韌性。[53] 單斜晶在 11000C 時會經由產生劇烈的相變化而轉變成立方晶 相,此種相變化時常會導致整體材料地粉碎。若加入一些添加物形成 具有螢石結構的立方相時,則不會有巨大應變產生。而使得力方晶相 穩定在低溫下的材料,稱為(CSZ)立方穩定相氧化鋯,這種立方相 穩定合金氧化物,事實上是把相變化的移轉溫度移到較低的溫度。且 如果調整穩定劑的含量可製造出同時具立方晶相和單斜晶相或正方 晶混合的微結構,這些材料稱為(PSZ)部分穩定氧化鋯。[53] Claussen 提出,ZrO2也能以第二相方式被加入 Al2O3中,而形成 顆粒形式的複合材料,Claussen 認為韌性的增加是由於應力誘導
(Stress Induced)微裂痕。事實上,工業界早已使用融合 Al2O3、ZrO2
相製作研磨的砂輪片,可見其具有優越的機械性質。在反應燒結的過 程中,可以在微結構中觀察到 mullite 基材與 ZrO2 或 Al2O3 基材與
mullite 和 ZrO2成核。而在Al2O3基材與mullite 和 ZrO2成核的機制中,
如果 mullite 是以針狀的方式成核,則會在基材中有另一種橋連 (bridging)的韌化機制。而利用燒結的方式有一缺點,因 ZrSiO4分
解反應會致使孔洞的增加而使得機械性質下降。但此問題可藉以燒結 過程中先施以緻密化有效改善。[54]
而氧化鋯的添加,也對於氧在複合材料中擴散的速度有著極大的 影響,Liu 等人[34]提出 SiC/ZrO2/mullite 的氧化速率較 SiC/mullite 快,
因為 ZrO2 的存在降低了 SiO2 結晶化的速率。Lin 等人[46,47]發現
SiC/ZrO2/mullite 的氧化速率遠高於 SiC/mullite,是因為 ZrO2具有較
高的擴散速率,提供一個快速的氧分子擴散路徑。Tsai 和 Lin [50]更指 出,氧化溫度 1000℃和 1200℃時,ZrO2的含量皆存在一個臨界體積 百分比,當 ZrO2的含量高於20vol%時,氧化速率急速增加。基地的 成分會影響複合材料的氧化速率[49-51]Tsai[50]指出當氧化鋯含量低於 20vol%時,氧在基地的擴散係數小於氧在 SiO2的擴散係數時,氧化 深度較小、SiO2的厚度變化較大,氧化速率較慢,此種氧化模式稱為
模式Mode I 之示意圖。當氧化鋯含量高於 20vol%時,氧在基地的擴 散係數大於氧在 SiO2 的擴散係數,氧氣容易穿過基地向內擴散將內
部的碳化矽氧化,因此氧化深度較大、SiO2的厚度變化較小,氧化速
率較快,此種氧化模式稱為Mode II, Fig. 2-3 為 SiC 氧化模式 Mode II 之示意圖。 2.6 氧化矽的相變化行為 氧化矽的變態行為一直都是大家特別感興趣的對象。在室溫下 最安定的氧化矽形態是低溫型石英 (1ow quartz ),當溫度達到 573℃ 時,低溫型石英將經由移位變態而成為高溫型石英 (high quartz )。到 850℃時,石英將以極緩慢的速率變態(即重組變態)為鱗石英,事實 上,有若干實驗結果可證明,若無其他雜質存在,石英幾乎無法變成 鱗石英。在850 到 1470℃間,鱗石英是安定的形態;等溫度超過 1470 ℃,鱗石英又將經由另一次重組變態而成為白矽石。在從高溫冷卻下 來的途中,鱗石英和白矽石都會發生移位變態;其中高溫型白矽石 ( high cristobalite ) 會在 200 到 270℃間藉著扭曲結構的方式變成低溫 型白矽石( low cristobalite ),而高溫型鱗石英( high tridymite ) 除了在 160℃時,會變成中溫型白矽石 ( middle tridymite ) 外,還會在 105 ℃時變成低溫型白矽石 ( low tridymite )。以上這些變態總共造成了七
種不同的氧化矽同素異形體及三種基本結構。這三種基本結構間的變 態皆為重組變態,即使能夠發生,其速率也相當慢;若希望變態在較 短的時間內完成,唯一的辦法,就是添加其它能擔任溶劑角色的物 質。相反地,各基本結構的高溫與低溫型態間的移位變態則發生得很 快,不受任何外力的干擾。 [56] 2.7 Al2O3和 SiO2的二元相圖 由文獻可得知莫來石(mullite)和白矽石(cristobalite)的共晶反應 發生在1580℃,而形成一含約 95mol%的液體。莫來石與氧化鋁間的 固相線溫度是在1828℃處。[56] 在此兩相混合的過程中,固熔反應機制發生在α-Al2O3&SiO2 擴 散介面,SiO2分解 Al2O3分子,此反應的驅動力是因為當氧化鋁分子 被混合入液相的原子結構,使得熔融的氧化矽內部自由能的減少。 Al2O3 分子可自結晶的α-Al2O3 藉著打斷鍵結的方式溶解出來,但需 要足夠的能量。這些能量是來自於Al2O3溶解所造成矽質液態中內能 的釋放。總和這些步驟可得到反應如下:[57]
step I : SiO2 +xAl2O3 =SiO2⋅xAl2O3 (2-4) step II: α −Al2O3 = xAl2O3 +(1−x)α −Al2O3 (2-5) step III: SiO2 +α −Al2O3 =SiO2 ⋅xAl2O3 +(1−x)α− Al2O3 (2-6)
此系統的共晶成分很接近相圖氧化矽的這一端。結果,雖然只 有添加少量的氧化鋁,當溫度高於 1600℃時,液體的數量及大幅增 加。也就是這個緣故,矽磚的發展過程中人們發現,矽磚中只要含少 量的氧化鋁,其性質就會劣化。但目前已有一種添加少量氧化鋁經過 生料特選及處理所製成的氧化鋁量及低的超矽磚,被用在需要承受高 溫的結構物上。[56] 此外,在 1587℃以下,其平衡相是莫來石與氧化矽,由於這兩 種相的相對數量因成分而異;在黏土質耐火磚中(含有 35 至 55%的氧 化鋁)的性質也有對應的變化,當溫度超過 1600℃時,其所含的液體 數量系由氧化矽和氧化鋁的比例所決定。在這些的運用上,人們喜歡 含氧化鋁量高的耐火磚。氧化鋁的含量越多,磚的耐高溫性質就越 好,因為莫來石所佔的分率增多。氧化鋁含量超過72wt%後者磚將全 部都是莫來石或莫來石與純氧化鋁的混合物。在這種情況下,除非是 溫度高於 1828℃,否則不會產生液體。有些實際的運用就需要熔融 莫來石,因為它在高溫下對腐蝕和變形的抵抗力均極優異。而純氧化 鋁是本系統中耐高溫性最佳者。 而在其他相關系統中,長石-黏土-燧石系統的三元系統中的文獻 也提到;其共晶溫度是990℃,熔點是 1140℃。隨著溫度昇高,平衡 的液相(固相是莫來石)數量逐漸增多。在溫度略高於 1200℃的情形
