國
立
交
通
大
學
生物資訊及系統生物研究所
博
士
論
文
蛋白質-配體結合模式預測與其結合區域定性研究
A study for predicting protein-ligand binding modes and
characterizing protein-ligand binding sites in structure-based drug
design
研 究 生:陳彥甫
指導教授:楊進木 教授
蛋白質-配體結合模式預測與其結合區域定性研究
A study for predicting protein-ligand binding modes and characterizing
protein-ligand binding sites in structure-based drug design
研 究 生:陳彥甫 Student:Yen-Fu Chen
指導教授:楊進木 Advisor:Jinn-Moon Yang
國 立 交 通 大 學
生物資訊及系統生物研究所
博 士 論 文
A Thesis Submitted to Institute of Bioinformatics and Systems Biology National Chiao Tung University in partial Fulfillment of the Requirements
for the Degree of Ph.D. in Bioinformatics and Systems Biology
September 2010
Hsinchu, Taiwan, Republic of China
i
蛋白質-配體結合模式預測與其結合區域定性研究
研究生:陳彥甫 指導教授:楊進木博士 國立交通大學 生物資訊及系統生物研究所 博士班摘 要
隨著蛋白質結晶結構的快速增加,以結構為基礎之藥物設計與虛擬藥物篩選(virtual screening)在先導藥物開發過程日漸重要。目前一系列的分子對接(protein-ligand docking) 以及虛擬藥物篩選方法已經被應用到先導藥物發展中,並且已獲得數個成功的藥物開發 案例。即使如此,目前由巨量的虛擬藥物篩選資料中找出真正具有活性的先導藥物仍然 是一個困難的挑戰。其問題肇因於目前對蛋白質-配體之間的結合機制了解仍然有所不 足,使得已發展的蛋白質-配體結合計分方程式不夠周全。 針對上述議題,我們已提出了以藥物孔洞為基礎的計分方程式(pharmacophore-based scoring function)與虛擬藥物篩選共通計分方法應用準則(consensus scoring criteria)之研 究。其中,共通計分方法是透過結合數個計分方程式的共同處,相較於單一計分方法可 以有更好的虛擬藥物篩選準確性。然而虛擬藥物篩選的計分方程式通常無法辨識蛋白質 -配體間的關鍵結合特性[例如: 藥效基團熱點(pharmacophore hotspot)],而這些關鍵特性 卻通常是觸發或抑制目標蛋白質對其調控的生物反應必要條件。雖然應用藥物孔洞方法 與相關計分方程式可以找出關鍵結合特性,但是這些方法需要一系列已知的活性配體, 這些資料必須由實驗取得,使應用性受到限制。因此,對於虛擬藥物篩選過程發展更好 的篩選後分析(post-screening analysis)與關鍵特性之發現方法,將對於藥物發展具有重要 價值。在本研究中,我們已經發展出 site-moiety map (簡稱 SiMMap)方法,並且將其延伸
應用到辨識與定性垂直同源蛋白質(ortholog)的共通結合環境 (orthologous SiMMap)研究 之中。SiMMap 透過統計對目標蛋白質與一群對其預測或共結晶之配體所產生的交互作 用,推測位於目標蛋白質結合區域內之錨點(anchor),並用以描述分布在結合區域中的 配體官能基偏好(moiety preference)以及物化特性集合。每一個錨點具有三個基本構成要 件:1)由具一致交互作用之殘基構成的結合袋點(binding pocket);2)複數個虛擬配體構 成的官能基組成;3)結合袋點與官能基之交互作用關係(包含靜電力、氫鍵及凡德瓦力交 互作用)。實驗證據已顯示錨點通常是蛋白質-配體結合區域中的熱點。同時 site-moiety map 也可提供將官能基團(靜電力、氫鍵及凡德瓦力特性之官能基)之組合最佳化的建 議,有助於設計潛在先導藥物。實驗結果也證實當小分子化合物與 site-moiety map 描述 的錨點特性高度相符時,通常有高度潛力成為目標蛋白質的抑制劑或促進劑。SiMMap 已提供全球服務,網址為 http://simfam.life.nctu.edu.tw/。我們相信我們對於藥物孔洞為 基之計分方程式、虛擬藥物篩選之共通計分方法應用準則的成果、以及 site-moiety map 之研究,將對藥物發現與了解蛋白質-配體機制有所幫助。
ii
A study for predicting protein-ligand binding modes and characterizing
protein-ligand binding sites in structure-based drug design
Student : Yen-Fu Chen Adviser : Dr. Jinn-Moon Yang Institute of Bioinformatics and Systems Biology
National Chiao Tung University
ABSTRACT
As the number of protein structures increases rapidly, structure-based drug design and virtual screening approaches are becoming important and helpful in lead discovery. A number of docking and virtual screening (VS) methods have been utilized to identify lead compounds, and some success stories have been reported. However, identifying lead compounds by exploiting thousands of docked protein-compound complexes is still a challenging task. The major weakness of virtual screenings is likely due to incomplete understandings of ligand binding mechanisms and the subsequently imprecise scoring algorithms.
To address these issues, we have proposed a pharmcophore-based scoring function approach and a consensus strategies among different scoring methods in VS. The consensus scores would improve the performance and, on average, the performance of the combined method performs better than the average of the individual scoring functions. Nevertheless, the approaches generally cannot identify the key features (e.g., pharmacophore spots) that are essential to trigger or block the biological responses of the target protein. Although pharmacophore techniques have been applied to derive the key features, these methods require a set of known active ligands that were acquired experimentally. Therefore, the more powerful techniques for post-screening analysis to identify the key features through docked compounds and to characterize the binding site provide a great potential value for drug design.
Recently, we have developed the site-moiety map (SiMMap) method and extended to characterize the consensus binding environments (i.e., anchors) of orthologous targets (orthSiMMap). SiMMap statistically derived anchors from the interaction profiles between query target protein and its docked (or co-crystallized) compounds, and then described the relationship between the moiety preferences and physico-chemical properties of the binding site. Each anchor includes three basic elements: a binding pocket with conserved interacting residues, the moiety composition of query compounds, and pocket-moiety interaction type (electrostatic, hydrogen-bonding, or van der Waals). Experimental results showed that an anchor is often a hot spot and the site-moiety map can be helpful to assemble potential leads by optimal steric, hydrogen-bonding, and electronic moieties. When a compound highly agrees with anchors of site-moiety map, this compound often activates or inhibits the target protein. The SiMMap web server is available at http://simfam.life.nctu.edu.tw/. We believe that our evolutionary approach with pharmacophore-based scoring functions, consensus scoring criteria for virtual screening, and the method of site-moiety map are useful for drug discovery and understanding biological mechanisms.
