行政院國家科學委員會專題研究計畫 期中進度報告
C 型肝炎病毒非結構蛋白 2 之功能性研究(2/3)
計畫類別: 個別型計畫
計畫編號: NSC94-2320-B-006-081-
執行期間: 94 年 08 月 01 日至 95 年 07 月 31 日
執行單位: 國立成功大學醫學檢驗生物技術學系
計畫主持人: 楊孔嘉
共同主持人: 張定宗
計畫參與人員: 陳靜如 侯永儀
報告類型: 精簡報告
報告附件: 出席國際會議研究心得報告及發表論文
處理方式: 本計畫可公開查詢
中 華 民 國 95 年 5 月 29 日
行政院國家科學委員會補助專題研究計畫
▓ 期 中
進度報告
計畫名稱: C 型肝炎病毒非結構蛋白 2 之功能性研究(2/3)
計畫類別:▓個別型計畫
計畫編號:NSC
94
-
2320
-
B
-
006
-
081
-
執行期間:94 年 8 月 1 日至 95 年 7 月 31 日
計畫主持人:
楊孔嘉
共同主持人:張定宗
計畫參與人員: 陳靜如 侯永儀
成果報告類型(依經費核定清單規定繳交):
▓
精簡報告
本成果報告包括以下應繳交之附件:
□赴國外出差或研習心得報告一份
□赴大陸地區出差或研習心得報告一份
□出席國際學術會議心得報告及發表之論文各一份
□國際合作研究計畫國外研究報告書一份
處理方式:除產學合作研究計畫、提升產業技術及人才培育研究計畫、
列管計畫及下列情形者外,得立即公開查詢
□涉及專利或其他智慧財產權,□一年□二年後可公開查詢
執行單位:
國立成功大學醫學檢驗生物技術學系
中
華
民
國
95
年
5
月
25
日
中文摘要: C 型肝炎病毒的非結構蛋白 2 (NS2)已知會改變基因表現,但是它的致病機轉目前仍
不清楚。在本篇研究中,我們利用DNA 微陣列(microarray)檢測不同基因表現的差異,主要看
到在表現NS2 的 Huh-7 細胞中,四穿膜蛋白(tetraspanin)之 CD9,glutaredoxin,PACE4 和 ERp72
的RNA 會被調升。因為 tetraspanin 是主要細胞膜上的組成物,會促進細胞融合(cell fusion)和病
毒擴散(viral spread),所以我們更進一步探討 tetraspanin 與 NS2 之相關調控。而且不論是在肝癌 細胞或是非肝癌細胞株中,NS2 都可以促進細胞表面的 CD9 和 CD81 表現。在表現 NS2 的細 胞中,細胞膜表面的CD9 和 CD81 增加,形成類似一個 tetraspanin web。並且藉由突變 NS2 片 段,發現主要是NS2 的 C 端部分調控 CD9 和 CD81 之表現。再藉由小兒麻痺病毒感染的模式, 更可以知道tetraspanins 的調升會使細胞對於病毒感染的敏感性增加。NS2 和 NS2 的 C 端部分, 都可以增加小兒麻痺病毒所產生的溶菌斑的大小和數量,但是若是突變後去掉C 端則不會有影 響。所以,NS2 會藉由 CD9 和 CD81 的增加而促進小兒麻痺病毒的感染和擴散。同時,小兒麻 痺病毒在表現NS2 的細胞中所產生的溶菌斑,會被 CD9 的抗體所抑制。由本篇研究結果推知, C 型肝炎病毒的 NS2 蛋白可以藉由細胞表面四穿膜家族之 CD9 和 CD81 的增加,進而促進細胞 對於病毒感染的敏感性。
英文摘要: The non-structural (NS) 2 protein of hepatitis C virus (HCV) has been suggested to alter gene expression, but its pathogenic effects are currently unknown. In this study, we examined the differential gene profile using a complementary DNA microarray, resulting in up-regulation RNA levels of tetraspanin CD9, glutaredoxin, PACE4 and ERp72 in NS2-expressing Huh7 cells. We further characterized the NS2-mediated modulation of tetraspanins because those proteins are the major membranous components which may potentially induce cell fusion and viral spread. The NS2 could cause inductons of surface CD9 and CD81 in cell lines from both hepatic and non-hepatic cell origins. An increased density of CD9- and CD81-containing spots on the plasma membrane resembling tetraspanin web was observed in the NS2-expressing cells. Subsequently, the NS2 deletion mutants showed that their C-terminal domain mediated the up-regulation of CD9 and CD81. The effect of elevated tetraspanins on the cells’susceptibility to virus infection was investigated using the poliovirus infection model. We showed that the NS2 and its deletion mutant containing C-terminal domain could increase the size and the number of the poliovirus plaques, but the NS2 mutant with the C-terminal domain deleted could not. Thus, the functional NS2 by being capable of up-regulating CD9 and CD81 may enhance poliovirus infection and spread. In addition, the poliovirus plaque formation in the NS2-expressing cells was significantly reduced by the anti-CD9 antibody. In conclusion, the HCV NS2 protein can increase the surface expressions of tetraspanin CD9 and CD81 which may facilitate the infections of susceptible viruses.
關鍵字:
C 型肝炎病毒 非結構蛋白 2 四穿膜蛋白家族
前言: Hepatitis C virus (HCV)1, a small enveloped virus belonging to the Hepacivirus genus in the
Flaviviridae family, is a major etiology worldwide for the cause of liver diseases, from mild to severe
hepatitis, cirrhosis and hepatocellular carcinoma (1). The 9.6-kb single-stranded RNA genome of HCV encodes a polyprotein precursor, encompassing 10 individual viral proteins ordered from the amino terminus as follows: core, envelope (E) 1, E2, p7 and non-structural (NS) proteins 2, 3, 4A, 4B, 5A and 5B. The process of the HCV polyprotein precursor requires three proteolytic enzymes, which are the host signal peptidase to separate the core, E1, E2 and p7 (2), the HCV NS2/3 protease to cleave between the NS2 and NS3 (3-5), and the HCV NS3-4A protease to release the rest of the NS proteins (6,7). The mature HCV proteins may individually, in conjunction with other viral and host factors, contribute to establish a persistent HCV infection and to elicit hepatic pathogenesis during chronic HCV infections.