下,此系統平衡狀態變化不大。因此,在該溫度下做長時間的燒結所 所得到的結果和在較高的溫度下做短時間燒結所得到的應極為相 似。[56] 在燒製期間,長石顆粒首先在 1140℃時熔化,但因它們的粘度甚 高,在溫度達到 1200℃之前,它們的形狀不會改變。到了 1250℃左 右,尺寸小於 10μm 的長石顆粒將因和周圍的粘土起反應而消失, 其它較大的顆粒也開始和粘土起反應 ( 鹼金屬離子自長石中擴散出 來,莫來石也在玻璃中形成)。此時,粘土相開始收縮而出現裂縫。 針狀的小莫來石顆粒在 1000℃左右即已出來,隨著溫度昇高,莫來 石晶體逐漸長大。到了1400℃左右,莫來石已長成長約 0.01mm 的稜 柱形晶體。在溫度達到 1250℃以前,石英顆粒幾乎不發生變化;過了 1250℃後,小的石英顆粒之稜角逐漸消失。隨著溫度昇高,各石英晶 粒四周的高矽玻璃( high-silica glass )溶環( solution rim )範團逐漸增 大。到了 1350℃時,尺寸小於 20μm 的石英顆粒均已全部熔化;到 了 1400℃,石英顆粒已所剩無幾,整個瓷體幾乎全是由莫來石和玻 璃所組成。[56]
在 Muller and Schneider et. (1993) [57]的研究中分別以 Quartz、 Cristobalite、SiO2 glass 和α-Al2O3作用形成 mullite 的機制作研究,
例,其起始反應始於≤1100℃下 Quartz 因溶入鹼性金屬氧化物(Al2O3)
而使固熔點轉低,產生融化(melting)的現象,接著,藉富含 SiO2-融化
物滲透入α-Al2O3 晶粒聚集處而作用,在 1470℃左右,有一誘發
mullite 成核的機制形成,應是液相促進其反應所致。至 1550℃左右, 有越來越多的mullite 自其外環的 glassy silicate 中形成;且有一部份 的 SiO2 自 完 全 無 結 晶 的 glass 變 成 幾 乎 不 具 反 應 性 的 結 晶 相
cristobalite,可能在 SiO2和α-Al2O3接觸的介面造成擴散的阻礙。但
到了高溫 1650℃,mullite 藉著控制其結晶化的液相發生,熔融了殘 留的cristobalite 和α-Al ≥ 2O3而完全生成mullite。[57] 2.8 韌化機制簡介 Krishan K. Chawla 在文獻中提到[64]陶瓷基複合材料(CMC)的討論 中提到韌化機制有以下常見幾種方式: 1.Matrix microcracking 裂縫傳遞時被導引至其他可能路徑而消耗裂 縫能量。
2.Fiber/matrix debonding leading to crack deflection 添加物和基地相 之間的鍵結力失效而成為裂縫偏折前進路線。
3.Fiber pullout 添加物為鬚晶時出現,添加物在被拔出時會消耗能量。 4.Phase transformation toughening 相變化常伴隨著體積變化而在裂縫
尖端產生壓應力,裂縫的傳遞需消耗能量才能繼續前進。
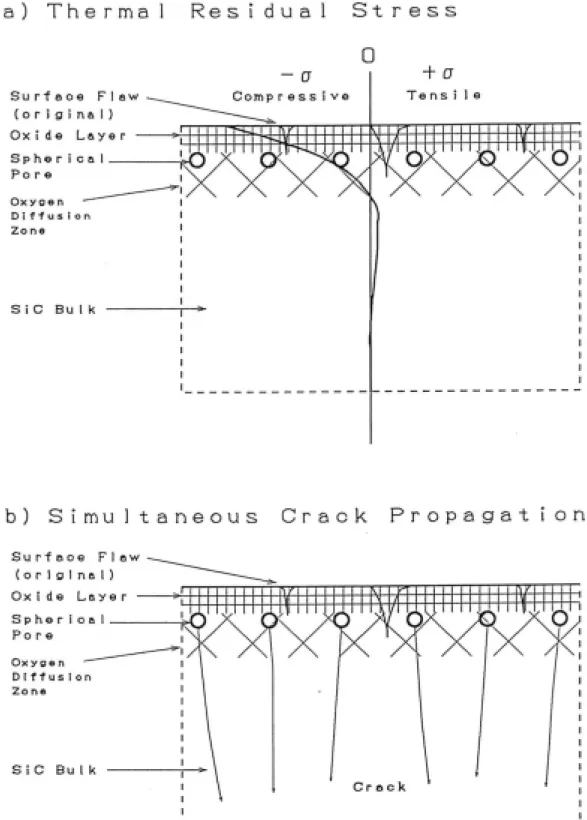
試片破壞韌性可定義為應力-應變圖中曲線下的面積,也就是試片在 斷裂前所吸收的能量,曲線下的面積越大表示試片的韌性越佳。上述 各機制其原理在於透過能量消耗(energy-dissipating),使裂縫在傳遞 過程中能量被抵消掉,而裂縫前進的程度會減少,藉此提高試片的破 壞韌性及強度。C.Kaya[65]在SiC/Al2O3/Y-TZP 複合材料中提到存在一
種韌化機制,稱為Thermal residual stress 熱殘餘應力。起因於試片基 地相和添加物之間的熱膨脹係數不相同,從高溫降至室溫過程中會因 熱脹冷縮程度相異而產生內應力。由於碳化矽的熱膨脹係數較小因此 會受到壓應力,在碳化矽周圍發現呈放射狀分佈的微裂縫。微裂縫除 了有韌化效果外,也能提高其他韌化機制發生的機率,如 grain bridging 晶粒橋接等韌化機制。此外,由於不同成分的體積變化不同, 將 會 使 基 地 相 與 添 加 物 間 的 鍵 結 變 弱 , 而 這 將 使 韌 化 機 制 debonding/sliding 有作用。文中提到尚有另一種韌化機制 modulus-load transfer。在 modulus-load transfer 中,裂縫前端的壓力會被轉向,沿 著複合材料中較高Young’s modulus 的顆粒向遠離裂縫平面處前進。 經此作用,在裂縫前端的有效剪應力強度被減弱,試片破壞韌性直接 受到提升,不過此時氧化鋯的應力誘發相變韌化機率也同時降低。
第三章
實驗步驟
本實驗以熱壓法製備Al2O3/SiC/ZrO2複合材料,有關本實驗的步
驟如Fig. 3-1 所示,包括分散實驗、熱壓試片、熱壓試驗、氧化實驗、 四點抗折實驗、X 光繞射分析、Weight gain 分析及 SEM 和 TEM 斷 面觀察分析。
3-1 粉體之特性
本實驗採用α-Al2O3 粉體(AKP53,Sumitomo.,Tokyo, Japan),
3mol% Y2O3 partially stabilized Zircon 粉體(TZ-3Y,Tosoh corporation,
Tokyo, Japan),和α-SiC 粉體(Cerac Inc.,Milwaukee, WI U.S.A)。其中 粉 體 粒 徑 量 測 方 法 為 沉 降 法 (SediGraph5100, Mcromeritics, GA, USA),比表面積量測測量利用 BET 方法,理論密度則使用 pycnometer (Multivolume pycnometer 1305, Micromeritics, GA, USA) 量測,而化 學成分則由廠商提供。
3-2 分散實驗
量取1g 的粉末,以 20ml 的甲醇、乙醇兩種溶劑,利用 OH-、H+ 離子調整不同的PH 值使粉末帶電,並加入 NH4+、NO3-當電解質,使
粉末表面產生擴散電偶層而分開,在利用超音波震盪(Model XL-2020, SONICATOR, Heat Systems Inc., Farmingdale, NY)5-10 分鐘將凝聚分
子打散,即形成旋濁的溶液,將其置放24 小時後,在將 20ml 的溶劑 稀釋為 1000ml,則完成分散實驗之溶液。然後將測試的溶液置於介 面電位分析儀(Model Zetaplus, Brovkaven, USA)之電極中,測量溶液 中粉末粒子於外加電場下的移動速率,利用溶液性質和電泳速率即可 算出介面電位值。 3-3 熱壓試片的製備 3-3-1 SiC/ZrO2/Al2O3複合材料粉末的製備 將欲混合的SiC、ZrO2、Al2O3三種粉末分別以我們欲混合的比例 秤重後並加以混和,以界面電位量測出來較高的去離子水為溶劑,且 利用 NH4OH 調整 PH 值為 10.5,接下來以超音波震盪,使其成為均 勻分散的懸浮液。將以均勻分散的懸浮液加熱攪拌將其快速揮發,再 將其放入真空烘箱內以150℃上下烘乾。烘乾後再以研缽島碎,最後 利用 80mesh 篩網過篩,加以收集,即完成粉末的製備工作。不同粉 末的編號和成分如Table I 所示,基地的組成以 Al2O3和ZrO2的相對 體積百分比為計算標準。 3-3-2 SiC/ZrO2/Al2O3複合材料熱壓試片 將混合製備好之粉末倒入石墨模具中,且在粉末與石墨間以
