國立臺中教育大學科學教育與應用學系
科學教育碩士學位暑期在職進修專班碩士論文
指導教授:張嘉麟 博士
1-氟化萘光電子光譜與磷光光譜的
理論研究
A theoretical study of the photoelectron
and phosphorescence spectroscopy of
1-fluoronaphthalene
謝誌
時光荏苒,四年的暑期班轉瞬間便到了尾聲,能夠再次當回學生,追求自己 的理想,是幸福的。儘管大學從自然科學系畢業之後,在學校能教授自然課的機 會並不多,我仍然希望繼續在自然領域上有所精進,避免久未接觸而日漸生疏, 為自然教育盡一份心力。 在這四年進修期間,最感謝的是指導教授張嘉麟老師。量子力學算是我在大 學時概念較模糊的一塊,藉由這次進修的機會,除了自己查找相關的科學史脈絡、 增加背景知識,老師的講解總是清楚、明瞭,讓我很快能進入狀況,尤其在我遭 遇困難時,實質及心理上都給了我很大的幫助和鼓勵。 另外,感謝賴金宏教授與陳錦章教授撥冗閱讀論文,提拱許多寶貴的建議, 使這本論文更具有參考價值,也使我獲益良多。同時,非常感謝黃瓊慧博士在最 後這個暑假的協助,我才能及時完成論文初稿的修改。非常幸運地,我和黃瑞陽、 郭文信成為同組同學,一同研究功課、互相幫忙,度過美好的求學時光。 在此也感謝將軍國小及外垵國小的同事們,在我學業、工作兩頭燒時,所給 予的協助。最後,感謝我的家人和親愛的朋友:函頴、詩婷、柏村,這期間我雖 然失去了一點點,但我沒有失去理想,你們的陪伴是我堅強的後盾,心中著實溫 暖!摘要
本 研 究 的 目 的 在 於 藉 由 量 子 化 學 計 算 的 方 法 , 研 究 1- 氟 化 萘 (1-fluoronaphthalene, C10H7F)分子基態、離子基態和最低能階三重激發態的平衡結 構、諧和振動頻率和光電子光譜及磷光光譜。我們利用密度泛函理論之 B3LYP、 B3PW91 泛函數,與 6-311++G(d, p)、6-311++G(2d,p)、aug-cc-pVTZ 三個基底函數 組來做計算。我們使用本研究室開發的法蘭克康登因子計算方法,模擬出 1-氟化 萘的光電子光譜及磷光光譜。我們也使用完備基組(Complete basis set, CBS)極限的 計算方法(CBS-4M and CBS-QB3),計算 1-氟化萘的絕熱游離能和三重態的激發 能。 結果發現計算的分子平衡結構與振動頻率和實驗值相符。藉由法蘭克康登因 子模擬的光電子光譜與實驗光譜非常吻合。使用 CBS-4M 方法計算 1-氟化萘的絕 熱游離能與實驗值僅相差 507 cm-1,計算的絕熱游離能與實驗值的相對差值為 0.77%;使用 CBS-QB3 方法計算 1-氟化萘的激發能與實驗值僅相差 432 cm-1,相 對差值為 2.05%。 關鍵詞:法蘭克-康登因子、1-氟化萘、光電子光譜、磷光光譜、游離能、激發能II
Abstract
The equilibrium geometries, harmonic vibrational frequencies and normal modes of 1-fluoronaphthalene together with its cation and the lowest triplet state were computed by using the density functional theory. The 6-311++G(d,p), 6-311++G(2d,p) and aug-cc-pVTZ basis sets were adopted in the calculations. The photoelectron and phosphorescence spectroscopy of 1-fluoronaphthalene were simulated by computing Franck-Condon factors with the approach developed by our group. The adiabatic ionization energy and the excitation energy of the triplet state were also calculated by the complete basis set method (CBS-4M and CBS-QB3).
The computed equilibrium geometries and vibrational frequencies are in agreement with experiment. Based on the computed Franck–Condon factors, the photoelectron spectra of 1-fluoronaphthalene were simulated and found to be in agreement with the experiment. The adiabatic ionization energies using CBS-4M method in the 1-fluoronaphthalene are in good agreement with the experiment within 507 cm-1. Compared with reported experiment values, the relative variations of computed ionization energies are 0.77 %. The excitation energy of 1-fluoronaphthalene calculated by using the CBS-QB3 method only deviates from the experimental value by 432 cm-1, the relative variations of computed excitation energies are 2.05 %.
Keywords: Franck-Condon factor; 1-fluoronaphthalene; photoelectron spectrum; phosphorescence spectrum; ionization energy; excitation energy
目次
摘要 ... I Abstract ... II 目次 ... III 表目次 ... IV 圖目次 ...V 第一章 緒論 ... 1 1.1 研究動機 ... 1 1.2 文獻探討 ... 2 1.2.1 1-氟化萘的研究 ... 2 1.2.2 法蘭克-康登因子的研究 ... 5 1.2.3 光電子光譜與磷光機制 ... 6 第二章 研究方法 ... 9 2.1 量子化學計算方法 ... 9 2.1.1 研究流程 ... 9 2.1.2 密度泛函理論 ... 9 2.1.3 泛函數 ... 10 2.1.4 基底函數組 ...11 2.1.5 完備基組極限 ... 12 2.2 法蘭克-康登積分 ... 13 2.3 光譜模擬 ... 17 第三章 結果與討論 ... 21 3.1 平衡結構 ... 21 3.2 振動頻率及振動模式 ... 32 3.3 光電子光譜與磷光光譜 ... 38 3.4 游離能與激發能 ... 55 第四章 結論 ... 57 參考文獻 ... 59 附錄 ... 65IV
表目次
表 3. 1 不同基組下 1-氟化萘分子基態的平衡鍵長 ... 25 表 3. 2 不同基組下 1-氟化萘離子基態的平衡鍵長 ... 26 表 3. 3 不同基組下 1-氟化萘三重態的平衡鍵長 ... 27 表 3. 4 不同基組下 1-氟化萘分子基態的平衡鍵角 ... 29 表 3. 5 不同基組下 1-氟化萘離子基態的平衡鍵角 ... 30 表 3. 6 不同基組下 1-氟化萘三重態的平衡鍵角 ... 31 表 3. 7 不同基組下 1-氟化萘分子基態的振動頻率 ... 35 表 3. 8 不同基組下 1-氟化萘離子基態的振動頻率 ... 36 表 3. 9 不同基組下 1-氟化萘三重態的振動頻率 ... 37 表 3. 10 1-氟化萘不同方法所計算之絕熱游離能與實驗值[7][4]對照表 ... 55 表 A- 1 1-fluoronaphthalene 的平衡結構 1... 65 表 A- 2 1-fluoronaphthalene 的平衡結構 2... 67 表 A- 3 1-fluoronaphthalene 的平衡結構 3... 69 表 A- 4 1-fluoronaphthalene 的平衡結構 4... 71 表 A- 5 1-fluoronaphthalene 的平衡結構 5... 73 表 A- 6 1-fluoronaphthalene 的平衡結構 6... 75 表 A- 7 1-fluoronaphthalene 的振動頻率 1... 77 表 A- 8 1-fluoronaphthalene 的振動頻率 2... 79 表 A- 9 1-fluoronaphthalene 的振動頻率 3... 81 表 A- 10 1-fluoronaphthalene 的振動頻率 4... 83 表 A- 11 1-fluoronaphthalene 的振動頻率 5 ... 85 表 A- 12 1-fluoronaphthalene 的振動頻率 6... 87 表 A- 13 1-fluoronaphthalene 的振動頻率 7... 89 表 A- 14 1-fluoronaphthalene 的振動頻率 8... 91 表 A- 15 1-fluoronaphthalene 的振動頻率 9... 93 表 A- 16 1-fluoronaphthalene 的振動頻率 10... 95 表 A- 17 1-fluoronaphthalene 的振動頻率 11... 97 表 A- 18 1-fluoronaphthalene 的振動頻率 12... 99圖目次
