國
立
交
通
大
學
生物資訊及系統生物研究所
碩
碩
碩
碩
士
士
士
士
論
論
論
論
文
文
文
文
以蛋白質-蛋白質交互作用家族為基礎建立模板導向
之同源模組
Template-based Homologous Modules through
Protein-protein Interaction Families
研 究 生:林怡瑋
指導教授:楊進木 教授
中
中
中
中
華
華
華
華
民
民
民
民
國
國
國
國
一
一
一
一
百
百
百
百
年
年
年
年
八
八
八
八
月
月
月
月
以蛋白質-蛋白質交互作用家族為基礎建立模板導向之同源模組
Template-based Homologous Modules through Protein-protein
Interaction Family
研 究 生:林怡瑋 Student:Yi-Wei Lin
指導教授:楊進木 Advisor:Jinn-Moon Yang
國 立 交 通 大 學
生物資訊及系統生物研究所
碩 士 論 文
A Thesis Submitted to Institute of Bioinformatics and Systems Biology College of Biological Science and Technology
National Chiao Tung University in partial Fulfillment of the Requirements for the Degree of Master in
Bioinformatics and Systems Biology
August 2011
Hsinchu, Taiwan, Republic of China
中
中
中
I
以蛋白質-蛋白質交互作用家族為基礎建立模板導向之同源模組
學生:林怡瑋 指導教授:楊進木 國立交通大學 生物資訊與系統生物所碩士班摘
要
在相同時間和空間尺度下,分子間精確地聚集且協同作用對於生物程序是不可或缺 的,例如細胞週期和轉錄作用。模組 (module) 是指一群具有高度連結並執行特定生物 功能的蛋白質所組成。就如同同源蛋白質 (homologous protein) 和同源蛋白質-蛋白質交 互作用 (homologous protein-protein interaction) 的概念,當一群模組來自一個共同的祖 先並且在不同物種中都執行相似的生物功能時,則這些模組被認為是同源模組 (homologous module)。以同源蛋白質-蛋白質交互作用家族為基礎,我們提出一個新概念: 「模組家族 (module family)」。模組家族包含一群同源模組,而同源模組是由一群同源 蛋白質-蛋白質交互作用家族所構成。從多物種的基因組 (genome) 來推論同源模組可提 供一個契機去了解模組的演化和蛋白質交互作用體 (protein interactome)。 在本研究中,透過推論模板導向的方法,將模組家族的概念驗證在 MIPS CORUM 資料庫所收集的模組模版 (module template)上。首先,透過同源蛋白質-蛋白質交互作用 家族,從 1,679 個物種定義出同源模組候選者。隨後,當同源模組候選者具備三個條件: 第一,蛋白質相似性 (E-values ≤ 10-10 );第二,蛋白質-蛋白質交互作用相似性 (joint E-values ≤ 10-40);第三,拓撲相似性 (蛋白質-蛋白質交互作用對齊比例 ≥ 0.3 和蛋白質 對齊比例 ≥ 0.5),則此模組被認為與它的模組模板相似並稱為模板導向之同源模組(template-based homologous module)。我們驗證模板導向之同源模組的特性,結果指出其
蛋白質間具有高度連結性,以及在 Gene Ontology 的註解上傾向執行相似的生物功能。 進一步分析模板導向之同源模組的組成特性,我們發現模組家族中的核心組成(core
component) 往往是生物體生存所需的必需蛋白質,核心組成乃指跨多物種及物種分群 (division group) 的同源蛋白質-蛋白質交互作用家族,亦即具有高分的蛋白質-蛋白質交
互作用演化程度 ( protein-protein interaction evolution score)。 實驗結果指出模組家族中 的核心組成在調控模組的生物功能上扮演重要的角色。綜合以上所述,顯示來自模板導 向之同源模組的蛋白質-蛋白質交互作用演化程度可以反映出模組家族之必需蛋白質。 我們相信同源模組對於了解生命的基本要素有所助益。
II
Template-based Homologous Modules through Protein-protein
Interaction Families
Student: Yi-Wei Lin Advisor: Dr. Jinn-Moon Yang Institute of Bioinformatics and System Biology
National Chiao Tung University
English Abstract
ABSTRACT
Precise assembling and cooperation between molecules in time and space scale are essential for biological processes, such as cell cycle and transcription. A module is a group of proteins that are highly connected and perform a certain kind of biological functions. The modules, which often share a common ancestor and perform similar biological functions across species, can be considered homologous modules, just as homologous proteins and homologous protein-protein interactions (PPIs). Based on PPI families, we proposed a new concept “module family”, which comprises a group of homologous modules consisting of a group of homologous PPIs across species. To infer homologous modules from multiple genomes provides an opportunity to understand the module evolution and protein interactome. In this study, we verified the concept through inferring template-based homologous modules from module templates provided by MIPS CORUM database. First, we identified candidates of homologous modules from 1,679 species through PPI families. Subsequently, the identified candidates were regarded as template-based homologous modules, constituting module families, if the modules are similar to their module template with (i) protein similarity (E-values ≤ 10-10), (ii) PPI similarity (joint E-values ≤ 10-40), and (iii) topology similarity (PPI aligned ratio ≥ 0.3 and protein aligned ratio ≥ 0.5). We examined the properties of the template-based homologous modules, and the results showed that the template-based homologous modules often contain the high connectivity and its protein members perform similar biological functions based on Gene Ontology terms.
We further analyzed the component properties of the template-based homologous modules. We found that the core components, which are the consensus of PPI families across multiple species and division groups (i.e. high PPI evolution score), of the module families are often essential proteins for the survival of an organism. Our results showed that the core components of module families play an important role to regulate biological functions of module. In conclusion, the experimental results reveal that the PPI evolution score derived from template-based homologous modules could reflect essential proteins of a module family. We believe that homologous modules are useful to understand essential elements of a life.
III
誌謝
誌謝
誌謝
誌謝
Acknowledgement 感謝 主耶穌,在兩年的碩士生涯中,在眾多良師益友的支持和協助下,怡瑋才得 以完成這本論文,謹藉著這小小的篇幅,致上我最深切的謝意。 首先要感謝我的指導教授楊進木老師,很幸運可以進入老師所帶領的實驗室,您嚴 謹的研究態度和寬廣的視野,以及實驗室良好的討論氣氛,讓怡瑋得以初探科學研究的 縝密邏輯及樂趣。在研究過程中,難免遭遇困難與瓶頸,而您總是不厭其煩地幫助學生 修正方向和提供生涯規劃的建議,更令怡瑋獲益良多。這樣的成長和變化,不但是在治 學上,也更是變化了怡瑋的人生態度。接著我要感謝我的口試委員,包含我的指導教授 楊進木教授、黃奇英教授、鄭添祿教授、彭慧玲教授。感謝每位教授在百忙之中抽空擔 任我的口試委員並且評鑑我的論文,以及在口試期間針對我的研究提供的寶貴意見,有 了他們的指導與建言才使得這本論文能更臻完美。 同時,感謝實驗室的學長姐和各位同學們,感謝研究上同一組的峻宇學長和尚文學 弟,因為能與你們有充分的討論還有系統程式方面的協助,使我的研究能夠順利進行。 感謝俊辰學長、宇書學長、怡馨學姊、章維學長、PIKI 學長、志達學長、一原學長和彥 修學長在研究上的種種幫助和論文修訂。也感謝敬立、力仁、超哥、韋帆、伸融、御哲 碩班學長、同學和學弟妹們在實驗室給予的協助,謝謝大家在研究上和生活上的關心與 幫助,怡瑋銘記於心。 我的父母親林國財先生、林月里女士一直是怡瑋最熱情溫暖的避風港,包容我的任 性,了解我的徬徨,容忍我的囉唆。謝謝妹妹姿甄、家慧和爽朗的老弟家豪,分擔我的 情緒,在忙碌的工作和課業中抽出時間關懷問候,彼此加油打氣,一起撐過最辛苦的四、 五年。 怡瑋何其幸運,竟然能得到這樣多的幫助、愛護。最後容我再說一次說:謝謝,感 恩您們。IV
Table of Contents
Chinese Abstract ... I English Abstract ... II Acknowledgement ... III Table of Contents ... IV List of Tables ... VI List of Figures ... VIIChapter 1 Introduction ... 1
1.1 Background ... 1
1.2 Motivation ... 2
1.3 Thesis overview ... 3
Chapter 2 Methods and Materials ... 5
2.1 Homologous module ... 5
2.2 Essential protein set and mapped essential protein set ... 6
2.3 Characteristics of modules and homologous modules of homologous module ... 7
