國 立 台 中 教 育 大 學 科 學 應 用 與 推 廣 學 系 碩 士 論 文
指導教授:張嘉麟 博士
二氟化硫碳光電子光譜的理論研究
Theoretical study of the photoelectron
spectra of thiocarbonyl fluoride
研究生:蔡世傑 撰
謝 誌
擔任教職多年之後,能重新再當一個學生,是一件既幸運、但卻又很辛苦的 事情。在台中教育大學二年半的在職進修,總算苦盡甘來,完成了這一本研究著 作。一路走來,要感謝的人很多,在每一門課程裡,每位老師都給予我不同的見 解與啟發。當然,影響最深、最要感謝的,莫過於我的指導教授-張嘉麟老師了。 除了「傳道、授業、解惑」外,他總是在學生遭遇學習瓶頸時,提供最適切的催 化劑,幫助學生度過難關,讓我在身為人師、人父、人夫、人子等分身乏術的情 況下,還能順利的去體驗身為學生的酸甜苦辣。無論如何,都要感謝張嘉麟老師 不辭辛勞的教導。 另外,我要感謝陸維作教授與葉聰文教授,撥冗閱讀論文,給我許多寶貴的 意見。同時也要感謝我的研究夥伴-瑋莉、祥印,讓我在學習的路途上,不但不 孤單,更從中獲得學習樂趣,並在研究過程中,互助合作、彼此關懷。 最後,要感謝的是家人。雖然我的學業總是造成他們許多的不便和犧牲,但 我仍然得到他們最多的關懷與支持,讓我即使疲累無助時,仍感溫馨。僅以此論 文完成的喜悅,分享我所感謝的每一個人。摘 要
本研究利用量子計算的方法,研究二氟化硫碳(F2CS)分子與離子的結構和光 電子光譜。我們利用密度泛函理論之B3LYP泛函數,以6-311+G(d)、6-311+G(2d)、 6-311+G(3d)、6-311+G(3df)、aug-cc-pVDZ、aug-cc-pVTZ等六種基組,計算出F2CS 分子及離子基態的平衡結構與振動頻率,並計算分子電離成正離子的法蘭克-康登 因子,進而模擬出F2CS的光電子光譜。結果發現分子電離成正離子時,CS鍵會伸 長,兩個CF鍵都會縮短,FCF鍵角則變大,且F2CS + 與F2CS一樣都屬於C2v點群; 但F2CS - 則變成非平面分子,屬於Cs點群。F2CS - 尚未有相關的實驗報告,本研究 預測此離子具有穩定的平衡結構。F2CS的模擬光電子光譜和實驗結果相當類似。 另外,我們也以CCSD和CCSD(T)方法,計算出F2CS的絕熱游離能和電子親和力, 並與實驗值做比對,結果發現絕熱游離能與實驗值的差距不到0.1eV;而電子親 和力預測值約在0.6~0.7eV之間。 關鍵字 關鍵字關鍵字 關鍵字:二氟化硫碳、光電子光譜、密度泛函理論、法蘭克-康登因子、絕熱游離 能、電子親和力Abstract
The study investigated the structures and photoelectron spectra of thiocarbonyl fluoride (F2CS) in the neutral and ionic ground states by using quantum calculation methods. The equilibrium geometries and harmonic vibrational frequencies of F2CS were calculated by means of density functional theory with the B3LYP functional and six basis sets of 6-311+G(d), 6-311+G(2d), 6-311+G(3d), 6-311+G(3df), aug-cc-pVDZ and aug-cc-pVTZ. Franck-Condon factors for the ionization of the neutral molecules were also computed in order to simulate the photoelectron spectra. It was found that the CS bond lengthens, the CF bond shortens, and the CFC angle opens when the molecule get ionized. However, both of F2CS
+
and F2CS belong to the same C2v point group. Conversely, F2CS
-
is a nonplanar molecule of the Cs point group. This study predicts that F2CS
-
has a stable equilibrium structure, awaiting for experimental verification. The simulated photoelectron spectra of F2CS are in accord with the experiment. Meanwhile, the adiabatic ionization energy and the electron affinity of F2CS were also calculated using the CCSD and CCSD(T) methods. The adiabatic ionization energy is in agreement with the experiment within 0.1eV. The electron affinity of F2CS is predicted to be about 0.6~0.7eV.
Keywords: thiocarbonyl fluoride; photoelectron spectra; density functional theory; Frank-Condon factors; adiabatic ionization energy; electron affinity
目 次
第一章 第一章第一章 第一章 緒論緒論緒論………...1 緒論 1.1 研究動機………...………1 1.2 文獻探討………...………3 1.2.1 原子物理的發展………….………3 1.2.2 分子結構的理論計算………...5 1.2.3 密度泛函理論的主要貢獻……….5 1.2.4 密度泛函理論的相關發展……….7 1.2.5 法蘭克-康登因子的相關發展………8 1.2.6 二氟化硫碳的相關研究……….9 1.3 論文架構………...…..………11 第二章 第二章第二章 第二章 研究方法研究方法研究方法………..………...13 研究方法 2.1 密度泛函數理論……….14 2.1.1 Hohenberg-Kohn 理論………..14 2.1.2 Kohn-Sham 理論……….…..15 2.1.3 泛函數………..18 2.1.4 基底函數組………...19 2.2 分子與離子的計算……….212.3 法蘭克-康登因子………23 2.4 光譜模擬……….26 第三章 第三章第三章 第三章 研究結果研究結果研究結果………..………...27 研究結果 3.1 平衡結構……….27 3.2 振動模式及振動頻率……….30 3.3 轉動常數、游離能及電子親和力……….36 3.4 法蘭克-康登因子………40 3.5 光電子光譜……….45 第四章 第四章第四章 第四章 結論結論結論………..………...49 結論 參考文獻………..………...53 附錄一:二氟化硫碳(F2CS)分子振動模式之計算………57 附錄二:執行 Gaussian 03 程式的一般做法………..59 附錄三:在 B3LYP/6-311+G(3df)計算下各振動模式的法蘭克-康登因子……….63
表 目 次
表 2.1 Pople 系列之基底函數組………21 表 3.1.1 二氟化硫碳分子及正離子在不同計算基組下的平衡結構……….28 表 3.1.2 二氟化硫碳負離子在不同計算基組下的平衡結構………..28 表 3.1.3 二氟化硫碳分子轉變成正離子時的平衡結構變化………..30 表 3.1.4 二氟化硫碳分子轉變成負離子時的平衡結構變化………30 表 3.2.1 二氟化硫碳分子的正規振動模式………31 表 3.2.2 二氟化硫碳正離子的正規振動模式………32 表 3.2.3 二氟化硫碳負離子的正規振動模式………33 表 3.2.4 二氟化硫碳分子在不同計算基組下的振動頻率………35 表 3.2.5 二氟化硫碳正離子在不同計算基組下的振動頻率………35 表 3.2.6 二氟化硫碳負離子在不同計算基組下的振動頻率………36 表 3.3.1 在不同計算基組下二氟化硫碳分子態的轉動常數………37 表 3.3.2 在不同計算基組下二氟化硫碳正離子態的轉動常數………38 表 3.3.3 在不同計算基組下二氟化硫碳負離子態的轉動常數………38 表 3.3.4 在不同計算基組下二氟化硫碳的絕熱游離能………39 表 3.3.5 在不同計算基組下二氟化硫碳的電子親和力………40 表 3.4.1 二氟化硫碳正離子相對於分子態之平衡結構位移∆Q 因子………..41表 3.4.2 二氟化硫碳正離子相對於分子態之 S 因子………41 表 3.4.3 在 aug-cc-pVTZ 計算基組下所計算出的 Duschinsky matrix….………42 表 3.4.4 二氟化硫碳正離子相對於分子之法蘭克-康登因子………...44
圖 目 次
圖 3.1 二氟化硫碳分子、正離子平衡結構圖示……….27
圖 3.2 二氟化硫碳負離子平衡結構圖示………...27
圖 3.5.1 二氟化硫碳分子電離成正離子基態的模擬光譜……….46
第一章 緒論