圖 3. 1 1-氟化萘分子基態的平衡結構及原子標號[B3LYP/aug-cc-pVTZ] ... 21 圖 3. 2 1-氟化萘離子基態的平衡結構及原子標號[B3LYP/aug-cc-pVTZ] ... 22 圖 3. 3 1-氟化萘三重態的平衡結構及原子標號[B3LYP/aug-cc-pVTZ] ... 22 圖 3. 4 1-氟化萘三重態的平衡結構及原子標號[B3LYP/6-311++G(d,p)] ... 23 圖 3. 5 不同基組下 1-氟化萘分子基態的平衡鍵長比較圖 ... 25 圖 3. 6 不同基組下 1-氟化萘離子基態的平衡鍵長比較圖 ... 26 圖 3. 7 不同基組下 1-氟化萘三重態的平衡鍵長比較圖 ... 27 圖 3. 8 不同基組下 1-氟化萘分子基態的平衡鍵角比較圖 ... 29 圖 3. 9 不同基組下 1-氟化萘離子基態的平衡鍵角比較圖 ... 30 圖 3. 10 不同基組下 1-氟化萘三重態的平衡鍵角比較圖 ... 31 圖 3. 11 不同基組下 1-氟化萘分子基態的振動頻率比較圖 ... 32 圖 3. 12 不同基組下 1-氟化萘離子基態的振動頻率比較圖 ... 33 圖 3. 13 不同基組下 1-氟化萘三重態的振動頻率比較圖 ... 33 圖 3. 14 模擬的 1-氟化萘光電子光譜圖及標定(計算方法:B3LYP/6-311++G(d,p)) 39 圖 3. 15 模擬的 1-氟化萘光電子光譜圖及標定(計算方法:B3LYP/6-311++G(2d,p)) ... 39 圖 3. 16 模擬的 1-氟化萘光電子光譜圖及標定(計算方法:B3LYP/aug-cc-pVTZ) . 40 圖 3. 17 模擬的 1-氟化萘光電子光譜圖及標定(計算方法:B3PW91/6-311++G(d,p)) ... 41 圖 3. 18 模擬的 1-氟化萘光電子光譜圖及標定(計算方法:B3PW91/6-311++G(2d,p))VI 圖 3. 20 不同基組模擬的 1-氟化萘光電子光譜比較圖(B3LYP)半高寬 30 cm-1 ... 43 圖 3. 21 不同基組模擬的 1-氟化萘光電子光譜比較圖(B3PW91)半高寬 30 cm-1 .... 43 圖 3. 22 不同基組模擬的 1-氟化萘光電子光譜比較圖(B3LYP)半高寬 400 cm-1 ... 44 圖 3. 23 不同基組模擬的 1-氟化萘光電子光譜比較圖(B3PW91)半高寬 400 cm-1 .. 44 圖 3. 24 模擬的 1-氟化萘光電子光譜與實驗光譜[7]比較圖,半高寬 400 cm-1 ... 45 圖 3. 25 模擬的 1-氟化萘磷光光譜圖及標定(計算方法:B3LYP/6-311++G(d,p)) ... 47 圖 3. 26 模擬的 1-氟化萘磷光光譜圖及標定(計算方法:B3LYP/6-311++G(2d,p)) . 47 圖 3. 27 模擬的 1-氟化萘磷光光譜圖及標定(計算方法:B3LYP/aug-cc-pVTZ) ... 48 圖 3. 28 模擬的 1-氟化萘磷光光譜圖及標定(計算方法:B3PW91/6-311++G(d,p)) 48 圖 3. 29 模擬的 1-氟化萘磷光光譜圖及標定(計算方法:B3PW91/6-311++G(2d,p)) ... 49 圖 3. 30 模擬的 1-氟化萘磷光光譜圖及標定(計算方法:B3PW91/aug-cc-pVTZ)... 49 圖 3. 31 不同基組模擬的 1-氟化萘磷光光譜比較圖(B3LYP) 半高寬 30 cm-1 ... 52 圖 3. 32 不同基組模擬的 1-氟化萘磷光光譜比較圖(B3PW91) 半高寬 30 cm-1 ... 52 圖 3. 33 不同基組模擬的 1-氟化萘磷光光譜比較圖(B3LYP) 半高寬 200 cm-1 ... 53 圖 3. 34 不同基組模擬的 1-氟化萘磷光光譜比較圖(B3PW91) 半高寬 200 cm-1 .... 53 圖 3. 35 模擬的 1-氟化萘磷光光譜與實驗光譜[4]比較圖,半高寬 200 cm-1 ... 54
第一章 緒論
本論文旨在運用密度泛函理論(density functional theory)對 1-氟化萘 (1-fluoronaphthalene)分子基態、離子基態及最低三重態進行理論計算的研究, 並以本實驗室發展的方法去計算法蘭克-康登因子(Frank-Condon factor, FCF), 模擬出 1-氟化萘的光電子光譜及磷光光譜,並與實驗值作比較;以及計算絕熱游 離能和三重態的激發能。 本論文分為四個章節,第一章為緒論,第二章為研究方法,第三章為結果與 討論,最後一章為結論。 本章分為兩節,第一節說明研究動機,第二節進行文獻探討,包括 1-氟化萘 的研究、法蘭克-康登因子計算、光譜及其理論研究。
1.1 研究動機
量子力學起源於十九世紀末,因在微觀尺度中觀測到的現象和理論的預測產 生矛盾的情形,無法以古典力學的角度來解釋,而逐漸發展成一新的理論體系, 在物理學上如電磁學、電子學、粒子物理、凝聚態物理以及宇宙學,材料科學、 醫藥化學、化學鍵結理論、密碼學等等都有量子力學為基礎的實際應用。 在量子化學上[1],我們盡力於解薛丁格方程式,求得分子的波函數,了解2 於是我們使用各種近似方法來求解。1998 年諾貝爾化學獎得主 John Pople 發展 出量子化學計算軟體(Gaussian),使得量子化學計算能以在各領域被廣泛運用。 光譜學也是量子理論重要的應用領域之一,它跨越了實驗操作上的困難,得到高 解析度的數據,隨著計算方法及硬體設備不斷的改善,不僅可做為實驗研究的初 探,輔助光譜標定的工作,精確的計算更可幫助我們達到實驗未竟之地。 本研究是藉由量子化學計算的方法,研究 1-氟化萘分子基態、離子基態和最 低三重激發態的平衡結構、諧和振動頻率,模擬出光電子光譜及磷光光譜。一些 單取代的萘分子已被各種實驗光譜法所研究,包括它們的基態跟電子激發態的特 性、游離能等,但離子態光譜及激發三重態(triplet state)返回基態的磷光光譜 研究並不多,本研究可幫助人們更加了解其分子的物理化學特性。
1.2 文獻探討
本研究的重點為計算 1-氟化萘的法蘭克-康登因子與模擬其光電子光譜及磷 光光譜,因此以下進行 1-氟化萘分子、法蘭克-康登因子及光譜理論研究的文獻 探討。1.2.1 1-氟化萘的研究
本研究分子 1-氟化萘(1-fluoronaphthalene),為萘的衍生物,萘分子是把兩個 苯環共用的兩個相鄰的碳原子稠合而成,在萘的結構式把萘環上一個α-位置的 碳所連結的氫,取代成氟。氟化萘可經由萘的鹵化親電子取代反應生成,依據氟在萘環上所取代氫的位置,可分為氟連接於α-碳(圖 3.1 的 C2、C5、C10、C11) 上面的 1-氟化萘(1-fluoronaphthalene, α-fluoronaphthalene),以及 β-碳(圖 3.1 的 C1、C6、C14、C15)上的 2-氟化萘(2-fluoronaphthalene, β-fluoronaphthalene)。在 萘環上,由於π電子的非定域(delocalized)並不像苯環那樣完全平均化,而是在 α-碳原子上的電子雲密度較高,β-碳原子上的次之,中間共用的兩個碳原子上 則更小,因此親電子取代反應一般發生在α-位,製備時 1-氟化萘的產量會高於 2-氟化萘。 1-氟化萘分子式 C10H7F,分子量 146.16,結構為平面分子,隸屬於 Cs 點群, 熔點-12℃,沸點 215℃,是一穩定分子,不相容於強氧化劑,常溫常壓下為無色 至淡黄透明液體,易燃,具刺激性毒性。 1- 氟 化 萘 可 當 作 有 機 合 成 原 料 , 用 在 以 叔 丁 基 鋰 介 導
(tert-Butyllithium-mediated)的方法合成 6-芳基啡啶(6-substituted phenanthridines), 此類化合物在醫學上因它的抗菌性而被研究與應用。 1-氟化萘也是製造藥物度洛西汀(R)-Duloxetine 的關鍵中間產物原料,此藥 物以商品名千憂解(Cymbalta™)販售,可抑制血清素及正腎上腺素的再回收機轉, 用來治療憂鬱症、焦慮症[2, 3]及治療糖尿病周邊神經病變引發的急性疼痛,以 及治療壓抑性小便失禁症。 另外,因為萘的衍生物分子對丟電子與接收電子的不同傾向,對光學材料
4
的不穩定性。在近幾年的薄膜電晶體液晶顯示器(Thin film transistor liquid crystal display, TFT -LCD)製程上有使用萘的氟化物分子作為液晶材料,有助於改善早期 液晶材料結構的不穩定性。 歷年來,對於 1-氟化萘的相關研究,條列如下: J. Ferguson 等人[4]於 1954 年研究萘及其簡單衍生物的磷光光譜實驗,繪製 出 1-氟化萘的磷光光譜同時測得其激發能。 T. Iredale 等人[5]於 1960 年研究以近紫外線蒸氣吸收光譜探討單鹵素取代的 萘分子電子態。 H. Gattermann 及 M. Stockburger 等人[6]於 1975 年做出萘分子三重激發態回 到基態的實驗值。 L. Kiasinc 等人[7]於 1983 年進行並苯之光電子光譜及電子結構與取代基效 應之實驗時,繪製出 1-氟化萘的光電子光譜,並測得其垂直游離能。 T. Iliescu 等人[8]於 1984 年研究在 77K 下 1-氟化萘和 2-氟化萘的晶格陣列的 吸收光譜、螢光光譜及磷光光譜。以及三重態回到基態的振動頻率。 Richard P. Rava 等人[9]於 1985 年研究 1-氟化萘和 2-氟化萘的二光子螢光蒸
氣激發光譜(Two-photon vapor fluorescence excitation spectra)。
Sheng Yuan Tzeng 等人[10]在 2012年以共振二光子(Resonant two-photon)游
離與質量解析臨界游離(mass-analyzed threshold ionization)光譜術研究 1-氟化萘
1.2.2 法蘭克-康登因子的研究
量子化學從「波函數」來描述物質的各種物理量,而電子躍遷的機率,正比 於波函數絕對值的平方。法蘭克-康登因子(Franck-Condon factor, FCF)[11-13] 即是分子的兩個電子態間,振動波函數重疊積分的平方,法蘭克-康登因子在分 子輻射與非輻射躍遷的研究上是重要的一環。 在法蘭克-康登因子計算方法的歷史發展上,1930 年 Hutchisson[14] 導出諧 和振子平衡位置改變與頻率改變之法蘭克-康登因子公式。1951 年 Manneback[15] 發展並應用遞迴法(recurrence method)去計算法蘭克-康登因子的積分。1959 年 Wagner[16],導出法蘭克-康登因子閉合的積分公式。1959 年,Ansbacher[17] 發 表一個更精確的方程式來算這些積分,他的方法被廣泛的應用於用來計算一維諧 和 振 子 的 法 蘭 克 - 康 登 因 子 。 Koide[18] 等 人 使 用 二 次 量 子 算 子 法 (Second quantization operator method),解出和 Ansbacher 的方程式類似的結果。Sharp 和Rosenstock 於 1964 年使用分析法,運用生成函數(generating function)導出多維度 法蘭克-康登積分(Franck-Condon Integrals, FCIs)的算式[19]。1999 年 Islampour 推導出多 維的諧 和振子 法蘭克 -康登 因子的積 分公式 ,他們 的一維 結果 和 Ansbacher 的公 式是 一樣 的 [20] 。 Mebel 等人 [21] 使 用 厄 米 多項 式 ( Hermite