2.3.1 Connectivity of homologous module ... 7
2.3.2 Biological function of homologous module ... 7
2.4 PPI evolution score ... 8
Chapter 3 Results and Discussion ... 9
3.1 Homologous modules ... 9
3.2 Characteristics of homologous modules ... 10
3.2.1 Connectivity of homologous modules ... 10
3.2.2 Consensus of biological function of homologous modules ... 11
3.3 Core components of homologous modules ... 12
3.4 Essential MF terms of GO ... 14
V
3.6 Example analysis ... 17
3.7 Application: Crystal structure-based homologous modules ... 19
3.8 Discussion ... 21
Chapter 4 Conclusions ... 23
4.1 Summary ... 23
4.2 Major contributions and future works ... 23
VI
List of Tables
Table 1. The list of the number of modules in TOP 20 organisms from KEGG MODULE
database ... 26
Table 2. The list of data sets using definition and verification of module family ... 27
Table 3. Modified division groups from NCBI taxonomy database ... 28
Table 4. The 181 essential GO molecular functions (MF) terms ... 29
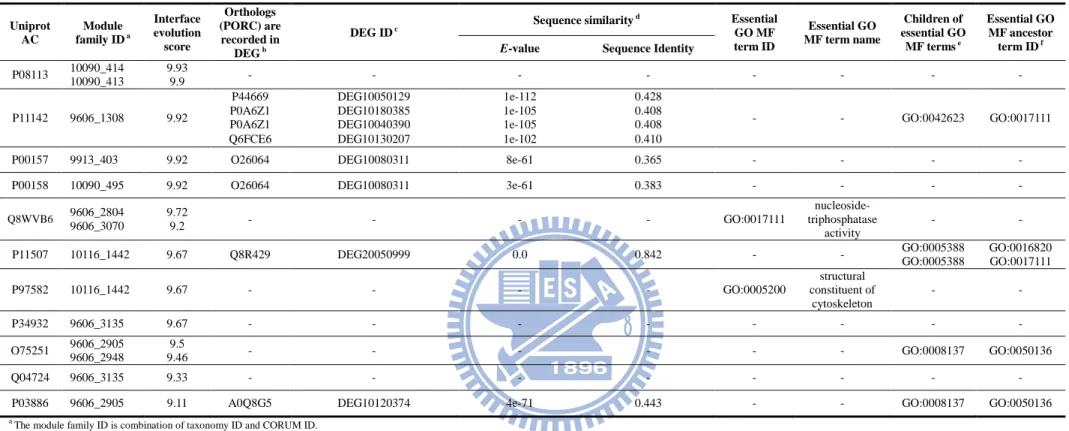
Table 5. Validation of unannotated protein in core components by the orthology database (PORC) and essential GO MF terms ... 39
VII
List of Figures
Figure 1. Assembling and cooperating between molecules in time and space scale are essential for transcription. ... 43 Figure 2. Module performs in a certain kind of process and relatively autonomous with
respect to other parts of the protein-protein interaction network. ... 44 Figure 3. Thesis framework ... 45 Figure 4. Overview of identifying homologous modules through protein-protein interaction
(PPI) families using F1-ATPase synthase-IF1 of B. taurus as the module template. ... 46
Figure 5. Evaluations of the topology similarity ... 47 Figure 6. Characteristics of modules and homologous modules. ... 48 Figure 7. The distribution between the fraction of modules and average RSS scores of GO
Biological Process (BP) and Cellular Components (CC). ... 49 Figure 8. Evaluations the PPI evolution scores using 1,578 module templates. ... 50 Figure 9. GO molecular function (MF) terms of essential proteins and core components. ... 51 Figure 10. The occur ratios of 181 essential GO MF terms between the essential proteins and
proteins of core components with interface evolution score ≥ 8. ... 52 Figure 11. The nucleosome remodeling and deacetylase (NuRD) module (CORUM ID: 614)
family and the core components. ... 54 Figure 12. The BRG1-based SEI/SNF chromatin remodeling module (CORUM ID: 2852)
family and the core components. ... 55 Figure 13. Overview of identifying module family for homologous modules search using
proteins RPB1, RPB2, and RPB8 of RNA polymerase II module (PDB code 3fki) in
Saccharomyces cerevisiae as the module template. ... 56
Figure 14. Molecular interfaces change analysis through binding models and multiple
sequence alignments of module family of RNA polymerase II. ... 57 Figure 15. The mechanisms of intra-module and inter-module (RNA polymerase II- MLL
1
Chapter 1 Introduction
1.1 Background
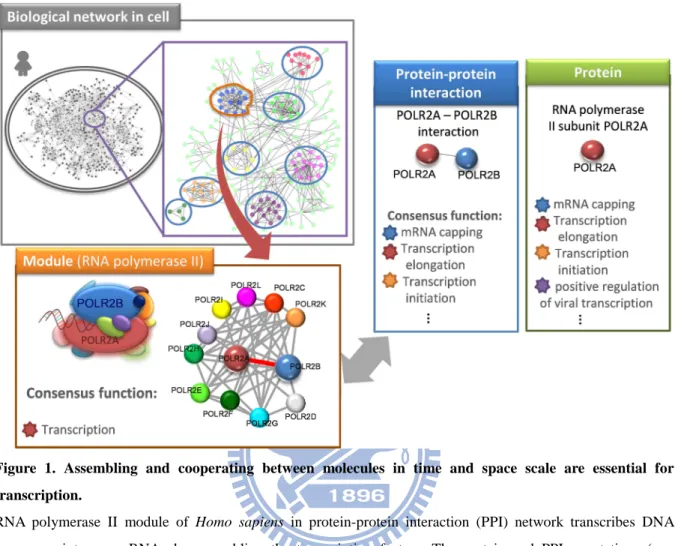
Assembling and cooperating between molecules in time and space scale are essential for biological processes, such as the cell cycle and transcription (Fig. 1) [1]. The organization of molecules is regarded as a module which is involving in a certain kind of process (e.g. natural variation, function, and development) and relatively autonomous with respect to other parts of the organisms (Fig. 2) [2, 3]. To identify and characterize the modules in a species, genome-scale module discovery approaches, such as gene expression and graph-based methods [3-5], have been proposed. Modules can provide insights of interactome evolution for two reasons. First, organizing biological systems into modules may permit changes and affect the evolutionary mechanisms within one module without perturbing other module. Second, modules can be combined and reused to create new biological functions [6-9]. The increasing number of complete genomes makes it useful for inferring modules in newly sequenced genomes and identifying the essential elements of modules through multiple species.
Recently, several databases provide modules across multiple species based on orthology (protein family), such as KEGG MODULE database [10, 11] and the online database resource Search Tool for the Retrieval of Interacting Genes (STRING) [10, 11]. KEGG organism-specific modules is defined as a tight functional unit and complexes in the pathway through a set of orthologs [11], which are classified into pathway modules, structural complexes, functional sets, and genomic signatures. TOP 20 organisms assigned with KEGG organism-specific modules are mainly bacteria commonly used in molecular research projects, such as Klebsiella pneumonia and Escherichia coli, and so on (Table 1). STRING database
2
provides functional networks through cross-genome homology searches to transfer functional interactions by mapping orthologous proteins. Orthologous protein sequences often provide the clues for understanding the functions of a newly sequenced gene [12]. Furthermore, a protein can be annotated the biological process and molecular functions by considering its interacting proteins in a protein-protein interaction (PPI) network [13]. Therefore, homologous PPIs (PPI family) provide new insights for understanding the functional organization of the proteomes (e.g. conservations of interacting domain-domain pairs and function pairs) [12, 14]. As the increasing number of PPIs become available, to identify homologous modules across multiple species via PPI families should be useful to understand the module evolution, functions, and characteristics which are critical to analyze PPI networks of biological systems.