The HCV NS2 protein, with a molecular weight of 23 kDa, contains four hydrophobic transmembrane domains (7). Membrane extraction experiments have shown that NS2, when expressed alone (7) or in combination with the preceding p7 protein (8), may be inserted into the endoplasmic reticulum (ER)-derived membrane, where the membranous components allow a protective compartmentalization for high local concentrations of HCV proteins in order to form replicase complexes for achieving efficient viral RNA synthesis (9-11). In addition to protein-protein interaction experiments having demonstrated substantial physical interactions between NS2 and NS5B RNA polymerase (12), it raises the possibility that NS2 might be one of the protein components in the HCV replicase complex located at the specific ER membranous site. However, subgenomic HCV replicons containing NS proteins from only 3-5B can display replicative capacity, suggesting that NS2 is not indispensable for HCV RNA synthesis (13,14). It remains unclear whether NS2 can modulate HCV RNA replication. However, inactivation of NS2/3 processing can dampen the NS3 protease activity and accelerate the proteosome-dependent degradation of the unprocessed fusion protein (15). Thus, NS2 might affect the HCV life-cycle according to the contributions to the enzymatic central structure of NS2/3 protease and the recognizing sequences that make a cleavage between NS2 and NS3.
At present, few studies elucidate the functions of mature HCV NS2 protein and the NS2-mediated alteration of cell phenotypes. A recent study demonstrated that this protein can interact with a liver-specific apoptosis-inducing factor, CIDE-B, to inhibit apoptosis by counteracting cytochrome c release in a caspase-dependent manner (16). Another study suggested that HCV NS2 can inhibit the expression of several cellular and viral genes by modulating their promoter activity (17), but the effects on cellular features are still unknown. To investigate the HCV NS2-induced alteration of cellular genes, we screened the differential gene profile by using complementary DNA (cDNA) microarray chips in NS2-expressing Huh7 cells. The results showed increased RNA levels of CD9, glutaredoxin, PACE4 and ERp72. The CD9 protein, a major structural component in the surface tetraspanin membranous web, is known to participate in membrane-remodeling events, such as virus-induced syncytia formation, myotube formation, sperm-egg fusion, cell adhesion and cell migration (18), whereas glutaredoxin, PACE4 and ERp72 are considered as stress-responsive genes. Subsequently, we conducted experiments to characterize NS2-mediated regulations of CD9 and its closest relative in the tetraspanin family, CD81. Our data showed that HCV NS2 could up-regulate CD9- and CD81- RNAs and that it also increased the surface expression quantity of these tetraspanin proteins. By using poliovirus as a model to test cell susceptibility to virus infection, our results demonstrated that HCV NS2 could enhance viral spread and infectivity and that the enhancement of viral susceptibility was associated with the NS2-mediated CD9 upregulation. These data suggested that HCV NS2 may increase the levels of functional tetraspanins, thus potentially making cells more sensitive to virus infections.
研究方法:
Materials R-phycoerythrin (PE)-conjugated monoclonal antibody (MoAb) recognizing human CD9 (clone M-L13) or CD81 (clone JS-81) and R-PE-conjugated mouse IgG were obtained from BD PharMingen (San Diego, CA). MoAb against CD9 (clone MM2/57) used in the plaque reduction assay was obtained from Serotec, Inc. (Oxford, UK). The poliovirus strain Sabin was isolated with cultivated RD cells from a patient in the clinical virology laboratory of National Cheng Kung University Hospital,
and the virus titer was determined before use. The Dual-GloTM Luciferase Assay System used in the
reporter assay was purchased from Promega Corp. (Madison, WI).
Construction of plasmids The vector pEGFP-N1 (Clontech, Palo Alto, CA) was used in plasmid construction for expressing HCV proteins fused with a C-terminal tag of enhanced green fluorescent protein (EGFP). HCV cDNA sequences were originated from a genotype 1b strain. The cDNA containing amino acids 827-1026 (nt 2823-3422) of the NS2 region were amplified by the polymerase chain reaction (PCR) and inserted into the pEGFP-N1 vector to generate pNS2(827-1026)-EGFP. Plasmids pNS2(827-926)-EGFP and pNS2(926-1026)-EGFP were constructed by inserting the corresponding PCR product from the amino-terminal (N-terminal) sequences of NS2 containing amino acids 827-926 (nt 2823-3122) and the carboxy-terminal (C-terminal) sequences containing amino acids 926-1026 (nt 3120-3422) into the pEGFP-N1 vector. For assessing the promoter activity, upstream promoter regions of human CD9 gene, ranging from nt -1088 to +110, and human CD81 gene, ranging
from nt -922 to +44 were prepared by PCR amplification with genomic DNA of Huh7 cells. The corresponding DNA fragment was then ligated with pGL3-Basic vector to generate pGL3-CD9 and pGL3-CD81 plasmids, which were used in the following luciferase reporter assay. The nucleotide sequences for all the plasmid constructs described above were confirmed by DNA sequencing.
Cell lines and transfection Three types of hepatic cell lines, Huh7, HepG2, PLC, and 2 types of non-hepatic cell lines, HeLa, and 293A, were used in the present study. Cells were grown at 37°C in
Dulbecco’smodified Eagle’smedium (GibcoBRL, Gaithersburg, MD) supplemented with 10% fetal
bovine serum. Transfection was carried out with the indicated plasmid DNA by LipofectAMINE 2000 reagent (Invitrogen, Carlsbad, CA). For transiently expressing EGFP-fused viral genes, experiments were performed at 48 h post-transfection. To establish stable transfectants, cells were transfected for 48
h followed by selection with 500 μg/mL G418 (Amresco Inc., Solon, USA). After 2-3 weeks, each
resistant colony with a positive signal of EGFP-tag fluorescence was grown individually, and the expression of EGFP-fused viral gene was confirmed by standard reverse transcription-PCR with appropriate primers.