iii
Acknowledgement
來到新竹交大十二個年頭,從一個懵懂的大學生一路念到了博士學位。回首這些生 命中的精彩日子,每一個畫面都充滿了無數的回憶。在這段學習的黃金時代,難免遇到 瓶頸與困難,感謝許多貴人的幫助讓我得以安然度過,並且學到如何面對壓力與思考問 題的能力。我認為這些能力將是未來面對下一階段的人生路程之重要基礎與助力。 第一個貴人是我的指導教授楊進木博士。在這段學習的過程中,我的指導教授總是 嚴格且不厭其煩的指出我的缺點與問題,並提出可能的方向讓我依循。雖然克服困難的 過程中,難免跌跌撞撞,但是這些解決問題的經驗、創新思考與邏輯歸納能力將是我未 來人生中的寶藏。 接著我要特別感謝的是實驗室同組的凱程、伸融與御哲,由於有了他們的配合與日 以繼夜的努力,我們才能在最短的時間之內突破困難的關卡,完成這本論文中的許多重 點工作。除此之外,指導教授、凱程與我之間的討論與合作過程,也是突破這些困難的 另外一個重要關鍵。許多的深入探討與解決困難的經驗,都是在這個時候得到啟發與進 展,並且讓我獲益良多。 口試期間承蒙所內黃鎮剛教授、趙瑞益教授與梁美智教授,及所外王雯靜教授、徐 祖安教授與謝興邦教授百忙之中不吝賜教並且提供許多寶貴建言,使得本文得以更加完 善。同時我也從口試委員以及指導教授身上看到頂尖學術研究人才所應具備的條件以及 視野,這將是我未來需要繼續努力的目標。 在本文的理論發展過程中,我的指導教授提供許多與國內外頂尖實驗室合作的機 會,使得我有機會可以與跨領域的研究人才合作與交流,這樣的交流過程中經常會激發 出許多新的想法與題材。同時在本中有引用到與這些頂尖實驗室(美國 Fordham 大學 D. Frank Hsu 教授、清大王雯靜教授、國衛院徐祖安教授與交大楊昀良教授)在合作過程中 所完成與提供寶貴實驗資料,特此感謝。 在這段辛苦而充實的研究生活中,實驗室的同學們也不時的給予我許多生活上的幫 助或研究討論的空間,像是已經畢業的再威、右儒與振寧,或是現在的宇書、章維、怡 馨、一原、志達…等許多同學,在這裡也要感謝他們。除此之外在論文的撰寫過程中, 感謝已經畢業的陳俊辰博士給予我許多的撰寫方面的建議,使我事半功倍。 最後我必須要特別感謝我的父母與家人。在許多困難的時候,他們總是默默的支持 著我,無怨無悔的付出。每當我覺得筋疲力盡的時候,我知道總是有一盞溫暖的燈火等 著我回家,隨時幫我加滿重新面對挑戰的能量。因此我將此論文獻給我的父母,希望他 們可以以我為榮。iv
Contents
摘 要 ... i ABSTRACT ... ii Acknowledgement ... iii Contents ... iv Chapter 1. Introduction ... 1 1.1 Background ... 1 1.2 Thesis overview ... 3Chapter 2. Related works ... 6
2.1 Pharmacophore-based scoring functions ... 10
2.2 Consensus scoring criteria ... 11
2.3 Combinative clustering analysis... 14
2.4 Summary ... 16
Chapter 3. Site-moiety map for recognizing interaction preferences between protein pockets and compound moieties ... 17
3.1 Introduction ... 17
3.2 Method ... 17
3.2.1 Definitions of site-moiety map, anchor and pocket ... 19
3.2.2 Constructing site-moiety map ... 19
3.3 Results ... 21
3.3.1 Web service ... 21
3.3.2 Thymidine kinase and estrogen receptor ... 23
3.4 Summary ... 26
Chapter 4. The application of site-moiety map for characterizing protein-ligand binding sites and discovering adaptive inhibitors for orthologous protein targets ... 28
4.1 Introduction ... 28
4.2 orthSiMMap methods ... 29
4.3 Results ... 31
4.3.1 orthSiMMap method ... 31
4.3.2 Orthologous SiMMaps and orthSiMMaps of SKs ... 33
v
4.3.4 Site-directed mutagenesis ... 41
4.3.5 Analogues assay and orthSiMMap ... 42
4.3.6 Structural mechanism of the inhibitor binding for shikimate kinases ... 43
4.3.7 Performance of the orthSiMMap method... 46
4.4 Summary ... 48 Chapter 5. Conclusion ... 51 5.1 Summary ... 51 5.2 Future works ... 52 Appendix A ... 55 List of publications ... 55 Journal papers ... 55 Conference paper... 55 References ... 56
vi
List of Figures
Figure 1.1. Overview of structure-based drug design and related works.. ... 3 Figure 1.2. The research framework for predicting protein-ligand binding modes and
characterizing protein-ligand binding sites in structure-based drug design. ... 5 Figure 2.1. Main procedure of structure-based virtual screening. ... 7 Figure 2.2. The influences of ligand structures and molecular weight on docking energy.. ... 8 Figure 2.3. The binding-site pharmacological consensuses are identified by overlapping the
docked conformations of (a) 10 known ER antagonists and (b) 10 known ER agonists against the reference proteins 3ert and 1gwr, respectively. ... 9 Figure 2.4. The main steps of GEMDOCK for virtual database screening, including the target
protein and compound database preparation, flexible docking, and post-docking analysis. 11 Figure 2.5. Rank/score curves of five methods for four virtual screening targets: (a) TK, (b)
DHFR, (c) ER-antagonist receptor, and (d) ER-agonist receptor. ... 12 Figure 2.6. The relationships between the GH-score improvement with (a) normalized value of
variance of rank/score graph and (b) normalized value of Pl/ Ph of 40 pairing combinations
of five methods for four virtual screening targets. (c) The GH-score improvements with normalized variances of rank/score graphs (R/Svar) and normalized relative performance
measurement (Pl/ Ph) of 40 RCS and SCS pairing combinations of five methods for four
virtual screening targets. (d) The positive and negative GH-score improvements are denoted with circle and cross, respectively. ... 13 Figure 2.7. The known active ligands of four VS targets, estrogen receptors (ER) of antagonists
(a) and agonists (b), (c) thymidine kinase (TK), and (d) human dihydrofolate reductase (DHFR).. ... 14 Figure 2.8. Overall process of the two-stage combinative cluster analysis.. ... 15 Figure 2.9. Designing a reference threshold of P-L interaction and atom-pair descriptors.. ... 15 Figure 3.1. Overview of the SiMMap server for the site-moiety map using herpes simplex virus type-1 thymidine kinase (TK) and 1000 docked compounds as the query. ... 18 Figure 3.2. The SiMMap server analysis results using estrogen receptor (ER) and 1000 docked
compounds as the query.. ... 22 Figure 3.3. The relationships between the site-moiety map and 22 co-crystallized ligands of ER. ... 24 Figure 4.1. Framework of the orthSiMMap method. ... 30 Figure 4.2. Shikimate kinase orthSiMMaps. ... 32
vii
Figure 4.3. The site-moiety maps of (a) HpSK and (b) MtSK. Each anchor represents one of three binding environments (electrostatic: blue; hydrogen-bonding: green; van der Waals: black).. ... 34 Figure 4.4. Structures of the 38 inactive compounds from the NCI and Maybridge databases. 37 Figure 4.5. Characterization of shikimate kinase inhibitors by enzyme assay, orthSiMMaps,
site-mutagenesis studies and analogues.. ... 40 Figure 4.6. Interaction profiles between selected anchor residues and 27 tested compounds.. . 43 Figure 4.7. Probing the affinity pockets in HpSK. ... 45 Figure 4.8. Performance of the orthSiMMap method on apo-form HpSK and MtSK.. ... 47 Figure 5.1. The NAD(P) and ATP related pathways play key roles in various biological
functions, such as aromatic amino acid synthesis, pyrimidine metabolism and TCA cycle regulation. ... 52 Figure 5.2. Preliminary result of the PathDrug on the shikimate pathway.. ... 53
viii
List of Tables
Table 3.1. The relationship between the anchors and moieties of 15 co-crystallized ligands for TK ... 24 Table 3.2. Comparing SiMMap with other methods on thymidine kinase and estrogen receptor
by false-positive rates ... 25 Table 3.3. The mapping between the anchors and active and typical compounds for ER ... 27 Table 4.1. Summary of 37 pairs of orthologous targets ... 35 Table 4.2. Summary of 12 inhibitors with inhibition assay, compound structures, docked poses,
and consensus anchors ... 38 Table 4.3. Properties of potent inhibitors for HpSK and MtSK a ... 39 Table 4.4. The ranks of active compounds using orthSiMMap, energy-bases, and combination
scoring methods for apo and closed forms of HpSK and MtSK ... 47 Table 4.5. Some selected top-ranked compounds using orthSiMMap, energy-bases, and
combination scoring methods for apo and closed forms of HpSK and MtSK ... 49
1
Chapter 1
Introduction
1.1 Background
Virtual screening (VS) of molecular compound libraries has emerged as a powerful and inexpensive method for the discovery of novel lead compounds for drug development 1-10.
Given the structure of a target protein active site and a potential small ligand database, VS predicts the binding mode and the binding affinity for each ligand and ranks a series of candidate ligands. There are four main reasons for the rapid acceptance and success of VS: 1) The availability of the growing number of protein crystal structures; 2) The advent of structural proteomics technologies; 3) The enrichment and speed of VS 2,11; and 4) The contribution of VS to the reduction in the cost of drug discovery.
Each VS computational method involves two basic critical elements: efficient molecular docking and a reliable scoring method. Scoring methods for VS should effectively discriminate between correct binding states and non-native docked conformations during the molecular docking phase and distinguish a small number of active compounds from hundreds of thousands of non-active compounds during the post-docking analysis. There are three general classes of scoring functions that calculate the binding free energy, including knowledge-based
12, physics-based 13, and empirical-based 14 scoring functions.