The discovery of sequence orthologs to a known protein often provides clues for understanding the function of a newly sequenced gene [12]. However, a protein could be annotated new functions by considering its interacting proteins in protein-protein interactions (PPIs) network [13]. Therefore, homologous PPIs (a PPI family) in multiple species networks could provide new insights for understanding the functional organization of the proteomes during evolution (e.g. conservations of interacting domain-domain pairs and function pairs) [12]. As an increasing number of PPIs become available, identifying homologous PPIs should be useful to understand the conserved and divergent intra-module interactions of modules across multiple species. Perhaps conserved proteins and PPIs of modules observed through PPI families are the core components for regulating biological function in the biological system.
1.2 Motivation
3
families [12, 14]. According to our knowledge, module family, which comprises a group of homologous modules, is the first approach that identifies homologous modules of the module template from a large complete genomic database (e.g. Integr8 [15]) through PPI families. Notably, the core components of a module family are the conserved PPIs across multiple division group and species. Our results show that homologous modules are highly connected and perform a certain kind of biological functions, and the core components are often the essential elements for survival of an organism according to essential gene database (DEG) [16] and Gene Ontology (GO) database [17]. We believe that the module families and core components are useful for understanding the module evolution and PPI networks of biological systems across multiple species.
1.3 Thesis overview
The thesis consists of the two studies “module family” and “core components of a module family”. Figure 3 and Table 2 show the thesis framework and data sets using in this study. This study infers homologous modules (a module family) from module templates (Section 2.1). First, we identified homologous module candidates of a module template from a large complete genomic database (Integr8) through PPI families. Subsequently, these module candidates were regarded as homologous modules (called module family) of this template, if the candidates are significantly similar to their module template: (i) protein similarity (E-values ≤ 10-10) [18, 19], (ii) PPI similarity (joint E-values ≤ 10-40) [12], and (iii) topology similarity (PPI aligned ratio ≥ 0.3 and protein aligned ratio ≥ 0.5) (Section 3.1).
We analyzed characteristics of modules and homologous modules, and our results show that the homologous modules often contain the high connectivity (Section 3.2.1) and perform consensus of biological functions (Section 3.2.2). Furthermore, we attempt to identify the core components of homologous modules through PPI families (Section 3.3), orthologs of
4
essential proteins, and essential molecular functions (Section 3.4 and Section 3.5). Furthermore, we applied our concept on the crystal structure to identify template-based homologous modules (Section 3.7). In summary, we could identify homologous modules (module family) across multiple species using PPI families. The module family should be useful for deriving the core components and annotating the modules in a newly sequenced genome. Moreover, the module family can offer the new insight to analyze the module evolution and functions.
5
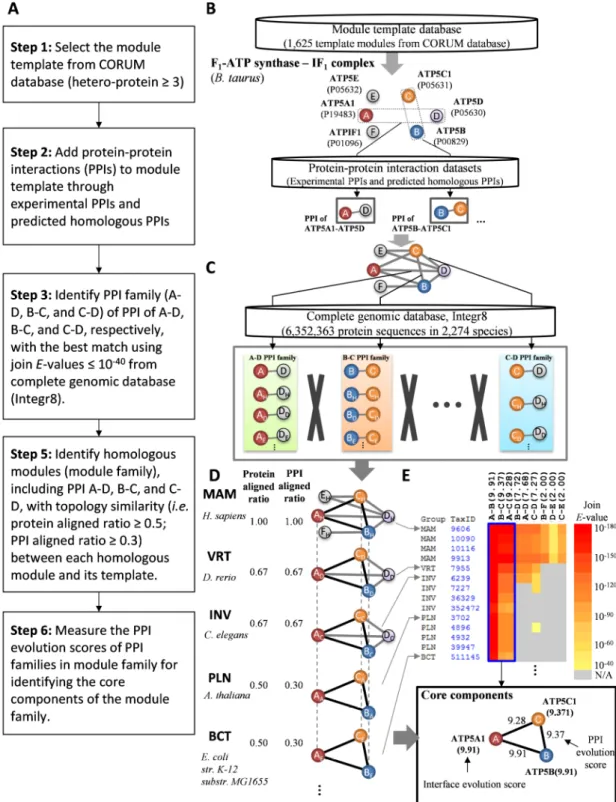
Chapter 2 Methods and Materials
Figure 4 shows the details of our method to identify the template-based homologous modules (module family) by the following steps (Fig. 4A): First, we select a module template database, which consists of 1,625 protein complexes (i.e. 1,165 in Homo sapiens, 268 in Mus
musculus, 157 in Rattus norvegicus, and 35 in Bos taurus), from comprehensive resource of
mammalian protein complexes database (CORUM; release 2.0) [20]. Then internal PPIs of a module template are added using template-based homologous PPIs, including experimental PPIs (i.e. IntAct [21], BioGRID [22], DIP [23], MIPS [24], and MINT [25]) and predicted homologous PPIs [12, 26] when the template is lack of intra-module interactions (Fig. 4B). For each PPI of a template, we derived its PPI family with joint E-value ≤ 10-40 [12] by searching from a complete genomic database (Integr8 version 103, containing 6,352,363 protein sequences in 2,274 species) using BLASTP [15](Fig. 4C). The homologous modules of a module template are derived from these searched PPI families, combined into homologous module candidates, according to the topology similarity between the module template and these candidates (Fig. 4D). For each module family, the module family profile is constructed to visualize the proteins and PPIs compositions across multiple species. Finally, for each module family, we derive GO biological process (BP), GO cellular component (CC), PPI and interface evolution scores and the core components (Fig. 4E).
2.1 Homologous module
Here, we use the module template M (including proteins A, B, C, D, E and F) with night interfaces A-B, A-C, A-D, B-C, B-D, B-F, C-D, C-E and D-E as an example to define the homologous module of M as follows: (1) A', B', C', D', E' and F' are the homologous proteins
6
of A, B, C, D, E, and F, respectively, with the significant sequence similarity (BLASTP
E-values ≤10-10) [18, 19]; (2) A'-B', A'-C', A'-D', B'-C', B'-D', B'-F', C'-D', C'-E' and D'-E' are
the template-based homologous PPIs of A-B, A-C, A-D, B-C, B-D, B-F, C-D, C-E and D-E, respectively, with significant joint sequence similarity (joint E-value ≤ 10−40) [12]; (3) A', B', C', D', E' and F' is the homologous module of template M with high topology similarity (here, defined as protein aligned ratio ≥ 0.5 and PPI aligned ratio ≥ 0.3). The protein and PPI aligned ratio are defined as the number of proteins (PPIs) in the homologous module divided by the number of proteins (or PPIs) in the module template, respectively. Here, protein aligned ratio
≥ 0.5 and PPI aligned ratio ≥ 0.3 are considered as topology similarity according to the
statistical analysis of 75,706 modules (370 reference modules) in 1,442 species based on KEGG MODULE database [10].
2.2 Essential protein set and mapped essential protein set
To validate the biological meaning of core components in module families, we collected 11,384 essential proteins in 25 species from DEG (version 6.5) database [16], including 8 eukaryotes (e.g. Homo sapiens and Saccharomyces cerevisiae) and 17 prokaryotes (e.g.
Escherichia coli and Bacillus subtilis).
The functions of essential genes (or proteins) were considered as an essential foundation for all cells [27]. Therefore, the homologous proteins of an essential protein might be also indispensable [16]. Here, the protein of module template was considered as mapped essential protein when this protein is homologs of an essential protein recorded in DEG database. For example, the SMARCB1 (SNF5 homolog) is a mapped essential protein which is a homologous protein of both essential proteins Cc (Snf5-related 1, D. melanogaster) and Cs
7
2.3 Characteristics of modules and homologous modules of homologous
module
2.3.1 Connectivity of homologous module
To validate a homologous module which is relatively autonomous with respect to the other parts in a PPI network, we quantified the connectivity (Ct) of a module and is defined as
n t C m C 2
= [28],where n and m are the protein and PPI numbers in a module. For example, Ct is
1 if the proteins are complete connections in a module.
2.3.2 Biological function of homologous module
This study applied the relative specificity similarity (RSS) [29] to define the average RSS (AvgRSS) score for measuring the BP and CC similarities based on GO terms between all proteins in a homologous module. The AvgRSS score is given as
i j C AvgRSS n n i n j ≠ =
∑∑
= = , j) RSS(i, 2 1 1 (1)where i and j are any pair proteins in a module; n is the protein number of a module.