Microarray hybridization of cDNA chips The MillenniaChipI cDNA microarray chips used in this
study were designed and manufactured as described previously (19-23). This chip contains 884 human cDNA fragments collected from sequence-verified IMAGE clones, which can be classified into kinases, proteases, cell surface proteins and receptors, tumor suppressors/oncogenes, apoptosis and cell cycle regulators, angiogenesis regulators, DNA repair components, cytokines and growth factors, transcriptional factors and stress responders. Thirteen control genes, including those from plant, bacteriophage and several human housekeeping genes were also spotted on the chip. The protocol for labeling followed that of Chen et al (20). Briefly, mRNA from Huh7 cells stably transfected with either pEGFP-N1 or pNS2(827-1026)-EGFP plasmid was reverse-transcribed in the presence of a random hexamer, control RNA, biotin-labeled dUTP, dNTP mixture, RNAsin and MMLV reverse transcriptase. The derived biotin-labeled cDNAs were then individually hybridized to two chips. After extensive washing, streptavidin-beta-galactosidase was applied and the beta-galactosidase substrate, x-gal, was then added for colorimetric development. The hybridization signals, visualized as blue spots on the chips, were analyzed using a high-resolution scanner (PowerLook 3000; UMAX, Taipei, Taiwan), and analyzed by ScanAlyse 2 (Patrick O. Brown, Stanford University). The control and housekeeping genes served to normalize signals from other tested genes. Those genes in paired samples showing up-regulation or down-regulation by more than 2.5 times were considered as significant differences. Dual-color flow cytometric analysis Dual-color flow cytometric analysis (FACSCalibur, Becton Dickinson, CA) was performed to measure the green fluorescence emitted by EGFP in transfected cells expressing either EGFP alone or NS2-EGFP fusion proteins and the red fluorescence emitted by R-PE-conjugated mouse MoAbs recognizing a cell surface epitope. Mock transfected cells served as negative control for EGFP fluorescence, and R-PE-conjugated mouse IgG antibody was used for background evaluation of PE fluoresence.
Florescence microscopic analysis Cultivated cells were seeded on sterile coverslips in 6-well plates and incubated at 37°C overnight. After being fixed with 4% paraformaldehyde, the cells were stained for nuclei with DAPI (Sigma, St. Louis, MO) and MoAbs recognizing CD9 or CD81 (PharMingen). The cells were washed with phosphate buffered saline containing 0.02% Triton x-100, and then incubated with Alexa-546 labeled goat anti-mouse secondary antibody (Alexa, San Francisco, CA). Afterwards, fluorescent images were inspected using a Leica confocal fluorescence microscope (Leica model TCS SP2, Germany).
Real-time PCR Total RNA was extracted from cultured cells with REzolTM C&T reagent (PROtech
Technologies, Taiwan) and reverse-transcribed with specific reverse primer pair for CD9, CD81, and poliovirus VP1. Reverse transcription was carried out with M-MLV reverse transcriptase (Promega Corp, Madison, WI) and the corresponding reverse primer at 42°C for 1 h. The generated cDNA of CD9, CD81 and poliovirus were subjected to real-time PCR on a LightCycler machine (Roche
Diagnostics)with theprimerpairsand probes,including forward primer:5’-CCA TCC ACT ATG CGT
TGA AC-3’,reverseprimer:5’-CTG AAG ATC ATG CCA AAT ATC ATG-3’,hybridization probes:
5’-TGT CGA AGA CCT CTT TGA TGG CAT C-FL and 5’-LC Red 640-GGA CAG GAC TTC ACG
5’-CAG GAT CAT CTC GAA GAT CAT G-3’,hybridization probes:5’-GTG GCA GTC CTC CTT
GAA GAG GTT-FL and 5’-LC Red 640-CTG ATG ATG TTG CTG CCC GAG G-PH for CD81;
forward primer: 5’-AGC ACT CAA CCC CAG AGT GT-3’,reverse primer: 5’-GAC AGG CCA ATC
ACT GGT TT-3’,and TaqMan probe:5’-6FAM-ATT AGC CGC ATT CAG GGG CCG GA-DB-3’for
poliovirus VP1. Programs of real-time PCR for CD9 and CD81 were performed with initial denaturation at 95ºC for 7 min, followed by 45 amplification cycles of 5 s at 95ºC, 15 s at 54ºC and 15 s at 72ºC, whereas that for poliovirus had an initial denaturation at 95ºC for 10 min, followed by 45 amplification cycles of 15 s at 95ºC, and 30 s at 60ºC. The absolute amounts of CD9-, CD81-, and poliovirus RNAs were calculated based on an external standard curve that was generated by analysis of standard samples, containing various amounts of the corresponding plasmid-carrying target template. Target RNA levels were then expressed by normalization to the quanty of glucose-6-phosphate dehydrogenase (G6PDH) RNA (LightCycler-h-G6PDH Housekeeping Gene Set, Roche Diagnostics) measured in parallel samples.
Plaque assay with poliovirus infection Plaque assay with poliovirus infection was performed with
transfectant Huh7 cells stably expressing EGFP or NS2-EGFP fusion proteins. Briefly, 106cells were
seeded into each well of six-well plastic plates, and grown at 37C overnight. Four hundred microliters of minimum essential medium (GibcoBRL, Gaithersburg, MD) containing serially diluted poliovirus stock were added to each tested well. After absorption for 1 h at 37C, the cell monolayers were washed and overlaid with 2 mL of 1% methylcellulose in minimum essential medium and further incubated for 2 days at 37C. Plaques were visualized after fixation with 10% formalin and stain with 1% crystal violet.
For the plaque reduction assay, cells were seeded into wells in the absence or presence of 20 microgram/mL of MoAbs against human CD9 (clone MM2/57) or CD81 (clone JS-81) or mouse IgG control. Then, the processes of poliovirus infection and plaque formation were performed following the same procedure as described above, but in the absence or presence of the indicated antibody. Plaques were visualized after incubation for 2 days followed by crystal violet staining.