6 子 FCI 的計算法[22-28],其中考慮了杜辛斯基效應,利用理論計算所得到的結 構和頻率,計算兩電子態間的法蘭克-康登因子並且應用至分子吸收光譜的研究 中。 複雜的多原子分子,不同電子態的位能面之間,可能發生位移(平衡結構不 同)、扭曲(振動頻率不同)、旋轉(座標系統不平行)的現象,因此,在計算其 重疊積分時,不同模式的座標會耦合(couple)在一起而無法分離,這種模式混 和的現象,就稱為杜辛斯基效應(Duschinsky effect)[29] ,此效應使得多維度 FCF 計算,成為一個難題。 張嘉麟教授在 2013 年進行了新的諧振子 FCI 公式推導,更開發了計算程 式,其目的不僅包含了杜辛斯基效應,且適用於任何維度的諧振子 FCI 計算[30]。 本研究即是利用 2013 年發表的方法,進行 1-氟化萘的光電子光譜與磷光光譜研 究。
1.2.3 光電子光譜與磷光機制
實驗中,根據光電效應的概念, Turner 等人(1962)發展出紫外線光電子 光譜術(Photoelectron spectroscopy, PES)[31],當分子被固定的低能量紫外線光 源照射後,電子會游離出來,因為給予分子的總能量已知,分子游離後電子的動 能可測量,將數據加以分析便可知離子基態的光電子光譜。臨界光電子光譜 (Threshold photoelectron spectroscopy, TPES)改用可調光源,改進 PES 使用固定波長光源造成電子動能過大的缺點,使得光電子能譜解析度從 80cm-1
升到 10cm-1
[33, 34]。Schlag 及 Müller-Dethlefs 等人(1984)進一步發展出零動 能光電子光譜(Zero kinetic energy photoelectron spectroscopy, ZEKE) [35],測 量零動能的電子來獲得離子光譜,因為電子不具動能,因此解析度大為提升。 Johnson 等人 (1991) 在 ZEKE 的 基礎上 發展 出質 量解 析臨 界游離 光 譜術 (Mass-analyzed threshold ionization spectroscoopy, MATI)[36] ,因為是經電場 游離產生的零動能離子,而能夠偵測分子質量的訊息,研究同位素分子、分子團 簇以及自由基等時就不會受到限制,是目前仍被廣泛使用的光譜術。
電子依照庖利不相容原理(Pauli Exclusion Principle),同一個軌域的兩個電
子自旋方向必定相反。假設當分子吸收入射光能後,其中的電子從基態 S0(通 常為單重態)躍遷至具有相同自旋多重度的激發態 S2,此時電子可以經由非常 快的(短於 10-12秒)內轉換過程無輻射躍遷至能量稍低並具有相同自旋多重度 的激發態 S1,接著經由系間跨越(intersystem crossing)過程無輻射躍遷至能量 較低且具有不同自旋多重度的激發態 T2(通常為三重激發態),再經由內轉換過 程無輻射躍遷至激發態 T1。當經由系統間跨越產生的 T1態經由放光方式釋出能 量而回到 S0,這種不同自旋重態間的放光過程稱為「磷光」(phosphorescence), 磷光因為違反自旋禁止躍遷規則(forbidden transition),所以其速率常數通常比 由 S1回到 S0放出的螢光小好幾個數量級(< 10-5 s),使得 T1的生命期極長(從 10-4秒到數分鐘)、磷光強度極弱。
第二章 研究方法
本章共分三節,第一節簡介量子計算方法,包括密度泛函理論的原理及能量 的計算,第二節敘述法蘭克-康登因子的計算方法,第三節說明光譜模擬的方法。2.1 量子化學計算方法
2.1.1 研究流程
首先我們利用 GaussView 畫出 1-氟化萘的分子結構,並且對各個原子標號, 輸出描述各原子間鍵長與鍵角的 Z 矩陣(Z-matrix)。使用 Gaussian 09 套裝軟體[37] 計算出分子最穩定的幾何結構與振動頻率。得到萘分子基態、離子基態與三重態 的最佳化平衡結構與振動頻率後,利用 FCF 計算公式,預測光譜的強度分布型 態,再用 OriginPro7 軟體繪製模擬的 1-氟化萘分子的光電子光譜圖及磷光光譜圖, 然後與實驗光譜圖比較分析。最後採用 CBS-QB3 及 CBS-4M 兩種方法來計算 1-氟化萘分子、離子及三重態的絕熱游離能(adiabatic ionizationenergy, AIE)和激發 能。2.1.2
密度泛函理論10 理 論 假 設 多 電 子 體 系 符 合 波 恩 - 奧 本 海 默 近 似 法 (Born-Oppenheimer approximation)[41],亦即一個多電子體系,其電子的運動和能量與原子核的運動 和能量,是可以相互分離的。傳統的全始計算上解薛丁格方程式包含 3×N 個變 數(N 為電子數),當分子的電子數大到一定程度,方程式就會變得複雜難解。 而 DFT 跟系統大小無關,電子密度可利用一個三維空間座標的函數 E[ρ(x,y,z)] 求得,其中 E 代表能量,ρ代表電子密度,x,y,z 代表位置,故計算效率較佳。 1965 年 Kohn-Sham 方法發表[42]為了求得更準確的動能,把多電子轉化成多個 單電子,將電子間的交互作用作為相關的動能與位能,進一步修改了 Hohenberg 與 Kohn 的理論中的誤差。
DFT 的方法在 Hartree-Fock 全始計算(ab initio calculation)的近似基礎上進一 步利用電子密度來描述電子間交互作用以及電子間的關聯性(electron correlation), 因此也更加準確的描述多電子系統,故所預測的能量及其他物理量較 HF 精確, 為近年來被廣泛使用的量子化學計算方法。
2.1.3 泛函數
本研究選用的泛函數有二,為混合型泛函數[43],如下:
B3LYP(Becke style three-parameter density functional theory with Lee-Yang-Parr
correlation functional)B3 代表三參數交換泛函數[44],並混合採用 LYP 非侷域相 關泛函數[45]。此種方法利用 Hatree-Fock (HF)的交換函數與密度泛函理論 (DFT)進行混和所得到的新函數,進行計算後所得到的修正能量為 HF 交換
能與 DFT 能混合後的結果,因此使用 B3LYP 計算法比單獨使用 HF 計算法, 更能得到精準的能量值,
B3PW91(Becke style three-parameter density functional theory with Perdew-Wang
1991 correlation functional) B3 代表三參數交換泛函數,PW91 是 1991 年由 Perdew-Wang 提出的非侷域(non-local)相關泛函數[46]。
2.1.4 基底函數組
基底函數組(basis set)是以數學形式表達分子軌域的特性,用來組合建構 波函數。選用不同的基底函數組會影響到計算結果的準確度和計算時間,折衷兩 者,本研究使用的基組為 Pople 等人[47]所提的 6-311++G(d, p)、6-311++G(2d,p) 與 Dunning 等人[48]提的 aug-cc-pVTZ 三種基組,分別與泛函數 B3LYP 及 B3PW91 搭配,共得到六個計算方式。所有的量子化學計算皆以 Gaussian 09 軟體進行。 基底函數以 6-311++G(d, p)為例:最前面的 6 代表描述內殼層軌域用 6 個初 始高斯函數線性組合;311 表示價殼層軌域是由 3 個基底函數所構成,且這些基 底函數各有 3、1、1 個初始高斯函數組合成。第一個+號代表氫以外的其他原子 加擴散函數,第二個+代表氫加擴散函數,這可讓電子分布於較大空間;(d, p)代 表加入了極化函數,也就是加入更高角動量的軌域型態,讓電子的分布型態更加 靈活,讓電子分布於不同位向,以求能更近似真實的軌域特性。基底函數以12
2.1.5 完備基組極限
Petersson 等人開發了許多完備基組極限的計算方法,用於非常精確且複雜 的能量計算,簡稱 CBS 法[49]。CBS-QB3 是 Petersson 等人[50]於 1998 年開 發的化學計算模型,使用密度泛函數 B3LYP 的幾何形狀和頻率來進行能量計算, 大幅提高能量計算結果的可靠性與精確性,此模型適用於化學反應的過渡態。 CBS-4M 是 Petersson 等人[51]於 1999 年開發的最新化學計算模型,使用最低 群體定位方法,雖然此法的精確度比其他模型低,但可適用於更大的系統,而且 修改了幾個問題後,更是提高了 CBS-4M 和 CBS-QB3 的計算可靠性。 本研究的絕熱游離能和激發能藉著 CBS-4M 及 CBS-QB3 方法計算,若經由 CBS 法所求得 1-氟化萘分子態的平衡結構能量為 E1CBS、1-氟化萘離子態的平衡 結構能量為 E2CBS、1-氟化萘三重態的平衡結構能量為 E3CBS,則分子的絕熱游離能(Adiabatic ionization energy, AIE)及激發能,公式為: 游離能 EI=E2CBS-E1CBS
激發能 EE=E3CBS-E1CBS
須注意的是,所有計算輸出的能量單位都是 Hartree(626.5095 kcal/mol),其與電 子伏特間可用 Eh = 27.21138505 eV 的公式做轉換。
2.2 法蘭克-康登積分
法蘭克-康登因子(Franck-Condon factor, FCF)為分子兩電子態振動波函數重 疊積分(亦即 FCI)的平方,可利用輸入分子的兩個不同電子態的平衡結構、不 同振動模式的角頻率等數據進行計算。由於法蘭克-康登因子即表示為當分子受 激發時,位於分子某電子態某一振動能階的電子,躍遷到另一個電子態的某振動 能階的發生機率,因此透過 FCF 的計算,可以進行分子的吸收、螢光、磷光、 與光電子等光譜訊號強度分布的預測。 本研究使用的法蘭克-康登因子公式,即是張嘉麟教授在 2013 年所發展出來 的[30],包括新的諧振子 FCI 公式推導及計算程式開發,此方法不僅包含杜辛斯 基效應(Duschinsky effect),且適用於任意維度的諧振子 FCI 計算,公式簡述如 下: n 維諧振子的 FCI 可以方程式表示為:
i i
i i i i i n i v i i vi n n vv v N H Q H Q Q Q dQ v v v i
2 2 1 2 1 2 1 2 1 2 1 exp (1) 其中 2 1 1 2 ! !