We defined the random module sets to measure BP and CC of a module family. Each module template constructed 50 random modules, which were selected randomly the same protein number from the genome of template's organism, and each random module was the same number of proteins with the module template. Among 1,625 template modules, the random data set consisted of 81,250 random modules.
8
2.4 PPI evolution score
Inferring homologous modules from multiple genomes provides an opportunity to understand the evolution, conserved functions, and core components of modules. Here, we measure the conservation of PPI family using PPI evolution score (PPIES). For evaluating
PPIES, we selected and clustered the division names of NCBI taxonomy database [30] into
five division groups, including mammals (MAM), vertebrates (VRT), invertebrates (INV), plants (PLN) and bacteria (BCT) (Table 3). For each PPI (z) of a module family, the PPI evolution score (PPIES) is defined as
B b P p I i V v M m DG PPIESz = + + + + + (2)
where DG is the number of division groups that contain at least one species in the module family; M, V, I, P, and B are the total numbers of species belong to MAM, VRT, INV, PLN, and BCT, respectively; and m, v, i, p, and b are the numbers of species belong to MAM, VRT, INV, PLN, and BCT for the PPI z, respectively. For each protein (k) in a module, we define its interface evolution score (IES) based on the maximum PPIES as
IESk= max1≤j≤g(PPIESj) (3)
where g is the number of proteins interacting to protein k. For example, the IES of protein
α-subunit (ATP5A1) is 9.91 in F1-ATP synthase-IF1 module (CORUM ID: 574 [31]) family
because of interacting with β-subunit (ATP5B; PPIES = 9.91), γ-subunit (ATP5C1; PPIES = 9.28), and δ-subunit (ATP5D; PPIES = 7.68) (Fig. 4E).
9
Chapter 3 Results and Discussion
In this study, we proposed a new concept (module family) and a method for inferring the module families and the essential elements of the life across multiple genomes through PPI families. Based on 1,625 module templates in MIPS CORUM database, we inferred 1,578 module families by searching the Integr8 database via 290,137 sequence-based PPI families and 86,252 structure-based PPI families. These homologous modules are often high connectivity and 89% and 96% module families have consensus BP and CC, respectively, based on GO terms. We further derived PPI and interface evolution scores to analyze the evolution and core components of a module family. According to PPI and interface evolution scores, the molecular functions (MF) of 808 proteins of core components are often for the survival of an organism and are highly correlated (Pearson's correlation=0.88) to the MFs of 8,364 essential genes in DEG database. Finally, we applied our concept “template-based homologous module” on the crystal structure (PDB code 3fki [32]) of RNA polymerase II in
Saccharomyces cerevisiae.
3.1 Homologous modules
To understand the functions and characteristics of module families, we collected 75,706 organism-specific modules of 370 reference modules in 1,442 organisms from KEGG MODULE database. According to the data set, the protein aligned ratios between ~56% (42,065) and ~82% (62,080) organism-specific modules and their respective reference modules were exceed 0.9 and 0.5, respectively (Fig. 5A). Currently, the PPIs of a module are often limited and not consistent for different databases. For example, KEGG organism-specific modules were lack of internal PPI annotations. To decide the topology
10
similar threshold between modules and module templates, we added module PPIs through three PPI databases: 1) 275,787 experimental PPIs in the annotated PPI database (IntAct, MIPS, DIP, MINT, and BioGRID); 2) 9,016 PPIs derived from PDB crystal structures [26]; and 3) our previous sequence-based and structure-based homologous PPIs with joint E-value
≤ 10-70 [12] and Z-score ≥ 4 [14], respectively. Among 75,706 organism-specific modules, 23,092 modules can be added at least one internal PPI and the internal PPIs of the reference module are determined by considering all PPIs of its organism-specific modules. The PPI aligned ratios of 65% organism-specific modules are more than 0.3 (Fig. 5B). Based on these observations, we set the protein aligned ratio and PPI aligned ratio of the topology similarity to 0.5 and 0.3, respectively.
Based on these results, we inferred template-based homologous modules of 1,625 high-quality module templates which were collected from MIPS CORUM database. The CORUM database provides manually annotated protein complexes, which assemble multiple proteins to perform biological functions, from mammalian organisms [20]. These 1,625 complexes include 1,165 (Homo sapiens), 268 (Mus musculus), 157 (Rattus norvegicus), and 35 modules (Bos Taurus) with at least three proteins. According to these 1,625 module templates, we identified 1,578 module families (including 53,529 modules in 1,679 species) via our previous 290,137 sequence-based PPI families and 86,252 structure-based PPI families [12, 14].
3.2 Characteristics of homologous modules
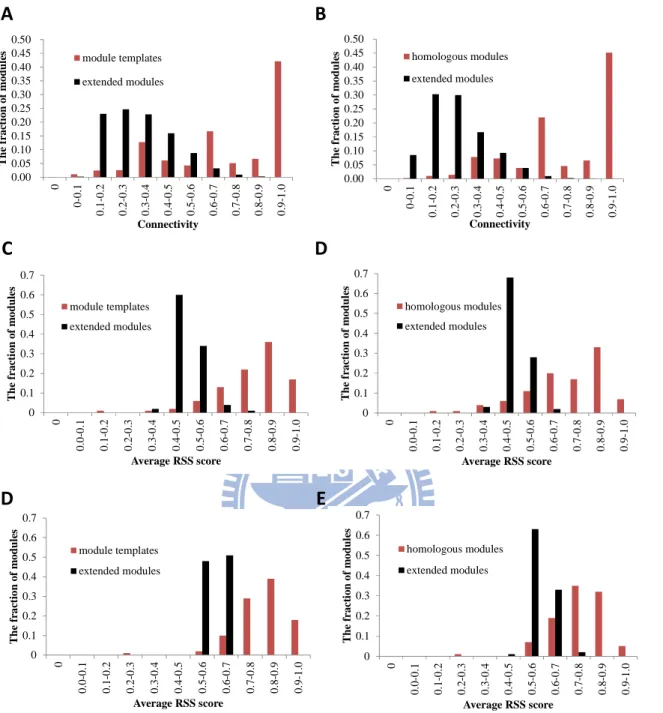
3.2.1 Connectivity of homologous modules
11
network, is often high connectivity in a PPI network. Figure 6A shows the relationships between the connectivity (Ct) of module templates and their respective extended modules, which extend one-layer PPIs and proteins for each protein in an original module. Among 1,625 module templates, connectivity values of 71% (1,114) templates are more than 0.6; conversely, 4% (71) extended modules are more than 0.6. In addition, 53,529 homologous modules and their extended modules are 78% (41,890) and 1% (752) with connectivity ≥ 0.6, respectively (Fig. 6B). 83% templates and 95% homologous modules have higher connectivity than their extended modules. In the F1-ATP synthase-IF1 module family, the
connectivity of this homologous module in Homo sapiens is 0.6 and the connectivity of its extended module decreases to 0.39. These results show that the homologous modules are relatively autonomous and high connectivity in a PPI network.
3.2.2 Consensus of biological function of homologous modules
Components of a module, assembling and cooperating in a PPI network, simultaneously preform a certain kind of biological functions. Here, we applied average RSS (AvgRSS) score to measure the consensus of biological functions (e.g. biological process similarity and location similarity) based on the GO terms of BP and CC. We compared the AvgRSS scores of BP and CC between the module templates and their respective extended modules. Among 1,625 module templates, the AvgRSS scores of BP (89% module templates) and CC (96% module templates) were more than 0.6 (Figs. 6C and 6E). Sequentially, The BP and CC
AvgRSS scores of 77% and 91% homologous modules, respectively, were more than 0.6(Figs.
6D and 6F). In contrast, only 2% extended modules of homologous modules have BP AvgRSS scores ≥ 0.6. The CC AvgRSS scores of 72% homologous module and 2% extended modules were more than 0.7.