Luciferase reporter assay Huh7 cells were cotransfected with a constant amount of one firefly luciferase reporter along with various amounts of the plasmid encoding, either EGFP or NS2(827-1026)-EGFP. The tested firefly luciferase reporters included pGL3-CD9 and pGL3-CD81, whereas pGL3-Control and pGL3-Basic reporters were served as positive and negative controls, respectively. The constitutive renilla luciferase reporter containing the herpes simplex virus thymidine kinase (HSV-TK) promoter (phRL-TK, Promega) was simultaneously delivered into cells and used to normalize transfection efficiency. After transfection for 48 h, cells were lysed in Reporter Lysis Buffer,
and the luciferaseactivitiesweredetermined according to themanufacturer’sinstructions(Dual-GloTM
Luciferase Assay System kit, Promega) using a luminometer (DLReady, Promega).
結果:
NS2-EGFP fused proteins were localized in the cytoplasm Huh7 cells were transfected with plasmids expressing either EGFP or various NS2-EGFP fusion proteins of different lengths, including NS2(827-1026)-EGFP, NS2(827-926)-EGFP and NS2(926-1026)-EGFP for 48 h. The EGFP signals were initially visualized with confocal fluorescent microscopy. The results showed that the EGFP protein was distributed throughout the whole cell, with a greater intensity in the nucleus than in the cytoplasm (Fig. 1A). In contrast, the three NS2-EGFP fusion proteins, NS2(827-1026)-EGFP (Fig. 1C), NS2(827-926)-EGFP (Fig. 1E) and NS2(926-1026)-EGFP (Fig. 1G) displayed punctate structures that were excluded from the nucleus, in a pattern resembling ER localization as described previously (7,16,24). Thus, our data confirmed the proper expression of those NS2-EGFP fusion proteins and their subcellular distributions in the transfected cells. Subsequent flow cytometric analysis also used the direct EGFP-tag fluorescence signals to gate cells that expressed the NS2-EGFP fusion proteins.
HCV NS2 protein up-regulated the RNA expression of stress responsive genes and tetraspanin CD9 and CD81 To determine whether HCV NS2 affects cellular gene expression, we examined the
change of RNA levels by stably expressed NS2(827-1026)-EGFP in Huh7 cells with MillenniaChipI
cDNA microarray chips. As expected, the internal control genes and house keeping genes showed no significant difference between cells expressing EGFP and NS2(827-1026)-EGFP (Fig. 2A). However,
the RNA levels of CD9 (Hs. 114286), glutaredoxin (Hs. 28988), PACE4 (Hs. 498494) and ERp72 (Hs. 93659) were significnatly higher in the NS2(827-1026)-EGFP cells than those in the EGFP cells, by induction folds of 4.0, 3.4, 3.1 and 2.9, respectively (Fig. 2A). CD9, a member of the tetraspanin family, is known as the main structural protein in the surface membranous web associated with cell membrane remodeling events, including virus-induced cell fusion and virus spread (25), whereas glutaredoxin (26,27) PACE4 (28) and ERp72 (29-33) are recognized as intracellular stress markers, and their induction may reflect cells facing stress when the HCV NS2 protein is ectopically expressed. Thus, this result suggests that HCV NS2 may cause stress responses and alter CD9-related membrane functions. Subsequent studies were concentrated on the NS2 modulation of CD9 and included CD81, the closest sequence relative and functional partner to CD9 in the tetraspanin family (25).
We then quantified the RNA levels of CD9 and CD81 by real-time PCR with specific primers and
fluorescentprobesasdescribed in “Experimental prodecures”.After normalization with G6PDH as an
internal control, the results showed that the Huh7 cells stably expressing NS2(827-1026)-EGFP up-regulated CD9-RNA and CD81-RNA by 4.5 and 4.0 folds, respectively, as compared to the EGFP control cells (Fig. 2B). These results showed corroborative evidence that HCV NS2 can induce the RNA expression of CD9 and as well CD81.
It must be noted that none of the other genes on the chip were apparently down-regulated by HCV NS2(827-1026)-EGFP expression in our cDNA microarray analysis.
HCV NS2 enhanced surface CD9 and CD81 proteins in hepatic and non-hepatic cell lines We then evaluate the modulation of CD9 and CD81 by HCV NS2 at the protein level by dual-color flow cytometric analysis on cells transiently transfected with EGFP or NS2(827-1026)-EGFP. The EGFP transfectants generally exhibited a higher percentage of green-fluorescence bright cells than did the NS2(827-1026)-EGFP cells, e.g. a positive rate of 56.4% vs. 24.8% for Huh7 cells in a representative experimental run (Fig. 3A). Nevertheless, the EGFP bright subpopulations, recognized as positive transfectants were gated for measurement of surface CD9 or CD81 by R-PE-conjugated MoAbs. The mean fluorescence intensity (MFI) of CD9 for the NS2(827-1026)-EGFP transfectants was elevated in three hepatic cell lines, Huh7, HepG2 and PLC as well as in two non-hepatic cell lines, HeLa and 293A, as compared to that for the corresponding EGFP control cells, by various extents between 3.0 to 6.0 folds (Fig. 3B). Similarly, the MFI of CD81 was also increased in the NS2(827-1026)-EGFP transfectants of 293A, HeLa, Huh7 and PLC cells by 2.5-4.5 folds (Fig. 3B). These results suggest that NS2 can up-regulate surface CD9 and CD81 expression in many cell types from both hepatic and non-hepatic origins. It must be noted that the untransfected HepG2 cells and their transfectants expressing either EGFP or NS2(827-1026)-EGFP tested CD81 negative in our flow cytometric measurement, indicating that HCV NS2 does not alter the CD81-null phenotype in these cells.