本研究的目的在 於 運 用 密 度 泛 函 理 論 (density functional theory), 對 二氟 化硫碳(F2CS)分子與 離 子 進 行 量 子 計 算 的 理 論 研 究 , 並 計 算 分子電離成正 離子的法蘭克-康登因子(Frank-Condon factor, FCF), 進而模擬出二氟化硫碳的光 電子光譜。本章共分為三節,第一節介紹研究動機和研究背景,第二節進行文獻 探討,第三節則將論文架構做簡要描述。 1.1 研究動機研究動機研究動機 研究動機 從量子力學基礎上發展出來的理論物理、量子化學及其相關的計算,讓我 們開闢了通往微觀世界的另一個途徑。以往我們只能在實驗室,透過實驗去了解 化學反應的過程與結果,或透過儀器設備檢測,追蹤化學反應的動態。現在藉由 理論化學計算,讓我們有能力去了解發生於瞬息之間的化學反應,或預測某些激 發態與過渡態的分子幾何構形,還有可能了解活性位上的電荷轉移。 隨著計算技術的飛速發展,計算機已進入各個化學實驗室。也因此刺激了 量子化學計算以及理論化學方法的快速發展。量子化學計算已不再是理論化學家 的專利,它已成為實驗化學、生物領域、藥物設計,材料研究等方面的有利工具。 雖然各種光譜工具為反應物及生成物的結構提供了多種參數,但這些來自於實驗
手段的研究結果,為反應機制所提供的信息仍稍嫌不足。因此,準確的理論計算 有助於了解分子的光譜學及光解動力學。例如在 Chang(張嘉麟教授)和 Chen 於 2002 年對氯乙烯的研究中[1],計算出氯乙烯五個低能量的單重激發態,並且 藉由法蘭克-康登因子的計算模擬其吸收光譜,其結果與實驗相當吻合。後來 Chang 進一步計算氯乙烯激發態的位能面,發現氯原子可經由兩個途徑光解:一 是在激發態經由(π, σ*)排斥位能面分解;另一個途徑是結構扭曲至(π, π*)態的位 能阱,再內轉化(internal conversion)至基態而分解。此一發現與氯乙烯的光解實驗 相符,因而使我們更了解氯乙烯分子的光解機制。 在模擬分子的吸收光譜時,需要計算法蘭克-康登因子,亦即,兩電子態振 動波函數重疊積分的平方。近年來,學術界對法蘭克-康登因子的計算頗有進展, 在 2005 年,Chang 推導出一個新的公式[2],可利用理論計算所得到的分子結構 和振動頻率,計算兩電子態間的法蘭克-康登因子,並應用至溴乙烯吸收光譜的 研究上[3]。結果發現π* ← π、3s ← π、和 3pz ← π具有較強的躍遷,模擬光譜有 助於解析實驗光譜的細微結構。 是故,化學已由只做實驗不計算,演變為先實驗再計算,也必將逐步演變為 先計算再實驗,亦即對於未知的分子,可先透過理論計算預測其性質,以做為實 驗的指引。理論計算的結果與實驗結果相比較,若有很高的一致性,勢必解決許 多實驗中無法解釋的問題。何況,近年來的計算技術已經使傳統化學發生深刻的
變化,隨著物理化學理論、計算方法、大型計算軟體、高性能計算機的發展,目 前各種需求的計算化學課題正在全世界蓬勃發展,由此而產生的計算化學也在化 學研究中佔有越來越重要的地位。例如:紅外、拉曼光譜實驗,雖可幫助我們測 定分子的結構及其周圍環境,但為了得到這些有用的信息,必須測定分子各種振 動所對應的各個吸收峰。以實驗觀點來講,那是件困難的事,因為即使是簡單分 子的光譜,都包含基頻、合頻、泛頻,並且有許多吸收峰彼此間的頻率差異極微 小,在目前實驗器材所能解析的限制下,很難分辨其差異。為解決這方面的困難, 我們可以用計算來模擬分子的振動,所得的頻率能幫助光譜實驗進行指認。於 是,本研究將繼氯乙烯和溴乙烯的研究之後,針對二氟化硫碳進行量子計算,並 模擬出二氟化硫碳的光電子光譜。 1.2 文獻探討文獻探討文獻探討 文獻探討 1.2.1 原子物理的發展 原子物理的傳統研究範疇,在於探討原子、分子、與其離子的結構,以及研 究電子、原子、分子與電磁場,彼此間交互作用所產生的物理現象。原子中電子 的分佈,及其與原子核之間的電磁作用,決定了所有化學與生命合成物的性質。 而這些電子與原子核之間的交互作用,也是液體及固體中親和力的來源。事實 上,它們也決定大部分日常生活中所見到的物理及化學現象。例如日光燈的螢
光、水的沸騰、電視影像管中電子束的軌跡、及各種化學反應等。因此,原子物 理的研究在自然科學領域中,具極重要的地位。新研究工具的發明,及原子物理 與應用科學的密切關係,促使原子物理的研究領域,在近年來急速地拓展。原子 物理在自然科學領域中具有舉足輕重的地位的另一個主要因素,是它與其他許多 基本及應用科學的密切關係。直接與我們日常生活有關的近代物理和化學,其主 要的理論革新在於量子力學的建立。 理論上,分子的電子結構皆可由薛丁格方程式表示。對於電子數少的系統, 藉由求解薛丁格方程式也確實能計算出非常精確的結果。一般而言,兩個粒子間 的相關效應,已能相當精確的計算出[4]。例如,1927 年 Heitler 和 London 應用量 子力學分析氫原子軌域結構[5],1932 年 Mulliken 的分子軌域概念,能精確計算 氫原子的鍵長和游離能[6]。但在多電子體系中,電子與電子間以及電子與原子核 間的複雜交互作用,在當時一直無法獲得具體的解釋。於是,多體系統動力理論 的確立,一直是物理學中最具挑戰性的問題之一。早期為大家所熟知及應用的近 似法有 Thomas-Fermi model [7]、Hartree-Fock theory [8]、微擾理論(perturbation theory) [9]等,更進一步發展出來的理論有密度泛函理論(density-functional theory) [10]、耦合法(coupling method) [11]等。而在實驗上,探討原子結構與特性的兩個 主要研究方向,是原子光譜及原子碰撞研究。原子光譜是對原子與光子交互作用
可以得到原子結構的許多資料,如能階、游離能等。 1.2.2 分子結構的理論計算 有關分子結構的理論計算,方法與技巧有很多,其主要目的,一方面可以得 到與實驗值相符合的理論計算值,另一方面,在缺乏實驗數據的情況下,可以用 理論計算值做為預測值,輔助相關研究的進行。而早期發展可分為兩類方法[12]: 一類將波函數以多組態基底展開,直接求解;另一類是微擾方法,逐步得到可靠 的答案。兩類方法其實經常交互使用,相輔相成。其中,第一類較偏重技術面, 原則上,我們假定原子的波函數可以由完備基底展開,只是由於它是無窮級數, 我們必須想辦法選取適當的基底做有限項的展開,再針對系統考慮如何計算。而 微擾方法,則比較強調系統化,如何逐步建立穩固的理論基礎,再往更準確的數 值推算[13]。事實上,為朝向精確計算結果的目標,這兩類方法也必須相互折衷, 相互擷取優點,漸漸地同時被應用,沒有明顯區隔。另一方面,針對某一些特定 系統,若有其他適合的理論或近似方式,物理學家也常用微擾理論的計算結果檢 驗該理論或方法的優劣。 1.2.3 密度泛函理論的主要貢獻 1920 年代,量子力學的巨大成就,使得偉大的物理學者 Dirac 宣稱化學的研 究發展已經走到了盡頭[14],因其內容已全部被包含在量子力學方程式中。 然而,事實並不全然如此,方程式雖然可被寫下來,但由於太過複雜,往往
無法解出,也因此,對於化學的了解並不能提供太多的內涵。時至 1998 年,諾 貝爾化學獎由 Walter Kohn 及 John Pople 兩人共同獲得,而 Kohn 的主要貢獻就在 於密度泛函理論(Density Functional Theory, DFT)的發展[15]。與量子力學的波函 數(wave function)計算相比較,Kohn 認為密度泛函理論對於多電子系統的研究, 有兩方面的貢獻[16]: 第一,是對於基本物理的了解。當我們遵循量子力學的原則,由波函數起始, 多電子系統的波函數必須用 Slater 行列式來描述,當電子數量增加時,此行列式 常變成非常之大,無法解出來。反觀密度泛函理論,其求解的是電子密度,這是 一個三維空間座標的函數,讓我們得以用電子密度為觀點去了解多電子系統的內 涵。 第二,是實用性方面。傳統波函數的處理方式,目前,較準確的方法只能到 數十個原子左右,再多就因計算量太大而不可行,例如 DNA、有機分子等大分 子,往往難以被傳統波函數的處理方式解決,而 DFT 甚至可以處理上千個原子 的系統[17]。
Kohn 的老師 J. H. Van Vleck 在很早期的論文中,也曾經討論到多體波函數的 問題。Kohn 斷言,當電子數目大約超過一千以後,多電子波函數是一個不合理 的科學概念[18]。所謂不合理,其意義是指波函數無法被準確的計算出,並正確 的記錄下來。如果再將電子交互作用也完全計入,一千個電子數目的估計則仍差
距頗大。 1.2.4 密度泛函理論的相關發展 早在 1927 年,Thomas 和 Fermi 就已各自提出以電子密度來表示能量的理論, 但這個早期的理論有點粗糙,更嚴重的是用在化學方面,甚至出現分子不會形成 鍵結的計算結果[19]。Thomas-Fermi 理論忽略了電子彼此間交互作用的交換能, 限制了計算的準確性,因此未受重視,也阻礙了 Thomas-Fermi 理論的推展。到 了 1960 年代,從電子密度出發的概念給了 Kohn 新的想法,1964 年 Hohenberg 與 Kohn 發表了後來極著名的 Hohenberg-Kohn 理論[20]。這理論進一步成為密度 泛理論的雛形,因為它提出多電子系統中的各項性質,可一併視為該系統的基態 電子密度的函數。 而提到密度泛函理論,也必須先提及 Hartree-Fock 理論[8],因為密度泛函理 論是建立在 Hartree-Fock 理論的基礎上。主要觀點在於 Hartree-Fock 理論假設多 電子體系符合波恩-歐本海默近似(Born-Oppenheimer approximation)[21],亦即, 一個多電子體系,電子的運動和能量與原子核的運動和能量,是可以相互分離 的。於是他們將多電子體系中的電子,視為在其他電子構成的平均勢場中運動的 粒子[22,23],並將多電子體系複雜的薛丁格方程式,簡化為單電子波函數的乘積 Ψ(1,2,3,…,n)=Ψ(1)Ψ(2)Ψ(3)…Ψ(n),再利用變分法求得波函數的解。