n i i i v v i i v v N i i (2) i i (3)14 ) ( polynomial Hermite : ) (x 厄米多項式 Hv ωi為第 i 個模式中的角頻率, 為普朗克常數 h 除以 2π。 若納入杜辛斯基效應的處理,則 Q可寫成[23, 27, 29]: D JQ Q (4) J : Duschinsky matrix(杜辛斯基矩陣) D : shift vector(位移向量) 在此 T L L J ' (5) L’表示起始態的位移矩陣(displacement matrix), LT表示激發態的位移矩陣的轉置矩陣(transpose of a matrix)
'
0 0 2 1 'M X X L D (6) M 為由原子量所構成的對角矩陣(diagonal matrix) X0、X’0分別表示激發態以及起始態的平衡結構笛卡爾座標(Cartesian coordinate)。 由以下兩式
n i j i j ij i i Q B Q C x 1 (7)
n i j i j ij i i x B x C Q 1 (8) 我們可得:
i i
i i n i j j ij n i v i n i j j ij vi n n vv v NE H a x d H a x d Ax dx v v v i 2 1 2 1 2 1 exp
(9) 其中
1 1 2 1 2 2 1 i k ki k n k ki k i i J A B A (10)
n k i k kj ki k kj ki k i ij J J A B B A B 1 1 1 2 1 1 (11)
n k i k k ki k ki k k i i D J A B C A C 1 1 1 2 1 1 (12) 1 , 1 , i ii i B B (13)
1 1 j i k kj ik ij ij B B B B , ji1 (14)
n i k k ik i i C B C C 1 (15) i ii a (16) ij i ij B a , ji (17) 1 1 i i i J (18)
1 1 j k kj ik ij i ij J J B , j1 (19) i i i C d (20)
n k i k ik i i J C D d 1 (21)16
n i n i i i i iD AC E 1 1 2 2 2 1 exp (22) 在式子(10)-(15)中,A、B、C 係數必須由較低 i 值計算到較高 i 值,而 B' 和 C'係數則由反向操作獲得。 接著,把式子(9)代入下列的厄米展開式
n k k k n n H d x k n d x H 0 2 (23) 其中 k n 是二項式係數。 展開後的積分式恰好為多重高斯積分的乘積,故再以高斯積分簡化:
2 1 2 2 2 ! ! 1 2 exp a a s dx ax x s s (24) 其中
2s1
!!135(2s1)是雙階乘。 得到 V HI v v v v v v n n 0 2 1 2 1 (25) 其中 2 1 1 0 00 0 00 0
n i i i i A E I (26) 為絕熱躍遷的 FCI。
12 1 ! ! 2
n i i i v v v v V i i (27)
ij ij ij ij u k u k F F F H 0 0 3 2 1 (28)式子(28)中 F1F2F3分別為
n i n j n j k ij ij ij k ij ij ij ij ij a k u a k u F 1 1 1 1 (29)
n i i m i m d H d H F i i 1 2 (30)
n j j K j K A F j 1 2 3 1!! 2 (31) 在此, in in i u k m (32) in in i u k m (33)
j i n i ij ij j k k 1 1 K (34) 且 K j的值必須是偶數。
j1 i p ip i ij v k u (35)
1 1 j p ip i ij v k u (36)在上式中,0≦kij≦uij(i =1~n, j = i ~ n);0≦k’ij≦u’ij(i, j = 1 ~ n)。 經由以上步驟所導出的任意維度 FCI 計算公式,優點在於每一個方程式的 運算式皆具有封閉形式的通解,有利於編寫成計算程式。若結合分子結構、振動 頻率、振動模式、激發能量及躍遷偶極矩等數據,則可模擬出各種光譜,對於預 測分子的光譜結構,具有高度價值與實用性。
18 振動模式標記法為 Mulliken notation[52]。1-氟化萘的點群為 Cs,依對稱性區分, 有 in-plane(A’)以及 out-of-plane(A’’)這二種對稱性,編排方法先從平面振動模式 (A’)依照振動頻率由高到低排列並給予 1、2、3…33 的編號,接著再換非平面的 振動模式(A’’)依照振動頻率由高到低排列,並接續 A’振動模式給予編號 34、35 一直到第 48 號。 本研究使用不同計算基組,計算出代表兩個電子態間振動躍遷機率的法蘭克 -康登因子之後,我們以 FCF 為光譜線的高度(強度),高斯函數(Gaussian function) 為線形,將所有躍遷訊號疊加,即可得到模擬的光電子光譜。其計算方程式如下:
2 2 1 ' 2 ' ( , ) 2 ln 4 exp ) ( I (38) 其中 I 是相對強度,ω是相對於絕熱躍遷的激發能,
' , 是指 ' 能 階的振動能量,12是半高寬(full width at half maximum, FWHM),可依欲對照 的實驗光譜圖的解析度加以調整,以求盡量相符。在對照本研究模擬光譜與實驗光譜前,須先將文獻中的實驗光譜圖以繪圖軟 體作數位化處理,以便進行重新繪製,再利用 Origin Pro7 軟體與理論模擬光譜 圖進行圖形的疊合比對。我們必須先把紙本或電子檔的實驗光譜圖存成電子圖檔, 經由 Origin Pro7 軟體把圖形線條轉換成 xyz 座標數值列表,其中 x 對應於電子 動能,y 對應於訊號強度,z 為圖形的灰階顏色值。我們要的圖形資料為黑色或 接近黑色,所以顏色值須是 0(純黑色)或接近 0 的數值,經由軟體資料分析中的 篩選功能,設定選取 z 小於某數值(本研究設定 50),即可輸出成光譜的 xy 數值,
完成光譜圖形的數位化。 光電子光譜 x 軸為光電子的動能,y 軸為電子訊號強度,其中需注意的是, 模擬光譜圖的 x 軸光電子動能的單位是以波數(cm-1)表示,而文獻中的實驗光 譜圖 x 軸對應的單位為電子伏特(eV),所以必頇將電子伏特轉換成波數的單位 (1eV = 8065.644651cm-1),而我們採用相對能量為 y 軸,對齊理論與實驗光譜 的原譜帶後,進行兩者的疊合與比較。
第三章 結果與討論
本章主要呈現 1-氟化萘(C10H7F)的量子化學計算之研究結果與討論。本章分 為四節,第一節呈現 1-氟化萘分子基態、離子基態和三重態的最佳平衡結構,並 完成鍵長、鍵角的標定。第二節比較 1-氟化萘的振動頻率與振動模式。第三節依 據法蘭克-康登因子的計算結果,模擬出 1-氟化萘的光電子光譜及磷光光譜,除 了比較不同計算方法,也與實驗光譜進行結果比對。第四節探討 1-氟化萘分子的 游離能與激發能。3.1 平衡結構
圖 3. 1 1-氟化萘分子基態的平衡結構及原子標號[B3LYP/aug-cc-pVTZ]22
圖 3. 2 1-氟化萘離子基態的平衡結構及原子標號[B3LYP/aug-cc-pVTZ]
圖 3. 4 1-氟化萘三重態的平衡結構及原子標號[B3LYP/6-311++G(d,p)] 本研究採取泛函數 B3LYP 及 B3PW91 的方法,搭配 6-311++G(d, p)、 6-311++G(2d,p)、aug-cc-pVTZ 三種基底函數分別計算,共得到六個計算方式。 由於資料繁多,各基組間的計算結果差異不大,故在本章僅針對部分基組資料進 行說明,其餘資料列於附錄中。 由圖 3.1 可知 1-氟化萘分子的結構,是由 10 個碳原子組成 2 個苯環,外圍 接有 7 個氫原子和 1 個氟原子,氟原子是接在一個α-位置的碳上(本研究標號為 C11),所有的原子皆位於同一平面,對稱性隸屬於 Cs 點群,共有 19 個鍵長,30 個鍵角。
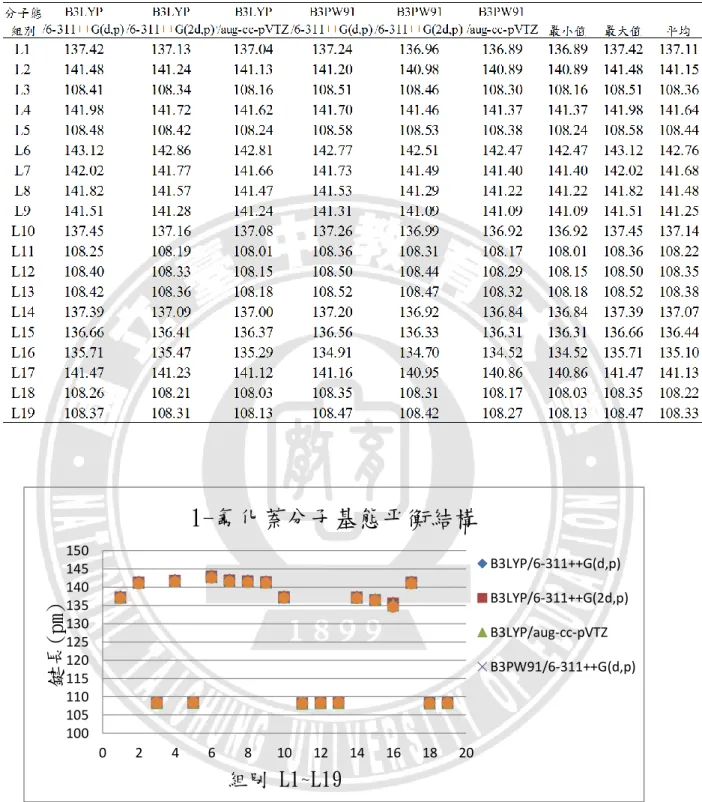
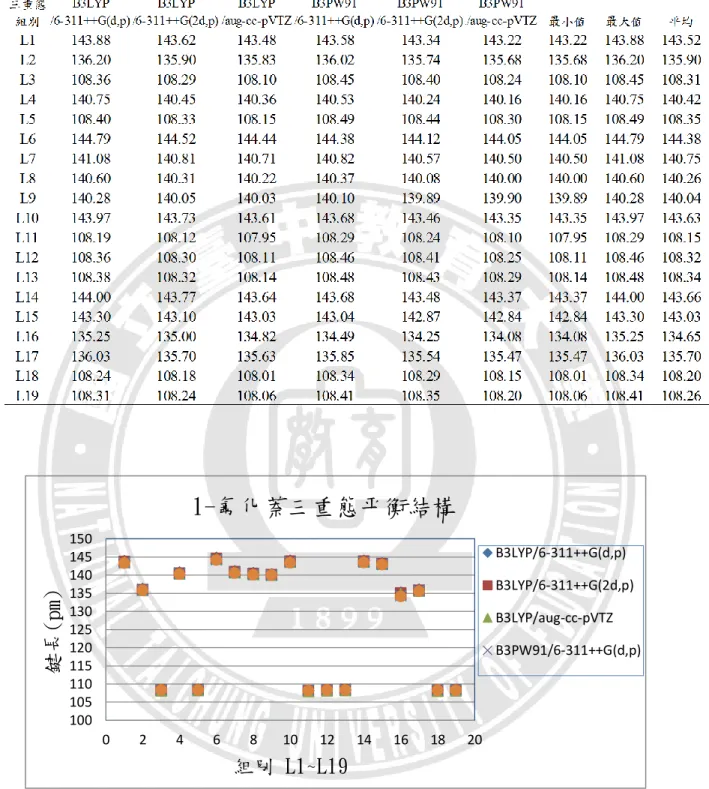
24 分析討論,資料如附錄表 A-3。在鍵長部分,L1:C1=C2、L10:C5=C6、L14: C10=C15 這三組雙鍵的鍵長落在 137.00~137.08 pm 之間,L15:C11=C14 雙鍵為 136.37 pm。L2:C1=C6、L4:C2=C3、L7:C3=C10、L8:C4=C5、L9:C4=C11、 L17:C14=C15這些共振雙鍵的鍵長落在 141.12~141.66 pm 之間,L6:C3=C4鍵長 稍長,為 142.81 pm。在碳碳之間因雙鍵的原子間作用力較大,鍵能比共振雙鍵 強,鍵長較共振雙鍵短,又 L15:C11=C14的這組雙鍵較其他三組雙鍵短,推論 為 F 接在 C11上之故。L16:C11-F18單鍵鍵長為 135.29 pm。7 組 C-H 鍵長落 在 108.01~108.24 pm。 以下將附錄表 A1~表 A6 中,1-氟化萘分子基態、離子基態、三重態的鍵長 資料依序整理成表 3.1、表 3.2、表 3.3,並繪製如圖 3.5、圖 3.6、圖 3.7,進行六 種基組計算結果的比較。以相鄰兩原子為一組,可分為 19 組鍵長,即為橫坐標 的 L1~L19,縱座標則表示該組別的鍵長長度。
表 3. 1 不同基組下 1-氟化萘分子基態的平衡鍵長 圖 3. 5 不同基組下 1-氟化萘分子基態的平衡鍵長比較圖 100 105 110 115 120 125 130 135 140 145 150 0 2 4 6 8 10 12 14 16 18 20
鍵長
(pm)
組別 L1~L19
1-氟化萘分子基態平衡結構