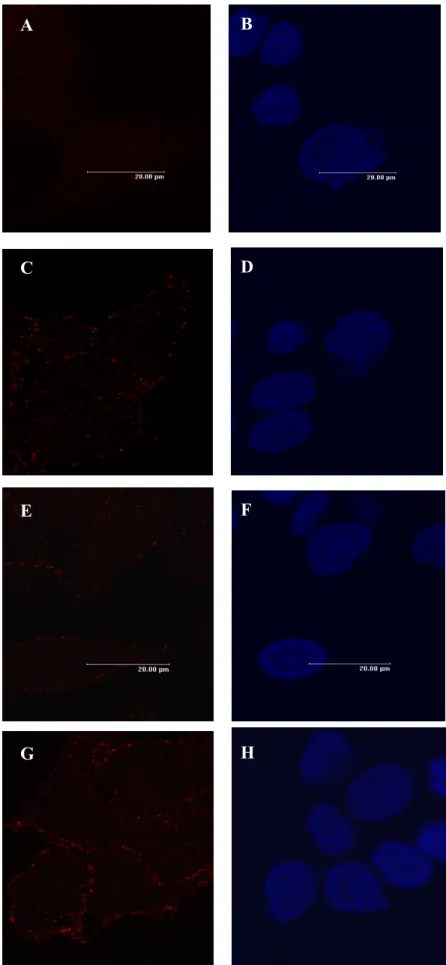
To determine whether HCV NS2 can modulate the tetraspanin membranous web, MoAbs recognizing surface CD9 or CD81 were incubated with Huh7 cells stably expressing EGFP or NS2(827-1026)-EGFP. Microscopic observation visualized the tetraspanin complexes as punctate spots on plasma membrane. As shown in figure 4, the quantities of CD9- (C vs. A) and CD81- (G vs. E) containing spots were higher in the NS2(827-1026)-EGFP cells than in the EGFP control cells. Again, this result suggests that HCV NS2 causes a quantitative elevation of CD9 and CD81 complexes at the membranous site, thus possibly altering membrane functionality.
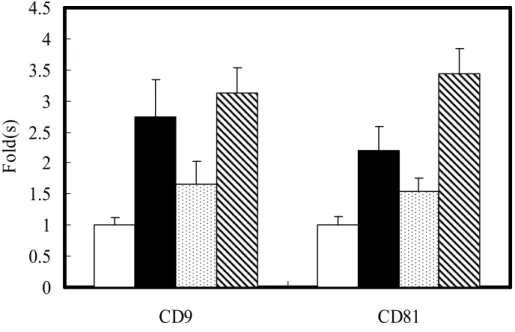
The C-terminal half of HCV NS2 was responsible for CD9 and CD81 up-regulation Next, we identified the functional NS2 domain for induction of CD9 and CD81 expression by exploiting the N-terminal half mutant, NS2(827-926)-EGFP, and the C-terminal half mutant, NS2(926-1026)-EGFP, in transfected Huh7 cells. In comparison to the EGFP control cells, the NS2(926-1026)-EGFP significantly stimulated up-regulation of surface CD9 and CD81 as did the NS2(827-1026)-EGFP cells (Fig. 5). In contrast, the NS2(827-926)-EGFP cells appeared to have only a slightly higher CD9 and CD81 expression than the EGFP control cells. Our results suggest that the NS2 C-terminal region is largely responsible for the modulation of CD9 and CD81 expression.
HCV NS2 increased cell susceptibility to poliovirus infection via CD9 up-regulation Tetraspanin CD9 protein has been shown to participate in the processes of virus infections, such as virus uptake by target cells, formation of cell syncytia, and production and release of progeny virus (34-36) We
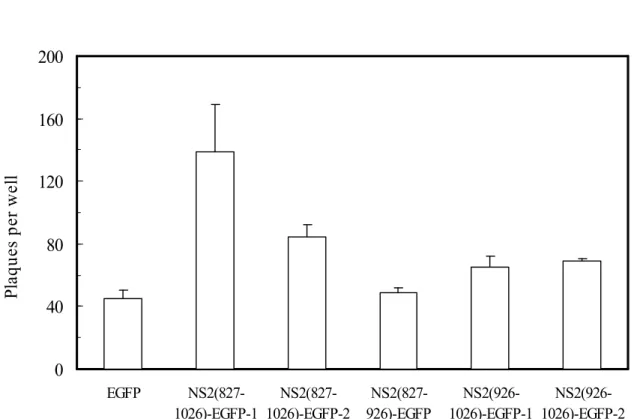
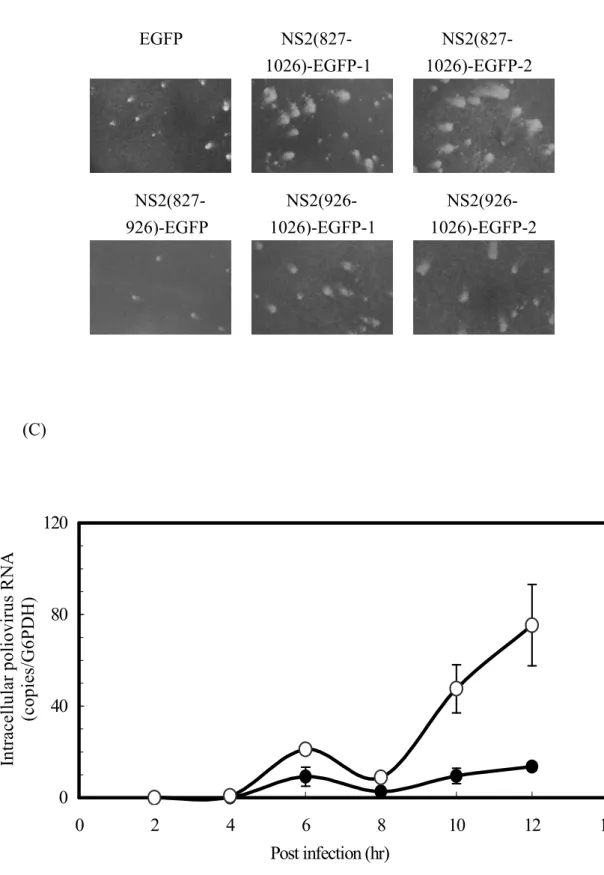
pathogens due to the elevation of tetraspanin CD9 and/or CD81. However, since at present there is no efficient cultivated cell system to evaluate HCV infection, we used poliovirus to analyze the NS2-mediated effects on viral infection by plaque formation assay. Poliovirus usually causes pathological lesions to human neurons leading to paralysis, but other cells from liver, spleen and pancreas can also support the infection and multiplication of this virus (37). We compared the numbers of poliovirus plaques formed on monolayer Huh7 cells with stable expression of EGFP, NS2(827-1026)-EGFP, NS2(827-926)-EGFP or NS2(926-1026)-EGFP. The results showed that the two independent clones of full-length NS2, NS2(827-1026)-EGFP-1 and NS2(827-1026)-EGFP-2, and the other two independent clones of NS2 C-terminal half mutant, NS2(926-1026)-EGFP-1 and NS2(926-1026)-EGFP-2, yielded more plaques than did the EGFP control by 3.1, 1.9, 1.5 and 1.5 folds, respectively (Fig. 6A). Furthermore, significantly larger plaques were formed on the monolayer NS2(827-1026)-EGFP and NS2(926-1026)-EGFP cells than on the EGFP control (Fig. 6B). On the contrary, the NS2(827-926)-EGFP cells showed no apparent differences in numbers and sizes of poliovirus plaques to those from the EGFP control cells (Fig. 6, A and B). Combined, the results shown in figures 5 and 6, indicate that the NS2 constructs that induced the CD9 and CD81 up-regulations were able to promote poliovirus plaque formation, and that the NS2 deletion mutant unable to enhance the tetraspanin expression did not affect the poliovirus infection. Thus, the NS2-enhanced CD9 and CD81 coincided with the NS2-enhanced virus susceptibility.