到 的 結 果 也 比 從 Thomas-Fermi 理 論 所 得 到 的 結 果 更 準 確 ; 而 另 一 方 面 , Hohenberg-Kohn 變分法求得的能量卻比從 Hartree 方程式求得的更準確,也獲得 更準確的電子軌道方程式,於是他們發表了 Kohn-Sham 方程式[24]。相關理論的 進一步內容陳述,則在第二章研究方法中另做說明。 1.2.5 法蘭克-康登因子的相關發展 法蘭克-康登因子的理論發展,最早由Condon根據Frank之推論,設定電子在 激發過程中,原子核並未移動,進而推導出法蘭克-康登因子的雛型[25,26]。接著, Hutchisson提出Franck-Condon factors可藉由電子躍遷後,分子結構所產生的位移 與扭曲計算推得[27]。在計算上,Manneback使用數學上的遞迴公式來計算法蘭克 -康登因子的積分[28],Wagner則推導出一種計算法蘭克-康登因子的積分公式 [29]。而Ansbacher也推導出一個求解法蘭克-康登因子積分的計算公式,這個方法 在解決一維諧振子時非常有效[30]。事實上,早期法蘭克-康登因子的計算,由於 牽涉艱深的理論應用與數學計算,在推廣與應用經常受到侷限。並且,在計算振 動波函數重疊積分的過程中,不同振動模式間的座標又會產生交互作用,因此, 也使得計算多模式混合的法蘭克-康登因子變的非常困難。為處理這方面的困境, Sharp和Rosenstock以Hutchisson計算一維諧振子的方法為基礎,推導出了一個數 學展開式,其展開式的係數可用於計算多重模式混合的法蘭克-康登因子[31]。 Kikuchi等人則以Sharp和Rosenstock的方法為基礎,推導出了一個較為簡單的多維
度法蘭克-康登因子計算公式[32]。另外,為便於電腦計算,Dotorov等人推出一個 重疊積分的遞迴方程式[33,34],只要準確計算出原帶的數值,便可利用遞迴公式 推算出其它振動態的法蘭克-康登因子,所以被廣為採用,但其缺點是計算時間較 長,或是需要大量的記憶體。後續有許多研究者則發展出不同的演算法以改善遞 迴法的計算效率。然而,使用遞迴法無法避免的問題是,當原帶的計算值偏差較 大時,此誤差會累積影響後續的計算,使誤差進一步擴大。本研究所採用的計算 公式則是由Chang所推導出[2],在 計 算 上 並 不 採 用 遞 迴 函 數 的 趨 近 , 計 算 過 程 巧 妙 運 用 了 厄 米 多 項 式 的 展 開 式 , 簡 化 了 繁 複 的 計 算 過 程 , 並 縮 短 了 計 算 時 間。雖然其假設經過簡化,但當分子在兩個電子態之間的結構差異 不大時,所計算出的法蘭克-康登因子,在應用上與實驗值的差距非常小,同時也 解決了複雜的計算過程,使得應用更為簡便,因此,對於分析複雜的多原子分子 (polyatomic molecules)的光譜,是一個很好的工具。藉由法蘭克-康登因子的計算, 本研究才能將所得的數據轉化為模擬光譜。有關計算公式及方法的敘述,則在第 二章研究方法中詳細說明。 1.2.6 二氟化硫碳的相關研究 二氟化硫碳(F2CS),在Middleton等人[35]多年的研究下,已知它是tetrafluoro 1,3 dithietane (SCF2SCF2)的主要熱裂解產物。早期由於該分子的製備困難,或能 取得的純度不高,因此針對該分子所從事之相關研究並不多。直到1972年,Kroto
和Suffolk[36]首度採用其特有的熱裂解流體技術,從純度大於95%的SCF2SCF2取 得二氟化硫碳分子,並偵測出二氟化硫碳的粗略光電子光譜,從光譜中發現有四 條較強的頻帶,由第一頻帶得知其絕熱游離能為10.45±0.01eV,並能標定其游離 出的電子是來自於S原子的3p軌域。而Downs[37]則以紅外線光譜測得二氟化硫碳 中性分子六個振動模式的振動頻率,分別為ν1=1368cm-1、ν2=787cm-1、ν3= 526cm-1、ν4=622cm-1、ν5=1189cm-1、ν6=417cm-1。隨後並由Careless等人[38] 藉由微波光譜測量出該分子的許多化學特性及數據,所測得二氟化硫碳分子的基 態平衡結構參數為r(CF)=1.315ű0.01、r(CS)=1.589ű0.01、∠FCF=107.1°±1.0, 而基態轉動常數則為A=11892.6±0.5、B=5133.03±0.03、C=3580.32±0.03MHz。 Kroto和Suffolk[36]也將其研究中所得到的二氟化硫碳光電子光譜與先前所做的 硫甲醛(H2CS)光電子光譜做結果比較,他們認為,藉助於氟同位素的特性,能更 有效的呈現出光電子光譜的標定證據,並有助於實驗的處理。此類的替代比較, Robin等人[39]也同樣在其研究中,指出更多二氟化硫碳及硫甲醛的化學特性資 料。而Clouthier等人[40]更在其研究中指出,比起其他類似結構的碳化物,二氟 化硫碳在某些方面,能在光物理和光化學過程中提供更有效的特性探測。由此可 知針對二氟化硫碳分子及其光電子光譜研究的重要性。 由於二氟化硫碳的實驗及理論研究並不多,光電子光譜的相關研究也很少, 因此,本研究進行二氟化硫碳分子與正、負離子的量子計算,探究其光電子光譜
的結構,並針對其平衡結構、振動模式及振動頻率、轉動常數、游離能等,與實 驗值做可能的比較與分析。 1.3 論文架構論文架構論文架構 論文架構 本論文共分四章,除了本章緒論外,第二章介紹本研究所採用的研究方法, 包括密度泛函理論、法蘭克-康登因子以及光譜模擬的說明。第三章呈現二氟化硫 碳分子、離子的計算結果,並將理論計算出的模擬光譜與實驗光譜作比對。第四 章為結論及對後續研究的建議。
第二章 研究方法
本研究利用 J. Pople (1998 諾貝爾化學獎得主)等人所發展之 Gaussian 03 套裝 軟體[41],計算二氟化硫碳分子與離子的平衡結構和正規振動模式。Gaussian 03 套裝軟體已廣泛地被化學家、化學工程師、生物化學家、物理學家等所使用。從 最基本的量子力學原理開始,Gaussian 03 可以預測分子的能量、結構、振動頻率, 和由計算得來的許多分子特性。Gaussian 03 在很多情況下可以用來作為解讀分子 和化學反應的工具;包含穩定性、難以藉由實驗觀察的混合物、出現時間很短的 中間產物、和過渡時期的分子結構。 為計算出二氟化硫碳分子與離子的平衡結構和正規振動模式,本研究使用密 度泛函數理論(density functional theory)的 B3LYP 泛函數,基組(basis set)為 6-311+G(d) 、 6-311+G(2d) 、 6-311+G(3d) 、 6-311+G(3df) 、 aug-cc-pVDZ 、 aug-cc-pVTZ。雖然二氟化硫碳分子的基態為平面分子,隸屬於 C2v點群,但我們 假設其離子態的對稱性為未知數,因此計算採用完全幾何優選 (full geometrical optimization) 策略,亦即捨棄平面對稱性,計算的初始結構設定為 C1點群。 本章共分為四節,第一節介紹密度泛函理論以及選用的基組,第二節介紹分 子和離子的幾何優選以及正規振動模式的計算方法和流程,第三節介紹法蘭克-康登因子的假設、推導和計算方法。第四節說明光譜模擬的方法。2.1 密度泛函數理論密度泛函數理論密度泛函數理論 密度泛函數理論 傳統量子力學的方法,對處理單電子系統的問題,理論上可以得到相當準確 的結果。但對於多電子系統而言,因為電子與電子間,存在著許多複雜的交互作 用,因此,使用薛丁格方程式去求解多電子系統的波函數,是件相當困難的事 [42,43]。然而,許多物理性質的探討卻都離不開多電子系統的範圍。密度泛函理 論使得分子基態的電子能量、波函數及其它性質,可以由只包含三種變數之電子 機率密度(probability density)決定,故能提高量子計算的效率[44]。密度泛函理 論與從始計算的不同處,就在於傳統的從始計算,對於一個系統,只要知道其所 含的原子核及電子數,就可以藉由薛丁格方程式而知道這個系統的性質。但薛丁 格方程式具有3N個變數(N為電子數),因此,方程式會變得相當複雜。而密度 泛函理論中的電子密度,只需有三個變數,且與系統大小無關,問題變得單純化 許多,所需計算時間也相對較少。另外,對於分子的平衡結構和振動頻率,應用 密度泛函理論的計算結果通常與實驗值相當接近[45]。因此,對於多電子系統, 密度泛函理論可說是解決其波函數具複雜交互作用的好方法。 2.1.1 Hohenberg-Kohn 理論 1964 年,Hohenberg 和 Kohn 說明了密度泛函理論中的兩個重要結論[46]:首 先,在多電子體系中,電子基態的能量可以用電子密度函數 ρ(r)的泛函表示。其
次,運用變分法,計算出以電子密度函數 ρ(r)為變數的能量泛函 EGS[ρ(r)]之最小 值,亦即求其最低能量狀態,便能得到基態能量 EGS。其原理簡述如下: 我們將一個粒子的總能量,以不含時(time-independent)薛丁格方程式表示為
( )
r E( )
r Hψ = ψ , 以 漢 彌 爾 頓 算 子 (Hamiltonian operator) 表 示 為 ψ ψ ψ V E m∇ + = − 2 2 h , 其 中 2 2 2 2 2 2 2 z y x ∂ ∂ + ∂ ∂ + ∂ ∂ = ∇ 。 雙 電 子 系 統 則 可 以 表 示 為(
1 2) (
1 2)
(
1 2)
2 2 2 1 , , , 2 2m ψ m ψ V r r ψ r r = Eψ r r + ∇ − ∇ − h h ,式中V(
r1, r2)
是位能函數。為 解決上述問題,Hohenber-Kohn 指出,可將基態的總能 EGS[Ψ(r1,r2,r3,……,rN)]寫成 電荷密度函數的泛函 EGS[ρ(r)],這使得計算過程的自由度得以降低。其次,若代 入的數值不是基態的電荷密度函數 ρGS(r),則總能量 EGS 可表示為 EGS[ρ(r)]≥ EGS[ρGS(r)]。從 Hohenber-Kohn 理論可以知道,若要計算出最低的基態能量,必 須藉由變分法,將不同的 ρ(r)代入 EGS[ρ(r)],直到求得最小值,進而獲得基態能 量。該理論提出,在多電子系統中,所有的特性可一併視為系統基態電子密度函 數 ρ(r)的函數,也是現今密度泛函理論的雛形。 2.1.2 Kohn-Sham 理論 1965 年,Kohn 和 Sham 提供另一種新的計算模式[47],此方法利用具備等效 位勢(effective potential)的獨立電子系統來取代原來的多體問題,而其中有關電子 間之多體效應,則以交換相關(exchange-correlation)泛函[48,49]來描述。其理論推 導如下:以ρ(r)為變數的多電子系統的基態總能泛函EGS[ρ(r)]可表示為
EGS[ρ(r)]=Tm[ρ(r)]+Eee[ρ(r)]+Eext[ρ(r)] (1)
其中 Tm[ρ(r)]為多電子動能,可表示為
[ ]
(
)
(
, ,..)
