Controlled synthesis of highly dispersed platinum nanoparticles in
ordered mesoporous carbons{
Shou-Heng Liu,
aRong-Feng Lu,
bShing-Jong Huang,
aAn-Ya Lo,
aShu-Hua Chien
bcand Shang-Bin Liu*
a Received (in Cambridge, UK) 26th May 2006, Accepted 19th June 2006First published as an Advance Article on the web 6th July 2006 DOI: 10.1039/b607449a
A novel route has been developed to fabricate ordered carbon mesoporous materials with well-dispersed, highly stable Pt nanoparticles of ca. 2–3 nm on the pore walls using platinum acetylacetonate as the co-feeding carbon and Pt precursor. Recent developments in the fabrication of porous carbon supports with high surface areas, controlled porosity, and hence a propensity towards lowering the metal catalyst loading have received considerable attention in the R&D of electrode catalysts for fuel cells.1–9Among them, precious-metal incorporated carbon
mesoporous materials (CMMs)10,11have been extensively studied particularly for their applications as carriers for hydrogen storage12
and as catalyst supports for fuel cells.3–9It is well-known that the properties of the incorporated metals depend strongly on their particle size, shape, and dispersion. Conventional techniques for embedding platinum or mixed-metal nanoparticles in the porous supports typically invoke post-synthesis treatment either by impregnation,5–7,10f adsorption,2b or ion-exchange13 methods. However, these aforementioned methods normally lead to uncontrolled growth of metal particle size and shape and aggregation/sintering at elevated temperatures due to minimization of the surface chemical potential.
Here we report a novel procedure for synthesizing a new mesoporous platinum–carbon nanocomposite based on the pyrolysis of carbon and Pt precursors in a mesoporous silica, such as SBA-15.14However, unlike previous attempts by which
Pt(0) clusters with an average diameter of 10–40 nm were found to be either buried in the glassy carbon matrix15or on the exterior
surfaces of the carbon rod,4,5 our samples prepared by direct replicated synthesis using organometallic compounds as the Pt precursor and co-feeding secondary carbon source result in nanosized Pt(0) particles with an average diameter of ca. 2–3 nm studded on the interior pore walls of the tubular CMM. As will be discussed later, the Pt-CMMs so fabricated possess highly stable, well-dispersed Pt nanoparticles that are exposed to the environ-ment and hence render applications not only as adsorbents for hydrogen storage but also as carbon monoxide-tolerant electro-catalytic materials for direct methanol (DMFC) and proton exchange membrane fuel cells (PEMFC).
The SBA-15 mesoporous silica template was synthesized according to the procedures reported earlier.14 Further direct replication of SBA-15 material into Pt-CMMs were carried out by the following steps: (i) about 0.5 g of calcined SBA-15 was dehydrated at 673 K for 4 h under vacuum; (ii) varied amounts of platinum acetylacetonate (Pt(acac); 98%, Acros) were dispersed in the furfuryl alcohol (FA; 98%, Acros) and trimethylbenzene (TMB; 98%, Acros) under ultrasonication; (iii) oxalic acid (98%, Acros) was used as the acid catalyst for polymerization of the FA solution; (iv) the mixture solution infiltrated in SBA-15 by incipient wetness impregnation at room temperature, followed by polymerization at 333 K then at 353 K, each for 12 h in air; (v) the resultant composite was treated at 423 K for 3 h, ramped to 573 K with a heating rate of 1 K min21, then to 1073 K with a heating rate of 5 K min21and maintained at that temperature for 3 h; (vi) the carbonization procedure was performed under vacuum; (vii) finally, the resultant black powders were leached with HF (1 wt%) aqueous solution for at least 24 h to remove the silica template, washed with distilled water and alcohol, then dried at 373 K to obtain the Pt-CMMs. Samples were characterized by various analytical and spectroscopic techniques, as described in the ESI.{
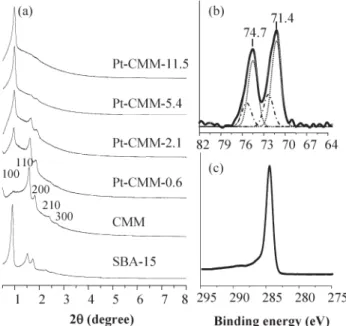
As shown in Fig. 1a, the XRD pattern observed for CMM synthesized in the absence of organometallic Pt(CH(COCH3)2)2
aInstitute of Atomic and Molecular Sciences, Academia Sinica, Taipei
10617, Taiwan. E-mail: [email protected]; Fax: +886-2-2362-0200; Tel: +886-2-2366-8230
bInstitute of Chemistry, Academia Sinica, Taipei 11529, Taiwan c
Dept. of Chemistry, National Taiwan University, Taipei 10617, Taiwan {Electronic supplementary information (ESI) available: Characterization methods, Pt dispersion measurements, CO tolerance tests and evaluation of electrocatalytic performances. See DOI: 10.1039/b607449a
Fig. 1 (a) Powder XRD patterns of SBA-15, CMM, and various Pt-CMMs samples; (b) platinum XPS spectrum of Pt-CMM-0.6; (c) carbon XPS spectrum of Pt-CMM-0.6.