However, the performance of these scoring functions is often inconsistent across different systems from a database search. The inaccuracy of the scoring methods, i.e., inadequately predicting the true binding affinity of a ligand for a receptor, is probably the major weakness for VS. It has been reported that fusion among different scoring methods in VS would improve the performance and, on average, the performance of the combined method performs better than the average of the individual scoring functions. More recently, the same phenomena has been previously reported in information retrieval (IR) and in molecular similarity measurement. Charifson et al. (1999)15, presented a study in which they used an intersection-based consensus approach to combine scoring functions. The evidences showed an enrichment in the ability to discriminate between active and inactive enzyme inhibitions for three different enzymes (p38 MAP kinase, inosine monophosphate dehydrogenase, and HIV protease) using two different
2
docking methods (DOCK16 and GAMBLER) and thirteen scoring functions. Then, Bissantz et
al. (2000)17 , Stahl and Rarey (2001)18, and Verdonk et al. (2004)19 et.al., also reported their
works for consensus scores (CS) improving VS. Wang and Wang (2004)20 presented an idealized computer experiment to explore how consensus scoring works based on the assumption that the error of a scoring function is a random number in a normal distribution. They also studied the relationship between the hit-rates and the number of scoring functions and the performance of several ranking strategies (the rank-by-score, the rank-by-rank, and the rank-by-vote strategy) for consensus scorings.
These reported results seem to depend on the method of combination (by rank, by score, by intersection, by MIN, by MAX, and by voting) and the number and nature of individual scoring functions involved in the combination. While researchers focus to realize the benefit of method combination and consensus scorings, the major issues of how and when these individual scoring functions should be combined remain a challenging problem not only for researchers but also perhaps more importantly, for practitioners in virtual screening.
In addition, some of these VS methods are capable of identifying so-called “pharmacological preference” that is often the important interactions or binding-site hot spots typically evolved from known active ligands and the target protein 21-22. These preferences might improve screening accuracy and guide the design and selection of lead compounds for subsequent investigation and refinement during lead discovery and lead optimization processes. However, identifying lead compounds by exploiting thousands of docked protein-compound complexes is still a challenging task, too. The major weakness of virtual screenings is likely due to incomplete understandings of ligand binding mechanisms and the subsequently imprecise scoring algorithms 2,6,9.
Most of docking programs16,23-24 use energy-based scoring methods which are often biased toward both the selection of high molecular weight compounds and charged polar compounds. These approaches25-26 generally cannot identify the key features (e.g., pharmacophore spots) that are essential to the biological responses of the target protein. Although pharmacophore techniques27 have been applied to derive the key features, these methods are restricted by a set of known active ligands that were acquired experimentally. Therefore, the more powerful techniques for post-screening analysis to identify the key
3
features through docked compounds and to understand the binding mechanisms provide a great potential value for drug design.
1.2 Thesis overview
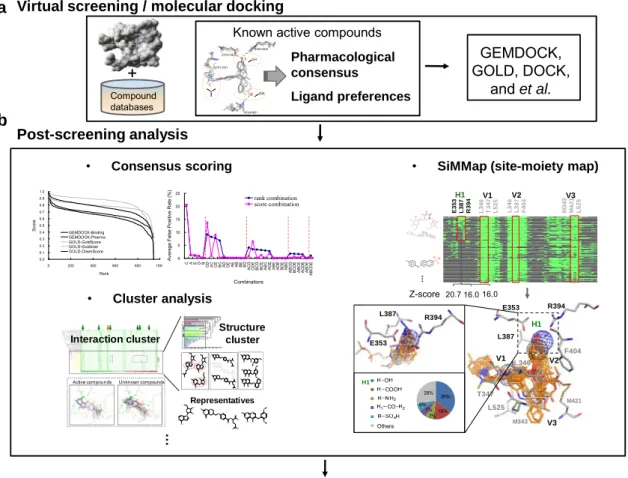
For addressing above issues, some studies have been reported (Fig. 1.1). Three of our related studies were briefly described in Chapter 2. The study of the pharmacophore-based scoring function proposed a target-specific scoring function by utilizing the protein-ligand interactions and physic-chemical properties of known actives to improve the accuracy and precision for the ranking of VS data (Fig. 1.1a). The studies of consensus scoring and cluster
Compound databases
GEMDOCK, GOLD, DOCK,
and et al.
Virtual screening / molecular docking a
Bioassay and identify active ligands Post-screening analysis b 20 … E3 5 3 L3 8 7 R3 9 4 L3 4 6 T3 4 7 L5 2 5 L 346 L 387 F 404 M 343 M 421 L5 2 5 H1 V1 V2 V3 H1 V1 V2 R394 T347 F404 M421 M343 L525 E353 L387 L346 V3 Others 36% 16% 7% 7% 6% 28% H1 R394 E353 L387 16.0 20.7 16.0 Z-score
• SiMMap (site-moiety map)
+ OH A N D351-OD1 E353-OE2 R394-NH2 H524-ND1 OH B C
Known active compounds
Pharmacological consensus Ligand preferences … Representatives N NN N N O O N N O N OH N NH2 OH I
Active compounds Unknown compounds
Interaction cluster N N O N OH N NH2 OH I N N O N OH N NH OH OH C H3 CH3 N+ C H3 S N NH2 N OH C H3 Structure cluster N N N N O N N O NO O O O N NN O N N O N O O OO N NN N N N O O N NN N N O O N NN N N N N N NN O N N O N O O OO • Cluster analysis • Consensus scoring 0.0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1.0 0 200 400 600 800 1000 Rank Sc or e GEMDOCK-Binding GEMDOCK-Pharma GOLD-GoldScore GOLD-Goldinter GOLD-ChemScore 0 5 10 15 20 25 C A E D BCD AC CE BC AD DE AE AB BE BD AC D CD E BC D BC E AB C AC E ABE AD E BD E AB D ABC D BC D E ABC E AC D E ABD E ABC D E Combinations A verage Fal se P os iti ve R at e (% ) rank combination score combination
Figure 1.1. Overview of structure-based drug design and related works. The major steps of structure-based drug design include (a) virtual screening and (b) post-screening analysis and following bioassay.
analysis addressed the issues of improving enrichment for the post-screening analysis stage (Fig. 1.1b). Furthermore, we also applied these methods on the inhibitor discoveries of the
4
dengue virus E protein and the influenza virus neuraminidase. Although some of novel inhibitors were discovered in these researches, we still found the drawbacks of these previous studies. Firstly, the pharmacophore-based scoring function is limited by the consensus of known active compounds. Second, the consensus scoring criteria and cluster analysis are helpful for improving the enrichment of VS, but these methods does not use the protein-ligand interaction data and ligand structures produced in the VS process for investigating the key environment of the protein-ligand binding site.
To address these issues, we developed the SiMMap approach to infer the key features by a site-moiety map describing the relationship between the moiety preferences and the physico-chemical properties of the binding site in Chapter 3 (Fig. 1.1b). The further application and validation of SiMMap was presented in the Chapter 4. According to our knowledge, SiMMap is the first public server that identifies the site-moiety map from a query protein structure and its docked (or co-crystallized) compounds. The server characterizes a binding site by pocket-moiety interaction preferences (anchors) including binding pockets with conserved interacting residues, moiety preferences, and interaction type.
In Chapter 4, we further extended SiMMap to orthologous SiMMap. We derived the orthologous site-moiety maps (orthologous SiMMap) from identifying consensus binding environments of orthologous proteins; orthologous SiMMap represents the conserved binding environment or "hot spots" among orthologous targets in an aim to investigate the protein-ligand interface family and apply for discovering potential leads across multiple species. Finally, Chapter 5 described some conclusions and future perspectives.
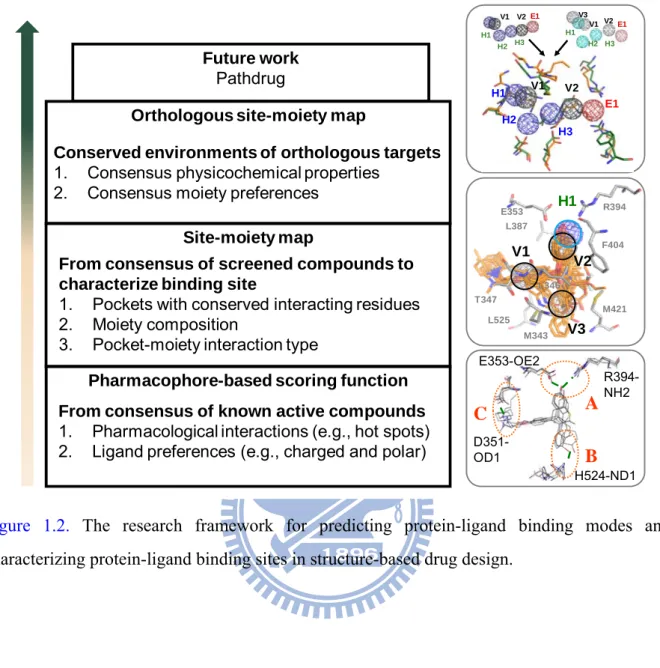
The research framework of this thesis is shown as Figure 1.2. The concept of the research of pharmacophore-based scoring function is that utilizing the consensus of known active compounds identifies the key feature of binding site. However, such approach needs the known active compounds and prefers the compounds similar with the known set. To address these limitations, we extract the consensus of screening compounds to characterize the binding site and further validate on the inhibitor discovery of orthologous shikimate kinases.