2 ,.. , Tm 1 2 2 r1 r2 m r r iψ ψ ρ = − h∑
∇ , (2) 其密度泛函的形式未知。 Eee[ρ(r)]是電子與電子間的交互作用能,可表示為[ ]
(
, ,..)
(
1, 2,..)
2 2 1 r r r r e r r E ij i j ee ρ ψ∑
ψ − = (3) 其密度泛函的形式也是未知,Eext[ρ(r)]是外界施加的位勢,對此粒子密度分佈所 獲得的能量,表示為[ ]
=∫
V( ) ( )
r r d r Eext ext 3 ρ ρ (4) 其中Vext( )
r 是已知的函數,可隨著不同的問題帶入不同的函數。 Kohn-Sham 方法從式(2)、(3)中將電子獨立運動的總動能 TS[ρ(r)],與電子間 的庫倫位能 EH[ρ(r)]分離出來。剩下的部分則視為電子間的交換相關能 Exc[ρ(r)], 所以動能 EGS[ρ(r)]可重新表示成EGS[ρ(r)]=TS[ρ(r)]+EH[ρ(r)]+Exc[ρ(r)]+Eext[ρ(r)] (5)
如此,除了交換相關能 Exc[ρ(r)]之外,所有的泛函項皆有明確的公式可表示,其
[ ]
=−∑∫
( )
∇( )
i i i s r r d r m T * 2 3 2 2 ψ ψ ρ h (6) 而古典靜電分佈的庫倫位能EH[ρ(r)]可表示為[ ]
∫
( ) ( )
=∫
( ) ( )
− = d r V r r d r r r r r EH 3 ' H 3 ' ' ρ ρ ρ ρ (7) 最後,Kohn-Sham 方法中,雖然無法求得交換相關能 Exc[ρ(r)]的精確值,但可定 義趨近的位能εXC[
ρ( )
r]
,由局部密度近似法[50]求得近似解,其公式可表示成[ ]
[
( )
r]
( )
r d r EXC XC 3 ρ ρ ε ρ =∫
(8) 其中針對交換相關能再以變分法取得近似,所以可以表示成( )
[
r]
XCLDA[
( )
r]
XC ρ ε ρ ε = (9) 這代表著,原本需要知道整個ρ(r)方位分佈,才知道空間中各點的εXC大小,而現 在只要給定位置r0代入ρ(r),便可以得到該位置的ρ0,也就可以得到該位置的εXCLDA 值。所以εXC大小只跟該位置的電荷密度大小有關。 最後我們在式(5)總能泛函中,藉由變分原理求得各泛函數的導數,並將各個 波函數在滿足歸一化條件下,將 Kohn-sham 方程式表示成 i i i xc ext H r V r V r E V m ψ = ψ + + + ∇ − ( ) ( ) ( ) 2 2 2 h (10) 而計算過程則是利用迭代法,以初始電荷密度ρ(r),得到有效位能Vext(r), 再代入Kohn-Sham方程式求得各能階及對應的軌域,而計算出新的電荷密度。若 新的電荷密度與初始電荷密度不相同,則重新計算一個新的電荷密度,重複這樣consistent field)計算[51]。 2.1.3 泛函數(Functional) 在密度泛函數理論模型中,計算方法的名稱是以交換相關泛函數的廣義公式 代替。也就是說,密度泛函數理論名稱來源,是由交換泛函數及相關泛函數的名 稱組合而成。其公式如下: XC
E (B3LYP)=A*EXSlater+(1-A)* HF X E +B* Becke X DE + VWN C E
+C* LeeYang Parr X DE , , 其 中 , Slater X E 是 Slater 之 均 勻 密 度 (local-density) 交 換 泛 函 數 , HF X E 是 Hartree-Fock之 交換 泛函數 , DEXBecke為交換泛函數之修正項, VWN C E 是在均勻
(uniform) 電 子 雲 之 相 關 泛 函 數 , 而 DEXLeeYang Parr
, , 則 對 相 關 泛 函 數 做 不 均 勻 (non-local, gradient)修正,因此,公式已同時做了均勻及梯度之考量。而為適用於 像二氟化硫碳這樣的小分子或離子之計算特性,我們採用B3LYP的參數[52],A = 0.80、B = 0.72、C = 0.81,去計算較準確的游離能、電子親和力等。 其次,所謂的B3LYP,是指使用Becke之三參數交換泛函數[53]與LYP相關泛 函數[54,55]做計算,其中LYP是描述由Lee、Yang、Parr三人所提出的相關泛函數。 而Becke之三參數交換泛函數是Becke利用Hatree-Fork的exchange function與密度 泛函數理論的exchange-correlation-energy function進行混合所得的新函數方法。其 計算所得到的修正能量為HF exchange energy與DFT energy混合後的結果,所在計
算上比HF方法更能夠準確的計算出能量值,也因此B3LYP為本研究採用的交換相 關泛函數。 2.1.4 基底函數組(basis set) 在進行密度泛函理論計算時,需要選擇一基底函數組(basis set,簡稱基組)用 來表達分子軌域之波函數。基組是描述一個系統中每個電子可能存在的軌域,用 數學式的方式來描述,可進一步結合近似全部的電子波函數以進行理論計算。基 組必需符合每個電子不同的邊界條件,亦即每個電子只能出現在空間中的特定區 域。較大的基組群對電子的空間所在限制條件較少,更能符合量子理論中電子在 空間中任何位置皆可能出現的假設,因此,較大的基組群能得到更近似分子軌域 性質的計算結果,但是相對的所需計算時間也會較長。 通常基組是利用高斯函數的線性組合(linear combination)來描述分子軌域,基 組是由一群基底函數組合而成,而基底函數則是由許多初始高斯函數(primitive Gaussin)組合而成。使用適當的基底函數做計算,通常會在所需時間與所需結果 精確度取一折衷點,這對於分子軌域的理論計算是很重要的。本研究以Pople和 Dunning兩人所提的基底函數組,分別以四個及二個基組做計算,並比較研究結 果。 為能適用於二氟化硫碳分子與離子,在Pople等人[56]為代表所提的基底函數 組中,本研究選用6-311+G(d)、6-311+G(2d)、6-311+G(3d)、6-311+G(3df)四基組。
這些型態的基底函數組,其形式及要件整理如表2.1(見下頁)。 為求得更精確的計算結果,可加入擴散函數(diffuse function)[57]來涵蓋距離 原子核較遠處的電子。以本研究中使用的 6-311+G(3d)為例,「+」號表示加入擴 散函數,擴散函數與標準的價層軌域函數的大小相比而言,為一個擁有 s 軌域型 態或 p 軌域型態的放大函數。因此,加入擴散函數之後,軌域會擁有更大的空間, 可以供給電子佔據。 另外,在分子結構的計算中,為使每個原子可調的位置更具彈性,可在基組 中加入極化函數(polarization functions)[58]。極化性基底函數的特色,是在對每個 原子軌域的描述中,加入較其在基態所需要,或含有較高角動量的軌域型態,使 整個基組更能接近真正的分子軌域性質。以 6-311+G(3d)為例,「d」表示加入極 化函數, 亦即重原子中加入更高階的角動量函數,而 3d 則表示對重原子加入三 組 d 形式的極化函數。
表 2.1 Pople 系列之基底函數組 Pople 系列 例如:6-311++G(d,p)、6-311+G(2d,p)、6-311+G(3d) 6 內殼層的每個原子軌域用一個基底函數來表示,此基底函數由 6 個初始 函數線性組合而成 311 價殼層的三個基底函數組合,每一基底函數分別由 3、1、1 個初始函數 所構成 一個+ 重原子加入擴散函數 二個+ 重原子加入擴散函數、氫原子加入擴散函數 (2)d (兩個)重原子加入極化函數 (2)p (兩個)氫原子加入極化函數 以 Dunning 等 人 [59-62] 為 代 表 所 提 的 基 底 函 數 組 中 , 本 研 究 選 用 aug-cc-pVDZ、aug-cc-pVTZ。這兩個基底函數組已刪除了與Pople系列重複的函 數,並做了數學轉換以便增加計算效率,並且這兩個基底函數組也已經包含極化 函數。而在cc-pV*Z基底函數關鍵字之前,加上aug-前置字串,表示增加擴散函數。 也就是說,除了極化函數,還加上擴散函數。 2.2 分子與離子的計算分子與離子的計算分子與離子的計算 分子與離子的計算 本研究選用Gaussian 03套裝軟體中含電子相關之DFT/B3LYP方法並配合 6-311+G(d)、6-311+G(2d)、6-311+G(3d)、6-311+G(3df)、aug-cc-pVDZ、aug-cc-pVTZ