graphite foil 隔離以避免熱壓後試片和模具的沾黏情形發生,接下來以 石墨模具放入熱壓爐中,(熱壓爐的型號為 Model HP50-HTG-7010, Thermal Technol. Inc., USA),先施以 5Mpa 的壓力將粉末預壓成型, 成型後釋放壓力,然後抽真空至2x10-4Torr,在通入 Ar,確保爐體內 沒有空氣後開始加熱,於溫度 3000C 之前以每分鐘 10℃較慢的速度 加熱,並保持真空,使得粉末間的水氣能夠完全的釋出,於溫度300 ℃之後在通入氬氣將水氣排出,且維持一大氣壓以避免氧化產生,並 施壓至5MPa,當溫度升至 1000℃時,壓力升至 10MPa,然後再達到 最高溫度之前,線性增加壓力至 32MPa。 接下來在溫度下降過程中 壓力漸漸釋放,直到溫度 1100℃時,壓力全部釋放完畢,待試片完 全的冷卻之後再從石墨模具中取出。 3-4 試片加工 熱壓完之試片,首先除去表面的graphite foil,並利用平面磨床將 兩面各研磨 1mm,再以研磨拋光機拋光至 3μm,最後切割成 10×6× 3mm 小試片。 3-5 氧化試驗 3-5-1 氧化試驗前準備 因為我們是以單位面積之重量變化(△W/A)做氧化分析,所以在
氧化前先利用游標卡尺測量試片的尺寸並計算其表面積,再以丙酮及 超音波洗淨並由電子天秤測量其氧化前重量。
3-5-2 氧化試驗
將高溫爐(Model 51333, Lingberg, WI)以每分鐘 15℃的升溫速度 升溫至欲氧化的溫度後,將試片置於坩堝後放入爐子中,置入試片時 爐體溫度會稍微的下降,待升到所設定的溫度後,再開始依我們所設 定的時間計時;而在本實驗中氧化溫度為 1350℃,氧化時間分別為 9、50、200 小時,達到時間後再將試片由爐子中取出。但由於在溫 度極高時將試片放入爐子,會因為試片內材料的熱膨脹係數不同而會 有crack 的情況產生,因此在做 1350℃的氧化時,我們將試片在 1000 ℃左右時就先放入爐子內,避免因為試片crack 或彎曲而影響氧化結 果。 此外取出試片時採用爐冷方式,將高溫爐溫度降至室溫再取出,避免 溫度差極大而產生crack。 3-6 單位面積之重量變化(△W/A)分析 由高溫爐內取出試片,在試片冷卻之後以電子天秤量測其重量, 把氧化前後的重量變化除以試片的總表面積,即可作為氧化速率的表
示方法。
3-7 四點抗折測試
本實驗韌性試片採用單邊缺口樑(Single Edge Notched Beam,SENB) 方法來製備,所有步驟依照陶瓷技術手冊內述準則。試片基本尺寸為 40mm×4mm×3mm(長×高×寬)。在切缺口前會先用鑽石盤,粒徑為 45μm, 作試片邊緣導角處理,並使用粒徑 3μm 的鑽石砂紙作表面拋光處 理。最後使用鑽石刀片在試片正中處往試片內部切至試片高度(4mm) 的 1/3 處為止。試片製備後如 Fig. 3-2 所示。鑽石刀片厚度為 0.02 ㎜,試片缺口寬度應略大於 0.02 ㎜。試片使用丙酮清洗並使用超音 波震盪。使用電子天平秤重記錄氧化前重量,有效準確度為小數點以 下四位。經過氧化處理後取出試片記錄氧化後重量。四點抗折實驗前 使用游標卡尺量測紀錄氧化處理後的試片長寬高尺寸。採用四點抗折 儀器(型號為Instron 8511.40)來測量機械性質,上支點跨距為 20mm, 下支點的跨距為 40mm。測試時預切的缺口向下置放,而與儀器衝頭 接觸面寬為 3mm。經過四點抗折測試後,將斷裂的試片使用鑽石刀片 沿平行斷面方向切下厚約 1 mm 的試片,依氧化鋯含量及氧化處理時 間的不同作分組。強度試片基本尺寸和韌性試片相同,不同的是強度 試片不用預切缺口,測試時與儀器衝頭的接觸面寬為 4mm。之後的氧
化處理條件和韌性試片相同。經過抗折測試後同樣使用鑽石刀片切出 所需的試片及分組。 3-8 X-RAY 繞射分析 利用 X-繞射線儀(Siemens Mode 5000)對氧化前後的熱壓試片進 行 分 析 , 來 初 步 鑑 定 其 所 生 成 的 氧 化 物 相 別 。 儀 器 設 定 電 流 為 200mA、電壓為 50KV,所收取的繞射角度定在 100~800;掃瞄為速度 每分鐘 6°,每隔 2θ=0.01°自動記錄 x-ray 的強度之後,再將分析所 得到的結果在與JCPDF 卡相互比對,以判定相別。 3-9 掃描式電子顯微鏡(SEM/EDS)
以掃描式電子顯微鏡(Model JXA-8600SX, JEOL, Tokyo, Japan)之 二次電子成像,以line scan 或 x-ray mapping 模式進行成分定性分析 及波長散佈分析儀(WDS)對試片進行定量分析。在觀察前以蒸鍍方法 鍍上一層碳或白金以增加導電性。在本實驗中我們觀測試片斷面,藉 以觀察斷裂面的晶粒分佈和表面型態,粗步瞭解crack 的行為。
3-10 掃描穿透式電子顯微鏡觀察(STEM/EDS)
應後之試片,試片製作如Fig. 3-2 所示,以明視野像(Bright Field Image, BFI)觀察介面組織及擇區繞射(Selected Area Diffraction Pattren, SADP) 作 為 辦 定 相 別 的 依 據 , 能 量 分 散 光 譜 儀(Energy Dispersive Spectrometer, EDS)鑑定各個相的組成元素。
另外可使用軟體CaRInc crystallography,輸入 Space group、晶格 常數、原子之相對位置及其相關之晶體資料,可模擬分析晶體結構的 繞射圖形,與實際繞射圖形相互比對鑑定。以上資料可藉由Pearson’s handbook of crystallographic data for intermetallic phase[58]查知。
第四章 實驗結果與討論
4.1 氧化重量增加分析 Fig. 4-1 是在 1350℃氧化溫度下不同氧化鋯含量試片之氧化重量增加 量平方對氧化時間的關係圖。計算方法是測量試片的氧化前後重量差 除以試片總表面積,單位是µg/㎜2。氧化過程中重量的增加主要來自 於碳化矽的氧化,試片氧化程度可以重量增加量來判斷。AZ0/SiC 和 AZ10/SiC 試片兩者氧化曲線相似,試片的氧化重量增加量不明顯, 表示氧化程度輕微。而 AZ20/SiC 氧化曲線介於 AZ0/SiC、AZ10/SiC 和AZ50/SiC 中間,是一氧化過渡相,由 Lin[46-48]的文獻中得知試片基 地相中的氧化鋯含量有一臨界值為 20%,氧化鋯含量在臨界值以上 時氧化速率快氧化深度大,如 AZ50/SiC;若在臨界值以下其氧化速 率慢氧化深度小,如 AZ0/SiC、AZ10/SiC。AZ50/SiC 試片的氧化重 量增加量明顯,表示氧化程度嚴重。 AZ50/SiC 氧化曲線在一開始時就急速上升,但 9 小時後及發現其曲 線斜率漸漸變小,此現象對照於x-ray 的分析結果發現,在 9 小時後 zircon 就急速的生成,逐漸降低了氧化鋯的含量,因而降低了氧化速 率,造成氧化曲線斜率漸漸變小的情形。