5
Future work
Pathdrug
Pharmacophore-based scoring function From consensus of known active compounds
1. Pharmacological interactions (e.g., hot spots) 2. Ligand preferences (e.g., charged and polar)
Site-moiety map
From consensus of screened compounds to characterize binding site
1. Pockets with conserved interacting residues 2. Moiety composition
3. Pocket-moiety interaction type
Orthologous site-moiety map
Conserved environments of orthologous targets
1. Consensus physicochemical properties 2. Consensus moiety preferences
A D351-OD1 E353-OE2 R394-NH2 H524-ND1 B C H1 V1 V2 R394 T347 F404 M421 M343 L525 E353 L387 L346 V3 E1 V2 H2 H3 V1 H1 E1 V2 H2 H3 V1 V3 H1 H1 V1 H2 E1 H3 V2
Figure 1.2. The research framework for predicting protein-ligand binding modes and characterizing protein-ligand binding sites in structure-based drug design.
6
Chapter 2
Related works
Virtual screening (VS) of molecular compound libraries has emerged as a powerful and inexpensive method for the discovery of novel lead compounds for drug development 2-3 (Fig. 2.1). The VS computational method involves two basic critical elements: efficient molecular docking and a reliable scoring method. Scoring methods for VS should effectively discriminate between correct binding states and non-native docked conformations during the molecular docking phase and distinguish a small number of active compounds from hundreds of thousands of non-active compounds during the post-docking analysis. The scoring functions that calculate the binding free energy mainly include knowledge-based12, physics-based13, and empirical-based 14 scoring functions.
In addition, some of these VS methods are capable of identifying so-called “pharmacological preference” that is often the important interactions or binding-site hot spots typically evolved from known active ligands and the target protein21-22 (Fig. 2.1b). These preferences might improve screening accuracy and guide the design and selection of lead compounds for subsequent investigation and refinement during lead discovery and lead optimization processes. However, the pharmacological preferences for each protein target and corresponded ligands are limited by the demand of pre-studied bioassays or structure data.
Currently, the screening quality of docking methods using energy-based scoring functions alone is often influenced by the molecular weight and the structure of the ligand being screened (e.g., the numbers of charged and polar atoms) (Fig. 2.2). These methods are often biased toward both the selection of high molecular weight compounds (due to the contribution of the compound size 28-29) and charged polar compounds (due to the pair-atom potentials of the electrostatic energy and hydrogen-bonding energy).
7
Compound databases
GEMDOCK, GOLD, DOCK, and et al.
Virtual screening / molecular docking
Prepare target protein
Prepare compound database
a
Bioassay and identify active ligands Virtual screening results from
multiple scoring methods Post-screening analysis
Select representatives and improve hits
Cluster analysis
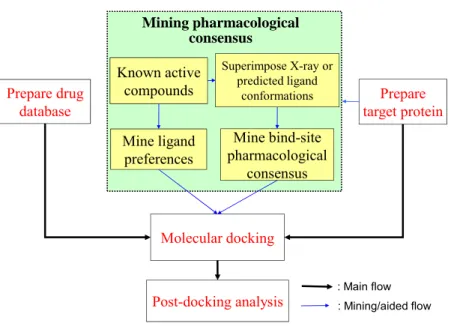
Pharmacophore-based scoring function
Known active compounds Mine ligand preferences Mine bind-site pharmacological consensus Mining pharmacological consensus
Superimpose X-ray or predicted ligand conformations OH A N D351-OD1 E353-OE2 R394-NH2 H524-ND1 OH B C
Consensus scoring criteria
0.0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1.0 0 200 400 600 800 1000 Rank Sc or e GEMDOCK-Binding GEMDOCK-Pharma GOLD-GoldScore GOLD-Goldinter GOLD-ChemScore 0.00 0.10 0.20 0.30 0.40 0.50 0.60 0.70 0.80 0.90 1.00 0.00 0.20 0.40 0.60 0.80 1.00 g (Pl/Ph) g (R/ Svar ) 0 5 10 15 20 25 C A E D B CD AC CE BC AD DE AE AB BE BD ACD CD E BC D BC E AB C AC E AB E AD E BD E AB D AB CD BC DE AB CE AC DE AB DE AB CD E Combinations A ver age F al se P os iti ve Rat e ( % ) rank combination score combination variances Representatives N NN N N O O N N O N OH N NH2 OH I
Active compounds Unknown compounds
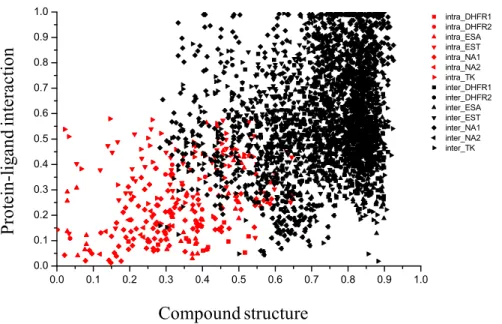
Interaction cluster N N O N OH N NH2 OH I N N O N OH N NH OH O H C H3 CH3 N+ C H3 S N NH2 N OH C H3 Structure cluster N N N N O N N O NO O O O N NN O N N O N O O OO N NN N N N O O N NN N N O O N NN N N N N N NN O N N O N O O OO 0.00.10.20.30.40.50.60.70.80.91.0 0.0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1.0 intra_DHFR1 intra_DHFR2 intra_ESA intra_EST intra_NA1 intra_NA2 intra_TK inter_DHFR1 inter_DHFR2 inter_ESA inter_EST inter_NA1 inter_NA2 inter_TK Compound structure (t) Pr ot ei n-li ga nd in te ra ct io n( t ) Protein-screening ligands b c d
Figure 2.1. Main procedure of structure-based virtual screening. (a) The major steps of structure-based virtual screening, including virtual screening, post-screening analysis, and bioassay. (b) Pharmacophore-based scoring function for virtual screening step. Post-screening analysis step is usually utilized for improving including (c) consensus scoring and (d) cluster analysis.
In the meanwhile, the performance of these scoring functions is often inconsistent across different systems from a database search 17-18. The inaccuracy of the scoring methods, i.e., inadequately predicting the true binding affinity of a ligand for a receptor, is probably the major weakness for VS. Furthermore, the application of VS2,30, to the drug discovery process invariably produces a large number of potential lead candidates. These prospective ligands need to be filtered in order to reduce their number for more precise and labor-intensive studies. Hence, it is urgent that the utilizations of post-analysis to minimize the number of false positives in the selection list and to propagate the true hits to the top of the list. (Fig. 2.1a, 2.1c
8 and 2.1d) Group B Group A O O O N O O Group A Group B ESA01 EST03 R394-NH2 E353-OE2 H524-ND1 O O O O O O A:ESA01 (-91.32) B: ESA01-CH(-76.86) 3 C: ESA01-COO -(-99.64) a b
Figure 2.2. The influences of ligand structures and molecular weight on docking energy. (a)
The fraction of polar atoms in ESA01-C is the smallest among these 3 ligands, whereas that of ESA01COO is the largest. The docked positions are similar, but the docking energies differ: -91.32 for ESA01, -76.86 for ESA01-CH3, and -99.64 for ESA01-COO. (b) ESA01 (blue) and EST03 (yellow) have a common group A, and EST03 has an additional substructure group B. The docked conformations (into reference protein 3ert) are similar, and the docking energies are -82.82 for ESA01 and -127.27 for EST03.
It has been reported that fusion among different scoring methods in VS would improve the performance and, on average, the performance of the combined method performs better than the average of the individual scoring functions.15,18-20,31 These reported results are significant and potentially robust in that the performance results of these consensus scoring (CS) methods seem to be independent of the target receptor and the docking algorithm. The reported results seem to depend on the method of combination (by rank, by score, by intersection, by MIN, by MAX, and by voting) and the number and nature of individual scoring functions involved in the combination. While researchers have come to realize the advantage and benefit of method combination and consensus scorings, the major issues of how and when these individual scoring functions should be combined remain a challenging problem not only for researchers
9
but also perhaps more importantly, for practitioners in virtual screening.