六種 基組 進行 計算 。首 先進 行二 氟化 硫 碳分 子與 離子 的幾 何優 選(geometry oprimization),進行幾何優選的過程中,分子的幾何形狀會持續調整,直到達成 位能曲面上的穩定點為止,而幾何優選可採用的策略分為完全幾何優選(full oprimization)及部分幾何優選(partial optimization)計算。完全幾何優選是對所有指 定的變數進行優選計算以找出能量最低的結構;部分幾何優選則只針對其中的一 些變數進行幾何優選。 本研究採用完全幾何優選的策略進行計算。在鍵長、鍵角的初值方面,由於 輸入數值越接近實驗值或該分子的能量最低點,計算越容易達到收斂,因此我們 採用實驗值作為初始輸入數值。根據幾何優選的計算結果,我們可以找到位能曲 面 上 的 穩 定 點 , 但 是 這 些 穩 定 點 除 了 可 能 是 區 域 中 的 最 低 能 量 點 (local minimum),也可能是一個鞍點(saddle point),因此我們根據幾何優選結果,計算 出二氟化硫碳的振動正規模式(vibrational normal modes)頻率,以頻率值作為判斷 依據,若頻率為正值,則代表此種分子結構為位曲面上的極小值,也就是分子的 一個平衡結構;若頻率值為一虛數(imaginary value),則代表這個幾何結構為位能 曲面上的一個鞍點。
本研究中游離能計算,是利用耦合簇(coupled cluster)理論[63,64]來計算絕熱 游離能(adiabatic ionization energy)。耦合簇理論是進行 Hartree-Fock 方法計算後, 接著對多電子相關能(electron correlation)進行的一種高精確計算方法,常用在中
小型分子。耦合簇理論是 Kümmel 等人在 1980 年提出[65],本來用在核子物理光 譜,而經過修正後,變成用在分子和原子對電子相關能的計算。本研究為求得更 精確的游離能,採用 CCSD 和 CCSD(T)兩種方法,CCSD 是指單重激態能量和雙 重激態能量的耦合簇集計算;而 CCSD(T)是除了單重激態能量和雙重激態能量的 耦合簇集計算,還加入微擾理論(perturbation theory)的非迭代式三重激態能量計算 [66]。本研究經 Gaussian 03 程式,計算出二氟化硫碳分子和離子的 CCSD 和 CCSD(T)能量。將離子的能量減去分子能量,即可獲得絕熱游離能。 2.3 法蘭克法蘭克法蘭克-法蘭克---康登因子康登因子康登因子康登因子 從量子力學的觀念中,物體的動量、角動量、能量等,皆可由物體的波函數 Ψ求得,並且,該物體出現的機率正比於波函數Ψ的絕對值平方Ψ2。法蘭克-康 登因子代表的是,位於分子中某一能階的電子,躍遷至不同能階時,其電子振動 躍遷之發生機率,亦即,法蘭克-康登因子是將分子在兩個不同能階的振動波函數 做重疊積分再取平方,
<
|
′
>=
∫
∞∗
;
=
|
<
|
′
>
|
2 ∞ − ′d
x
FCF
v
v
v
v
ψ
vψ
v 。為了將理 論計算的結果模擬成光譜圖,需要計算兩電子態之間的法蘭克-康登因子,因為法 蘭克-康登因子正比於兩電子態間振動躍遷的機率,故可預測或解釋光譜強度的分 布。本研究採用Chang的方法[2]計算法蘭克-康登因子,其公式考慮了位能面的位 移和扭曲,並做了以下兩個假設:(1) 分子態和離子態的位能面都是諧振子(harmonic oscillator)。 (2) 激發態與基態的振動模式有一對一的對應關係,忽略振動模式之間的混 合(mode-mixing),亦即忽略杜辛斯基效應(Duschinsky effect)。 基於以上兩個假設,可以推導出每個振動模式由分子態 v 到離子態 v′的法蘭 克-康登因子如下: 首先,諧振子的波函數可表示為 2 2 1
)
(
x v v vN
H
x
e
αα
ψ
=
− (1) 其中 2 1 ! 2 =π
α
v N v v 為歸一化常數(normalization constant)( )
x Hv 為厄米多項式(Hermite polynomial),例如( )
x H0 =1( )
x
H
1 =2 x( )
x
H
2 =4 x 2 -2( )
x
H
3 =8 x 3 -12x …等。 於是法蘭克-康登因子為( )( )
( )
( )
(
) (
)
( )
2 0 ' 0 ' ' ' ' ' ' ' 22
2
'
!
'
!
2
'
′
=
∑∑
= = − − + − ν ν ν ν ν ν ν να
α
ν
ν
ν
ν
k k k k k k k k sK
I
b
H
b
H
Ae
其中'
'
2
α
α
αα
+
=
A
(2)α
α
αα
′
+
∆
=
2)
(
'
Q
S
(3)( )
kk
!
(
!
k
)!
−
=
ν
ν
ν (4)'
'
α
α
α
α
+
∆
−
=
Q
b
(5)'
'
'
α
α
α
α
+
∆
=
Q
b
(6)h
ω
α
=
(7)h
'
'
ω
α
=
(8) ω為分子態之振動頻率,ω′為離子態之振動頻率,h為蒲朗克常數。(
)
kK
K
I
'
!
)!
1
2
(
)
(
α
α
+
−
=
(9) 其中 K = k + k′, K 限定為偶數,(2K - 1)!!為雙階乘(double factorial),其值為 1×3×5×…× (2K – 1)。∆Q 代表幾何結構的改變量,計算公式為R
M
L
Q
=
e∆
∆
1/2 (10) 其中 Le是離子態的位移矩陣,M 1/2 是原子質量開根號的對角矩陣,∆R 是離子態 與分子基態間直角座標的差。 綜言之,我們利用量子計算所得到的平衡結構(R)、振動頻率(ω)、和位移矩 陣(L),以公式(10)計算∆Q,以公式(7)和(8)得到α和α′,再代入其他各式,便可計 算由分子基態躍遷至離子基態的法蘭克-康登因子。 2.4 光譜模擬光譜模擬光譜模擬 光譜模擬 在計算出電子態的法蘭克-康登因子後,我們使用羅倫茲函數(Lorentzian line-shape function)來進行光譜模擬。我們假設每一個躍遷的譜線,都可以用一個 羅倫茲函數g(ν)來表示,其公式如下: g(ν)=1/[1+4(w
v
v
−
0 )2]其中,ν為頻率,ν0為躍遷的中心頻率,w為吸收峰的半高寬(full width at half
maximum)。
將每個躍遷的貢獻加起來,即可得到整個分子躍遷的模擬光譜。在模擬光譜
中,每根譜線的高度皆設定為法蘭克-康登因子,其半高寬預設為 50 cm-1;若要
和實驗值相比對,半高寬再視實驗的解析度加以調整。為能與實驗光譜做更貼切 -1
第三章 研究結果
本章以五小節呈現二氟化硫碳(F2CS)的理論計算結果,並與實驗值做比對。 第一節探討二氟化硫碳分子、正離子、負離子的平衡結構。第二節探討二氟化硫 碳分子的振動模式,並計算出分子、正離子、負離子的振動頻率。第三節則計算 出二氟化硫碳分子、正離子、負離子的轉動常數與κ值,及二氟化硫碳的絕熱游 離能、電子親和力。第四節介紹二氟化硫碳的法蘭克-康登因子。第五節為本研 究最終目的,將二氟化硫碳分子電離成正離子基態的模擬光電子光譜與實驗光譜 做比較。 3.1 平衡結構平衡結構平衡結構 平衡結構 圖 3.1 及圖 3.2 呈現二氟化硫碳分子、正離子、負離子的結構及原子編碼, 計算所得之平衡結構的參數數據,詳列於表 3.1.1 及表 3.1.2(見下頁)。 圖 3.1 二氟化硫碳分子、正離子平衡結構圖示 圖 3.2 二氟化硫碳負離子平衡結構圖示C
S
F
1F
2C
S
F
1F
2表 3.1.1 二氟化硫碳分子及正離子在不同計算基組下的平衡結構a 分子態 正離子態 CS CF ∠SCF ∠FCF CS CF ∠SCF ∠FCF 實驗值b 158.9 131.5 107.1 6-311+G(d) 159.9 132.0 126.4 107.2 167.2 126.6 122.2 115.6 6-311+G(2d) 159.7 131.7 126.4 107.2 166.9 126.5 122.2 115.6 6-311+G(3d) 159.4 131.7 126.4 107.2 166.6 126.4 122.2 115.6 6-311+G(3df) 159.5 131.4 126.4 107.2 166.6 126.3 122.2 115.6 aug-cc-pVDZ 160.7 132.5 126.4 107.2 167.8 127.3 122.2 115.6 aug-cc-pVTZ 159.8 131.7 126.4 107.2 167.1 126.5 122.2 115.6 a 鍵長單位為 pm,鍵角單位為度 b 實驗值資料取自參考文獻[38] 表 3.1.2 二氟化硫碳負離子在不同計算基組下的平衡結構a 負離子態 CS CF ∠SCF ∠FCF 二面角 6-311+G(d) 170.5 140.3 118.7 103.2 133.7 6-311+G(2d) 170.4 139.9 118.9 103.3 134.3 6-311+G(3d) 169.7 140.0 118.9 103.3 134.2 6-311+G(3df) 169.7 139.3 119.0 103.5 134.7 aug-cc-pVDZ 171.1 141.0 118.7 103.1 133.6 aug-cc-pVTZ 170.3 139.6 118.9 103.4 134.4 a 鍵長單位為 pm,鍵角單位為度