12
Relative to the extended modules, 96% (1,451) and 97% (1,493) module templates have higher BP and CC AvgRSS scores. Similarly, 91% (27,569) and 95% (21,092) homologous modules have higher BP and CC AvgRSS scores than their extended modules. For instance, the BP and CC AvgRSS scores of F1-ATP synthase-IF1 homologous module in Homo sapiens
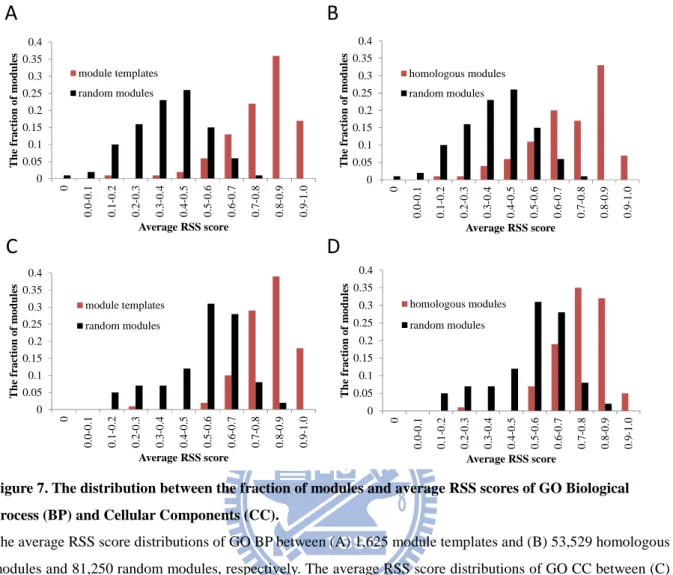
are 0.83 and 0.86, but the AvgRSS scores of BP and CC decrease to 0.45 and 0.61 for its extended module, respectively. Additionally, the AvgRSS scores of BP and CC of module templates (Figs. 7A and 7C) and homologous (Figs. 7B and 7D) modules are significantly greater than random modules. These results reveal that the homologous modules are high consensus in BP and CC.
3.3 Core components of homologous modules
We identified the core components of homologous modules by observing the relationship between IES values and 8,553 proteins in 1,578 module templates. These proteins were divided into two groups, unannotated and mapped essential proteins. Among 3,740 mapped essential proteins, the IES values (Equation 3) of 81% and 36% proteins are more than 6 and 8, respectively (Figs. 8A and 8B). To analyze the relationship between IES values and 3,740 mapped essential proteins, we defined the accuracy of each IES value interval as the number of mapped essential proteins divided by the total number of proteins. The correlation between accuracies and IES values is highly correlated (Pearson's correlation = 0.98). According to the annotation reliability (i.e. the number of homologs (recorded as essential proteins) of a template protein), the mapped essential proteins were divided into "mapped≥1 species" and "mapped≥2 species" groups. To compare with "mapped≥1" group, 98% and 62% "mapped≥2 species" essential proteins have IES ≥ 6 and ≥ 8, respectively, among 962 proteins (Fig. 8C). Here, we regarded the proteins (IES ≥ 8) and PPIs (PPIES ≥ 8) are core components of a module family.
13
Here, we used F1-ATP synthase-IF1 module family as an example to describe the core
component and IES scores (Fig. 4). During the process of oxidative phosphorylation, the chemical bond energy of ATP is produced by F1Fo ATP synthases through converting energy
stored in an electrochemical gradient of H+ or Na+ across the membrane into mechanical rotation [33]. Three subunits α- (ATP5A1), β- (ATP5B), and γ-subunits (ATP5C1) and their PPIs were considered as core components of F1-ATP synthase-IF1 module family through PPI
families (IES ≥ 9.28). For example, the PPI family of β- and γ- subunits across 1,372 species was constructed by the template interface chain D (β- subunits) and G (γ-subunits) of F1 ATP
synthase (PDB code: 2jdi [34]) of Bos taurus. Based on the profile of the F1-ATP synthase-IF1
module family in the organisms commonly used in molecular research projects (Fig. 4E), the PPI families of α-, β-, and γ- subunits are more conserved than others, such as ε-subunit (ATP5E) and ATPase inhibitor (ATPIF1). The ATP hydrolysis occurs in the α3β3 drives
rotation of the γ-subunit, which inserts long coiled-coil helices into central cavity of the α3β3
cylinder [35]. In addition, ATP hydrolysis activity was inhibited by the ε subunit of ATP synthase with C-terminal α-helical domain [33]. The protein sequences of ε subunit in mammals are different with other division groups, such as invertebrates, plants, and bacteria. On the other hand, the natural inhibitor of F1 ATP synthase regulates ATP synthase activity
with the N-terminal inhibitory sequences [36], but no homolog of ATPIF1 has been found in either chloroplasts or bacteria [33].
For the F1-ATP synthase-IF1 module family, the homologous proteins of ATP5A1,
ATP5B and ATP5C1 were the essential proteins in 11, 9 and 9 species (e.g. D. melanogaster and M. tuberculosis), respectively. In contrast, none homologous proteins of other components (e.g. ATPIF1) were essential proteins. These experimental results demonstrate that the core components of a module family preferred to be the essential elements for the survival of an organism.
14
3.4 Essential MF terms of GO
The GO terms provide the descriptions of BP, CC and MF of a protein (gene), such as catalysis and binding [17]. According to the modification of TF-IDF (term frequency–inverse document frequency) scoring scheme [37], we identified 181 essential GO MF terms to describe functional relationships of essential proteins and core components of module families (Table 4). First, we collected 8,364 essential proteins (called EP8364 set) from DEG database and 160,598 proteins (called CG27 set) in 27 completed genomes (25 species in DEG database and 2 species in module template set). These proteins in these two sets consist of at least one GO MF or GO BP terms. The occur ratio (CRt) of a GO MF term (t) is defined as
CRt=Pt/T, where Pt is the number of proteins with term t and T is the total number of proteins in sets EP8364 (8,364 proteins) or CG27 (160,598 proteins). For example, the occur ratio of the term "rRNA binding" is 0.0497 while Pt = 416 and T = 8,364 for the EP8364 set. The distributions of occur ratios of GO MF terms between the proteins in core components and the essential proteins are significantly similar (Pearson's correlation=0.88). In contrast, the Pearson's correlation of GO BP terms is 0.28 because the BP terms often describe a series of events accomplished by one or more ordered assemblies of molecular functions. The MF and BP terms are suitable for a protein and a module, respectively.
Sequentially, we developed "unique ratio (UR)" to statistically measure the GO MF term importance (specificity) to a protein by modifying the TF-IDF scoring scheme [37]. The unique ratio of a GO MF term (t) is defined as URt=CRtEP/CRtCG, where CRtEP and CRtCG are the occur ratios of term t in sets EP8364 and CG27, respectively. For example, the unique ratio of term "rRNA binding" is 9.72 while CRtEP =0.0497 and CRtCG =0.0051. Finally, we statistically selected 181 essential GO MF terms, which are significant specificity to essential proteins and core components with UR≥2, to avoid selecting the terms of specific species (e.g. azobenzene reductase activity) and high usage without the specificity (e.g. protein binding).
15
To analyze characteristics and functions of core components, we classified clustered these 181 essential GO MF terms into 12 groups, such as Translation (30 terms, 16%), Transcription (12 terms, 7%), Carbohydrate (26 terms, 14%) and Lipid (14 terms, 8%) metabolisms, Amino acid metabolism (12 terms, 7%) and RNA degradation (6 terms, 3%), Purine (12 terms, 7%) and Pyrimidine (4 terms, 2%) metabolism, and Oxidative phosphorylation (5 terms, 3%) (Fig. 9A and Table 4). The largest percentage (16%) of the essential GO MF terms is Translation group, such as rRNA binding (UR=9.72), translation release factor activity, codon specific (UR=6.48), structural constituent of ribosome (UR=4.71), and tRNA binding (UR=8.38). In the process of transcription, the information contained in a section of DNA is transferred to a newly assembled piece of messenger RNA (mRNA), which is a part of central dogma. The central dogma of molecular biology, including DNA replication, transcription, and translation, is the fundamental of life for sequence information transfer [38]. Among 181 essential GO MF terms, 30% essential GO MF terms are involving in the central dogma (Fig. 9A). Furthermore, we also analyzed the percentage of GO MF groups in 3,441 essential proteins (Fig. 9B). 71% essential proteins were annotated with GO MF terms which are relative to the central dogma, such as translation (54%).