Another frequently used technique for post-screening analysis is cluster analysis. Clustering methods based on compound structural similarity or interacting profiles can group VS data, reduce complexity of observation, and improve the performance of the scoring function32-34. Through the cluster analysis, the enormous data produced by VS process is able to easily visualize and efficiently handle. However, most of researchers only consider the descriptors of protein-ligand interactions or compound structures individually. The combination of protein-ligand interactions and compound topology could provide more detail and pure classifications for following biological assay and refinement. Therefore, some of related studies are briefly introduced as following (Fig. 2.1b and 2.2d).
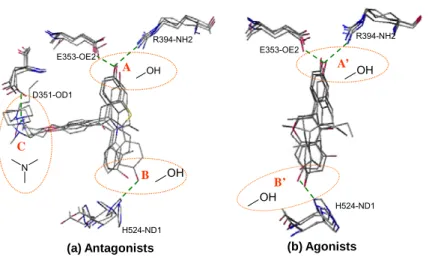
OH A N D351-OD1 E353-OE2 R394-NH2 H524-ND1 H524-ND1 OH R394-NH2 E353-OE2
(a) Antagonists (b) Agonists
A’ OH OH B’ B C
Figure 2.3. The binding-site pharmacological consensuses are identified by overlapping the docked conformations of (a) 10 known ER antagonists and (b) 10 known ER agonists against the reference proteins 3ert and 1gwr, respectively. (a) Four pharmacological interactions were identified and circled as A (phenolic hydroxyl group), B (phenolic hydroxyl group), and C (piperidine nitrogen). (b) Three pharmacological interactions were identified and circled as A (phenolic hydroxyl group) and B (phenolic hydroxyl group). The dashed lines indicate the hydrogen bonds formed between the ligand and the target protein. These pharmacological interactions are consistent with those evolved from X-ray structures.
10
2.1 Pharmacophore-based scoring functions
The screening quality of docking methods using energy-based scoring functions alone is often influenced by the molecular weight and the structure of the ligand being screened (e.g., the numbers of charged and polar atoms). These methods are often biased toward both the selection of high molecular weight compounds (due to the contribution of the compound size
28-29) and charged polar compounds (due to the pair-atom potentials of the electrostatic energy
and hydrogen-bonding energy).
A pharmacophore-based evolutionary approach for virtual screening was developed to address these issues. This tool, termed the Generic Evolutionary Method for molecular DOCKing (GEMDOCK), combines an evolutionary approach23,35-37 with a new pharmacophore-based scoring function. The former integrates discrete and continuous global search strategies with local search strategies to expedite convergence. The latter, integrating an empirical-based energy function and pharmacological preferences (binding-site pharmacological interactions and ligand preferences shown as Fig. 2.3), simultaneously serves as the scoring function for both molecular docking and post-docking analyses to improve screening accuracy (Fig. 2.4). We apply pharmacological-interaction preferences to select the ligands that form pharmacological interactions with target proteins, and use the ligand preferences to eliminate the ligands that violate the electrostatic or hydrophilic constraints. We assessed the accuracy of our approach using human estrogen receptor (ER) and a ligand database from the comparative studies of Bissantz et al.17 Using GEMDOCK, the average goodness-of-hit (GH) score was 0.83 and the average false positive rate was 0.13% for ER antagonists, and the average GH score was 0.48 and the average false positive rate was 0.75% for ER agonists. The performance of GEMDOCK was superior to competing methods such as GOLD and DOCK. We found that our pharmacophore-based scoring function indeed is able to reduce the number of false positives; moreover, the resulting pharmacological interactions at the binding site as well as ligand preferences are important for assigning confidence to the results of virtual screening experiments. These results suggest that GEMDOCK constitutes a robust tool for virtual database screening.
11 Prepare drug database Prepare target protein Molecular docking Post-docking analysis Known active compounds Mine ligand preferences Mine bind-site pharmacological consensus Mining pharmacological consensus : Main flow : Mining/aided flow Superimpose X-ray or predicted ligand conformations
Figure 2.4. The main steps of GEMDOCK for virtual database screening, including the target protein and compound database preparation, flexible docking, and post-docking analysis. GEMDOCK mines a pharmacological consensus from the target protein and known active ligands when available.
2.2 Consensus scoring criteria
The performance of these scoring functions is often inconsistent across different systems from a database search 18,31. The inaccuracy of the scoring methods, i.e., inadequately predicting the true binding affinity of a ligand for a receptor, is probably the major weakness for VS. It has been demonstrated that combining multiple scoring functions (consensus scoring) improves enrichment of true positives. Previous efforts at consensus scoring have largely focused on empirical results, but they are yet to provide theoretical analysis that gives insight into real features of combinations and data fusion for VS.
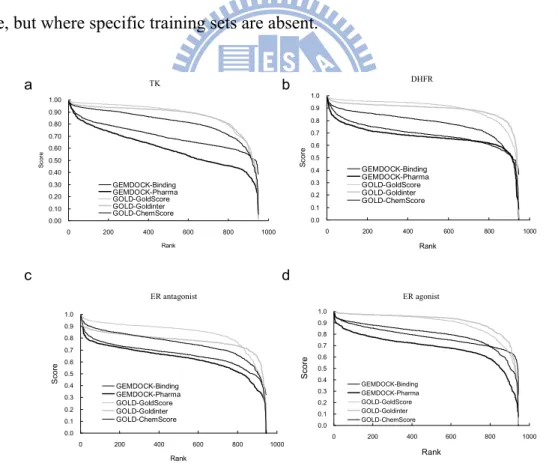
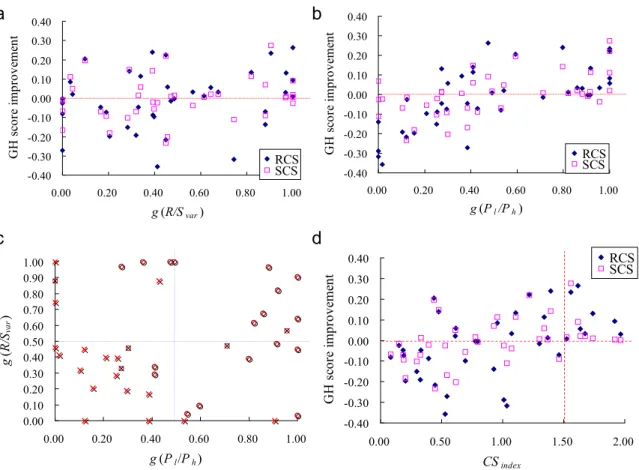
We explore consensus scoring (CS) criteria and provide a consensus scoring procedure for improving the enrichment in VS using data fusion and exploring diversity on scoring characteristics between individual scoring functions (Fig. 2.5). In particular, we demonstrate that combining multiple scoring functions improves enrichment of true positives only if (a) each of the individual scoring functions has relatively high performance, and (b) the scoring characteristics of each of the individual scoring functions are quite different (Fig. 2.6). These two prediction variables are also indicative criteria for the performance between rank
12
combination and score combination. Moreover our second criterion (b) using the rank/score characteristics as the scoring diversity is independent of the performance of the individual scoring function. It is therefore very useful in practical settings in the VS process when the performance of an individual scoring function (such as in criterion (a)) is not known or cannot be evaluated at the juncture. We have developed a novel CS system, available online http://gemdock.life.nctu.edu.tw/dock/download.php, which was tested for five scoring systems with two evolutionary docking algorithms on four targets, thymidine kinase (TK), human dihydrofolate reductase (DHFR), and estrogen receptors (ER) of antagonists and agonists (Fig. 2.7). Our procedure is computationally efficient, able to adapt to different situations, and scalable to a large number of compounds and to a greater number of combinations. Results of the experiment show a fairly significant improvement on the goodness-of-hit (GH) scores, false positive (FP) rate, and enrichment factors over average individual performance. This approach has practical utility for cases where the basic tools are known or believed to be generally applicable, but where specific training sets are absent.
0.00 0.10 0.20 0.30 0.40 0.50 0.60 0.70 0.80 0.90 1.00 0 200 400 600 800 1000 Rank Sc or e GEMDOCK-Binding GEMDOCK-Pharma GOLD-GoldScore GOLD-Goldinter GOLD-ChemScore TK DHFR 0.0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1.0 0 200 400 600 800 1000 Rank Sc or e GEMDOCK-Binding GEMDOCK-Pharma GOLD-GoldScore GOLD-Goldinter GOLD-ChemScore 0.0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1.0 0 200 400 600 800 1000 Rank Sc or e GEMDOCK-Binding GEMDOCK-Pharma GOLD-GoldScore GOLD-Goldinter GOLD-ChemScore ER antagonist ER agonist 0.0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1.0 0 200 400 600 800 1000 Rank Sc or e GEMDOCK-Binding GEMDOCK-Pharma GOLD-GoldScore GOLD-Goldinter GOLD-ChemScore a b c d
Figure 2.5. Rank/score curves of five methods for four virtual screening targets: (a) TK, (b) DHFR, (c) ER-antagonist receptor, and (d) ER-agonist receptor.