It is worth noting that the overall NS2-induction of CD9 and CD81 was much more correlated with the enlarged plaques than it was with the increased plaque number. These observations suggest that the increased tetraspanin proteins may significantly promote the virus spread to adjacent cells from a slightly increased number of infectious centers. To test this possibility, we quantified cell-associated poliovirus RNA at different time points postinfection. After the freeze-thaw procedure for three times, total RNA from the harvested cells was extracted and the poliovirus RNA was measured by real-time PCR. At 6 h postinfection, which is the most likely time for the initial detection of poliovirus RNA in the primary infected sites, the NS2(827-1026)-EGFP cells produced approximately a 2-fold higher viral titer than did the EGFP control cells (Fig. 6C). Consistent with the finding that NS2 slightly increases the plaque number shown above (Fig. 6A), this data also suggests that virus entry was moderately enhanced. After postinfection for 8 h, when the release and spread of the progeny virus from the original infected cells might have begun, the production of poliovirus RNA was accelerated in the NS2(827-1026)-EGFP cells, but not in the EGFP control cells (Fig. 6C). The results agreed with the plaque formation assay showing that HCV NS2 rendered cells more sensitive to viral spread or secondary infection, thus yielding enlarged plaques (Fig. 6B).
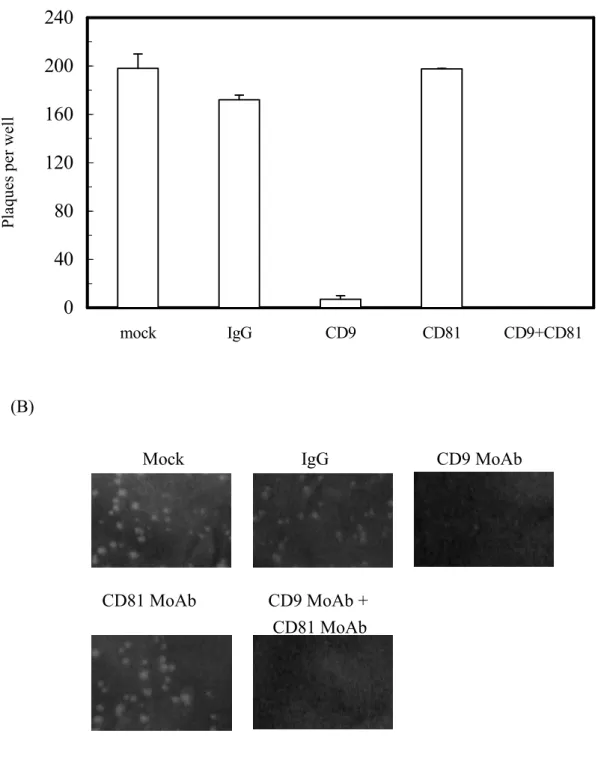
In order to elucidate whether the enlarged plaque size and increased plaque number in cells expressing NS2 after poliovirus infection was attributable to the induction of the surface tetraspanins, MoAbs recognizing CD9 or CD81 were used to block these proteins, followed by a plaque reduction assay. Our results showed that treatment with either anti-CD9 alone or with combined anti-CD9 and anti-CD81 reduced the number and size of poliovirus plaques in the NS2(827-1026)-EGFP cells. At the same time, there were no apperent effects on the plaque formation with the treatment of anti-CD81 or mouse IgG antibody, (Fig. 7A and 7B). Taken together the results shown above, HCV NS2 may increase cell susceptibility to poliovirus infection, at least in part, via CD9 up-regulation
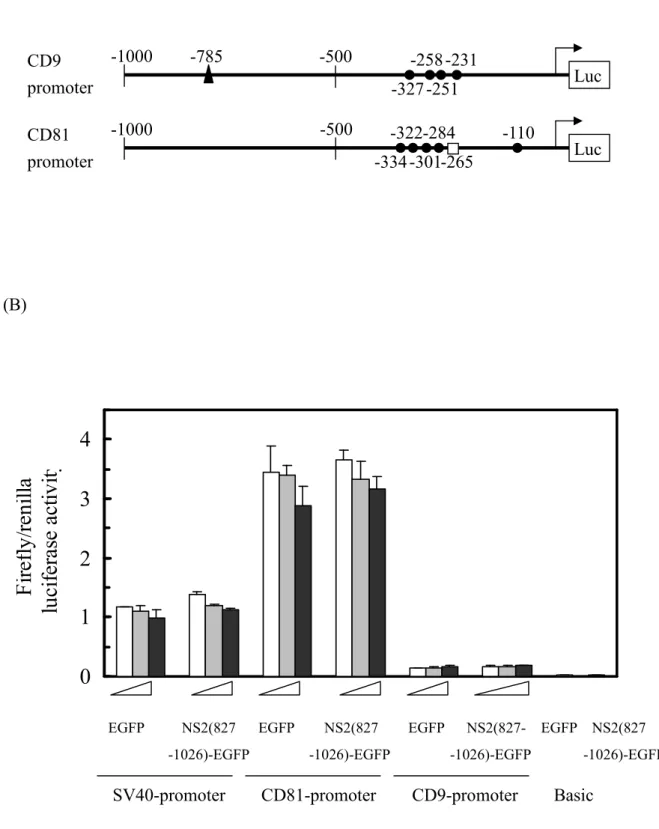
HCV NS2 protein did not alter the activities of putative CD9- and CD81-promoters To determine whether HCV NS2 protein directly enhances transcription activities from human CD9 and CD81 promoters, we evaluated the luciferase reporter driven by those putative promoters in the presence or
absence of HCV NS2. Approximately 1-kb 5’-flanking sequences from the translation start codon of the