B3LYP/6-311++G(d,p) B3LYP/6-311++G(2d,p) B3LYP/aug-cc-pVTZ B3PW91/6-311++G(d,p)26 表 3. 2 不同基組下 1-氟化萘離子基態的平衡鍵長 圖 3. 6 不同基組下 1-氟化萘離子基態的平衡鍵長比較圖 100 105 110 115 120 125 130 135 140 145 150 0 2 4 6 8 10 12 14 16 18 20
鍵長
(pm)
組別 L1~L19
1-氟化萘離子基態平衡結構
B3LYP/6-311++G(d,p) B3LYP/6-311++G(2d,p) B3LYP/aug-cc-pVTZ B3PW91/6-311++G(d,p)表 3. 3 不同基組下 1-氟化萘三重態的平衡鍵長 圖 3. 7 不同基組下 1-氟化萘三重態的平衡鍵長比較圖 100 105 110 115 120 125 130 135 140 145 150 0 2 4 6 8 10 12 14 16 18 20
鍵長
(pm)
組別 L1~L19
1-氟化萘三重態平衡結構
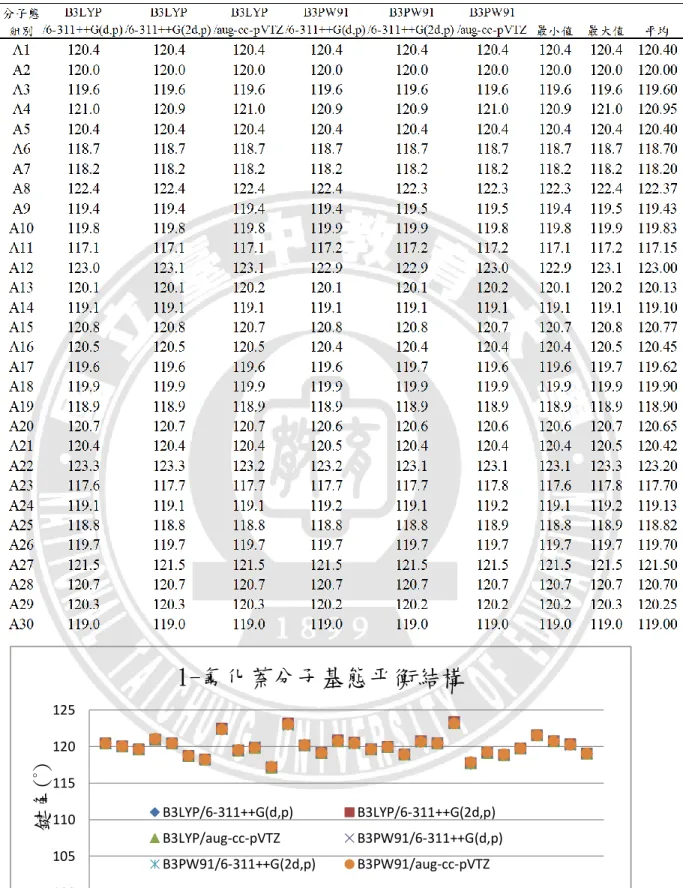
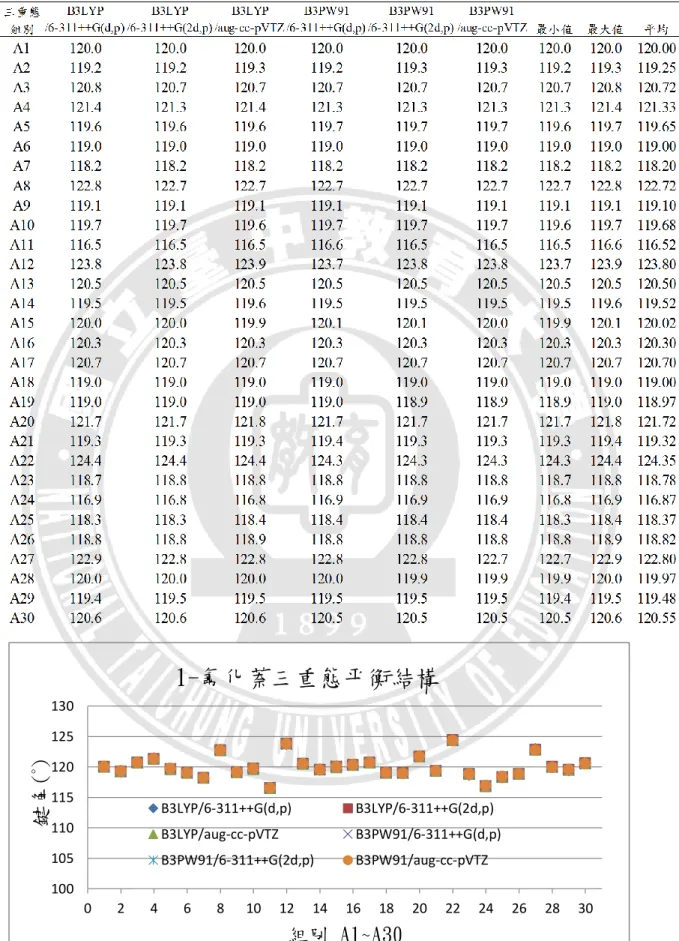
B3LYP/6-311++G(d,p) B3LYP/6-311++G(2d,p) B3LYP/aug-cc-pVTZ B3PW91/6-311++G(d,p)28 根據圖 3.5、圖 3.6、圖 3.7 所示,可以發現 1-氟化萘的分子、離子和三重態, 在平衡鍵長的比較圖上,同一種狀態使用不同基組計算的結果間並無顯著之差異。 平均來看,碳與碳之間三重態的鍵長最長,離子態次之,基態鍵長最小,依序是 141.03 pm、140.26 pm、139.90 pm;碳與氫之間則是基態的鍵長最長,離子態次 之,三重態鍵長最小,但差異甚小,依序是 108.33 pm、108.29 pm、108.28 pm, 這是由於電子是由π鍵所游離,不牽涉到 C-H 鍵的緣故。L16(C-F)在六種計算方 法的結果皆為分子態的鍵長最長,三重態次之,離子態最小,平均值分別為 135.10 pm、134.65 pm、131.37 pm,推測是因為 F 電負度大的關係。 在鍵角部分,以下將附錄表 A1~表 A6 中,1-氟化萘分子基態、離子基態、 三重態的鍵角資料依序整理成表 3.4、表 3.5、表 3.6,並繪製如圖 3.8、圖 3.9、 圖 3.10,進行六種基組計算結果的比較。橫坐標的 A1~A30 代表 30 組鍵角,縱 座標則表示該組別的鍵角角度。 六個基組所計算出來的鍵角沒有顯著差異,三種狀態中,最大鍵角都在 A22(∠C4C11C14),最小鍵角則都位於 A11(∠C3C4C11)。 以上對於 1-氟化萘的分子、離子和三重態的結構分析,以分子軌域的角度來 觀察平衡結構的變化,結構差異並不甚大。觀察 1-氟化萘三種狀態的最佳化平衡 結構,鍵長的部分都是 C─H 單鍵最短、接著是 C─F 單鍵、再來為 C=C 雙鍵, 而碳與碳的共振雙鍵最長。推測造成鍵長或鍵角變化的因素有:F 原子的強拉電 子現象;C=C 雙鍵的形成或打開;氫、碳、氟三種原子大小的立體空間效應。
表 3. 4 不同基組下 1-氟化萘分子基態的平衡鍵角 100 105 110 115 120 125 0 2 4 6 8 10 12 14 16 18 20 22 24 26 28 30
鍵角
(
°)
1-氟化萘分子基態平衡結構
B3LYP/6-311++G(d,p) B3LYP/6-311++G(2d,p) B3LYP/aug-cc-pVTZ B3PW91/6-311++G(d,p) B3PW91/6-311++G(2d,p) B3PW91/aug-cc-pVTZ30 表 3. 5 不同基組下 1-氟化萘離子基態的平衡鍵角 圖 3. 9 不同基組下 1-氟化萘離子基態的平衡鍵角比較圖 100 105 110 115 120 125 0 2 4 6 8 10 12 14 16 18 20 22 24 26 28 30
鍵角
(
°)
組別 A1~A30
1-氟化萘離子基態平衡結構
B3LYP/6-311++G(d,p) B3LYP/6-311++G(2d,p) B3LYP/aug-cc-pVTZ B3PW91/6-311++G(d,p) B3PW91/6-311++G(2d,p) B3PW91/aug-cc-pVTZ表 3. 6 不同基組下 1-氟化萘三重態的平衡鍵角 100 105 110 115 120 125 130 0 2 4 6 8 10 12 14 16 18 20 22 24 26 28 30
鍵角
(
°)
1-氟化萘三重態平衡結構
B3LYP/6-311++G(d,p) B3LYP/6-311++G(2d,p) B3LYP/aug-cc-pVTZ B3PW91/6-311++G(d,p) B3PW91/6-311++G(2d,p) B3PW91/aug-cc-pVTZ32
3.2 振動頻率及振動模式
1-氟化萘分子(C10H7F)是平面分子,屬於 Cs 點群,根據非線性分子的振 動自由度 3N-6(N 為原子數,N=18)的計算式,可知 1-氟化萘分子具有 48 種 振動模式。經由 Gaussian 09 套裝軟體計算後,所得到的 48 組振動頻率皆為正 值(附錄表 A-7 至 A18),可以證實理論計算後的平衡結構,是能量穩定的結構, 並非過渡態。 為了呈現各基組間振動頻率計算結果的差異,故將本研究的六種計算方法之 數據進行疊合比較,結果如圖 3.11、圖 3.12、圖 3.13 所示: 圖 3. 11 不同基組下 1-氟化萘分子基態的振動頻率比較圖 0 500 1000 1500 2000 2500 3000 3500 0 2 4 6 8 10 12 14 16 18 20 22 24 26 28 30 32 34 36 38 40 42 44 46 48 50振動頻率
(c
m
-1)
振動模式 ν
1~ν
481-氟化萘分子基態
B3LYP/6-311++G(d,p) B3LYP/6-311++G(2d,p) B3LYP/aug-cc-pVTZ B3PW91/6-311++G(d,p) B3PW91/6-311++G(2d,p) B3PW91/aug-cc-pVTZ圖 3. 12 不同基組下 1-氟化萘離子基態的振動頻率比較圖 圖 3. 13 不同基組下 1-氟化萘三重態的振動頻率比較圖 由圖 3.11、圖 3.12、圖 3.13 可見,1-氟化萘分子基態、離子基態、三重態, 個別使用六種計算基組所得振動頻率並無顯著差異。同時我們也將不同基組下分 子基態、離子基態、三重態的振動頻率數據整理成下表 3.7、表 3.8、表 3.9。在 0 500 1000 1500 2000 2500 3000 3500 0 2 4 6 8 10 12 14 16 18 20 22 24 26 28 30 32 34 36 38 40 42 44 46 48 50
振動頻率
(c
m
-1)
振動模式
ν
1~ν
481-氟化萘離子基態
B3LYP/6-311++G(d,p) B3LYP/6-311++G(2d,p) B3LYP/aug-cc-pVTZ B3PW91/6-311++G(d,p) B3PW91/6-311++G(2d,p) 0 500 1000 1500 2000 2500 3000 3500 0 2 4 6 8 10 12 14 16 18 20 22 24 26 28 30 32 34 36 38 40 42 44 46 48 50振動頻率
(c
m
-1)
振動模式 ν
1~ν
481-氟化萘三重態
B3LYP/6-311++G(d,p) B3LYP/6-311++G(2d,p) B3LYP/aug-cc-pVTZ34
值都很接近。而理論計算所花費的時間,不論是 B3LYP 或 B3PW91 的計算法, 都是 6-311++G(d,p)的基組耗時最短、6-311++G(2d,p)次之,而 aug-cc-pVTZ 所耗 費的時間最長,基於各基組間計算的結果差異不大,6-311++G(d,p)基組最具時間 上的優勢。
36
38
3.3 光電子光譜與磷光光譜
以下呈現 1-氟化萘的光電子光譜及磷光光譜的研究成果。在本研究使用的程 式中,光譜是以法蘭克康登因子正比於高度為 y 軸,相對能量為 x 軸,最後將光 譜數據利用 Origin Pro7 進行繪圖。本研究程式設定的半高寬(full-width at half maximum, FWHM)為 30 cm-1,但是文獻中的實驗光譜圖因年份較久,解析度通 常不是很高,因此必須調整理論模擬的半高寬。我們參考本研究室之前所作的相 關研究,調整出數個不同的半高寬,與實驗光譜進行比對,分別選出最合適的半 寬高。在光電子光譜部分,調整半高寬為 400 cm-1,以及使用原本的半高寬 30 cm-1, 再將兩種半高寬圖形重疊繪製;在磷光光譜部分,調整半高寬為 200 cm-1與實驗 光譜最相似。疊合時,我們將二者相對能量的數值對齊,調整光譜訊號的相對強 度,使二者位於
0
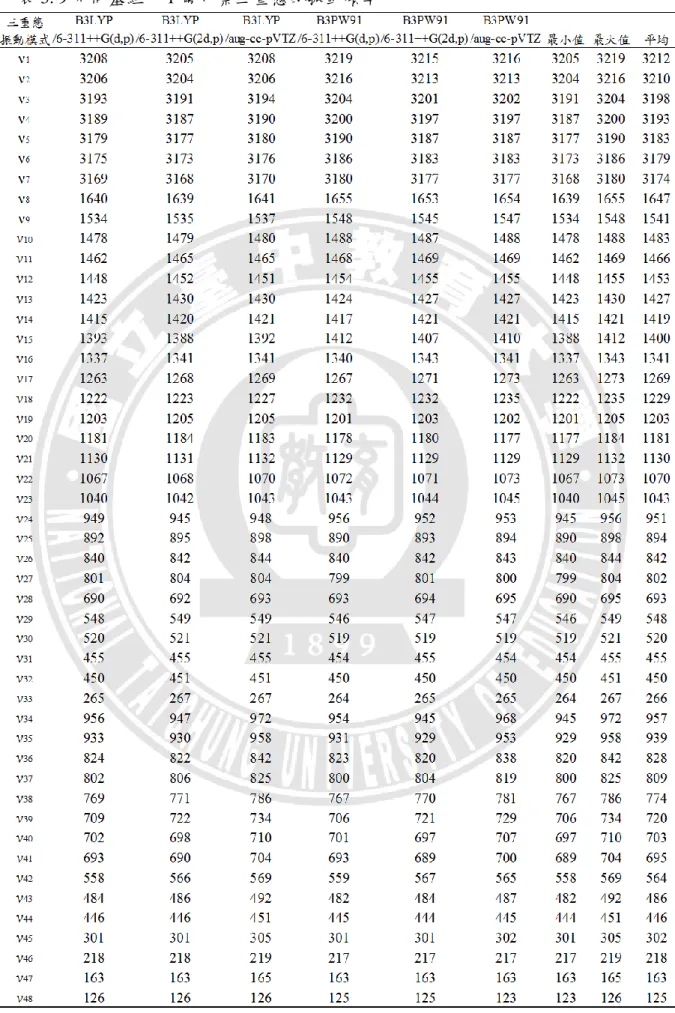
00的訊號強度一致,讓模擬光譜的圖形與實驗光譜圖盡量吻合, 以便和實驗光譜圖進行比較。 圖 3.14 至圖 3.19 為利用 B3LYP/6-311++G(d,p) 、B3LYP/6-311++G(2d,p) 、 B3LYP/aug-cc-pVTZ 、 B3PW91/6-311++G(d,p) 、 B3PW91/6-311++G(2d,p) 、 B3PW91/aug-cc-pVTZ 六種不同方法,所模擬出 1-氟化萘的光電子光譜圖。其中 黑色訊號為高解析度之半高寬 30 cm-1,而紅色訊號則是將半高寬調整為低解析 度 400 cm-1,以配合實驗光譜圖。我們仍可從不同的半高寬,看到躍遷譜帶所呈 現的訊號強弱關係。圖 3. 20、圖 3. 21 則是將六種基組所模擬的光電子光譜圖進 行疊合與比較,調整半高寬後之六張光譜圖的互相比較,呈現於圖 3.22、圖 3. 23。圖 3. 14 模擬的 1-氟化萘光電子光譜圖及標定(計算方法:B3LYP/6-311++G(d,p))
-1000 0 1000 2000 3000 4000 5000 6000