13 -0.40 -0.30 -0.20 -0.10 0.00 0.10 0.20 0.30 0.40 0.00 0.20 0.40 0.60 0.80 1.00 g (R/Svar) G H s cor e im pr ove m en t RCS SCS -0.40 -0.30 -0.20 -0.10 0.00 0.10 0.20 0.30 0.40 0.00 0.20 0.40 0.60 0.80 1.00 g (Pl/Ph) G H s cor e i m pr ov em en t RCS SCS a b 0.00 0.10 0.20 0.30 0.40 0.50 0.60 0.70 0.80 0.90 1.00 0.00 0.20 0.40 0.60 0.80 1.00 g (Pl/Ph) g (R/ Sva r ) -0.40 -0.30 -0.20 -0.10 0.00 0.10 0.20 0.30 0.40 0.00 0.50 1.00 1.50 2.00 CSindex GH s core im prov em en t RCS SCS c d
Figure 2.6. The relationships between the GH-score improvement with (a) normalized value of variance of rank/score graph and (b) normalized value of Pl/ Ph of 40 pairing combinations of
five methods for four virtual screening targets. (c) The GH-score improvements with normalized variances of rank/score graphs (R/Svar) and normalized relative performance
measurement (Pl/ Ph) of 40 RCS and SCS pairing combinations of five methods for four virtual
screening targets. (d) The positive and negative GH-score improvements are denoted with circle and cross, respectively.
14 O N N O O O O O N N O O I O O N N O O O N N O O I O O O N N N N O O I O O O N N O O O O N N N N O N O O N N N N O N O O O N N N N O N O O TK09 1kim_THM TK08 1ki7_ID2 TK10 2ki5_AC2 TK05 1ki2_GA2 TK06 1ki3_PE2 TK02 1e2m_HPT TK07 1ki6_AHU TK03 1e2n_RCA TK01 1e2k_MCT N N O O O O TK04 1e2p_CCV S O O O N O O O N O N O O O O N O O O N O O O O N O O O O N N O O N O O O S F F F F F O O O O O N EST01 1err.RAL EST06 EST02 3ert.OHT EST03 1hj1.AOE EST04 EST05 EST08 EST09 EST07 EST10 O O O O O O O O O O O ESA02 1l2i_ETC ESA03 1qkm_GEN ESA01 1gwr_EST ESA04 3erd_DES O O O O O O O O O O O O ESA05
ESA08 ESA09 ESA10 ESA06 O O O O ESA07 DHFR01 1boz.PRD N N N N N N O O N N N N N O O DHFR02 1dlr.MXA N N N N N N N O N O O O O DHFR03 1dls.MTX N N N N O N N O N O O O O DHFR04 1drf.FOL N N N N N N N O N O O N O O O DHFR09 1ohj.COP N N N N O N O N O O O O DHFR05 1hfr.MOT N N N N N N N DHFR06 1kms.LIH N N N N N O O DHFR07 1kmv.LII N N N N N N O O O DHFR08 1mvs.DTM N N N O N N O N O O O O DHFR10 2dhf.DZF a b c d
Figure 2.7. The known active ligands of four VS targets, estrogen receptors (ER) of antagonists (a) and agonists (b), (c) thymidine kinase (TK), and (d) human dihydrofolate reductase (DHFR). The ligand data set from the comparative studies of Bissantz et al. 17was used to evaluate the screening accuracy of different CS on TK, DHFR, ER, and ERA. For each target protein, the ligand database included 10 known active compounds and 990 random compounds.
2.3 Combinative clustering analysis
The increasing numbers of 3D compounds and protein complexes stored in databases contribute greatly to current advances in biotechnology, being employed in all kinds of pharmaceutical and industrial applications. However, screening and retrieving appropriate candidates as well as handling false positives presents a challenge for all post-screening analysis methods employed in retrieving therapeutic and industrial targets.
Using the combinative clustering method (Fig. 2.8), virtually screened compounds were clustered based on their protein-ligand interactions then structure clustering employing physical-chemical features was done to retrieve the final compounds. Based on the protein-
15 Virtual screening Top ranks (100) N N N N N N O O N N N N N O O N N N N N N N N N N N O N N O N O O O O N N N O N N O N O O O O Structure clustering O O OH OH O H O CH3 O O OH O H OH O O H CH3 O O OH O O OH O H OH OH O H N N O N OH N NH2 OH I N N O N OH N NH OH O H C H3 CH3 N+ C H3 S N NH2 N OH C H3
Active compounds Unknown compounds Unknown Active: old
compounds
Active: new Unknown compounds interaction cluster (45)
Select lowest energy conformations as representative structures N N N N N O O O O OH O O OH O H OH OH O H N N O N OH N NH2 OH I Interaction clustering (H-bond, Elect, vdW) 45 Similar structure cluster (4) 10 35 a b
Figure 2.8. Overall process of the two-stage combinative cluster analysis. (a) First stage clustering using protein-ligand interactions generated via GEMDOCK. (b) Second stage clustering of first stage results done using physical-chemical features.
0.0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1.0 0.0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1.0 intra_DHFR1 intra_DHFR2 intra_ESA intra_EST intra_NA1 intra_NA2 intra_TK inter_DHFR1 inter_DHFR2 inter_ESA inter_EST inter_NA1 inter_NA2 inter_TK Compound structure Pr ot ei n-lig and int er ac tion
Figure 2.9. Designing a reference threshold of P-L interaction and atom-pair descriptors. The complementation between atom-pair descriptor and the protein-ligand interaction descriptor is also show in this figure. The distance threshold of atom-pair descriptor was 0.55 (tanimoto coefficient). The threshold of distance of protein-ligand interaction descriptor was 0.39 (correlation coefficient).
16
ligand interaction profile (first stage), docked compounds can be clustered into groups with distinct binding interactions. Structure clustering (second stage) grouped similar compounds obtained from the first stage into similar structures clusters; the lowest energy compound from each cluster being selected as a final candidate. By representing interactions at the atomic-level and including measures of interactions strength (Fig. 2.9), better descriptions of protein-ligand interactions and a more specific analysis of virtual screening was achieved. The two-stage clustering approach enhanced our post-screening analysis by revealing accurate performances in clustering, mining and visualizing compound candidates, thus, improving virtual screening enrichment.
2.4 Summary
As the number of protein structures increases rapidly, structure-based drug design and virtual screening approaches are becoming important and helpful in lead discovery1-2,6. A number of docking and virtual screening methods 16,23-24,35 have been utilized to indentify lead compounds, and some success stories have been reported 1-2,4-5,7-8,10. However, identifying lead compounds by exploiting thousands of docked protein-compound complexes is still a challenging task. The major weakness of virtual screenings is likely due to incomplete understandings of ligand binding mechanisms and the subsequently imprecise scoring algorithms . In the related works, several studies were proposed for improving the accuracy and precision in the VS processes. First, the scoring function of GEMDOCK evolves the pharmacological preferences from a number of known active ligands to take advantage of the similarity of a putative ligand to those that are known to bind to a protein’s active site, thereby guiding the docking of the putative ligand. In the post-screening analysis process, the consensus scoring strategy using data fusion and exploring diversity on scoring characteristics between individual scoring functions for improving VS is proposed. When the huge amount of VS data needs to be interpreted, the combinative cluster analysis is applied for effectively mining the representatives and easily visualizing the VS data. Although we have been successfully applied these methods on the VS studies of two important virus targets, dengue virus and influenza virus, some shortcomings are needed to be addressed.
17
Chapter 3
Site-moiety map for recognizing interaction preferences
between protein pockets and compound moieties
3.1 Introduction
Most of docking programs16,23-24 use energy-based scoring methods which are often
biased toward both the selection of high molecular weight compounds and charged polar compounds in the top ranks. Meanwhile, these approaches generally cannot identify the key features (e.g., pharmacophore spots) that are essential to trigger or block the biological responses of the target protein. Although pharmacophore techniques27 have been applied to derive the key features, these methods require a set of known active ligands that were acquired experimentally. Therefore, the more powerful techniques for post-screening analysis to identify the key features through docked compounds and to understand the binding mechanisms provide a great potential value for drug design.