然而,觀察 AZ20/SiC 的曲 線卻無此情況而呈現線性的氧化曲線;相同的對照 x-ray 的結果發現雖然此試片也在2 小時後就出現 zircon 生成的情況,但是在大約在 9 個小時後,逐漸的有一部份的 zircon 和氧化鋁作用生成 mullite 和氧 化鋯,使得氧化鋯的含量在此作用下逐漸再增加,因此氧化鋯的體積 百分比不至降低太多而改變其氧化模式,仍是維持一開始的氧化模 式;因此AZ20/SiC 氧化至 200 小時時仍維持其線性氧化曲線情形。 氧化過程中重量增加主要是碳化矽的氧化,碳化矽的氧化過程可由下 列步驟簡單表示,步驟 1:氧由空氣中擴散接近試片表面;步驟 2: 氧擴散進入試片表面;步驟 3:氧在基地相中擴散;步驟 4:氧擴散 入碳化矽外圍之氧化矽層;步驟 5:氧與碳化矽產生交互作用。而在 這個過程中,主要控制氧化速率的步驟為步驟 3 與 步驟 4,因此, 氧在基地相中與氧化矽中的擴散速率對氧化的重量增加量造成很大 的影響。由 Lin[46-48]的文獻上得知,氧在氧化鋯的擴散速率較氧在氧 化矽中的擴散速率快,當氧化鋯在基地相的含量低於某一臨界值時, 氧化鋯在試片中分散,無法加快氧的擴散,此時氧在基地內的擴散速 率慢,氧在基地相的擴散係數遠小於氧在 silica 層的擴散係數,此時 的氧化速率是由氧在基地的擴散速率所控制,此氧化模式稱為 Mode I,AZ0/SiC、AZ10/SiC 試片即是這種氧化模式。而 AZ20/SiC 及 AZ50/SiC 試片其氧化鋯含量大於此一臨界值,氧化鋯在基地中分佈 範圍增加,使氧在基地相中擴散較快,氧化速率是由氧在 silica 中的
擴散速率所控制,是為Mode II 模式。Mode II 的氧化模式中,氧擴 散深入試片內部,使更多碳化矽顆粒氧化,造成氧化重量增加量提 高,故AZ20/SiC、AZ50/SiC 試片之氧化程度較 AZ0/SiC、AZ10/SiC 試片嚴重。Fig 2-2 是氧化矽層示意圖,氧由外界向試片內部擴散接觸 碳化矽顆粒,進而產生交互作用生成氧化矽層。Fig 2-3 及 2-4 則分別 為Mode I 和 Mode II 氧化型態圖示,氧化模式 Mode I 的試片氧化深 度淺碳化矽顆粒氧化層厚;氧化模式Mode II 試片則是氧化深度大碳 化矽顆粒氧化層淺。 4.2 X 光繞射分析 ( XRD ) 4.2.1 複合材料氧化表面之 X 光繞射分析 Fig. 4-2 是 AZ0/SiC 試片未氧化前以及在 1350℃經 9、50、200 小時 氧化後的試片表面 XRD 繞射光譜圖,未氧化前試片的 XRD 分析中 只有出現氧化鋁及α-SiC 的繞射峰;在 9 和 50 小時氧化後的情形也 沒有發生太大的變化。然而,在經過200 小時後,偵測到氧化矽結晶 相(cristobalite)及 mullite ( 3Al2O3.2SiO2 )的產生。Mullite 的生成有兩
種方式,一種是碳化矽氧化所生成的氧化矽在溶入氧化鋁後降低熔 點,變成液相型態(liquid state)的氧化矽,且在經過與氧化鋁長時間的 作用後生成液相 aluminosilicate,待鋁成分的擴散到達臨界值即會生
成結晶相mullite(3Al2O3.2SiO2)。 Fig. 4-3 是 AZ10/SiC 試片未氧化前以及在 1350℃經 9、50、200 小時 氧化後的試片表面XRD 繞射光譜圖。在經過 9 個小時之後,碳化矽 繞射峰幾乎消失,結晶相的氧化矽 ( cristobalite )的繞射峰仍存在著, 而zircon 也逐漸生成所以其繞射峰漸漸增強,此外又新生成了 mullite 相。在 50 小時的結果中發現,氧化矽因為逐漸地和氧化鋯及氧化鋁 交互作用而消耗殆盡,而氧化鋯及氧化鋁的繞射峰也逐漸的減弱,而 其所產生的 mullite 及 zircon 的相逐漸增加。在 200 小時後,zircon 也因為和氧化鋁作用而分解,生成mullite 及 ZrO2。在XRD 分析中,
可以見到zircon 和 Al2O3的繞射峰逐漸減弱而t-ZrO2和mullite 的繞射
峰逐漸增強。mullite 可藉由兩種方式而生成,一為由氧化鋁和氧化矽 直接生成;另一種方法則是先由氧化鋯及氧化矽生成zircon 過渡相, 再由zircon 與氧化鋁形成 mullite,zircon 的生成降低了 mullite 成核的 活化能,有利於mullite 的生成。
Fig. 4-4 是 AZ20/SiC 試片未氧化前以及在 1350℃經 9、50、200 小時 氧化後的試片表面 XRD 繞射光譜圖,從 XRD 的分析圖中可發現, AZ20/SiC 與 AZ10/SiC 相生成的情形大致相同,但因 AZ20/SiC 的氧 化鋯的含量佔基地相中的體積百分比20%,所以氧在基地中的擴散較
快;在 9 小時後,氧化矽和氧化鋯逐漸作用生成 zircon,因此 zircon 的繞射峰強度增強,而在此時mullite 也已經生成。在 50 個小時過後, zircon 的繞射峰強度開始減弱,同樣的是因為 zircon 和氧化鋁產生作 用,生成mullite 及 ZrO2的緣故。 Fig. 4-5 是 AZ50/SiC 試片未氧化前以及在 1350℃經 9、50、200 小時 氧化後的試片表面 XRD 繞射光譜圖;由於 AZ50/SiC 的氧化鋯的含 量佔基地相中的體積百分比達50%,所以氧在基地中的擴散極快,在 氧化 9 個小時後,碳化矽的繞射峰已完全消失,皆因其氧化成氧化 矽,且與氧化鋯形成zircon 的緣故,因此 zircon 的繞射峰強度逐漸增 強。50 個小時後,發現氧化鋁的繞射峰強度開始減弱而漸漸地生成 mullite。為何 AZ50/SiC 之所以會生成較多的 zircon 而只有微量的 mullite,是因為生成 zircon 和 mullite 皆需是氧化鋯和氧化鋁分別和氧 化矽作用,AZ50/SiC 試片中的氧化鋯含量多,因此氧化矽接觸到氧 化鋯的機會大,而且生成 zircon 所需的活化能較生成 mullite 所需的 活化能較小,因此,氧化矽會優先與氧化鋯生成zircon,氧化矽與基 地相中的氧化鋁作用的機會相對減少,因此mullite 的生成數量少。 4.2.2 複合材料破壞斷面之 X 光繞射分析 Fig. 4.6 是 AZ10/SiC 於 1350℃不同氧化時間之試片斷裂面 x 光繞射
光譜圖。氧化處理後的試片均在2θ 約 22°左右有一個氧化矽結晶相 的繞射峰出現。這是因為隨著氧化鋯的添加,使得氧在基地相中的擴 散速率逐漸變快,氧在擴散入碳化矽顆粒後生成amorphous 氧化矽, 而原本為 amorphous 之氧化矽在長時間高溫下會 devitrified (去玻化) 而形成結晶相之氧化矽 ( cristobalite )。 AZ10/SiC 試片在不同氧化處理下的 x 光繞射圖其存在晶相及繞射峰 角度位置都一致。原因是此時x 光繞射分析以試片斷裂面為主,和往 常分析試片氧化表面不相同。由於氧化行為是氧由外界向試片內部擴 散,且試片AZ10/SiC 的氧化模式是 mode I,氧化層生成於試片表面, 試片內部未受氧化影響。
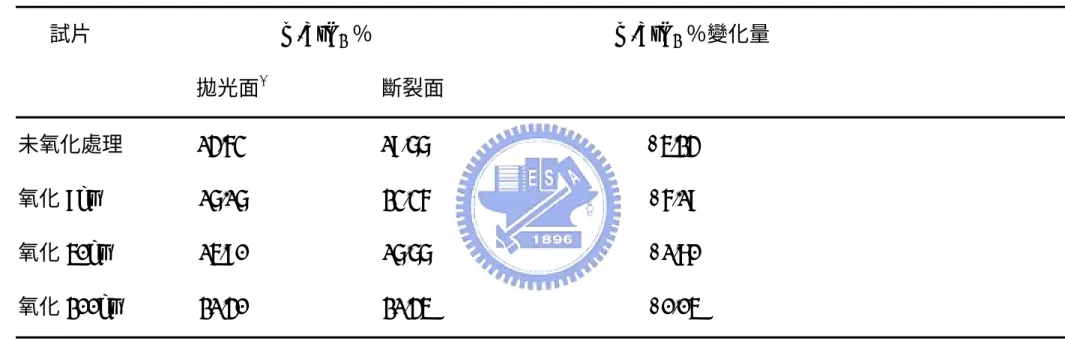