1510
171
0
The simulated photoelectron
spectroscopy of 1-fluoronaphthalene (B3LYP/6-311++G(d,p)) 101 0 1020 161 0 301 0 321 0 R e la ti ve I n te n si ty Relative Energy(cm-1) 00 0 10101610 -1000 0 1000 2000 3000 4000 5000 6000 151 0 171 0
The simulated photoelectron
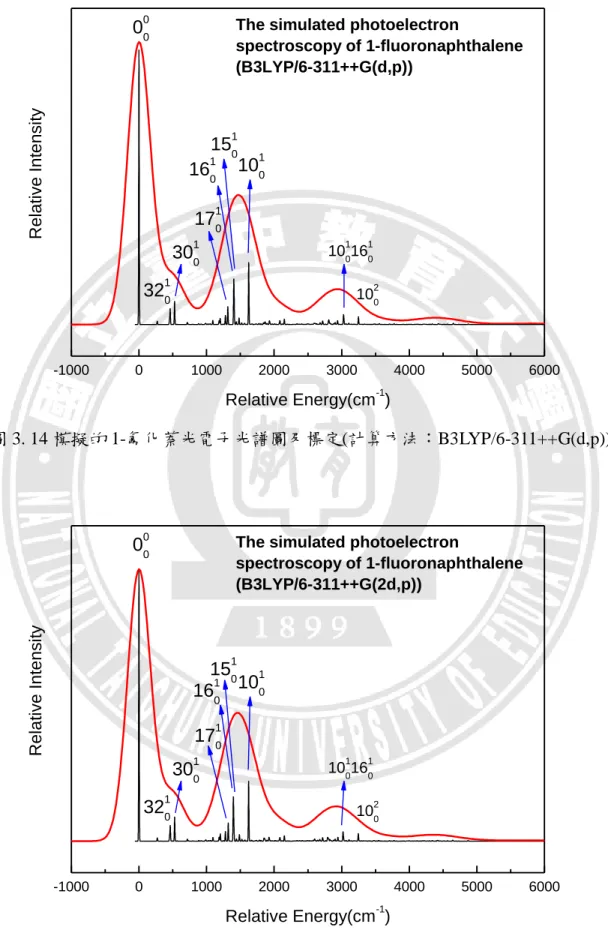
spectroscopy of 1-fluoronaphthalene (B3LYP/6-311++G(2d,p)) 1010 1020 161 0 301 0 321 0 R e la ti ve I n te n si ty Relative Energy(cm-1) 00 0 101 016 1 0
40 圖 3. 16 模擬的 1-氟化萘光電子光譜圖及標定(計算方法:B3LYP/aug-cc-pVTZ) 由圖 3.14 至圖 3.16 可看出光譜皆呈現三個明顯的躍遷譜帶,訊號最強的主 要是由原譜帶(
0
00)組成;其次由ν10、ν16、ν30、ν15、ν17、ν32 所組成。 而ν10 為 1623~1624 cm-1、ν16 為 1396~1402 cm-1、ν30 相對能量為 527~528 cm-1、ν15 為 1404~1408 cm-1、ν17 為 1315~1324 cm-1、ν32 為 462~463 cm-1 第三個明顯譜帶主要由ν10 與ν16 所組合而成還有1
0
20組成。以計算方法 B3LYP/aug-cc-pVTZ 為例,最大的 FCF 對應於能量在 1624 cm-1的躍遷,FCF 值 為 0.0963,標定為1
0
10;第二強的對應於能量在 1400 cm-1的躍遷,FCF 值為 0.0578, 標定為16
10;第三強的對應於 528 cm-1的躍遷,FCF 值為 0.0376,標定為 1 030
。 0 2000 4000 6000 151 0 1710The simulated photoelectron
spectroscopy of 1-fluoronaphthalene (B3LYP/aug-cc-pVTZ) 161 032 1 0 101 032 1 0 101 030 1 0 161 030 1 0 101 0 1020 161 0 301 0 321 0 R e la ti ve I n te n si ty Relative Energy(cm-1) 00 0 101 016 1 0
圖 3. 17 模擬的 1-氟化萘光電子光譜圖及標定(計算方法:B3PW91/6-311++G(d,p)) 圖 3. 18 模 擬 的 1- 氟 化 萘 光 電 子 光 譜 圖 及 標 定 ( 計 算 方 法 : -1000 0 1000 2000 3000 4000 5000 6000 1610 171 0
The simulated photoelectron
spectroscopy of 1-fluoronaphthalene (B3PW91/6-311++G(d,p)) 101 0 102 0 151 0 301 0 321 0 R e la ti ve I n te n si ty Relative Energy(cm-1) 00 0 10101510 0 2000 4000 6000 161 0 171 0
The simulated photoelectron
spectroscopy of 1-fluoronaphthalene (B3PW91/6-311++G(2d,p)) 101 0 102 0 151 0 301 0 321 0 R e la ti ve I n te n si ty Relative Energy(cm-1) 00 0 10101510
42 圖 3. 19 模擬的 1-氟化萘光電子光譜圖及標定(計算方法:B3PW91/aug-cc-pVTZ) 由圖 3.17 至圖 3.19 亦可看出光譜呈現三個明顯的躍遷譜帶,訊號最強的主 要是由原譜帶(
0
00)組成;其次由ν10、ν15、ν30、ν17、ν16、ν32 所組成。 而ν10 為 1637~1640 cm-1、ν15 為 1423~1429 cm-1、ν30 相對能量為 525~526 cm-1、ν17 為 1321~1328 cm-1、ν16 為 1416~1422 cm-1、ν32 為 462 cm-1。第 三明顯譜帶主要由ν10 與ν15 所組合而成還有10
20組成。 -1000 0 1000 2000 3000 4000 5000 6000 161 0 171 0The simulated photoelectron
spectroscopy of 1-fluoronaphthalene (B3PW91/aug-cc-pVTZ) 101 0 1020 151 0 301 0 321 0 R e la ti ve I n te n si ty Relative Energy(cm-1) 00 0 101 015 1 0
圖 3. 20 不同基組模擬的 1-氟化萘光電子光譜比較圖(B3LYP)半高寬 30 cm-1 0 1000 2000 3000 4000 5000
Re
lative
i
n
te
n
s
it
y
Relative energy (cm
-1)
B3LYP/6-311++G(d,p) B3LYP/6-311++G(2d,p) B3LYP/aug-cc-pVTZ Photoelectron spectroscopy of 1-fluoronaphthalene 0 1000 2000 3000 4000 5000Re
lative
i
n
te
n
s
it
y
B3PW91/6-311++G(d,p) B3PW91/6-311++G(2d,p) B3PW91/aug-cc-pVTZ Photoelectron spectroscopy of 1-fluoronaphthalene44 圖 3. 22 不同基組模擬的 1-氟化萘光電子光譜比較圖(B3LYP)半高寬 400 cm-1 圖 3. 23 不同基組模擬的 1-氟化萘光電子光譜比較圖(B3PW91)半高寬 400 cm-1 -2000 0 2000 4000 6000 800010000 B3LYP/6-311++G(2d,p) B3LYP/aug-cc-pVTZ
Re
lative
i
n
te
n
s
it
y
Relative energy (cm
-1)
Photoelectron spectroscopy of 1-fluoronaphthalene B3LYP/6-311++G(d,p) -2000 0 2000 4000 6000 800010000Re
lative
i
n
te
n
s
it
y
Relative energy (cm
-1)
B3PW91/6-311++G(d,p) B3PW91/6-311++G(2d,p) B3PW91/aug-cc-pVTZ Photoelectron spectroscopy of 1-fluoronaphthalene由圖 3. 20 至圖 3. 21 的對照比較可以發現,B3LYP 泛函數搭配的三種基組 的高解析度光譜之躍遷訊號量較 B3PW91 泛函數搭配的三種基組稍多,而 B3PW91 泛函數搭配的三種基組比 B3LYP 泛函數躍遷訊號強度強;在整體外觀 上,由圖 3. 22、圖 3. 23 可知六種計算方法的低解析度光譜皆呈現三個主要的譜 帶,在訊號強度的分布上也十分相似,不同基組所模擬出的光電子光譜,彼此之 間的差異不大。由於資料眾多,在此僅呈現 B3LYP/aug-cc-pVTZ 基組所模擬的 光譜圖與實驗光譜圖[7]的疊合比較,即為圖 3.24。
-4000 -2000
0
2000 4000 6000 8000 10000
1-fluoronaphthalene experiment B3LYP/aug-cc-pVTZR
el
at
ive
int
ensi
ty
46 從圖 3.24 中可發現,模擬的光電子光譜與實驗光譜相似,皆具有三個明顯 的躍遷譜帶,且相對能量位置相仿,惟第二和第三個譜帶相對位置高度比實驗值 略低,可見本研究室所研發的計算方法,用來模擬 1-氟化萘的光電子光譜,具有 良好的可靠性。 接下來呈現 1-氟化萘的磷光光譜研究成果。圖 3.25 至圖 3.30 為利用 B3LYP/6-311++G(d,p) 、 B3LYP/6-311++G(2d,p) 、 B3LYP/aug-cc-pVTZ 、
B3PW91/6-311++G(d,p) 、B3PW91/6-311++G(2d,p) 、B3PW91/aug-cc-pVTZ 六 種不同方法,所模擬出 1-氟化萘的磷光光譜圖。其中黑色訊號為高解析度之半高 寬 30 cm-1,而紅色訊號則是將半高寬調整為低解析度 200 cm-1,以配合實驗光譜 圖。我們仍可從不同的半高寬,看到躍遷譜帶所呈現的訊號強弱關係。圖 3. 31、 圖 3. 32 則是將六種方法所模擬的磷光光譜圖進行疊合與比較;而調整半高寬為 200 cm-1後之六張光譜圖的互相比較,呈現於圖 3.33、圖 3. 34。
圖 3. 25 模擬的 1-氟化萘磷光光譜圖及標定(計算方法:B3LYP/6-311++G(d,p)) 0 2000 4000 6000 8000 10000 12000 8103210 15103210 82 015 1 0 81 015 1 0 301 0 152 0
The simulated phosphorescence spectroscopy of 1-fluoronaphthalene (B3LYP/6-311++G(d,p)) 820
15
108
10 321 0R
e
la
ti
ve
I
n
te
n
si
ty
Relative Energy(cm
-1)
0