To address these issues, we presented the SiMMap method to infer the key features by a site-moiety map describing the relationship between the moiety preferences and the physico-chemical properties of the binding site. This method also provides the web server for public access. According to our knowledge, SiMMap is the first public server that identifies the site-moiety map from a query protein structure and its docked (or co-crystallized) compounds. The server provides pocket-moiety interaction preferences (anchors) including binding pockets with conserved interacting residues; moiety preferences; and interaction type. We verified the site-moiety map on three targets, thymidine kinase, and estrogen receptors of antagonists and agonists. Experimental results show that an anchor is often a hot spot and the site-moiety map is useful to identify active compounds for these targets. We believe that the site-moiety map is able to provide biological insights and is useful for drug discovery and lead optimization.
3.2 Method
Figure 3.1 presents an overview of the SiMMap server for identifying the site-moiety map with anchors, describing moiety preferences and physico-chemical properties of the binding site, from a query protein structure and docked compounds. The server first uses checkmol (http://merian.pch.univie.ac.at/~nhaider/cheminf/cmmm) to recognize the compound moieties
18 a b … O S HO O S OO HO S HOO O R 222 H5 8 R 222 E 225 W8 8 Y 172 R 163 Q 125 E1 H1 V1 H2 c d
Step 1: Query a target protein
structure and its docked (co-crystallized) compounds
Step 2: Generate
protein-compound interaction profiles and identify compound moieties
Step 3: Derive an anchor
candidate by identifying a pocket with significant interacting residues and moieties with Z-score ≥ 1.645
Step 4: Determine anchors by
grouping neighbor anchor candidates with same type. For each anchor, identify its binding pocket, top-significant
interacting residues, moiety preferences, and anchor type
Step 6: Output graphically
site-moiety map; anchors with moiety structures and
compositions; and pocket-moiety interactions.
Step 5: Determine site-moiety
map with anchors and rescore compounds
Figure 3.1. Overview of the SiMMap server for the site-moiety map using herpes simplex virus type-1 thymidine kinase (TK) and 1000 docked compounds as the query. (a) Main procedure; (b) The merged protein-compound interaction profile; (c) The pocket-moiety interaction preferences of the anchors: H (hydrogen-bonding). Each anchor consists of a binding pocket with conserved interacting residues, the moiety composition and anchor type; The site-moiety map has one hydrogen-bonding (H) and three van der Waals (V) anchors for ER. Each anchor contains the moiety structures and composition, anchor type, and key residues in the binding pocket.
19
and utilizes GEMDOCK35 to generate a merged protein-compound interaction profile (Fig. 3.1b), including electrostatic (E), hydrogen-bonding (H) and van der Waals (V) interactions. According to this profile, we infer anchor candidates by identifying the pockets with significant interacting residues and moieties with Z-score ≥ 1.645. The neighbor anchor candidates, which are the same interaction type and the distances between their centers are less than 3.5Ǻ, are grouped into one anchor. These anchors form the site-moiety map describing interaction preferences between compound moieties and the binding site of the query (Figs. 3.1c and 3.1d). Finally, this server provides graphic visualization for the site-moiety map; anchors with moiety structures and compositions; pocket-moiety interactions; and the relationship between anchors and moieties of query compounds.
3.2.1 Definitions of site-moiety map, anchor and pocket
The anchor (pocket-moiety interaction preference) is the core of a site-moiety map. An anchor possesses three essential elements: (1) a binding pocket with conserved interacting residues and specific physico-chemical properties; (2) moiety preferences of the pocket; (3) pocket-moiety interaction type (E, H, or V). An anchor can be considered as "key features" for representing the conserved binding environment element or a "hot spot" which involves biological functions. In addition, we regard a binding pocket, which consists of several residues significantly interacting to compound moieties, as a part of the binding site. The binding pocket often possesses specific physico-chemical properties and geometric shape to bind preferred moieties. The site-moiety map, which can help to assemble potential leads by optimal steric, hydrogen-bonding, and electronic moieties, is useful for drug discovery and understanding biological mechanisms.
3.2.2 Constructing site-moiety map
The SiMMap server performs six main steps for a query (Fig. 3.1a). Here, we used TK as an example for describing these steps.
Generating protein-compound interaction profiles and identification of compound moieties
First, users input a protein structure and its docked compounds. The server used checkmol to identify moieties of docked compounds and GEMDOCK to generate E, H and V interaction profiles. For each profile, the matrix size is N×K where N and K are the numbers of
20
compounds and interacting residues of query protein, respectively. An interaction profile matrix P(I) with type I (E, H, or V) is represented as
⎥ ⎥ ⎥ ⎥ ⎦ ⎤ ⎢ ⎢ ⎢ ⎢ ⎣ ⎡ = NK N N K K p p p p p p p p p I P L M O M M L L 2 , 1 , 2 2 , 2 1 , 2 1 2 , 1 1 , 1 ) (
where pi,j is a binary value for the compound i interacting to the residue j (Fig. 4.2B). For H
and E profiles, pi,j is set to 1 (green) if an atom pair between the compound i and the residue j
forms hydrogen-bonding or electrostatic interactions, respectively; conversely, the interaction is set to 0 (black). For van der Waals (vdW) interaction, an interaction is set to 1 when the energy is less than -4 (kcal/mol).
SiMMap identified consensus interactions between residues and compound moieties with similar physical-chemical properties through the profiles. For each interacting residue (a column of the matrix P(I)) (Fig. 3.1b), we used Z-score value to measure the interacting conservation between this residue and moieties. The standard deviation (σ) and mean (μ) were derived by random shuffling 1,000 times in a profile. The Z-score of the residue j is defined as
σ
μ f
Zj = j−
, where fj is the interaction frequency and given as
∑
== N i j i j N p f 1 .
We treated protein-compound interactions as a binomial distribution, and then consensus interactions with statistical significance could be identified by their normal approximation. Statistically, a binomial distribution is approximated by a normal distribution when either
p≤0.5 and np>5 or p>0.5 and n(1 − p)> 5, where n is the number of trials and p is the
probability of success. Here, n is the number of selected compounds and p is the probability of forming an interaction between a protein and a compound, that is, pi,j=1. Typically, the p values
ranged between 0.01 and 0.03 in this study. While the binomial distribution is a normal approximation, at least 500 compounds should be selected for constructing an interaction profile matrix.
Deriving anchor by identifying a pocket with significant interacting residues and moieties
Spatially neighbor interacting residues and moieties with statistically significant Z-score ≥ 1.645 were referred as an anchor candidate. Neighbor anchor candidates, which are spatially overlapped and the same anchor type, were clustered as an anchor and the anchor center is the
21
weighted geometric center of their interacting compound moieties. Here, two anchors were merged if the distance of two anchor centers is less than 3.5 Ǻ. In each anchor, top three residues with the highest Z-score values were regarded as key residues forming a binding pocket. For each anchor, we identified its moieties of docked compounds according to the moiety library derived from checkmol, and calculated the moiety composition (Fig. 3.1c). These anchors form the site-moiety map (Fig. 3.1d) of the query.
Outputting graphically site-moiety map and identifying active compounds
SiMMap can be applied to identify active compounds for structure-based virtual screening. One of weaknesses of virtual screening is likely incomplete understanding of the chemistry involved in ligand binding and the subsequently imprecise scoring algorithms. When a compound highly agrees with the anchors of the site-moiety map, this compound often activates or inhibits the target. The SiMMap server scores a compound by combining predicted binding energy of GEMDOCK and the anchor score between the map and the compound. The SiMMap score, S(i), for a compound i is defined as
∑
= + − = na a M i E i AS i S( ) 1 ( ) ( 0.001) (0.5) (1)where ASa(i) is the anchor score of compound i in the anchor a, n is the number of anchors, E(i)
is the docked energy of compound i, and M is the atom number of compound i. The anchor score is set to 1 when the compound i agrees the moiety preference of the anchor a. Here, the anchor score and the term M0.5 are useful to reduce the deleterious effects of selecting high molecular weight compounds26. Based on SiMMap scores, we can obtain new ranks of query compounds.