綜合六種基組的計算結果:二氟化硫碳分子C=S鍵所得的平均鍵長為159.8 pm(實驗值158.9 pm),兩C-F鍵所得的平均鍵長為131.8 pm(實驗值131.5 pm), 與實驗值比較,鍵長差距均小於1 pm。而六種計算基組所得的鍵角∠FCF均為 107.2°(實驗值107.1°),兩鍵角∠SCF則均為126.4°,與實驗值比較,鍵角差距 約0.1°。 分別比較六種基組的計算結果與實驗值之差異:C=S鍵長,在6-311+G(3d) 時與實驗值最接近,為0.5 pm;在aug-cc-pVDZ時與實驗值差距較大,為1.8 pm。 兩C-F鍵長,在6-311+G(3df)時與實驗值最接近,為0.1 pm;在aug-cc-pVDZ時與 實驗值差距較大,為1 pm。但整體而言,鍵長的差距皆小於0.6%。 利用六種計算基組,我們也計算出二氟化硫碳正離子、負離子的平衡結構, 並比較分子與離子結構上的改變(表3.1.3~表3.1.4,見下頁)。 在正離子相對於分子的結構變化:鍵長方面,C=S鍵向外伸長約7.2 pm,兩 C-F鍵則縮短約5.2 pm。鍵角方面,∠FCF擴張約為8.4°,兩個∠SCF則縮小約 4.2°。同時,正離子基態的結構與分子基態一樣,同屬於平面的C2v點群。 在負離子相對於分子的結構變化:鍵長方面,C=S鍵向外伸長約10.5 pm, 兩C-F鍵也向外伸長約8.2 pm。鍵角方面,∠FCF縮小約為7.5°,兩個∠SCF也縮 小約3.9°。同時,負離子基態的結構則與分子基態有相當大的不同,不再屬於平 面的C2v點群,而是具有二面角約134. 2°的Cs點群。
表 3.1.3 二氟化硫碳分子轉變成正離子時的平衡結構變化 結構 變化 C=S CS鍵伸長(約 7.2 pm) (CS鍵之π鍵失去一電子, 鍵結能力減弱) C-F 兩 CF鍵都縮短(約 5.2 pm) (電負度大之F更接近正離子時之 C) ∠SCF 鍵角變小(約 4.2°) ∠FCF 鍵角變大(約 8.4°) (正離子時 S 之孤電子對效應減弱) 表 3.1.4 二氟化硫碳分子轉變成負離子時的平衡結構變化 結構 變化 C=S CS鍵伸長(約 10.5 pm) C-F 兩 CF鍵都伸長(約 8.2 pm) ∠SCF 鍵角變小(約 3.9°) ∠FCF 鍵角變小(約 7.5°) SCF1與 SCF2 由同一平面轉為具二面角(約 134. 2°) 3.2 振動模式及振動模式及振動模式及振動振動模式及振動振動振動頻率頻率頻率頻率 二氟化硫碳分子為一平面結構,屬於 C2v對稱,且其為一非線性分子,該 對稱性具有 3N-6 個振動模式(N 為原子數),因此二氟化硫碳分子(N=4)有 6 個振 動模式。這 6 個振動模式分別屬於 A1、B1、B2,且 A1對稱有 3 個,B1有 1 個, B2有 2 個。其中 A1、B2為平面內(in-plane)對稱模式,B1對稱則為平面外(out-of-plane) 對稱模式。表 3.2.1~表 3.2.3 列出二氟化硫碳分子、正離子、負離子的六種振動 模式示意圖。
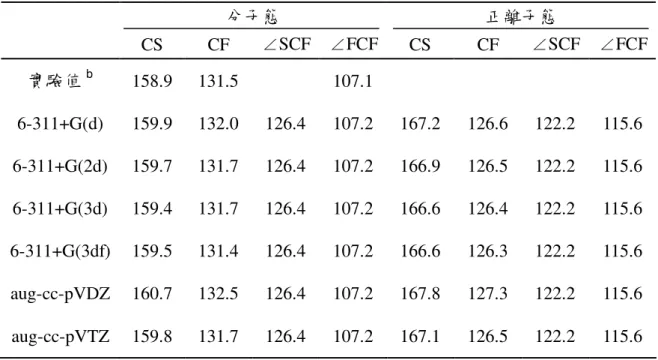
表 3.2.1 二氟化硫碳分子的正規振動模式
Mode Symmetry type Approximate description 圖示
ν1 A1 C=S stretch ↔ ν2 A1 C-F symmetric stretch ↔ ν3 A1 CF2 symmetric bend ↔ ν4 B1 out-of-plane bend ↔ ν5 B2 C-F asymmetric stretch ↔ ν6 B2 CF2 asymmetric bend ↔
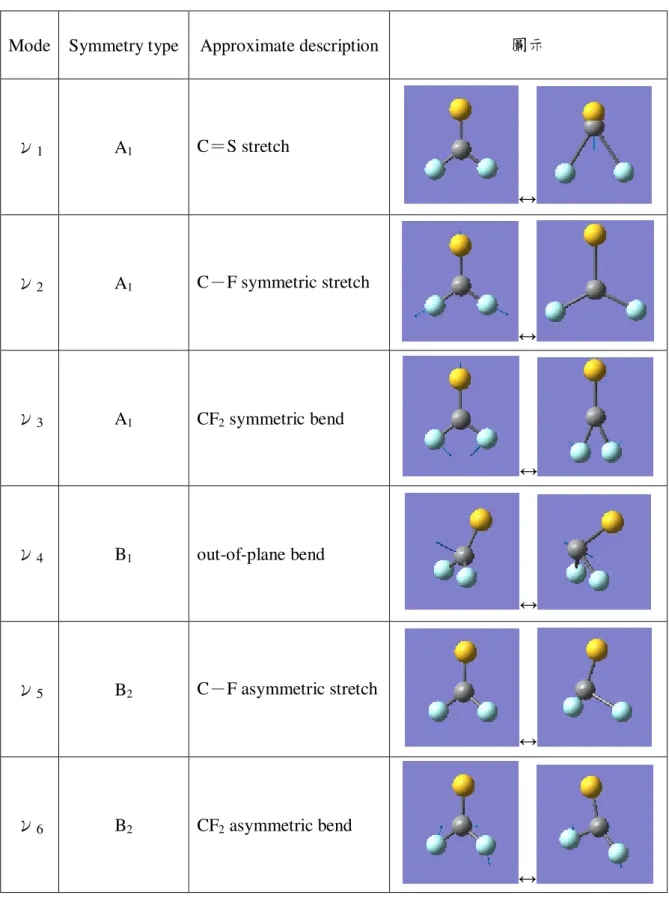
表 3.2.2 二氟化硫碳正離子的正規振動模式
Mode Symmetry type Approximate description 圖示
ν1 A1 C=S stretch ↔ ν2 A1 C-F symmetric stretch ↔ ν3 A1 CF2 symmetric bend ↔ ν4 B1 out-of-plane bend ↔ ν5 B2 C-F asymmetric stretch ↔ ν6 B2 CF2 asymmetric bend ↔
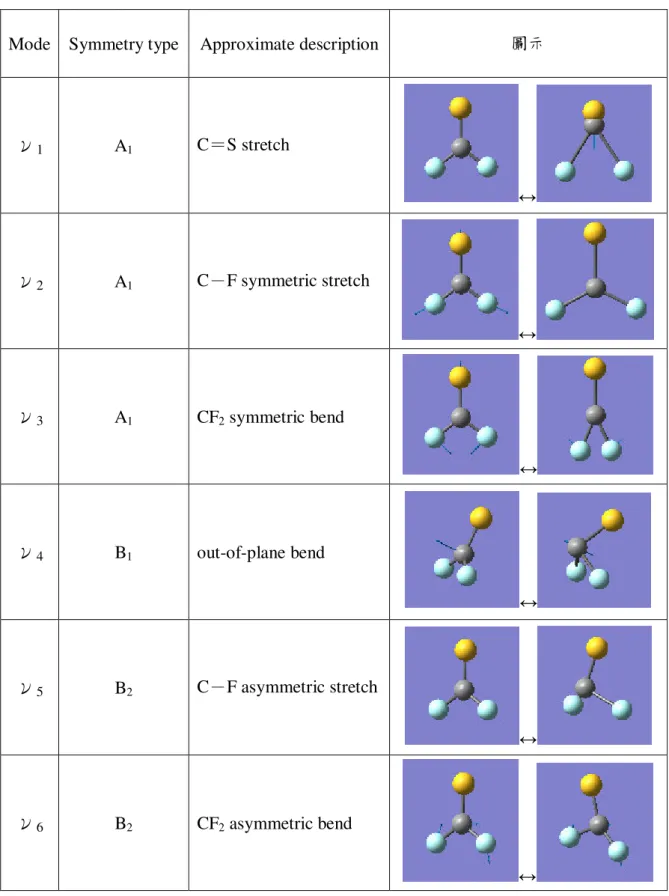
表 3.2.3 二氟化硫碳負離子的正規振動模式
Mode Symmetry type Approximate description 圖示
ν1 A’ C=S stretch
↔
ν2 A’ C-F symmetric stretch
↔
ν3 A’ CF2 symmetric bend
↔
ν4 A’ out-of-plane bend
↔
ν5 A” C-F asymmetric stretch
↔
ν6 A” CF2 asymmetric bend
在 A1對稱中,ν1振動模式為 C=S 伸縮(C=S stretch),ν2振動模式為 C-
F 對稱伸縮(C-F symmetric stretch),ν3振動模式為 CF2對稱彎曲(CF2 symmetric
bend)。在 B1對稱中,ν4振動模式為平面外彎曲(out-of-plane bend)。在 B2對稱中,
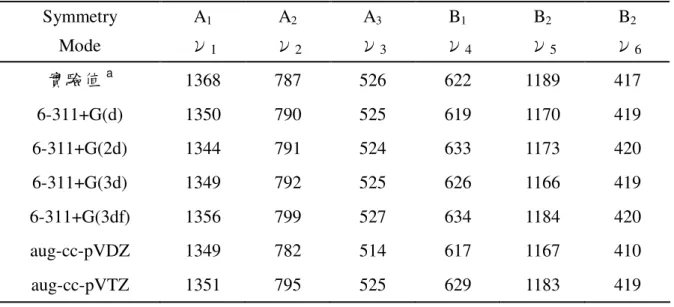
ν5振動模式為 C-F 不對稱伸縮(C-F asymmetric stretch),ν6振動模式為 CF2 不對稱彎曲(CF2 asymmetric bend)。 關於振動頻率的部分,從表3.2.4~表3.2.6中可知:六種基組計算出的振動頻率, 彼此間差異不大。若以分子態的振動頻率與實驗值相較,可發現低頻率的振動模 式與實驗值更為吻合,此外,6-311+G(3d)、6-311+G(3df)、aug-cc-pVTZ三種基組 的計算結果與實驗值更是一致,而aug-cc-pVDZ與實驗值的差距相對來講則較大。 綜合六種基組的計算結果:二氟化硫碳分子之振動頻率的平均值,ν1為 1350cm-1(實驗值1368cm-1),ν 2為792cm-1(實驗值787cm-1),ν3為523cm-1(實 驗值526cm-1),ν 4為626cm-1(實驗值622cm-1),ν5為1174cm-1(實驗值1189cm-1), ν6為418cm-1(實驗值417cm-1),各種振動頻率與實驗值比較,差距在0.2%到1.3% 之間。二氟化硫碳正離子之振動頻率的平均值,ν1為1389cm-1(較分子態大), ν2為791cm-1(與分子態差異甚小),ν3為486cm-1(較分子態小),ν4為633cm-1 (較分子態大),ν5為1437cm-1(較分子態大),ν6為332cm-1(較分子態小)。 而二氟化硫碳負離子之振動頻率的平均值,ν1為 998cm-1,ν2為721cm-1,ν3為 547cm-1,ν4為417cm-1,ν5為878cm-1,ν6為351cm-1。
表 3.2.4 二氟化硫碳分子在不同計算基組下的振動頻率(cm-1 ) Symmetry A1 A2 A3 B1 B2 B2 Mode ν1 ν2 ν3 ν4 ν5 ν6 實驗值a 1368 787 526 622 1189 417 6-311+G(d) 1350 790 525 619 1170 419 6-311+G(2d) 1344 791 524 633 1173 420 6-311+G(3d) 1349 792 525 626 1166 419 6-311+G(3df) 1356 799 527 634 1184 420 aug-cc-pVDZ 1349 782 514 617 1167 410 aug-cc-pVTZ 1351 795 525 629 1183 419 a 實驗值資料取自參考文獻[37] 表 3.2.5 二氟化硫碳正離子在不同計算基組下的振動頻率(cm-1 ) Symmetry A1 A2 A3 B1 B2 B2 Mode ν1 ν2 ν3 ν4 ν5 ν6 6-311+G(d) 1387 792 489 625 1438 336 6-311+G(2d) 1387 791 486 641 1434 333 6-311+G(3d) 1388 793 487 633 1433 333 6-311+G(3df) 1396 797 490 641 1446 333 aug-cc-pVDZ 1382 781 477 622 1428 325 aug-cc-pVTZ 1392 793 488 637 1445 333
表 3.2.6 二氟化硫碳負離子在不同計算基組下的振動頻率(cm-1
)
Symmetry A’ A’ A’ A’ A” A”