COMMUNICATION www.rsc.org/chemcomm | ChemComm
This journal is ß The Royal Society of Chemistry 2006 Chem. Commun., 2006, 3435–3437 | 3435
Published on 06 July 2006. Downloaded by National Kaohsiung University of Applied Sciences on 09/10/2014 08:58:17.
mesoporous carbon CMK-5, which may be achieved in the absence of a Pt precursor (route from Fig. 3a to 3f),10fthe synthetic route (from Fig. 3a through 3d) reported here results in uniform formation of Pt nanoparticles 2–3 nm in size in the inner pore walls of the tubular carbons. Consequently, two types of pores with respective diameters of 2.3 and 4.0 nm were identified especially for samples with low Pt loading, e.g., Pt-CMM-0.6. Upon increasing the Pt loading (>0.6 wt%), Pt-CMMs with structures resembling that of rod-like CMK-310c–e (Fig. 1a) but studded with well-dispersed Pt nanoparticles (Fig. 3e) were generated. As such, only one type of mesopore with a diameter of 2.3 nm was found. It is noted that the Pt metal particles in these Pt-CMMs are highly stable and sustain repeated oxidation and reduction cycles.
The dispersions of Pt nanoparticles in various samples obtained from H2chemisorption studies{ are also depicted in Table 1. The
Pt dispersion observed for the Pt-CMM-0.6 sample was found to increase by about four-fold compared to that of the Pt-CMM-0.6I prepared by the conventional impregnation method. For Pt-CMMs, a gradual decrease in Pt dispersion with increasing Pt loading was observed, as expected. Separate hydrogen uptake experiments performed under 113.3 kPa at 77 K showed that the H2 adsorption capacity for Pt-CMM-0.6 amounts to 2.06 wt%
corresponding to an increase of ca. 25% compared to that of CMM-0.6I (not shown). To investigate the CO tolerance of Pt-CMMs, we conducted additional pulsed chemisorption studies by competitive adsorption of CO with H2.{ Accordingly, a metallic
surface area of ca. 190 m2g(Pt)21for the Pt-CMM-0.6 sample was
deduced in which ca. 78% remained active after pre-adsorption of 500 ppm of CO. In the case of Pt-CMM-0.6I, however, nearly all Pt surfaces were inactivated after exposure to the pre-adsorbed CO.
To evaluate the electrocatalytic activity of Pt-CMMs during methanol oxidation reaction, we also performed cyclic voltam-metry (CV) in sulfuric acid.{ Specifically, we compared the performances of our Pt-CMM-11.5 sample with a commercial Pt/C catalyst (Johnson-Matthey; 20 wt% Pt on Vulcan XC-72). It was found that our sample exhibits a catalytic activity surpassing that of the commercial catalyst. In particular, a lower oxidation
peak potential was observed for the Pt-CMMs. In terms of the ratio of the forward anodic peak current density (If) to the reverse
anodic peak current density (Ib), the If/Ibvalue obtained for the
Pt-CMM-11.5 was 4.23, which amounts to about a four-fold increase compared to that of the commercial Pt/C catalyst, indicating that the latter is more vulnerable to coking by carbonaceous deposits18 and less tolerant towards CO poisoning. Further studies are being undertaken to investigate the detailed mechanism of CO tolerance of Pt-CMMs and to fabricate various metal and bi-metal supported CMMs.
Notes and references
1 (a) A. Roucoux, J. Schulz and H. Patin, Chem. Rev., 2002, 102, 3757; (b) J. M. Thomas, B. F. Jhonson, R. Raja, G. Sankar and P. A. Midgley, Acc. Chem. Res., 2003, 36, 20; (c) K. Y. Chan, J. Ding, J. Ren, S. Cheng and K. Y. Tsang, J. Mater. Chem., 2004, 14, 505.
2 (a) G. Wang, G. Sun, Z. Zhou, J. Liu, Q. Wang, S. Wang, J. Guo, S. Yang, Q. Xin and B. Yi, Electrochem. Solid-State Lett., 2005, 8, A12; (b) R. Ubago-Pe´rez, F. Carrasco-Marı´n and C. Moreno-Castilla, Appl. Catal., A, 2004, 275, 119.
3 (a) G. S. Chai, S. B. Yoon, J. S. Yu, J. H. Choi and Y. E. Sung, J. Phys. Chem. B, 2004, 108, 7074; (b) G. S. Chai, S. B. Yoon, J. H. Kim and J. S. Yu, Chem. Commun., 2004, 2766.