分析試片氧化前後的 m-ZrO2比例變化可觀察氧化鋯相變韌化是否對
試片破壞韌性有幫助。Kaya[65]提到SiC/Al2O3/Y-TZP 複合材料中氧化
鋯的各相比例可由下列公式計算得知: Vm=
( )
( )
( )
( )
( )
100 1 11 111 111 1 11 111 × + + + m m t m m I I I I I (4-2) Vt=1-Vm (4-3) Vm:試片中的 m-ZrO2比例 Vt :試片中的 t-ZrO2比例式(4-2)中的Im
(
111)
、Im( )
111 、It( )
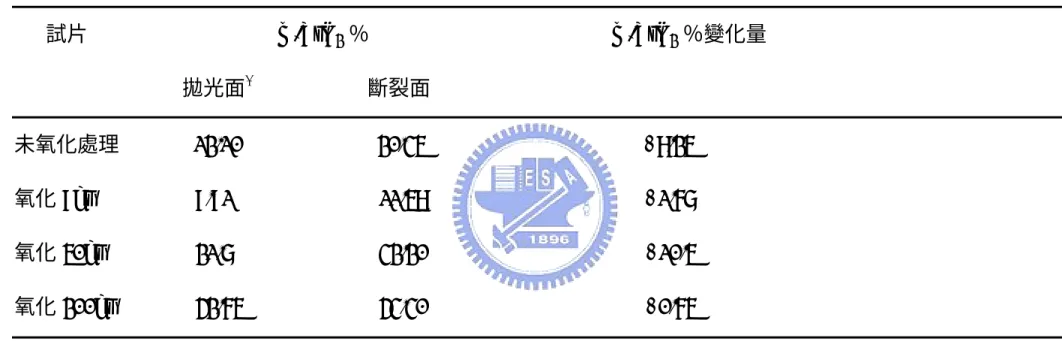
111 分別指氧化鋯的 Monoclinic 相 及 tetragonal 相在(111)繞射面之繞射峰強度,在 XRD 圖表上對應 的2θ 角度各為 29.07°、31.53°及 30.24°。量測機械性質測試前後的 斷裂面得到對應的Vm,並比較斷裂前後 Vm的比例差,藉此作為是否 發生氧化鋯相變韌化的依據之一。AZ10/SiC 試片測試結果表示於 Table 2,可發現氧化後試片的 m-ZrO2的比例變化皆在實驗誤差範圍 內,猜測是在斷裂面上的大部分 t-ZrO2 尺寸低於臨界尺寸(critical size),無法經由應力誘發產生相變韌化,所以斷裂前後的 Vm比例差 異小。 Fig. 4-7 是 AZ20/SiC 試片不同氧化時間下的 x 光繞射分析。氧化 9 小 時已有些微的 zircon 生成,是由氧化矽和氧化鋯反應而成,而在 50 小時氧化後有mullite 的生成,在經歷更長暴露時間後,zircon 會與氧 化鋁作用形成氧化鋯與 mullite,所以可以發現 200 小時的 X 光繞射 圖示中zircon 的量減少,而 mullite 的量較為增加。AZ20/SiC 試片測 試結果表示於 Table 3,表中未氧化試片的 m-ZrO2 比例變化較AZ10/SiC 高,表示氧化鋯含量增加會提高氧化鋯相變韌化機率。而 氧化 50 小時的試片出現機率提高,可推測是生成的 zircon 與氧化鋁 反應生成 mullite 與氧化鋯,因此使 t-ZrO2比例提高而增加斷裂前後
Fig. 4-8 是 AZ50/SiC 不同氧化時間下的 x 光繞射分析。和 AZ0/SiC、 AZ10/SiC 及 AZ20/SiC 試片不同,氧化後試片主要結晶相是 zircon 和 微量的mullite,可推斷大部分的氧化鋯與氧化矽反應形成 zirocn,這 將使氧化鋯相變韌化機率減低。其中zircon 是氧化矽向外擴散,與基 地中的氧化鋯反應生成。AZ50/SiC 試片測試結果表示於 Table 4。未 氧化試片的前後比例差異最大,破壞強度和破壞韌性測試結果也是最 佳,氧化鋯提供顯著強化效果。此外,可發現氧化後試片的 m-ZrO2 比例極高,而且氧化後的試片前後比例變化是零,猜測是氧化處理降 溫過程中的熱漲冷縮使試片產生大量體積變化,氧化鋯熱膨脹係數比 氧化矽及氧化鋁大,氧化鋯顆粒受到的是拉應力,使試片中的 t-ZrO2 轉變成m-ZrO2,如此一來氧化後的 AZ50/SiC 試片將不會有氧化鋯相 變韌化作用。 4.3 複合材料斷裂面微觀結構分析 Fig. 4-9 是 AZ0/SiC 試片於 1350℃不同氧化時間之斷裂面微觀結構分 析圖示,圖(a)至圖(d)分別為未氧化及氧化 9、50、200 小時的 SEM 圖示。AZ0/SiC 氧化模式為 Mode I,氧化速率慢氧化深度小,故不同 氧化時間試片的斷裂面型態相似,各試片斷裂面的起伏程度小,推對 破壞模式為穿晶破壞,不同氧化時間試片差異不大,惟在長時間氧化
後,斷裂面會趨於以穿晶破壞形式出現,此情形於氧化 200 小時試片 最是明顯,整個試片破斷面相當平坦,而且被撕裂的晶粒尺寸也越 大,出現了晶粒成長現象。由於破壞模式為穿晶破壞,試片氧化前後 破壞韌性相近。 Fig. 4-10 是 AZ10/SiC 試片於 1350℃不同氧化時間之斷裂面微觀結構 分析圖示,圖(a)至圖(d)分別為未氧化及氧化 9、50、200 小時的 SEM 圖示。可發現AZ10/SiC 試片斷裂面比 AZ0/SiC 試片粗糙,除了穿晶 破壞尚有沿晶破壞的產生,試片破壞韌性應會有所提高。但測試結果 顯示氧化前後破壞韌性相近,亦即氧化造成的負面效應和韌化機制的 正面效應相近。AZ10/SiC 試片添加了基地相中 10%含量的氧化鋯, 氧化速率比 AZ0/SiC 快,圖(b)是氧化 9 小時斷裂面圖示,此時試片 表面有氧化層的生成,其厚度也隨時間增加而往內增加,在氧化 50 小時後已達 20μm 之深,其中也有因碳化矽氧化而生成的孔洞。氧 化 200 小時試片的孔洞位置及數量比氧化 50 小時試片深入內部且 多,所以對試片破壞韌性的降低也更為明顯。 Fig. 4-11 是 AZ20/SiC 試片於 1350℃不同氧化時間之斷裂面微觀結構 分析圖示,圖(a)至圖(d)分別為未氧化及氧化 9、50、200 小時的 SEM 圖示。AZ20/SiC 試片氧化鋯含量的增加使試片氧化速率變快氧化深
度大,氧化9 小時的斷裂面已有氧化現象,而長時間如 50 或 200 小 時的氧化則能明顯觀察到氧化層。AZ20/SiC 長時間氧化後試片斷裂 面情形和AZ10/SiC 相似,試片生成厚度大的氧化層及穿晶破壞型態 的平坦斷裂面,氧化的負面效應降低試片破壞韌性。 Fig. 4-12 是 AZ50/SiC 試片於 1350℃不同氧化時間之斷裂面微觀結構 分析圖示,圖(a)至圖(d)分別為未氧化及氧化 9、50、200 小時的 SEM 圖示。由於氧化鋯添加量達基地相中的 50%,氧化速率極快氧化深 度大,其氧化後的斷裂面呈現極明顯的氧化,氧化後的斷裂面清楚觀 察 到 孔 洞 及 裂 縫 的 存 在 , 試 片 破 壞 強 度 與 韌 性 受 影 響 而 降 低 。 AZ50/SiC 試片的孔洞除了是碳化矽氧化成為氧化矽過程中產生的 CO 氣體逸散外,另一形成原因是氧化矽向外擴散與氧化鋯顆粒反應 生成zircon,原先的氧化矽顆粒位置則會變成孔洞,因此試片的孔隙 度會大幅提高,對試片的機械性質產生負面效應。 4.4 四點抗折測試分析 4.4.1 破壞強度分析 Fig. 4-13(a)是不同氧化鋯含量試片於 1350℃氧化溫度之破壞強度圖 示。圖中未氧化試片顯示出試片破壞強度隨氧化鋯含量的增加而提 升,表示添加氧化鋯能增加試片破壞強度,歸功於氧化鋯添加劑的破