Mode ν1 ν2 ν3 ν4 ν5 ν6 6-311+G(d) 993 723 547 417 871 351 6-311+G(2d) 992 716 546 416 877 352 6-311+G(3d) 997 721 547 418 868 353 6-311+G(3df) 1012 725 552 421 894 355 aug-cc-pVDZ 991 717 538 409 864 345 aug-cc-pVTZ 1004 721 551 419 891 352 3.3 轉動常數轉動常數轉動常數、轉動常數、、、游離能游離能游離能游離能及及及及電子親和力電子親和力電子親和力電子親和力 利用 Gaussian 03 套裝軟體,我們除能計算出二氟化硫碳分子與離子的平衡 結構和振動頻率外,亦能計算二氟化硫碳的轉動常數,表 3.3.1~表 3.3.3 分別列 出分子態、正離子態、負離子態的計算結果。 利用轉動常數A、B、C的數值,我們也可計算出不對稱參數(asymmetric parameter) κ值,計算公式如下: κ=(2B-A-C)/(A-C) 其中A、B、C為分別為以a、b、c軸旋轉之轉動常數(A≧B≧C)。若κ值為1,表示 該分子為扁圓形對稱陀螺(oblate symmetric top, A=B>C);若κ值為-1,表示該分 子為扁長形對稱陀螺(prolate symmetric top, A>B=C)。計算κ值可以瞭解它與對 稱陀螺間的關係。
表3.3.1中,二氟化硫碳分子的A常數平均為11815.40 MHz,約比實驗值小 77.20 MHz;B常數平均為5086.38 MHz,約比實驗值小46.65 MHz;C常數平均為 3555.70 MHz,約比實驗值小24.63 MHz。二氟化硫碳正離子的A常數平均為 11815.40 MHz;B常數平均為5086.38 MHz;C常數平均為3555.70 MHz。二氟化 硫碳負離子的A常數平均為11815.40 MHz;B常數平均為5086.38 MHz;C常數平 均為3555.70 MHz。 概括而言,各基組間所計算出之分子、正離子、負離子的轉動常數值差異不 大,分子的平均κ值為-0.63,與實驗值符合;正離子、負離子的平均κ值分別為 -0.60、-0.62。相對來講,分子、正離子、負離子都屬於較接近扁長型對稱陀螺的 型態。 表 3.3.1 在不同計算基組下二氟化硫碳分子態的轉動常數(MHz) A B C κ 實驗值a 11892.60 5133.03 3580.32 -0.63 6-311+G(d) 11787.59 5080.74 3550.42 -0.63 6-311+G(2d) 11834.82 5093.50 3560.93 -0.63 6-311+G(3d) 11835.39 5109.03 3568.57 -0.63 6-311+G(3df) 11888.24 5114.18 3575.88 -0.63 aug-cc-pVDZ 11710.07 5028.80 3518.01 -0.63 aug-cc-pVTZ 11836.28 5092.05 3560.36 -0.63 a 實驗值資料取自參考文獻[38]
表 3.3.2 在不同計算基組下二氟化硫碳正離子態的轉動常數(MHz) A B C κ 6-311+G(d) 11594.75 5199.28 3589.63 -0.60 6-311+G(2d) 11607.80 5213.73 3597.77 -0.60 6-311+G(3d) 11611.86 5232.39 3607.04 -0.59 6-311+G(3df) 11665.50 5230.49 3611.29 -0.60 aug-cc-pVDZ 11475.25 5153.25 3556.24 -0.60 aug-cc-pVTZ 11616.05 5204.95 3594.37 -0.60 表 3.3.3 在不同計算基組下二氟化硫碳負離子態的轉動常數(MHz) A B C κ 6-311+G(d) 10653.45 4755.81 3353.83 -0.62 6-311+G(2d) 10713.19 4762.38 3360.82 -0.62 6-311+G(3d) 10702.41 4786.85 3371.93 -0.61 6-311+G(3df) 10788.62 4798.88 3384.57 -0.62 aug-cc-pVDZ 10566.05 4717.63 3327.23 -0.62 aug-cc-pVTZ 10744.98 4773.23 3368.72 -0.62 另外,我們也以CCSD及CCSD(T)的計算方法求出二氟化硫碳在不同計算基 組下的絕熱游離能(adiabatic ionization energy)。表3.3.4呈現其計算結果,相較於
實驗值,差距約在0.9%~4.0%。綜合六種基組的計算結果,CCSD(T)的方法顯然 比 CCSD 的 方 法 較 接 近 實 驗 值 。 其 中 , 又 以 CCSD(T)/6-311+G(3df) 、 CCSD(T)/aug-cc-pVTZ兩者與實驗值最為接近,差距分別為0.15eV、0.10 eV。 表 3.3.4 在不同計算基組下二氟化硫碳的絕熱游離能(eV) CCSD 比對實驗值之差距 CCSD(T) 比對實驗值之差距 6-311+G(d) 10.03 0.42 (4.0%) 10.07 0.38 (3.5%) 6-311+G(2d) 10.12 0.33 (3.1%) 10.18 0.27 (2.5%) 6-311+G(3d) 10.13 0.32 (3.0%) 10.21 0.22 (2.3%) 6-311+G(3df) 10.21 0.24 (2.3%) 10.30 0.15 (1.4%) aug-cc-pVDZ 10.18 0.27 (2.5%) 10.25 0.20 (1.9%) aug-cc-pVTZ 10.25 0.20 (1.9%) 10.35 0.10 (0.9%) 實驗值a 10.45 a 實驗值資料取自參考文獻[36] 同時,我們也用 CCSD 及 CCSD(T)的計算方法求出二氟化硫碳在不同計算基 組下的電子親和力(electron affinity),表 3.3.5 呈現其計算結果。而電子親和力尚 無實驗值,以最高基組 6-311+G(3df)和 aug-cc-pVTZ 估計,計算的預測值約在 0.6 ~0.7 eV 之間。
表 3.3.5 在不同計算基組下二氟化硫碳的電子親和力(eV) CCSD CCSD(T) 6-311+G(d) 0.51 0.45 6-311+G(2d) 0.58 0.52 6-311+G(3d) 0.65 0.62 6-311+G(3df) 0.63 0.60 aug-cc-pVDZ 0.79 0.75 aug-cc-pVTZ 0.71 0.69 3.4 法蘭克法蘭克法蘭克-康登因子法蘭克 康登因子康登因子康登因子 利用表3.1.1及表3.2.2的平衡結構與振動頻率,我們能計算出二氟化硫碳正離 子相對於分子的平衡結構位移∆Q因子(參見表3.4.1,見下頁) 以及S因子(參見表 3.4.2,見下頁)。從S值的大小,可了解分子電離成正離子之模擬光譜的主要細微 結構。具有較大S值的振動模式,代表電子從分子躍遷至正離子基態這兩個振動 能階的躍遷機率較大,因此其振動躍遷的訊號也較大。若所有模式的S值皆接近 零,則代表光譜中的原帶是最強的訊號。本研究中,二氟化硫碳在各計算基組中, 都以ν1的S值為最大(約為1.429),代表ν1振動模式(C=S stretch)的吸收強 度為最強;ν3的S值為次大(約為0.247),代表ν3振動模式(CF2 symmetry bend) 的吸收強度為次強。 至於其它振動模式,ν2 S值小於0.1,而ν4、ν5、ν6的S值
甚至都幾乎為0。 表 3.4.1 在不同計算基組下二氟化硫碳正離子相對於分子之平衡結構位移∆Q 因子 ν1 ν2 ν3 ν4 ν5 ν6 6-311+G(d) 0.26988 -0.08420 0.17735 0 0 0 6-311+G(2d) 0.26594 -0.09103 0.18190 0 0 0 6-311+G(3d) 0.26689 -0.08579 0.18548 0 0 0 6-311+G(3df) 0.26085 -0.08999 0.17761 0 0 0 aug-cc-pVDZ 0.26545 -0.08491 0.19285 0 0 0 aug-cc-pVTZ 0.26270 -0.09341 0.17498 0 0 0 表 3.4.2 在不同計算基組下二氟化硫碳正離子相對於分子之 S 因子 ν1 ν2 ν3 ν4 ν5 ν6 6-311+G(d) 1.47778 0.08315 0.23632 0 0 0 6-311+G(2d) 1.43160 0.09722 0.24734 0 0 0 6-311+G(3d) 1.44518 0.08653 0.25772 0 0 0 6-311+G(3df) 1.38831 0.09581 0.23773 0 0 0 aug-cc-pVDZ 1.42626 0.08356 0.27289 0 0 0 aug-cc-pVTZ 1.40343 0.10275 0.22966 0 0 0 另外,我們也可檢視杜辛斯基效應的影響,藉由 Duschinsky matrix,能判斷
哪些振動模式彼此之間會互相影響,及其影響的大小。根據表 3.4.3,由計算結果 的 Duschinsky matrix,可以看出各種振動模式彼此之間的模式混合影響。
表 3.4.3 在 aug-cc-pVTZ 計算基組下所計算出的 Duschinsky matrix