00 81 015 2 0 0 2000 4000 6000 8000 10000 12000 8103210 15103210 82 015 1 0 81 015 1 0 301 0 152 0The simulated phosphorescence spectroscopy of 1-fluoronaphthalene (B3LYP/6-311++G(2d,p)) 820
15
108
10 321 0R
e
la
ti
ve
I
n
te
n
si
ty
0
0 0 81 015 2 048 圖 3. 27 模擬的 1-氟化萘磷光光譜圖及標定(計算方法:B3LYP/aug-cc-pVTZ) 圖 3. 28 模擬的 1-氟化萘磷光光譜圖及標定(計算方法:B3PW91/6-311++G(d,p)) 0 2000 4000 6000 8000 10000 12000 151 030 1 0 8103010 8201520 8103210 15103210 8201510 81 015 1 0 3010 1520
The simulated phosphorescence spectroscopy of 1-fluoronaphthalene (B3LYP/aug-cc-pVTZ) 820
15
1 08
1 0 321 0R
e
la
ti
ve
I
n
te
n
si
ty
Relative Energy(cm
-1)
0
0 0 81 015 2 0 0 2000 4000 6000 8000 10000 12000 8101010 1010 81 032 1 0 141 032 1 0 8201410 8101410 301 0 1420The simulated phosphorescence spectroscopy of 1-fluoronaphthalene (B3PW91/6-311++G(d,p)) 82 0
14
108
1 0 321 0R
e
la
ti
ve
I
n
te
n
si
ty
Relative Energy(cm
-1)
0
0 0 81 014 2 0圖 3. 29 模擬的 1-氟化萘磷光光譜圖及標定(計算方法:B3PW91/6-311++G(2d,p)) 0 2000 4000 6000 8000 10000 12000 81 032 1 0 151 032 1 0 82 015 1 0 81 015 1 0 301 0 152 0
The simulated phosphorescence spectroscopy of 1-fluoronaphthalene (B3PW91/6-311++G(2d,p)) 82 0
15
108
1 0 321 0R
e
la
ti
ve
I
n
te
n
si
ty
Relative Energy(cm
-1)
0
00 81 015 2 0 0 2000 4000 6000 8000 10000 12000 8103110 15103110 8201510 8101510 3010 1520The simulated phosphorescence spectroscopy of 1-fluoronaphthalene (B3PW91/aug-cc-pVTZ) 820
15
108
10 311 0R
e
la
ti
ve
I
n
te
n
si
ty
0
0 0 81 015 2 050 由圖 3.25 至圖 3.30 可看出原譜帶的訊號強度最強,代表三重態的結構與基 態的結構差異性不大。光譜皆呈現幾個明顯的躍遷譜帶,訊號最強的主要是由原 譜帶(
0
00)組成;其次由ν8、ν15 及ν8 與ν15 的組合所組成。 B3LYP 泛函數搭配的三種基組ν8 為 1610~1612 cm-1、ν15 為 1395~1401 cm-1、ν8 與ν15 的組合相對能量為 3006~3014 cm-1。第三個明顯譜帶主要由 2 08
、 2 015
、ν32、ν30 所組合而成。以計算方法 B3LYP/aug-cc-pVTZ 為例,最大的 FCF 對應於能量為 1399 cm-1,FCF 為 0.0422,標定為15
10;次強的是ν8,為 1612 cm-1,FCF 為 0.0417,標定為8
10;第三強的對應於 3011 cm-1的躍遷,FCF 值為 0.0370,標定為8
1015
10。 與其他五種方法較不同的是,B3PW91/6-311++G(d,p)方法中訊號最強的主要 是由原譜帶(0
00)組成;其次由ν8、ν14 及ν8 與ν14 的組合所組成。但ν14 與其他五種方法的ν15 相對能量相近。B3LYP 泛函數搭配的三種基組ν8 為 1625~1629 cm-1、 ν 15(6-311++G(d,p) 以 ν 14) 為 1417~1422 cm-1、 ν 8 與 ν 15(6-311++G(d,p)以ν8 與ν14)的組合相對能量為 3042~3051 cm-1。以計算方法 B3PW91/aug-cc-pVTZ 為例,最大的 FCF 對應於能量為 1627 cm-1,FCF 為 0.0428, 標定為8
10;次強的是ν15,為 1419 cm-1,FCF 為 0.0426,標定為 1 015
;第三強 的對應於 3046 cm-1的躍遷,FCF 值為 0.0369,標定為 1 08
15
10。普遍來看,B3PW91 泛函數搭配的三種基組比 B3LYP 泛函數躍遷相對能量來得高。由圖 3. 31、圖 3. 32 的對照比較可以發現,B3PW91/6-311++G(d,p)模擬出的 躍遷訊號強度比其他計算方法稍弱些,B3PW91 泛函數搭配的三種基組比 B3LYP 泛函數躍遷訊號強度強;在整體外觀上,由圖 3. 33、圖 3. 34 可知六種計算方法 的低解析度光譜皆呈現三個主要的躍遷譜帶,在訊號強度的分布上也十分相似, 不同基組所模擬出的光電子光譜,彼此之間的差異不大。由於資料眾多,在此僅 呈現 B3LYP/aug-cc-pVTZ 基組所模擬的光譜圖與實驗光譜圖[4]的疊合比較,即 為圖 3.35。
52 圖 3. 31 不同基組模擬的 1-氟化萘磷光光譜比較圖(B3LYP) 半高寬 30 cm-1 圖 3. 32 不同基組模擬的 1-氟化萘磷光光譜比較圖(B3PW91) 半高寬 30 cm-1 0 4000 8000 12000
Re
lative
i
n
te
n
s
it
y
Relative energy (cm
-1)
B3LYP/6-311++G(d,p) B3LYP/6-311++G(2d,p) B3LYP/aug-cc-pVTZ Phosphorescence spectroscopy of 1-fluoronaphthalene 0 4000 8000 12000Re
lative
i
n
te
n
s
it
y
Relative energy (cm
-1)
B3PW91/6-311++G(d,p) B3PW91/6-311++G(2d,p) B3PW91/aug-cc-pVTZ Phosphorescence spectroscopy of 1-fluoronaphthalene圖 3. 33 不同基組模擬的 1-氟化萘磷光光譜比較圖(B3LYP) 半高寬 200 cm-1 0 4000 8000 12000 Phosphorescence spectroscopy of 1-fluoronaphthalene B3LYP/ 6-311++G(d,p) B3LYP/ 6-311++G(2d,p) B3LYP/ aug-cc-pVTZ
Re
lative
i
n
te
n
s
it
y
Relative energy (cm
-1)
0 4000 8000 12000 Phosphorescence spectroscopy of 1-fluoronaphthalene B3PW91/ 6-311++G(d,p) B3PW91/ 6-311++G(2d,p) B3PW91/ aug-cc-pVTZRe
lative
i
n
te
n
s
it
y
54 圖 3. 35 模擬的 1-氟化萘磷光光譜與實驗光譜[4]比較圖,半高寬 200 cm-1 在圖 3. 35 中,J. Ferguson 等人於 1954 年實驗所得的磷光光譜[4],訊號以藍 色線條呈現,其光譜所測得的能量轉換成相對能量後,從 0 記錄到-3280 cm-1。 模擬與實驗光譜圖相較的結果,在 0〜 -1000 cm-1的訊號強度分布上,可以觀察 到峰線相當類似,其中有 2 個最明顯的躍遷譜帶,轉換成相對能量後,分別在 0 cm-1、-465 cm-1、-1399 cm-1、-1612 cm-1等位置,顯示了本研究使用的計算方 法,不僅可用於光電子光譜,對於磷光光譜也具有相當可靠性。
0
-3000
-6000
-9000
-12000
phosphorescence spectroscopy of 1-fluoronaphthalene experiment B3LYP/aug-cc-pVTZR
el
at
ive
int
ensi
ty
Relative energy (cm
-1
)
3.4 游離能與激發能
我們也使用完備基組(Complete basis set, CBS)極限的計算方法(CBS-4M and CBS-QB3),計算 1-氟化萘的絕熱游離能和三重態的激發能。 以 CBS-4M 及 CBS-QB3 這兩種方法所計算出來的絕熱游離能及激發能,兩 者計算的結果均略大於實驗值。使用 CBS-4M 方法計算 1-氟化萘的絕熱游離能 與實驗值僅相差 507 cm-1,相對差值為 0.77%。使用 CBS-QB3 方法計算 1-氟化 萘的激發能與實驗值僅相差 432 cm-1,相對差值為 2.05%。 表 3. 10 1-氟化萘不同方法所計算之絕熱游離能、激發能與實驗值[7][ 4]對照表 能量 狀態 計算方法 energy (at 0 K) AIE (hartree) AIE (cm-1) Experiment (cm-1) 分子態 CBS-4M -484.406299 CBS-QB3 -484.319079 離子態 (游離能) CBS-4M -484.102392 0.303907 66701 66194 cm-1 [7] CBS-QB3 -484.015005 0.304074 66737 三重態 (激發能) CBS-4M -484.294070 0.112229 24632 21062 cm-1[4] CBS-QB3 -484.221147 0.097932 21494 游離能實驗值資料取自參考文獻[7],激發能實驗值資料取自參考文獻[4] 綜合以上所有的計算結果,本研究對於 1-氟化萘分子的平衡結構、振動頻率、 絕熱游離能和激發能的理論計算,與實驗值的差距都不大,而模擬出的光電子光 譜圖與磷光光譜圖,相較實驗光譜圖都有極高的符合程度,顯示本研究所使用的