壞強度比基地相佳(ZrO2 Strength = 600~700MPa),對試片破壞強度的 提升有相當的幫助。測試結果可簡單區分為兩類型;AZ0/SiC 和 AZ10/SiC 試片氧化速率慢,氧化前後的試片破壞強度相近;而 AZ20/SiC 和 AZ50/SiC 試片氧化速率快,氧化後試片破壞強度顯著變 差,其中AZ50/SiC 下降幅度更達 9 倍之多。兩試片差別在於 AZ20/SiC 得降幅較為緩和,破壞強度呈現逐漸降低趨勢;而 AZ50/SiC 則是氧 化 9 小時的破壞強度已降低為氧化前的 1/3,但後續的長時間氧化並 未持續大幅降低,推測是 AZ50/SiC 氧化比 AZ20/SiC 更為快速及深 入,在短時間已對試片造成嚴重氧化影響。試片抗折強度定義為試片 在張應力面產生破斷所需之最大應力,其測試結果受試片本身缺陷影 響極大;而缺陷類型分為試片表面及內部:表面缺陷如熱處理後表面 缺陷的改變,像是孔洞的增加或減少;而內部缺陷則像是孔隙度、因 受熱冷卻產生的內應力等。孔洞生成來源之一為試片中碳化矽的氧 化,反應式如下所示: CO SiO O SiC+ 2 → 2 + 2 3 (4-1) 由上式(4-1)知道碳化矽氧化過程會生成 CO 氣體,此氣體會固溶於基 地相中,碳化矽隨時間氧化會逐漸增加基地相中CO 氣體的濃度,達 到飽和濃度後會開始成核成長,最後在試片內部或表面成為孔洞,氧
化時間的增加會促進氣體的聚集造成孔洞的成長。氧化模式Mode II 的 AZ20/SiC 及 AZ50/SiC 由於氧化速率快,試片破壞強度明顯受到 影響,氧化後破壞強度都劇幅降低。而AZ10/SiC 雖然是氧化模式 I, 氧化程度比 AZ20/SiC 及 AZ50/SiC 輕微,但氧化後仍會在表面形成 孔洞缺陷,對試片破壞強度造成負面效應。 Fig. 4-14(b)是不同氧化鋯含量試片於 1350℃之氧化前後破壞強度比 較圖,比較後可得到試片氧化前後破壞強度的變化,結果顯示只有 AZ0/SiC 試片在 9 以及 50 小時氧化後出現破壞強度提高。而其餘的 AZ10/SiC、AZ20/SiC 和 AZ50/SiC 試片在不同氧化時間後的破壞強度 都是降低。AZ0/SiC 短時間氧化變佳的原因推測是液相組織的幫助所 致,碳化矽氧化所生成的氧化矽在溶入氧化鋁後降低熔點,生成液相 的 氧 化 矽 , 且 在 經 過 與 氧 化 鋁 長 時 間 的 作 用 後 生 成 液 相 aluminosilicate,此小範圍液相組織能減少試片表面的缺陷處,試片 破壞強度也因此能獲得提升。而且氧化 9 小時後已經有氧化矽的生 成,氧化矽與基地相會互相作用形成化學鍵結,不同相之間的鍵結力 會因此提高,對試片的破壞強度也會有幫助。Fig. 4-14 是 AZ0/SiC 經 1350℃氧化溫度 9 及 50 小時的 BEI 微觀結構,在氧化 9 小時後在試 片表面出現液相組織,隨氧化時間增加液相組織擴大範圍。比較圖(a) 及圖(b)的試片氧化表面,可發現圖(b)的液相組織範圍比圖(a)大的
多,此液相組織為 amorphous 結構,其破壞強度比結晶相的碳化矽 低,所以氧化 50 小時的破壞強度出現降低。 4.4.2 破壞韌性分析 韌性測試所得數值單位為MPa,經公式換算後可得 KIC,指材料之破 壞韌性,單位為 MPa.m1/2。 KIC= Y b d a M 2 2 / 1 6 (4-4) M= − 2 2 2 1 L L P 式(4-4)中 d 為試片寬度,b 為試片厚度,a 為缺口深度,P 為荷重(單 位為牛頓),L1為下支點跨距,L2為上支點跨距,Y 為無單位參數,
依 a/b 比值而定;Y=1.99-2.47(a/b)+12.97(a/b)2-23.17(a/b)3+ 24.80(a/b)4。在實驗中 a/b=1/3,經計算後 Y=2.0558。
Fig. 4-15(a)是不同氧化鋯含量試片於 1350℃氧化溫度之破壞韌性圖 示,未氧化試片同樣顯現出破壞韌性隨氧化鋯含量的增加而提高,推 斷是氧化鋯強化作用,而斷裂面的 m-ZrO2比例變化也隨氧化鋯含增
加而提高,表示氧化鋯提供相變韌化作用。Fig. 4-15(b)是不同氧化鋯 含量試片於 1350℃之氧化前後破壞韌性比較圖。和破壞強度測試結
果類似,氧化模式 Mode I 的 AZ0/SiC 和 AZ10/SiC 氧化前後破壞韌性 相近,僅氧化 50 小時後出現破壞韌性提高。而氧化模式 Mode II 的 AZ20/SiC 和 AZ50/SiC 氧化後的破壞韌性均出現下降。
在 AZ0/SiC 試片方面,Kaya 及 Yoshimura[65,67]提出幾種因氧化行為
所引致的強化及韌化機制,如 crack deflection、grain bridging、 debonding/sliding 等,透過這些韌化機制能夠提升試片破壞韌性。檢 視 AZ0/SiC 試片發現破壞韌性提升的最可能原因是添加的碳化矽顆 粒所引致的crack deflection。此外,在非立方晶結構之複合材料,破 壞韌性會隨晶粒大小而增加,達最大值後隨晶粒尺寸增加而降低。觀 察氧化後的 AZ10/SiC 斷裂面試片,發現晶粒隨氧化時間增加而出現 成長,而破壞韌性測試結果也是呈現先提高再下降趨勢。而氧化200 小時試片破壞韌性下降原因可推測氧化矽生成造成的影響,氧化矽形 成後會與基地相互相作用產生化學鍵結,兩相之間的鍵結力會提高, 因此預期的crack deflection 韌化作用會消失,破壞韌性出現下降。 AZ10/SiC 試片添加了基地相 10 %含量的氧化鋯,提供應力誘發之氧 化鋯相變韌化作用(Stress induced Tetragonal → Monoclinic,伴隨 9.7%體積增加)。Table 2 是 AZ10/SiC 試片於 1350℃不同氧化時間後 之 m-ZrO2比例變化表。由於比例變化在實驗誤差範圍內,可假設氧
化鋯相變韌化並非是主要韌化機制,推測是大部分t-ZrO2尺寸低於臨 界尺寸(critical size),無法經由應力誘發產生相變韌化。 AZ10/SiC 試片於 1350℃/50hr 出現破壞韌性提高,文前提到碳化矽氧 化過程會生成CO 氣體,這 CO 氣體最終會在試片內部或表面形成孔 洞,一般來說孔洞是缺陷對機械性質有不良影響,Fig. 4-16 是 AZ10/SiC 於 1350℃/50hr 之試片斷裂面微觀結構。在圖片下方可以清 楚發現分佈許多細小的孔洞。Lin[72]提到在碳化矽複合材料諸多韌化 機制中,孔洞也能提升試片破壞韌性。若孔洞的分佈情形是小而一致 時對試片破壞韌性是有助益的,反之若是大而散亂分佈則會降低試片 破壞韌性;其原因是小而均勻分佈的孔洞會分散消耗裂縫能量,裂縫 無法應力集中造成破壞;反之,大而散亂的空洞除了是缺陷處也會降 低試片緻密度,會使裂縫的傳遞更容易。推測此機制對氧化 50 小時 試片破壞韌性的提升也有幫助。
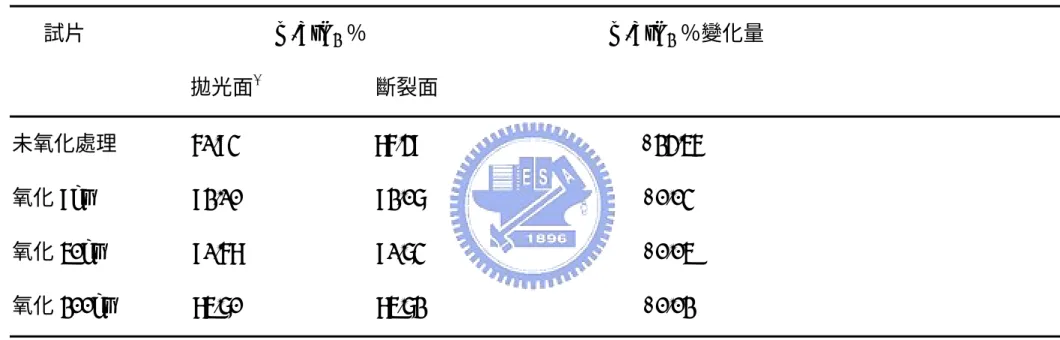
氧化模式Mode II 的 AZ20/SiC 和 AZ50/SiC 試片在氧化後的破壞韌性 都 是 下 降 的 , 兩 者 差 別 在 於 AZ20/SiC 破 壞 韌 性 的 下 降 幅 度 比 AZ50/SiC 輕微,其中 AZ50/SiC 試片氧化前後的破壞韌性最多出現達 10 倍的降幅。兩試片的氧化後外觀呈現均勻白色,並且伴隨著大量 體積變化。AZ20/SiC 試片截面四周出現因嚴重氧化而生成的白色氧