Among 181 essential GO MF terms, 40 terms (e.g. acetyl-CoA carboxylase activity,
UR=9.25) are recorded in Carbohydrate and lipid metabolisms, which are for the energy
balance of organisms and for various biochemical processes responsible for the formation, breakdown and interconversion [39, 40]. 22% essential GO MF terms are participated in carbohydrate and lipid metabolisms. 18 essential GO MF terms are included in Amino acid metabolism (e.g. cysteine desulfurase activity, UR=6.89) and RNA degradation (e.g. 3'-5' exonuclease activity, UR=5.27), which play an important role of the energy balance in reuse of RNA and amino acids. Purine (ATP-dependent RNA helicase activity, UR=5.04) and pyrimidine (thymidylate kinase activity, UR=6.98) metabolisms are regarded as a modular
16
minimal cell model [41]. The generation of the biological energy occurs mainly in oxidative phosphorylation group [42]. These results show that most of these 181 essential GO MF terms are indispensable for the survival of an organism.
3.5 Core components and Essential MF terms
To analyze the characteristics and functional annotations of core components, we compared the essential proteins and the proteins of core component using derived 181 essential GO MF terms (Figs. 9B and 9C). The distributions of occur ratios in 181 essential GO MF terms were significantly similar between the core component set (i.e. 808 proteins of 1,578 template modules) and the essential protein set (i.e. 8,364 essential proteins) (Fig. 10). Both sets have four GO MF terms with high occur ratios, including structural constituent of ribosome, ATPase activity, nucleoside-triphosphatase activity, and identical protein binding. Interestingly, the occur ratio of the MF term chromatin binding (in cell cycle group) in the core component set is much higher than the one in the essential protein set (Fig. 9C). Chromatin is a condensed structure in eukaryotic cells, but prokaryotic cells do not possess histones to form chromatin [43]. Relative to all module templates belong to mammals (eukaryote), most of essential proteins in DEG database are collected from prokaryotes. Therefore, the essential protein set has few chromatin binding annotation.
On the other hand, the essential protein set contains two terms related with translation (i.e. rRNA binding and tRNA binding) with high peaks but not in the core component set (Fig. 9C). Since these two terms are prokaryote specific in GO database, this is the reason, that the core component set has low occur ratios in rRNA binding and tRNA binding annotations. Moreover, Figure 9B shows the percentages of 246 proteins of core components and 3,441 essential proteins in 12 groups of 181 essential GO MF terms. We found that 14% proteins of
17
core components related with cell cycle but 3% in essential proteins due to chromatin binding. Similarly, rRNA binding and tRNA binding (prokaryote-specific annotation) in translation are the causes of higher percentage in essential proteins (54%) than ones in the core components (25%). Our results suggest that the proteins of core components are considered as the essential proteins due to the significantly similar distributions of the occur ratios in 181 essential GO MF terms.
To verify whether the unannotated proteins of core components implied potential essential proteins, we analyzed orthologous proteins and function annotations of essential proteins. The homologous or orthologous proteins of an essential protein could be considered to be essential [16]. Here, we used the orthologs in PORC database [15] and 181 essential GO MF terms to analyze unannotated proteins of core components. Among 400 unannotated proteins (IES ≥ 8) of core components, 146 proteins (37%) are the orthologous proteins of essential proteins or annotated at least one of 181 essential GO MF terms. Furthermore, the GO MF term, which is the child of essential GO MF terms could be considered as the essential GO MF terms. Therefore, 116 unannotated proteins (29%) possess the children annotations of 181 essential GO MF terms. Moreover, 73% unannotated proteins with IES ≥ 9 have at least one of these three aspects (Tables 5 and 6).
3.6 Example analysis
The nucleosome remodeling and deacetylase module (NuRD, CORUM ID: 614) of
Homo sapiens consists of histone deacetylase 1/2 (HDAC1/HDAC2), histone-binding protein
RBBP4 (RBBP4), chromodomain-helicase-DNA-binding protein 3/4 (CHD3/CHD4), metastasis-associated protein MTA1 (MTA1), and lysine-specific histone demethylase 1A (KDM1) (Fig. 11). The NuRD module was considered as a key modulator of ageing associated chromatin defects [44-46] and found widely in mammals, vertebrates, invertebrates,
18
and plants [47].
Using the NuRD module in Homo sapiens as a module template, its homologous modules across 233 species and 5 division groups involve in regulating negative regulation of gene-specific transcription from RNA polymerase II promoter (Fig. 11A). Eight PPI families (e.g. CHD3-CHD4, CHD3-HDAC2, and HDAC1-HDAC2) of this module family were regarded as core components due to their PPIES ≥ 8 (Fig. 11B). Among five predicted core proteins (i.e. HDAC1/2, RBBP4, and CHD3/CHD4), three proteins (i.e. HDAC1/2 and PBBP4) are the homologous proteins of essential proteins recorded in DEG database (Fig. 11C). CHD3 and CHD4 were annotated with several essential GO MF terms, such as chromatin binding and ATP-dependent DNA helicase activity (Fig. 11D). In addition, the CHD4, which possesses intrinsic ATP-dependent nucleosome-remodeling activity, can prevent accumulation of spontaneous DNA damage and increase ionizing radiation sensitivity [48]. These results show that proteins CHD3 and CHD4 should be core proteins of the NuRD module family.
Figure 11B shows that the PPI families of MTA1-HDAC1/2 and MTA1-CHD3/4 were conserved in mammals, vertebrates, and invertebrates. Metastasis-associated protein 1 (MTA1), the first gene found in the family of cancer progression-related genes, is widely upregulated in human cancers and plays an important role in tumorigenesis and tumor aggressiveness, such as tumor invasion and metastasis in breast cancer [49-51]. The MTA1 was lack of homologous proteins in Arabidopsis thaliana, Schizosaccharomyces pombe and
Saccharomyces cerevisiae, in homologene database of NCBI [30]. In addition, cancers in
plants (i.e. galls) grow locally rather than by metastasis [52]. Therefore, homologous PPIs of MTA1-HDAC1/2 and MTA1-CHD3/4 are not found in plants and fungi. On the other hand, RBBP4 is a conserved histone-binding protein and shares subunits of several multi-protein complexes involving in the establishment of heterochromatin [53, 54]. Because the
19
prokaryotic cells do not possess histones to package the DNA to form the chromatin [43], RBBP4-HDAC1/2 and RBBP4-CHD3/4 PPI families are highly conserved in eukaryotes but lack in bacteria (Fig. 11B).
In the second example, we used BRG1-based SWI/SNF chromatin remodeling complex (CORUM ID: 2852, regulating cell proliferation and differentiation [55]) as a module template to identify homologous modules and the core components (Fig. 12). Four proteins of this complex were considered as the core components because their IES values ≥ 8. In this complex, three proteins (ACTL6A, SMARCC1 and SMARCB1) are the mapped essential proteins based on the DEG database. The protein SMARCC2 should be a mapped essential protein according to its GO MF terms: chromatin binding, transcription coactivator activity and DNA binding. These results indicate that the proteins of core components often are essential proteins based on orthologs of essential proteins, the essential GO MF terms, and children terms of essential GO MF terms.
3.7 Application: Crystal structure-based homologous modules
We applied our concept, “template-based homologous module”, on crystal structures derived from PDB database. In the first step of gene expression in eukaryotic cells, RNA polymerase II and its associated factors form an elaborate protein module that transcribes DNA sequences into pre-mRNAs [56]. To study eukaryotic gene expression machinery, it is essential for understanding the mechanisms that regulate transcription via protein-protein interactions within the RNA polymerase II apparatus [32]. In our results, we used the crystal structure (PDB code 3fki [32]) of RNA polymerase II in Saccharomyces cerevisiae as a module template to identify the homologous modules (Fig. 13A). There are 12 proteins involving in this module, including DNA-directed RNA polymerase II subunit RPB1 (RPB1),
20
DNA-directed RNA polymerase II subunit RPB2 (RPB2) and DNA-directed RNA polymerase II subunit RPB8 (RPB8). Figure 13 shows the method and the searching result of RNA polymerase II module family which comprises seven homologous modules in Homo sapiens,
Mus musculus, Drosophila melanogaster and Saccharomyces cerevisiae. First, we identified
20 template-based PPI families (e.g. RPB1-RPB2 and RPB1-RPB8 PPI families of interface of chain A-B and A-H, respectively) with interface similarity Z-values ≥ 3 from Integr8 database (Fig. 13B). Next, we combined these PPI families to identify homologous modules (a protein module family), including three homologous modules of Homo sapiens, Mus
musculus and Drosophila melanogaster which comprise 12 proteins (proteins aligned ratio is
1.00) and 19 PPIs (PPI aligned ratio is 0.94) and a homologous module of Saccharomyces
cerevisiae which comprises 12 proteins (proteins aligned ratio is 1.00) and 20 PPIs (PPI
aligned ratio is 1.00) (Fig. 13B). These homologous modules are recorded in KEGG complex module (RNA polymerase II, eukaryotes; M00180) [57] for supporting our result. In addition, all proteins of four homologous modules in this module family have the same MF (e.g. DNA-directed RNA polymerase activity) and BP terms (e.g. transcription from RNA polymerase II promoter) in GO database. Similarly, interacting domain pairs [58] (e.g. RNA_pol_Rpb1_3–RNA_pol_Rpb8 of RPB1–RPB8) are conserved in the module family. Moreover, we provided the binding model to analyze the binding forces based on the template, such as hydrogen bonds, including Leu597-Tyr102 and Leu598-Arg25 of interface A-H and Glu846-Arg1135, Lys345-Asp1156 and Asp346-Arg1100 of interface A-B). Our results suggested all interacting residues forming the hydrogen bonds are often highly conserved and useful for observing the interface evolution across multiple species (Fig. 13A).