Q1 Q2 Q3 Q4 Q5 Q6 Q1’ 0.9786 0.2043 0.0258 0.0000 0.0000 0.0000 Q2’ -0.1979 0.9677 -0.1562 0.0000 0.0000 0.0000 Q3’ -0.0569 0.1477 0.9874 0.0000 0.0000 0.0000 Q4’ 0.0000 0.0000 0.0000 0.9995 0.0000 0.0000 Q5’ 0.0000 0.0000 0.0000 0.0000 0.9995 0.0169 Q6’ 0.0000 0.0000 0.0000 0.0000 -0.0168 0.9999 由 Duschinsky matrix 的對角線數值,若對角線數值愈接近 1,則不同模式之 間的影響愈小。例如,矩陣中 Q1’、 Q2’、 Q3’ 相對 Q4、Q5、Q6與 Q4’、 Q5’、 Q6’ 相對 Q1、Q2、Q3的數值都為 0,這顯示ν1、ν2、ν3振動模式與ν4、ν5、 ν6 振動模式彼此間並不互相影響,沒有振動模式混合的的情形。而矩陣左上方 及右下方有一個 3×3 及 2×2 矩陣,其數值分別為 0.9874 0.1477 0.0569 -0.1562 -0.9677 0.1979 -0.0258 0.2043 0.9786 和
0.9999 0.0168 -0.0169 0.9995 ,則說明ν1、ν2、ν3 振動模式彼此間有一定程度的模式混合 影響,尤其是ν2、ν1 模式間與ν2、ν3 模式間;ν4、ν5 振動模式彼此間也存 在些微的模式混合影響。然而,在 S 因子的分析中,我們也得知ν2振動模式對 光譜訊號的貢獻很小;ν4、ν5振動模式更是幾乎不產生訊號,因此,在後續的 模擬光譜與實驗光譜之比較,我們可以看到,即使忽略杜辛斯基效應的影響,模 擬光譜與實驗結果仍然相當吻合。 表3.4.4(見下頁)則顯示六種基組的法蘭克-康登因子計算結果,若其值越大, 則表示電子在這兩個振動能階間的躍遷機率越大。 以B3LYP/6-311+G(3df)之計算層級來說,二氟化硫碳從分子電離成正離子基 態的法蘭克-康登因子中,以
1
1 0為最大(約為0.24150),可知其為最強的躍遷能 帶;0
0 0為次大(約為0.17651),可知其為次強的躍遷能帶,其餘較明顯的躍遷, 依序為1
2 0、1
3 0、1
1 03
1 0、3
1 0、1
2 03
1 0、1
4 0、1
3 03
1 0。附錄三則完整列出各種振動模式 的法蘭克-康登因子,從計算結果,我們發現,單一振動模式的躍遷訊號相較於兩 種振動模式合成的躍遷訊號,其法蘭克-康登因子較大,因此可知其躍遷訊號較 強。同時,我們可得知單一振動模式的躍遷能帶總數有15個、兩種振動模式合成 的躍遷能帶總數有38個、三種振動模式合成的躍遷能帶總數有18個。並且,各法 蘭克-康登因子的數值總合達到0.9978,這顯示計算結果已涵蓋各振動能階間的所 有躍遷機率的99.78%。表 3.4.4 在不同計算基組下二氟化硫碳正離子相對於分子之法蘭克-康登因子 6-311+G(d) 6-311+G(2d) 6-311+G(3d) 標定 頻率 FCF 頻率 FCF 頻率 FCF 0 .16376 0 .16718 0 .16495 1387 .23866 1387 .23558 1388 .23492 2775 .17722 2774 .16970 2777 .17071 4162 .08935 4160 .08326 4165 .08435 5549 .03440 5547 .03128 5553 .03186 489 .04008 486 .04293 487 .04410 1876 .05842 1872 .06049 1875 .06281 3264 .04338 3259 .04357 3263 .04564 4651 .02187 4646 .02138 4652 .02255 6-311+G(3df) aug-cc-pVDZ aug-cc-pVTZ 標定 頻率 FCF 頻率 FCF 頻率 FCF 0 .17651 0 .16607 0 .17408 1396 .24150 1382 .23400 1392 .24066 2792 .16873 2763 .16770 2784 .16997 4188 .08021 4145 .08146 4176 .08170 5584 .02917 5526 .03016 5568 .03006 490 .04349 477 .04702 488 .04145 1886 .05951 1858 .06626 1880 .05731 3282 .04157 3240 .04748 3272 .04047 4678 .01976 4622 .02307 4664 .01946
3.5 光電子光譜光電子光譜光電子光譜 光電子光譜 承自前一節,我們將計算所得之法蘭克-康登因子用來模擬二氟化硫碳分子電 離成正離子基態的光電子光譜。光譜中躍遷訊號之強度正比於法蘭克-康登因子, 因此法蘭克-康登因子的數值大小可對應其光譜躍遷訊號之強度。在光譜模擬時, 我們假設每一個躍遷的譜線可以用一個羅倫茲函數來表示,因此,將每條譜線依 序連結就成為躍遷訊號強弱集合而成的光譜圖。 圖 3.5.1(見下頁)顯示二氟化硫碳在 6-311+G(d)、6-311+G(2d)、6-311+G(3d)、 6-311+G(3df)、aug-cc-pVDZ、aug-cc-pVTZ 基組下分子電離成正離子的模擬光譜。 為使光譜圖容易瞭解,而不至於讓解讀產生混淆,本研究只標定出較強的幾個振 動帶,並分別以半高寬 50 cm-1 、250cm-1 做出模擬光譜,便於和實驗光譜做比對。
(a)半高寬 50cm-1 (b)半高寬 250cm-1 圖 3.5.1 在不同計算基組下二氟化硫碳分子電離成正離子基態的模擬光譜
為能 將模 擬光 譜與 實驗 光 譜做 更貼 切之 比對 ,最 後我 們將 半高 寬 設為
350cm-1 ,並根據不同計算基組所得資料,分別做出二氟化硫碳分子電離成正離
圖 3.5.2 在不同基組下二氟化硫碳分子電離成正離子基態的模擬光譜與實驗光譜之比較
a
實驗值資料取自參考文獻[36];b 半高寬 350cm-1;c
第四章 結論
本研究以Gaussian 03套裝軟體為研究工具,進行二氟化硫碳(F2CS)分子和離 子的結構及光電子光譜等相關研究。在研究過程,我們採用密度泛函理論當中的 B3LYP 泛 函 數 和 6-311+G(d) 、 6-311+G(2d) 、 6-311+G(3d) 、 6-311+G(3df) 、 aug-cc-pVDZ、aug-cc-pVTZ共六種基組,計算出二氟化硫碳分子、正離子、負離 子的平衡結構、振動頻率、絕熱游離能和電子親和力,並與實驗值做比對。以 6-311+G(3df) 計算基組為例,在分子鍵長、鍵角的計算結果與實驗值比較,C-F 鍵長之差距小於0.1pm(小於0.1%),鍵角差距約0.1°(小於1%),可說與實驗 數據相當一致;而在分子振動頻率的計算結果,各種振動模式的振動頻率與實驗 值比較,最大差距為12cm-1 (小於0.9%)。其次,利用CCSD(T)方法,以aug-cc-pVTZ 計算基組為例,其絕熱游離能與實驗值差距不到0.1eV(小於0.9%),而電子親 和力尚無實驗值,以最高基組6-311+G(3df)和aug-cc-pVTZ估計,計算的預測值約 在0.6~0.7eV之間。這些計算結果,再次印證我們先前所言,理論計算的結果與 實驗結果相比較,若有很高的一致性,必能對許多實驗中難以解決的問題有所助 益,也突顯出理論計算的價值與重要性。並且,在缺乏實驗數據的情況下,我們 還能預測二氟化硫碳分子轉變成正離子或負離子時,其平衡結構、振動頻率的改 變情形。接著,我們利用計算出的平衡結構和振動頻率等數據,進一步計算法蘭克-康登因子,並模擬出二氟化硫碳分子電離成正離子基態的模擬光譜。在改變各種 不同半高寬後,最後將半高寬設為350cm-1,我們可以看出模擬光譜與實驗結果相 當吻合。此外,因為法蘭克-康登因子正比於兩電子態間振動躍遷的機率,故可 預測或解釋光譜強度的分布,也就是說,可用來了解二氟化硫碳分子電離成正離 子之模擬光譜的主要細微結構。根據本研究所計算出的法蘭克-康登因子,我們能 明確的辨認每一個躍遷訊號是來自於哪一個振動模式的躍遷能帶、或哪些振動模 式合成的躍遷能帶,並明確標定之。而從計算出的S值大小,我們也能了解,具 有較大S值的振動模式,代表電子從分子躍遷至正離子基態這兩個振動能階的躍 遷機率較大,因此其振動躍遷的訊號也較大。而就實驗觀點來講,這些都是困難 的事,因為即使是簡單分子的光譜,都包含基頻、合頻、泛頻,並且,許多峰彼 此間的頻率差異極微小,在目前實驗器材所能解析的限制下,很難分辨其差異。 為解決這方面的困難,本研究用計算來模擬分子的振動,所得的頻率能幫助光譜 實驗進行指認。 最後,當比較六個基組間的計算結果時,我們發現,不論針對二氟化硫碳分 子或離子,其平衡結構及振動頻率的計算結果,彼此間差異並不大。這可能是由 於二氟化硫碳的分子及離子並不大,相對來講,結構不複雜,故在計算小分子及 離子時,較難看出各基組間計算結果的優劣。但若以分子態的平衡結構與實驗值 相較,鍵長在6-311+G(3d)與6-311+G(3df)時與實驗值最接近,在aug-cc-pVDZ時