化層,隨著氧化時間增加氧化層厚度變厚;AZ50/SiC 試片則因為氧 化速率更快,試片截面內部和表面一樣都是白色的。試片破壞韌性不 佳原因之一推測是基地相和添加物熱膨脹係數不同,經長時間高溫氧 化處理後,在降至室溫過程中因體積變化不同而在試片內部產生大量 裂縫,這些裂縫能幫助負載外力時新生裂縫的傳遞,會使試片破壞韌 性降低。Fig. 4-17 是 AZ50/SiC 經 1350℃不同氧化時間之斷裂面 SEM 微觀結構,圖(a)至圖(c)分別為氧化 9、50 及 200 小時。由圖(a)即發 現試片斷裂面已出現氧化情形。而當氧化至 50 小時後,斷裂面已滿 佈孔洞,試片孔隙度高緻密度不佳,而且孔洞也互相連結成長,等到 氧化200 小時後,試片斷裂面滿佈裂縫。因此,氧化後試片破壞韌性 受氧化影響而比氧化前試片差。 4.4.3 韌化機制分析 AZ0/SiC 試片以碳化矽作為強化相,碳化矽本身破壞韌性比氧化鋁 好,可預期碳化矽能提升試片的破壞韌性。Fig. 4-18 是 AZ0/SiC 於 1350 /50hr℃ 之試片 TEM 圖示。圖(a)中箭頭標示處為裂縫,裂縫在前 進時遭遇碳化矽顆粒,碳化矽顆粒產生crack deflection 韌化作用,使 裂縫偏折沿碳化矽顆粒前進。圖(b)是碳化矽顆粒的 EDS 分析光譜。 而Fig. 4-19 是 AZ0/SiC 於 1350℃/50hr 之試片斷裂面 SEM 微觀結構,
同樣可以發現圖中的裂縫以 crack deflection 的方式前進。所以 AZ0/SiC 試片於 1350℃/50hr 氧化後破壞韌性提高的韌化機制推測是 碳化矽的crack deflection。
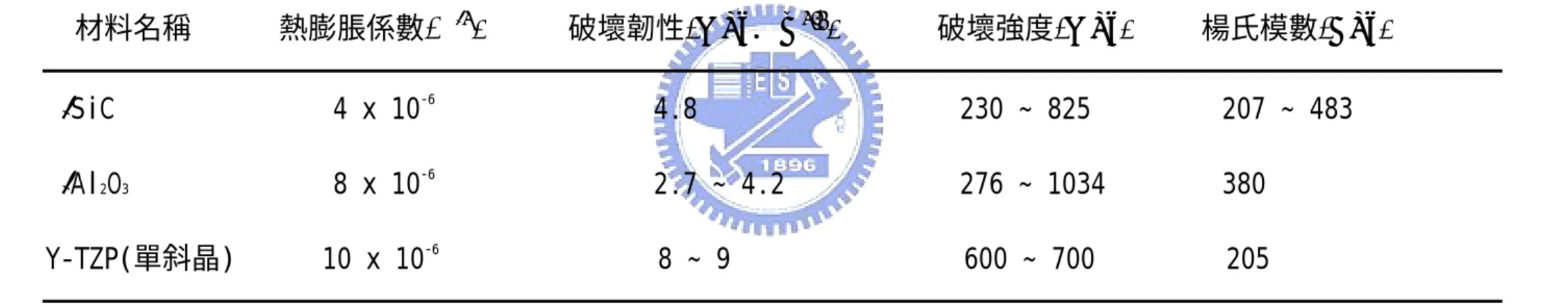
在 AZ10/SiC 試片方面,從Fig. 4-3 AZ10/SiC 於 1350℃不同氧化時間 之試片氧化表面 x 光繞射光譜圖中,發現試片在氧化 9 小時後已有 zircon 生成,而 zircon 是由氧化鋯和氧化矽兩者反應生成,因此試片 中的氧化鋯被消耗,使能產生有效相變韌化的氧化鋯是減少的。從 Table 2 中比較氧化 9 及 50 小時測試數據後,氧化 50 小時的比例變 化的確是減少的,所以此時氧化 50 小時試片破壞韌性的提高可能是 其他機制的幫助。 Kaya 在文獻中[65]提到複合材料添加物含量增加時將會使複合材料的 楊氏模數(Young modulus)增加,文中複合材料的楊氏模數從尚未強 化的 295 GPa 轉變至 40%碳化矽含量的 395 GPa,在此影響下試片會 發生 modulus-load transfer 現象。此現象下裂縫前端的剪應力會被轉 移沿組成物中高楊氏模數的顆粒前進並遠離裂縫平面。而裂縫前端的 剪應力下降,所以裂縫前進的能力降低,因此應力誘發的氧化鋯相變 韌化機率也會被降低。從Table 5 中知道碳化矽的楊氏模數較氧化鋯
來的高,且本實驗碳化矽含量為 30%,試片的楊氏模數達 360GPa, 表示上述韌化機制於本實驗中是可能的。
Fig. 4-20 是 AZ10/SiC 於 1350℃/50hr 之試片 TEM 圖示。從圖(a)可發 現氧化鋯相變韌化,箭頭標示處為氧化鋯顆粒,裂縫遭到氧化鋯的阻 擋而偏折前進;圖(b)是氧化鋯的 EDS 分析光譜。Fig. 4-21 是 AZ10/SiC 於 1350℃/50hr 之試片斷裂面微觀結構。圖中發現在晶粒周圍出現許 多微裂縫,推斷是試片中各成分的熱膨脹係數不同所導致的結果(碳 化矽:4 x 10-6;氧化鋁:8 x 10-6;氧化鋯:10 x 10-6)。熱脹冷縮 過程中熱膨脹係數較小的會受到壓應力,所以被微裂縫圍繞的晶粒應 是碳化矽顆粒。此微裂縫的生成會增加其他韌化機制的發生機率,如 crack deflection、grain-bridging 等。在 Fig. 4-22 AZ10/SiC 於 1350℃ /50hr 之試片斷裂面微觀結構圖示中,可以發現 crack deflection 現象, 此外,也發現斷裂面有液相組織生成,原先的裂縫被液相組織連結起 來,此 bridging 現象亦能提供韌化效果。所以AZ10/SiC 試片於 1350 ℃/50hr 氧化後破壞韌性提高的韌化機制推測是 modulus-load transfer 以及crack deflection。 AZ20/SiC 試片經氧化處理後機械性質變差,不論在破壞韌性或破壞 強度都是如此。這是因為 AZ20/SiC 試片是氧化模式 Mode II,試片
氧化速率快氧化深度大造成破壞強度與韌性低落,不過氧化 50 小時 的試片與氧化 9 小時試片相比較下卻出現破壞韌性提升,推測此時韌 化作用強過氧化負面效應。Fig. 4-23 是 AZ20/SiC 於 1350℃/50hr 之 試片BEI 微觀結構,箭頭標示處為氧化鋯顆粒,和 AZ10/SiC 試片情 形相似,試片產生氧化鋯相變韌化,圖中裂縫遭到氧化鋯的阻擋而偏 折前進。不過比起氧化前試片仍舊是破壞韌性降低。Fig. 4-24 是 AZ20/SiC 於 1350℃/50hr 之試片斷裂面 SEM 微觀結構,在 AZ20/SiC 試片斷裂面 XRD 圖示中,可以發現在氧化 50 小時後試片內部有 mullite 的 生 成 , 而 圖 中 箭 頭 標 示 處 就 是 mullite 在 發 生 debonding/sliding 後所產生的針狀凹痕,debonding/sliding 也是 一種韌化機制,能提供試片韌化效果。所以 AZ20/SiC 試片於 1350 ℃/50hr 氧化後破壞韌性提高的韌化機制推測是氧化鋯相變韌化及 debonding/sliding。
第五章 結論
1. SiC/Al2O3/ZrO2複合材料的氧化速率隨著氧化鋯的體積百分比而增
加。複合材料會因氧化鋯含量不同而呈現截然不同的氧化行為。 AZ0/SiC 和 AZ10/SiC 試片氧化速率較慢;AZ20/SiC 和 AZ50/SiC 試 片氧化速率急遽加快。 2.氧化後試片斷裂面型態因氧化鋯添加量不同而相異,AZ0/SiC 氧化 前後斷裂面相似,斷裂面平整且氧化現象不明顯,破壞模式為穿晶 破壞;AZ10/SiC 和 AZ20/SiC 氧化斷裂面相似,皆出現因氧化而生 成的氧化層及孔洞;AZ50/SiC 的氧化斷裂面則因氧化嚴重,滿佈 孔洞及裂縫。 3.AZ0/SiC 和 AZ10/SiC 的氧化程度輕微,試片氧化前後破壞強度與 韌性相近。AZ0/SiC 的破壞強度在 1350℃/50hr 氧化後提高,破壞 韌性在 1350℃/50hr 氧化後提高;AZ10/SiC 的破壞韌性在 1350℃ /50hr 氧化後提高。 4.氧化鋯添加量的提高會促使試片氧化速率加快,試片的機械性質因 此受到破壞;AZ50/SiC 的機械性質明顯低於 AZ0/SiC。
第六章 參考文獻
1. P. Wang, G. Grathwohl, F. Porz, F. Thummler“Oxidation Behavior of SiC Whisker-reinforced Al2O3/ZrO2 Composite” pmi ,23【6】(1991).