corresponding human CD9 and CD81 genes were positioned to drive a firefly luciferase reporter. Both constructs contained several consensus binding motifs for Sp1 (Fig. 8A), the known functional transcription factor on the CD9 promoter (38). It is worth noting that the CD9 construct contains one consensus binding motif for NFkappaB, a regulatory motif that has been shown to be suppressed by HCV NS2 (17). In this assay, the firefly luciferase activity was represented by its ratio against the expression of co-transfected renilla luciferase reporter, which served as a transfection efficiency control. Our data showed that transfection with pEGFP-N1 and pNS2(827-1027)-EGFP yielded comparable firefly luciferase activities under the control of upstream promoter sequences from either CD9 or CD81
genes (Fig. 8B), indicating that HCV NS2 modulation of CD9 and CD81 expression may not be due to the direct enhancement of the promoter efficiency.
討論: NS2 maturation occurs early in the cleavage process of HCV NS proteins. Initial membrane fractionation experiments suggested HCV NS2 as an integral cytoplasmic membrane protein (4,39), and later fluorescence microscopic visualization confirmed its ER-associated subcellular localization pattern (7,24). In agreement with the previous studies, our results showed that the
NS2(827-1026)-EGFP protein was enriched in the cytoplasm, and most likely localized at the ER membrane (Fig. 1C). A topological study revealed that HCV NS2 spans the ER membrane four times
via the predicted hydrophobic domains between amino acid stretches 810-835, 843-866, 872-919 and
928-956 (7). It is evident from the present study that both NS2(827-926)-EGFP (Fig. 1E) and NS2(926-1026)-EGFP (Fig. 1G) exhibit ER distribution patterns like that of NS2(827-1026)-EGFP. Our results indicate that either the first 3 hydrophobic domains between amino acids 810-835, 843-866 and 872-919 or only the fourth hydrophobic domain spanning amino acids 928-956 may be sufficient for ER membranous association of the NS2 protein. Although the NS2(827-926)-EGFP mainly showed an ER distribution pattern like NS2(827-1026)-EGFP, this N-terminal half of the NS2 protein could neither stimulate the CD9 or the CD81 expression, nor enhance the poliovirus susceptibility, suggesting that ER localization may not be a predominant factor leading to the tetraspanin up-regulation that makes cells more sensitive to virus infection. In addition, our deletion mapping results further showed that the functionally active portion was localized to the C-terminal half of the NS2 protein because the NS2 mutant containing C-terminus NS2(926-1026)-EGFP, has a comparable ability in up-regulating the tetraspanins and enhancing plaque formation to that of the intact HCV NS2, NS2(827-1026)-EGFP. However, the details of the functional amino acid sequences and active structure in the HCV NS2 C-terminus will require further identification. The study by Erdtmann et al (16) revealed that the region spanning amino acids 909-1027 of HCV NS2 is necessary and sufficient for binding with the
liver-specific pro-apoptotic protein CIDE-B, and that this interaction interferes with CIDE-B dependent cell-death. In the present study we identified a novel NS2 function of promoting tetraspanin expression and plaque formation, in addition to the known anti-apoptotic activity in the overlapping C-terminal domain.