第四章 結論
本研究使用密度泛函數 B3LYP 及 B3PW91 兩種計算法,搭配 6-311++G(d,p)、 6-311++G(2d,p)與 aug-cc-pVTZ 等基組,共計六組基組的組合進行幾何優 化 (geometry optimization)計算,得到 1-氟化萘之分子基態、離子基態與三重態的最 佳化平衡結構、振動模式、振動頻率及能量值等數據。 藉由本實驗室所開發的方法計算 FCF,繪製模擬的光電子光譜及磷光光譜圖 後,與實驗光譜圖進行比對,可以發現模擬光譜圖與實驗光譜圖在外形上非常吻 合,同時兩者的相對能量、訊號強度以及主要躍遷譜帶之形狀、數量,都相當類 似。 我們最後以 CBS-4M 及 CBS-QB3 的計算方法,計算 1-氟化萘的絕熱游離能 和激發能。以 CBS-4M 及 CBS-QB3 這兩種方法所計算出來的絕熱游離能,兩者 計算的結果均略大於實驗值。使用 CBS-4M 方法計算 1-氟化萘的絕熱游離能與 實驗值僅相差 507 cm-1,計算的絕熱游離能與實驗值最小的相對差異為 0.77%。 使用 CBS-QB3 方法計算 1-氟化萘的激發能與實驗值僅相差 432 cm-1,相對差值 為 2.05%。參考文獻
[1] I. N. Levine, Quantum Chemistry, 5th edition, Prentice-Hall, New Jersey, (1991).
[2] F. P. Bymaster, E. E. Beedle, J. Findlay, P. T. Gallagher, J. H. Krushinski, S. Mitchell, D. W. Robertson, D. C. Thompson, L. Wallace, D. T. Wong, Duloxetine (Cymbalta™), a dual inhibitor of serotonin and norepinephrine reuptake, Bioorganic & Medicinal Chemistry Letters, 13 (2003) 4477.
[3] D T. Wong, F. P. Bymaster, D. A. Mayle, L. R. Reid, J. H. Krushinski, D. W. Robertson, LY248686, a new inhibitor of serotonin and norepinephrine uptake, Neuropsychopharmacology. 8 (1993) 23.
[4] J. Ferguson, T Iredale, and J. A. Taylor, The phosphorescene spectra of naphthalene and some simple derivatives, J. Chem. Soc. (1954) 3160.
[5] T. Iredale, J. W. White, The electronic states of monohalogen substituted naphthalenes, Trans. Faraday Soc. 56 (1960) 1719.
[6] H. Gattermann, M. Stockburger, Spectroscopic studies on napththalene in the vapor phase. III. Phosphorescence and intersystem crossing yields of single vibronic levels, J. Chem. Phys. 63 (1975) 4541.
[7] L. Kiasinc, B. Kovač, and H. Güsten, Photoelectron spectra of acenes. electronic structure and substituent effects, Pure & Appl. Chem. 55 (1983) 289.
[8] T. Iliescu, I. Milea, P. M. Abdolrahman, The absorption, fluorescence and phosphorescence spectra of α and β-F, Cl, Br-naphthalenes in crystalline matrixes at 77 K, J. Mol. Struct. 115 (1984) 209.
60
[10] S. Y. Tzeng, J. Y. Wu, S. Zhang, W. B. Tzeng, Resonant two-photon mass-analyzed threshold ionization spectroscopy of 1-fluoronaphthalene and 2-fluoronaphthalene, J. Mol. Spectrosc. 281 (2012) 40.
[11] E. U. Condon, A theory of intensity distribution in band systems, Phys. Rev. 28 (1926) 1182.
[12] E. U. Condon, Nuclear motions associated with electron transitions in diatomic molecules, Phys. Rev. 32 (1928) 858.
[13] J. Franck, E. G. Elementary processes of photochemical reactions, Trans. Faraday Soc. 21 (1926) 536.
[14] E. Hutchisson, Band spectra intensities for symmetrical diatomic molecules, Phys. Rev. 36 (1930) 410.
[15] C. Manneback, Computation of the intensities of vibrational spectra of electronic bands in diatomic molecules, Physica 17 (1951) 1001.
[16] M. Wagner, Exakte Berechnung von Franck-Condon-Integralen, Z. Naturforschg. Teil A 14 (1959) 81.
[17] F. Ansbacher, A Note on the Overlap Integral of two Harmonic Oscillator Wave Functions, Z. Naturforschg. Teil A 14 (1959) 889.
[18] S. Koide, Ü ber die Berechnung von Franck-Condon-Integralen, Z. Naturforschg. Teil A 15 (1960) 123.
[19] T. E. Sharp, H. M. Rosenstock, Franck-Condon Factors for Polyatomic Molecules, J. Chem. Phys. 41 (1964) 3453.
[20] R. Islampour, M. Dehestani, S. H. Lin, A New Expression for Multidimensional Franck-Condon Integrals, J. Mol. Spectrosc., 194 (1999) 179.
[21] A. M. Mebel, M. Hayashi, K. K. Liang, S. H. Lin, Ab Initio Calculations of Vibronic Spectra and Dynamics for Small Polyatomic Molecules: Role of Duschinsky Effect, J. Phys. Chem. A 103 (1999) 10674.
![圖 3. 2 1-氟化萘離子基態的平衡結構及原子標號[B3LYP/aug-cc-pVTZ]](https://thumb-ap.123doks.com/thumbv2/9libinfo/7443849.109458/30.893.144.795.92.1065/圖321氟化萘離子基態的平衡結構及原子標號B3LYPaugccpVTZ.webp)
![圖 3. 4 1-氟化萘三重態的平衡結構及原子標號[B3LYP/6-311++G(d,p)] 本研究採取泛函數 B3LYP 及 B3PW91 的方法,搭配 6-311++G(d, p)、 6-311++G(2d,p)、aug-cc-pVTZ 三種基底函數分別計算,共得到六個計算方式。 由於資料繁多,各基組間的計算結果差異不大,故在本章僅針對部分基組資料進 行說明,其餘資料列於附錄中。 由圖 3.1 可知 1-氟化萘分子的結構,是由 10 個碳原子組成 2 個苯環,外圍 接有 7 個氫原子和 1 個](https://thumb-ap.123doks.com/thumbv2/9libinfo/7443849.109458/31.893.138.774.129.906/三重態及原子三種基底函數分別計算共得六個計算由於料列於附錄中.webp)