4 W. C. Choi, S. I. Woo, M. K. Jeon, J. M. Sohn, M. R. Kim and H. J. Jeon, Adv. Mater., 2005, 17, 446.
5 J. Ding, K. Y. Chan, J. Ren and F. S. Xiao, Electrochim. Acta, 2005, 50, 3131.
6 F. Su, J. Zeng, X. Bao, Y. Yu, J. Y. Lee and X. S. Zhao, Chem. Mater., 2005, 17, 3960.
7 (a) V. Raghuveer and A. Manthiram, Electrochem. Solid-State Lett., 2004, 7, A336; (b) V. Raghuveer and A. Manthiram, J. Electrochem. Soc., 2005, 152, A1504.
8 K. Wlkander, H. Ekstrom, A. E. C. Palmqvist, A. Lundblad, K. Holmberg and G. Lindbergh, Fuel Cells, 2006, 6, 21.
9 J. B. Joo, P. Kim, W. Kim, J. Kim and J. Yi, Catal. Today, 2006, 111, 171.
10 (a) R. Ryoo, S. H. Joo and S. Jun, J. Phys. Chem. B, 1999, 103, 7743; (b) M. Kaneda, T. Tsubakiyama, A. Carlsson, Y. Sakamoto, T. Ohsuna, O. Terasaki, S. H. Joo and R. Ryoo, J. Phys. Chem. B, 2002, 106, 1256; (c) S. Jun, S. H. Joo, R. Ryoo, M. Kruk, M. Jaroniec, Z. Liu, T. Ohsuna and O. Terasaki, J. Am. Chem. Soc., 2000, 122, 10712; (d) S. H. Joo, R. Ryoo, M. Kruk and M. Jaroniec, Chem. Commun., 2001, 349; (e) S. H. Joo, R. Ryoo, M. Kruk and M. Jaroniec, J. Phys. Chem. B, 2002, 106, 4640; (f) S. H. Joo, S. J. Choi, I. Oh, J. Kwak, Z. Liu, O. Terasaki and R. Ryoo, Nature, 2001, 412, 169.
11 (a) H. Darmstadt, C. Roy, S. Kaliaguine, S. J. Choi and R. Ryoo, Carbon, 2002, 40, 2673; (b) H. Darmstadt, C. Roy, S. Kaliaguine, T.-W. Kim and R. Ryoo, Chem. Mater., 2003, 15, 3300; (c) B. Sakintuna and Y. Yu¨ru¨m, Ind. Eng. Chem. Res., 2005, 44, 2893.
12 (a) J. Pang, J. E. Hampsey, Z. Wu, Q. Hu and Y. Lua, Appl. Phys. Lett., 2004, 85, 4887; (b) R. Gadiou, S.-E. Saadallah, T. Piquero, P. David, J. Parmentier and C. Vix-Guterl, Microporous Mesoporous Mater., 2005, 79, 121; (c) B. Z. Fang, H. S. Zhou and I. Honma, J. Phys. Chem. B, 2006, 110, 4875.
13 V. Lordi, N. Yao and J. Wei, Chem. Mater., 2001, 13, 733.
14 D. Zhao, J. Feng, Q. Huo, N. Melosh, G. H. Fredrickson, B. F. Chmelka and G. D. Stucky, Science, 1998, 279, 548.
15 (a) N. L. Pocard, D. C. Alsmeyer, R. L. McCreery, T. X. Neenan and M. R. Callstrom, J. Am. Chem. Soc., 1992, 114, 769; (b) H. D. Hutton, N. L. Pocard, D. C. Alsmeyer, O. J. A. Schueller, R. J. Spontak, M. E. Huston, W. Huang, R. L. McCreery, T. X. Neenan and M. R. Callstrom, Chem. Mater., 1993, 5, 1727.
16 (a) H. Darmstadt, C. Roy, S. Kaliaguine, S. J. Choi and R. Ryoo, Carbon, 2002, 40, 2673; (b) H. Darmstadt, C. Roy, S. Kaliaguine, T.-W. Kim and R. Ryoo, Chem. Mater., 2003, 15, 3300; (c) B. Sakintuna and Y. Yu¨ru¨m, Ind. Eng. Chem. Res., 2005, 44, 2893.
17 N. Toshima and K. Hirakawa, Polym. J. (Tokyo), 1999, 31, 1127. 18 J. Huang, Z. Liu, C. He and L. M. Gan, J. Phys. Chem. B, 2005, 109,
16644. Fig. 3 Schematic drawings of synthesis routes for (a) A (e) Pt-CMMs,
and (a) A (f) CMK-5.
This journal is ß The Royal Society of Chemistry 2006 Chem. Commun., 2006, 3435–3437 | 3437
Published on 06 July 2006. Downloaded by National Kaohsiung University of Applied Sciences on 09/10/2014 08:58:17.