3.3 Results
3.3.1 Web serviceSiMMap is an easy-to-use web server (Fig. 3.2). Users input a protein structure without ligands in PDB format and its docked or co-crystallized compounds in MDL mol, SYBYL mol2, or PDB format (Fig. 3.2a). These docked compounds should be generated by any external docking methods (e.g., DOCK, FlexX, GOLD and GEMDOCK) before users uploaded these compounds. Typically, the SiMMap server yields a site-moiety map within 5 minutes if the number of query compounds is less than 100. This server provides the graphic
22 a b d
…
…
cFigure 3.2. The SiMMap server analysis results using estrogen receptor (ER) and 1000 docked compounds as the query. (a) The user interface for uploading target protein structure and docked compounds. (b) The site-moiety map has one hydrogen-bonding and three van der Waals anchors for ER. Each anchor contains the moiety structures and composition, anchor type, and key residues in the binding pocket. (c) The details of moiety structures and residue-moiety interactions in the H1 anchor. (d) The SiMMap scores, ranks and the relationships between anchors and moieties of query compounds.
visualization of the site-moiety map and anchors elements, including a binding pocket with interacting residues, moiety compositions and structures, numbers of involved compounds, and anchor types (Fig. 3.2b). For each anchor, this server shows docked conformations of compounds and the detailed atomic interactions between pocket residues and moieties (Fig. 3.2c). In addition, SiMMap shows the new rank and compound moiety structures fitting the anchors for each query compound (Fig. 3.2d). SiMMap uses two open source tools for graphic visualization: Jmol (http://www.jmol.org/) for displaying three-dimensional protein and
23
compound structures with anchors and OASA (http://bkchem.zirael.org/oasa_en.html) for visualizing compound structures. The server allows users to download the anchor coordinates in the PDB format; interaction profiles; new ranks and anchor scores of query compounds.
3.3.2 Thymidine kinase and estrogen receptor
The SiMMap server inferred the site-moiety map of TK. This map consisted of four anchors (i.e., E1, H1, H2, and V1 (Fig. 3.1d) and the moiety composition and conserved interacting residues of each anchor (Fig. 3.1c). For example, the E1 anchor possesses a binding pocket with residue R222, and three moiety types (i.e., sulfuric acid monoester (40%), carboxylic group (35%) and phosphoric acid monoester (25%)) derived from 57 compounds. Meanwhile, the E1 includes the phosphate moiety of ATP and its residue R222 playing a major role to interact with the substrate 38-39. The preferred moiety types of an anchor are suitable groups interacting to conserved residues of the binding pocket. The moiety preference is able to guide the suggestion of functional group substitutions for lead structures.
We used estrogen receptor (ER), a therapeutic target for osteoporosis and breast cancer40, as the example. Based on 1000 docked compounds and ER, the SiMMap server identifies four anchors (H1, V1, V2, and V3) and provides moiety preferences and compositions in these anchors (Fig. 3.2b and 3.3). The H1 anchor comprises three residues (E353, L387, and R394) and five main moiety types: hydroxyl group (36%), carboxylic acid (16%), amine (7%), ketone (7%), and sulfuric acid monoester (6%) summarized from 319 compounds. Furthermore, three residues (L346, T347, and L525) and 839 compounds are involved in the V1 anchor, preferring five moiety types (i.e., aromatic ring (49%), heterocyclic group (22%), alkenes (11%), phenol (8%), and oxohetarene (4%)). The anchor V2 is a hydrophobic pocket containing L346, F404, and L387, and the former two re sidues are highly conserved41. These hydrophobic residues interact with aromatic ring (52%), heterocyclic group (23%), phenol (12%), alkenes (5%), and oxohetarene (3%). Finally, aromatic rings (55%), heterocyclic groups (17%), alkenes (11%), and phenols (9%) summarized from 560 compounds often form vdW contacts with the long side chains of M343, M421, and L525 in the anchor V3. The ring groups of antagonists are often stabilized by the side chains of M343, L346, T347, L387, M421, and L525. In this case, most selective estrogen receptor modulators of ER (e.g., EST_01 (raloxifene), EST_06 (LY-326315,) and EST_05 (EM-343)) agree with these four anchors (Fig. 3.2d and 3.3c). Anchors
24
Anchor SiMMap 22 ligands
a b c H1 V1 V2 R394 T347 F404 M421 M343 L525 E353 L387 L346 V3 Others Others Others Others 95% 5% 45% 41% 5%5% 5% 36% 16% 7% 7% 6% 28% 49% 22% 11% 8% 4% 6% 52% 23% 12% 5%3%4% 55% 17% 11% 9%2%6% PDB H1 V1 V2 V3 1xp1 1yim 1xqc 1xp6 1yin 1xp9 2r6w 2r6y 1sj0 3dt3 2qr9 2qh6 1uom 2ayr 2iog 2q6j 2ouz 1xpc 2iok 3ert 1err other 1r5k R4 R3 R1 R2 OH Aromatic moiety Heterocyclic moiety H1 V1 V2 V3 86% 14% 95%
Figure 3.3. The relationships between the site-moiety map and 22 co-crystallized ligands of ER. (a) The mapping between four inferred anchors (binding pocket with conserved interacting residues) and these 22 ligands in the active site. (b) The moieties of these 22 ligands in each anchor. Black cell presents that the moiety of the compound does not agree with the anchor H1. (c) The moiety compositions of 1000 docked compounds (SiMMap) and these 22 ligands.
Table 3.1. The relationship between the anchors and moieties of 15 co-crystallized ligands for TK
PDB code Compound structure Anchor
E1 H1 H2 V1
3vtk R-OH
1vtk R-OH
1p7c R-OH
25 1ki6 R-OH 1ki8 R-OH 1e2k R-OH 1ki7 R-OH 1ki4 R-OH 1kim R-OH 1e2p R-OH
1ki3 R-OH Aromatic moiety
1ki2 R-OH Aromatic moiety
1qhi R-OH R-NH2 Aromatic moiety
2ki5 R-OH R-NH2 Aromatic moiety
Table 3.2. Comparing SiMMap with other methods on thymidine kinase and estrogen receptor by false-positive rates
True positive (%) Thymidine kinase (TK) Estrogen receptor (ER)
SiMMap DOCK a FlexX a GOLD a SiMMap DOCK a FlexX a GOLD a
80 6.3b 23.4 8.8 8.3 1.1 13.3 57.8 5.3
90 6.8 25.5 13.3 9.1 1.1 17.4 70.9 8.3
100 6.8 27 19.4 9.3 7.5 18.9 NA 23.4
a Summarized from Bissantz et al.17
identified by the SiMMap server often contain key pockets and moieties. To initially validate the anchors for biological mechanisms (e.g., ligand binding and catalysis mechanisms), we selected 15 TK and 22 ER co-crystallized ligands (Table 3.1 and Fig. 3.3). The corresponding moieties of these co-crystallized ligands were highly matched the anchors derived from 1000 docked compounds (10 known active ligands and 990 randomly selected compounds described
26
in Data sets). The site-directed mutagenesis shows that the conserved interacting residues of the anchors are often essential for ligand binding and catalysis mechanisms. For ER target, 22 ER co-crystallized ligands contain three consistent moieties that are hydroxyl group and aromatic rings (Fig. 3.3b). The hydroxyl group forms hydrogen bonds with R394 and E353 in H1, and the aromatic ring yields vdW contacts with L346, L387, and F404 in V2. The other consistent aromatic ring forms vdW contacts with L346, T347, and L525 in V1. These results show that an anchor is often a hot spot and involved in biological functions.
To provide initial validation of the SiMMap server for virtual screening, we selected TK, ER, and ERA with 1000 compounds as test sets. First, we compared the accuracies of SiMMap with those of GEMDOCK on these three targets based on true positive rates. SiMMap, combining anchor scores and docking energies (Equation 1), outperforms GEMDOCK on these cases. We then compared SiMMap with other three programs (DOCK, FlexX, and GOLD) on TK and ER sets. All approaches were tested using the same proteins and compound sets (Table 3.2). When the positive rate was 90%, the false positive rates were 6.8% (SiMMap), 25.5% (DOCK), 13.3% (FlexX), and 9.1% (GOLD) for TK and were 1.1% (SiMMap), 17.4% (DOCK), 70.9% (FlexX), and 8.3% (GOLD) for ER.
The compound, which agrees with anchors of the site-moiety map, is often able to activate or inhibit the target protein (Tables 3.1 and 3.3). In addition, the anchor score (i.e. AS(i) defined in Equation 1) of SiMMap can be used to reduce the ill-effect of the energy-based scoring methods which are often biased toward both the selection of high molecular weight compounds and charged polar compounds25-26. For example, according to the SiMMap scores (Equation 1),
the top ranks of ER, MFCD0002206 (masoprocol) and MFCD00012748 were identified as the analogs of the active compounds (Table 3.3). The anchor score of SiMMap was helpful to reduce the highly polar compounds (e.g., MFCD00004690 and MFCD00013089 in ER) whose anchor scores are low. The anchor score of SiMMap can easily combine with other energy-based scoring functions.
3.4 Summary
The utility and feasibility of SiMMap method is demonstrated for statistically inferring the site-moiety map describing the relationship between the moiety preferences and physico-chemical properties of the binding site. The validation results show that the site-moiety map is