A tightly associated 10-subunit core and a heterodimeric subcomplex of subunits RPB4 and RPB7 assembled the dodecameric protein of RNA polymerase II [59]. For catalyzing RNA-chain polymerization, the 10-subunit core harbors the central transesterase activity, but
21
the proteins RPB4 and RPB7 enables promoter-dependent initiation by the polymerase and supports yeast growth under stress conditions [60, 61]. Based on above works, the homologous module in Saccharomyces cerevisiae comprised 20 PPIs but only 19 PPIs in
Homo sapiens, Mus musculus and Drosophila melanogaster. We found a PPI (i.e. RPB4-
RPB2) of homologous module in Saccharomyces cerevisiae does not exists in Homo sapiens,
Mus musculus and Drosophila melanogaster (Fig. 14). Through the binding model and
multiple species alignments from the template module, we found some contact residues in
Saccharomyces cerevisiae are changed (e.g. Tyr1217 of RPB2 in Saccharomyces cerevisiae to
Met1172 of POLR2B in Homo sapiens and Ser4 of RPB4 in Saccharomyces cerevisiae change to Gly4 of POLR2D in Homo sapiens) or absent (e.g. Arg1220 and Ser1221 of RPB2 and Arg12, Arg13, Arg14, Leu15 and Lys16 of RPB4) in Homo sapiens that result in the interaction losing (Fig. 14). For the programmed development of multicellular organisms and the homeostasis of cells, it is critical to regulate RNA polymerase II activity [62]. RPB4 involved in yeast growth under stress conditions, but resistance of stress in Homo sapiens,
Mus musculus and Drosophila melanogaster is more complicated. These results implied the
interactome are diverse between unicellular (e.g. Saccharomyces cerevisiae) and multicellular organisms (e.g. Homo sapiens, Mus musculus and Drosophila melanogaster).
3.8 Discussion
Modules could provide insights of a PPI network evolution for two reasons. First, organizing a biological system into modules permits the changes to affect the mechanisms within one module without perturbing other modules [6]. Second, the new biological functions can be created by the combination and reuse of modules [7, 63]. To identify and analyse homologous modules in the PPI networks across multiple species provide a new opportunity for exploring the evolutionary fundamentals of biological systems. Among 1,578
22
module families, we found that all proteins of 133 module families were recognized as core components. Interestingly, these module families were often involving in important biological processes, such as central dogma and cell cycle. This observation implied that these modules could be regarded as the essential modules of a life. For instance, BRG1-based SWI/SNF chromatin remodeling module family, which regulates cell proliferation and differentiation in eukaryotes, comprise four proteins which are regarded as core components (e.g. SMARCC1, SMARCC2, SMARCB1, and ACTL6A) (Fig. 12).
23
Chapter 4 Conclusions
4.1 Summary
This study proposes a new concept “module family” that consists of homologous modules derived from a large complete genomic database through a module template and PPI families. The experimental results show that homologous modules are highly connected and perform a certain kind of biological function. For a module family, its core components, which consist of conserved PPIs across multiple species and division groups, often forms the essential elements for the survival of an organism according to 181 essential GO MF terms. We believe that the module family and core components provide new insights for understanding module evolution and functions in the PPI networks of biological systems.
4.2 Major contributions and future works
According to our knowledge, module family, which comprises a group of homologous modules, is the first approach that identifies homologous modules of the module template from complete genomes through PPI families. We have developed a new method to identify homologous modules based on module templates of manually annotated protein complexes and crystal structures. Furthermore, the conserved and divergent internal PPIs of homologous modules provided clues to infer essential elements of modules.
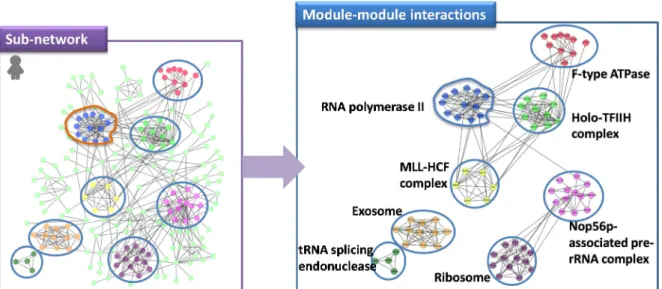
For the origination and diversity of novel phenotypes, we will focus on two issues: “What is (are) the essential element(s) of life” and “What is the formation of a new species”. Some modules are evolutionarily cohesive, in other words, these cohesive modules are conserved across multiple species [64]. The relationships between the connected modules allow construction of the module-module interaction network which is regarded as the
24
connection between different functional modules in the interactome [65]. Intra-module proteins have less widespread mutational effects but inter-module proteins, which integration occurs between modules, have higher rate of amino-acid substitutions [66, 67]. According to previous studies, inter-module interactions have more evolutionary modifications than intra-module interactions.
Inter-module interactions of RNA polymerase II module in human are mediated by protein-protein interaction, such as POLR2B-MEN1, POLR2B-WWOX, and POLR2B-GSK3B (Fig. 15). In other word, the inter-module proteins interacting with POLR2B, including MEN1, WWOX, and GSK3B, and participate other BP annotations of proliferation, steroid metabolic process, and glycogen metabolic process, respectively. Multiple endocrine meoplasia type 1 (MEN1) is a subunit of mixed-lineage leukemia (MLL) complex, a proto-oncogene with implication of development and leukemia pathogenesis [68, 69]. WWOX contains two WW domains at N-terminal and plays a role in regulating steroid metabolism [70]. Glycogen synthase kinase β (GSK3B) is a serine-threonine kinase with potent tumour suppressor qualities and regulates glucose storage and cell proliferation [71, 72]. In this section, we would propose a real case about the module-module interaction between RNA polymerase II module and MLL1 complex module.
The mechanism of RNA polymerase II module is involved in transcription that is the process of creating a complementary RNA copy of a sequence of DNA. MLL core complex uses a non-processive mechanism to catalyze multiple lysine methylations of histones, which is an important epigenetic indexing system for transcriptionally active and inactive chromatin domains in eukaryotic genomes [73]. Based on our concept of module family, we identified the module families of RNA polymerase and MLL complex. The module family of RNA polymerase was descripted above (Fig. 13 and 14). The MLL complex module in Homo
25
(MLL), menin (MEN1), Set1/Ash2 histone methyltransferase complex subunit ASH2 (ASH2L), rtinoblastoma-binding protein 5 (RBBP5), WD repeat-containing protein 82 (WDR82) and WD repeat-containing protein 5 (WDR5). In the MLL complex module family, there are two homologous modules (6 proteins and 15 PPIs) in Homo sapiens, one module (6 proteins and 15 PPIs) in Drosophila melanogaster and one module (5 proteins and 10 PPIs) in
Saccharomyces cerevisiae. Interestingly, we found histone-lysine N-methyltransferase MLL2
(MLL2) is the homologs of MLL1 in Homo sapiens and could replace the MLL1 to form the MLL complex. However, only one homologs histone-lysine N-methyltransferase trithorax (trx) and histone-lysine N-methyltransferase, H3 lysine-4 specific (SET1) is in Drosophila
melanogaster and Saccharomyces cerevisiae, respectively [74]. In addition, menin activates
the transcription of differentiation-regulating genes by covalent histone modification, and that this activity is related to tumor suppression by MEN1 [75-78]. Menin in the MLL complex associated with RNA polymerase II in Homo sapiens [79] and Drosophila melanogaster. However, there are no Menin homologs found in Saccharomyces cerevisiae genome. SET1 replaces the part of interaction between RNA polymerase II module and MLL complex module in Saccharomyces cerevisiae (Fig. 15). According to our results, we could find not only diversity of intra-module interactions but also diversity of inter-module interactions between different organisms. It is useful to homologous modules in across-genome scale and offer biologists to realize evolutions of module and behaviors of interactome.