2. 汪健民主編,”陶瓷技術手冊(上)”,粉末冶金協會,台灣,1999。 3. R. Porter, F. E. Lange, and A. H. Chokshi, "Proessing and Creep
Performace of SiC-Whisker-Reinforced Al2O3," Am. Ceram. Soc.
Bull., 66 [2] 343-47 (1987).
4. J. Homemy, W. L. Vaughn, and M. K. Ferber, "Processing and
Mechanical Properties of SiC-Whisker-Al2O3-Matrix Composites,"
Am. Ceram. Soc. Bull., 66 [2] 333-38 (1987).
5. T. N. Tiegs and P. F. Becher, "Sintered Al2O3- SiC-Whisker
Composites," Am. Ceram. Soc. Bull., 66 [2] 339-42 (1987).
6. R. Lundberg, B. Nyberg, K. Williander, M. Persson, and R. Carlsson, "Processing of Whisker-Reinforced Ceramics," Composites
(
Guilford, U. K.)
, 18 [2] 125-27 (1987).7. M. D. Sacks, Hae-Weon Lee, and O. E. Rojas, "Suspension Processing of Al2O3/ SiC Whisker Composites,"J. Am. Ceram. Soc., 71 [5]
370-79 (1988).
8. W. H. Rhodes, "Agglomerate and Particle Size Effects on Sintering Yttria-Stabilized Zirconia," J. Am. Ceram. Soc., 64 [1] 19-22 (1981). 9. M. Okuyama, G. J. Garvey, T. A. Ring, and J. S. Haggerty, "Dispersion
of Silicon Carbide Powders in Nonaqueous Solvents," J. Am. Ceram.
Soc., 72 [10] 1918-24 (1989).
10. 王立銘 和 韋文誠,"碳化矽的膠體製程研究," 陶業, 1993 年 7 月. 11. M. V. Parish, R. R. Garcia, and H. K. Bowen, "Dispersions of Oxide
Powders in Organic Liquids," J. Mater. Sci., 20 996-1008 (1985). 12. C. Baudin and J. S. Moya, "Oxidation of
Mullite-Zirconia-Alumina-Silicon Carbide Composites," J. Am.
Ceram. Soc., 73 [5] 1417-20 (1990).
13. P. Wang, G. Grathwohl, F. Porz, and F. Thummler, " Oxidation Behavior of SiC Whisker-reinforced Al2O3/ZrO2 Composites,"
Powder Metall. Int., 23[6] 371-5 (1991).
14. W. H. Rhodes, "Agglomerate and Particle Size Effects on Sintering Yttria-Stabilized Zirconia," J. Am. Ceram. Soc., 64 [1] 19-22 (1981). 15. P. J. Jorgensen, M. E. Wadsworth, and I. B. Cutler, "Oxidation of
Silicon Carbide," J. Am. Ceram. Soc., 42 [12] 613-16 (1959).
16. J. A. Costello and R. E. Tressler, "Oxidation Kinetics of Hot-Pressed and Sintered α-SiC," J. Am. Ceram. Soc., 64 [6] 327-31 (1981). 17. J. A. Costello and R. E. Tressler, "Oxidation Kinetics of Silicon
Carbide Crystals and Ceramics: I, In Dry Oxygen," J. Am. Ceram.
Soc., 69 [9] 674-81 (1986).
18. S. C. Singhal, "Oxidation Kinetics of Hot-Pressed SiC," J. Mater. Sci.,
11 1246-53 (1976).
20. R. C. A. Harris, "Oxidation Kinetics of 6H-αSilicon Carbide Platelets, " J. Am. Ceram. Soc., 58 [1-2] 7-9 (1975).
21. Z. Zheng, R. E. Tressler, and K. E. Spear, "Oxidation of Single-Crystal Silicon Carbide Part I. Experimental Studied, " J.
Electrochem. Soc., 137 [3] 854-58 (1990).
22. Z. Zheng, R. E. Tressler, and K. E. Spear, " Oxidation of Single-Crystal Silicon Carbide , " J. Electrochem. Soc., 1107-10 (1995).
23. C. E. Ramberg, G. Cruciani, K. E. Spear, and R. E. Tessler, " Passive-Oxidation Kinetics of High-Purity Silicon Carbide from 800 ℃ to 1100℃," J. Am. Ceram. Soc., 79 [11] 2987-2911 (1996). 24. P. S. Wang, S. M. Hsu, and T. N. Wittberg, " Oxidation of Silicin



![HPSH [ 氧化數平衡反應式係數 ]](data:image/gif;base64,R0lGODlhAQABAIAAAP///wAAACH5BAEAAAAALAAAAAABAAEAAAICRAEAOw==)