26
Tables
Table 1. The list of the number of modules in TOP 20 organisms from KEGG MODULE database
KEGG Taxonomy ID
NCBI
Taxonomy ID Organism Codes Organisms
No. of modules in KEGG MODULE database
T00772 507522 kpe Klebsiella pneumoniae 342 141 T00566 272620 kpn Klebsiella pneumoniae subsp. pneumoniae MGH 78578 141 T00910 484021 kpu Klebsiella pneumoniae NTUH-K2044 139 T01170 640131 kva Klebsiella variicola At-22 138 T01342 701347 esc Enterobacter cloacae SCF1 135 T00044 155864 ece Escherichia coli O157:H7 EDL933 134 T00672 439855 ecm Escherichia coli SMS-3-5 134 T00507 399742 ent Enterobacter sp. 638 133 T00784 409438 ecy Escherichia coli O152:H28 SE11 132 T00949 544404 etw Escherichia coli O157:H7 TW14359 132 T00831 585056 eum Escherichia coli O17:K52:H18 UMN026 132 T01422 741091 rah Rahnella sp. Y9602 132 T00778 444450 ecf Escherichia coli O157:H7 EC4115 131 T00338 364106 eci Escherichia coli O18:K1:H7 UTI89 131 T00829 585057 ect Escherichia coli O7:K1 IAI39 131 T00591 331112 ecx Escherichia coli O9 HS 131 T01098 573235 eoj Escherichia coli O26:H11 11368 131 T00068 316407 ecj Escherichia coli K-12 W3110 130 T00828 585034 ecr Escherichia coli O8 IAI1 130 T00048 386585 ecs Escherichia coli O157:H7 Sakai 130
27
Table 2. The list of data sets using definition and verification of module family
Data sets Comments
MIPS CORUM database [20] The CORUM database using as module template set provides manually annotated protein complexes, which assemble multiple proteins to perform biological functions, from mammalian organisms.
Annotated PPI database 275,787 experimental PPIs in the annotated PPI database (IntAct [21], BioGRID [22], DIP [23], MIPS [24], and MINT [25]) Predicted homologous PPI set Our previous sequence-based and structure-based homologous PPIs with joint E-value ≤ 10
-40 [12] and Z-score ≥ 3 [14], including 290,137 sequence-based PPI families and 86,252 structure-based PPI families
Integr8 database [15] A complete genomic database (Integr8 version 103, containing 6,352,363 protein sequences in 2,274 species)
KEGG MODULE database [11] KEGG organism-specific modules is defined as a tight functional unit and complexes in the pathway through a set of orthologs Gene Ontology (GO) database [17] We derive GO biological process (BP) to annotate homologous modules and GO molecular function (MF) to annotate core
components of module family.
Extended module data set Extending one-layer PPIs and proteins for each protein in an original module through homologous PPIs
Random data sets Each module template constructed 50 random modules, which were selected randomly the same protein number from the genome of template's organism, and each random module was the same number of proteins with the module template.
PORC ortholog database [15] PORC (putative orthologous clusters) are defined as orthologous families from Integr8 database.
Essential genes database (DEG) [16] We collected 11,384 essential proteins in 25 species from DEG (version 6.5) database, including 8 eukaryotes and 17 prokaryotes. EP8364 set We collected 8,364 essential proteins (called EP8364 set) from DEG database with at least one GO MF or GO BP terms.
CG27 set 160,598 proteins (called CG27 set) in 27 completed genomes (25 species in DEG database and 2 species in module template set) derived from Integr8 database
28
Table 3. Modified division groups from NCBI taxonomy database
Division group Division code a Division name b Number of species used in module family MAM PRI Primates 4 ROD Rodents MAM Mammals VRT VRT Vertebrates 3
INV INV Invertebrates 27
PLN c PLN Plants 42 BCT BCT Bacteria 1,596 N/A d PHG Phages 7 VRL Viruses SYN Synthetic UNA Unassigned ENV Environmental samples
a,b The division names and codes are derived from NCBI taxonomy database [30] (ftp://ftp.ncbi.nih.gov/pub/taxonomy/taxdump.tar.gz). c
The PLN division group includes plants and fungi (e.g. Saccharomyces cerevisiae).
d
. According to only 478 homologous modules (< 1 %) of 53,529 homologous modules (1,679 species) belong to phages, viruses, synthetic, unassigned, and environmental samples, therefore, we excluded these divisions.
29
Table 4. The 181 essential GO molecular functions (MF) terms
GO ID GO term Classification Number of proteins (CG27 a) Occur Ratio b (CG27; total 160,598 proteins) Number of essential proteins Occur Ratio (essential proteins; total 8,364 proteins) Unique ratio c (essential proteins) Number of proteins of templates (IES ≥ 8) d Occur Ratio (IES ≥ 8; total 808 proteins) Unique ratio (IES ≥ 8)
GO:0019843 rRNA binding Translation 822 0.0051 416 0.0497 9.7173 3 0.0037 0.7254 GO:0004820 glycine-tRNA ligase activity Translation 53 0.0003 24 0.0029 8.6948 0 0.0000 0.0000 GO:0000049 tRNA binding Translation 394 0.0025 172 0.0206 8.3822 2 0.0025 1.0089 GO:0004818 glutamate-tRNA ligase activity Translation 40 0.0002 17 0.0020 8.1605 1 0.0012 4.9690 GO:0004827 proline-tRNA ligase activity Translation 38 0.0002 16 0.0019 8.0847 1 0.0012 5.2305 GO:0004832 valine-tRNA ligase activity Translation 41 0.0003 17 0.0020 7.9614 0 0.0000 0.0000 GO:0004825 methionine-tRNA ligase activity Translation 38 0.0002 15 0.0018 7.5794 1 0.0012 5.2305 GO:0004814 arginine-tRNA ligase activity Translation 54 0.0003 21 0.0025 7.4671 1 0.0012 3.6807 GO:0004824 lysine-tRNA ligase activity Translation 49 0.0003 19 0.0023 7.4453 1 0.0012 4.0563 GO:0004826 phenylalanine-tRNA ligase activity Translation 88 0.0005 32 0.0038 6.9822 0 0.0000 0.0000 GO:0004823 leucine-tRNA ligase activity Translation 43 0.0003 15 0.0018 6.6981 0 0.0000 0.0000 GO:0016149 translation release factor activity, codon specific Translation 83 0.0005 28 0.0033 6.4775 1 0.0012 2.3947 GO:0004831 tyrosine-tRNA ligase activity Translation 45 0.0003 15 0.0018 6.4004 0 0.0000 0.0000 GO:0004822 isoleucine-tRNA ligase activity Translation 39 0.0002 13 0.0016 6.4004 1 0.0012 5.0964 GO:0004817 cysteine-tRNA ligase activity Translation 47 0.0003 15 0.0018 6.1280 0 0.0000 0.0000 GO:0004526 ribonuclease P activity Translation 71 0.0004 22 0.0026 5.9496 0 0.0000 0.0000 GO:0004829 threonine-tRNA ligase activity Translation 49 0.0003 15 0.0018 5.8779 0 0.0000 0.0000 GO:0004816 asparagine-tRNA ligase activity Translation 33 0.0002 10 0.0012 5.8185 0 0.0000 0.0000
a
The CG27 set (160,598 proteins annotated ≥ 1 GO MF terms) consists of 25 species in DEG and 2 species in module template set.
b The occur ratio of a GO MF term is defined as the number of proteins annotated this terms divided by the total number of proteins in the set. c The unique ratio of a GO MF term is defined as the occur ratio of a GO MF term divided by the occur ratio in 27 species genome set.