Both CD9 and CD81 belong to the tetraspanin superfamily, which displays the characteristic protein structure of four hydrophobic, transmembrane domains forming a small and a large extracellular loop, with the N- and C-terminal tails facing the cytosol. The HCV NS2 protein also has four transmembrane domains, but there are no known typical tetraspanin features in terms of conserved disulfide bonds and two distinct extracellular domains of unequal size. Tetraspanin proteins are widely expressed in the plasma membrane of many eukaryotic cell types. In general, those surface proteins in the tetraspanin superfamily are physically associated with primary partners, which include integrins and the immunoglobulin superfamily, and further combine through tetraspanin/tetraspanin interactions to build a higher-order network called the tetraspanin web (18,40), which is involved in a variety of cellular functions including penetration, invasion and fusion events (25). The mammalian tetraspanin family contains at least 32 members (25). Sequence alignment and phylogenetic analysis revealed that CD9 and CD81 are close relatives in this superfamily. Among the known functions, CD9 may participate in viral infection (34-36), cell mobility (41,42), cell adherence and fusion (43,44), and T cell activation (45), whereas CD81 may mediate signal transduction and cell adhesion in immune cell collaboration (46,47). Furthermore, CD81 has been identified as a putative HCV receptor (48) and as a requirement for hepatic infection by the Plasmodium sporozoite (49). In a tetraspanin web, CD9 can bind directly to CD81 on the plasma membrane, with palmitoylation of CD9 juxtamembrane cysteine residues stabilizing the complex (50). Our results demonstrated that the quantity of CD9- and CD81-containing (Fig. 4, A vs.C, E vs. G) hepatic tetraspanin complexes, which appeared as small globular masses at the cellular edge, were enhanced by HCV NS2. Thus, the NS2-mediated up-regulation of tetraspanin proteins might potentionally alter cellular functionality involving the membrane tetraspanin web. In this study, the plaque formation assay with infectious poliovirus strain demonstrated that Huh 7 cells expressing NS2(827-1026)-EGFP yielded a significantly increased number and enlarged size of plaques compared to the control cells expressing EGFP alone. In addition,
we demonstrated the essential link between NS2-mediated CD9 up-regulation and increased
susceptibility to poliovirus infection by the two following experiments.First, the enhancement of CD9
expression by the NS2 constructs, NS2(827-1026)-EGFP and NS2(926-1026)-EGFP, coincided with the ability to promote plaque formation. At the same time, the NS2 C-terminal deletion mutant, NS2(827-926)-EGFP was unable to increase CD9 and lost its capacity to increase plaques. Second, pre-incubation of the NS2-expressing cells with MoAb against CD9 led to plaque reduction. Previous studies reported that surface CD9 may not be the viral receptor per se but that it acts as a facilitator for virus-induced cell fusion for the feline immunodeficiency virus (34,35) and the canine distemper virus (36,51). As far as we know, the cell-to-cell spreading of viruses is an important mechanism in a variety of viral infections, and the cell surface CD9 molecule may probably be involved in post-infection syncytium formation and to commonly facilitate virus transmission. The present results further imply that the poliovirus is one of the human viruses exploiting CD9 as a cofactor for infection. Our data in this study therefore raises the possibility that, in general, HCV patients are more sensitive to other permissive viruses via cell-to-cell spread because of the elevated CD9 expression induced by the NS2 protein in HCV-infected tissues. To date no clinical studies have made such comparisons. It is worth noting that HCV NS2 can modulate surface CD9 and CD81, which are the two principal components of membranous tetraspanin web with a broad distribution on human cells. Therefore, cellular functions other than virus susceptibility may also be altered by the NS2 protein. It will be interesting to find out if there are distinct membrane-remodeling events involving NS2-mediated CD9/CD81 modulation in HCV susceptible cells other than hepatocytes.
In addition to CD9, three genes were significantly induced by HCV NS2 in our cDNA microarray analysis: (1) glutaredoxin (also known as thioltransferase), a cytosol protein acting as a glutathione-dependent hydrogen donor and as a thiol repair and dethiolating enzyme, can sense and maintain cellular redox status in a non-equilibrium steady state (52); (2) PACE4, a calcium-dependent serine endoprotease, belonging to the subtilisin-like proprotein convertase family which can process latent precursor proteins into their biologically active products (53,54); and (3) ERp72, an ER-resident molecular chaperone, binds specifically to nascent polypeptides and various denatured proteins to participate in disulfide isomerization during protein folding and assembly (55-58). Stress conditions, such as disturbance of the intracellular oxidative milieu, accumulation of misfolded/unglycosylated proteins and depletion of the ER calcium store, can induce the transcription of glutaredoxin (26), PACE4 (28) and ERp72 (29-33). This represents a survival response to protect cells from cytotoxic damage. Stimulation of these stress responsive genes indicates that ectopic expression of the NS2 protein may induce intracellular stress. The mechanism by which the NS2 protein up-regulated CD9 and CD81 could be associated with the intracellular stress responses. However, the HCV NS2 protein did increase the levels of reactive oxygen species, but our preliminary data showed no correlation between the inductions of oxidative stress and the elevations of tetraspanin CD9 and CD81. It has been shown that HCV NS2 modulates the transcriptional activity of various viral and cellular promoters (17) and that the NS2 protein does alter the RNA levels of CD9 and CD81 in our study. We therefore tested the possibility that HCV NS2 could act directly on the CD9- and CD81-promoters in the luciferase reporter assay. However, our studies here have shown that HCV NS2 protein does not act the regulatory functions on the putative CD9 and CD81 promoters with respect to transcriptional and/or translational efficiency. Further analysis is needed to investigate the mechanism by which HCV NS2 influences the CD9 and CD81 expression.
In conclusion, our data showed that HCV NS2 can cause induction of stress responsive genes, including glutaredoxin, PACE4, ERp72, as well as tetraspanin genes, including CD9 and CD81. The elevated levels of surface CD9 and CD81 proteins may alter the cellular membrane-remodeling functions, such as the cell-based evidences shown in this report, that NS2-mediated CD9 up-regulation may increase the susceptibility to virus infection, which is most likely due to the promotion of cell-to-cell viral spreading. Our results may reveal a novel pathogenic mechanism mediated by the HCV NS2 protein.
1. Hoofnagle, J. H. (1997) Hepatology 26, 15S-20S
2. Mizushima, H., Hijikata, M., Tanji, Y., Kimura, K., and Shimotohno, K. (1994) J Virol 68,
2731-2734
3. Grakoui, A., McCourt, D. W., Wychowski, C., Feinstone, S. M., and Rice, C. M. (1993) Proc
Natl Acad Sci U S A 90, 10583-10587
4. Santolini, E., Pacini, L., Fipaldini, C., Migliaccio, G., and Monica, N. (1995) J Virol 69,
7461-7471
5. Pieroni, L., Santolini, E., Fipaldini, C., Pacini, L., Migliaccio, G., and La Monica, N. (1997) J
Virol 71, 6373-6380
6. Reed, K. E., and Rice, C. M. (2000) Curr Top Microbiol Immunol 242, 55-84
7. Yamaga, A. K., and Ou, J. H. (2002) J Biol Chem 277, 33228-33234
8. Carrere-Kremer, S., Montpellier-Pala, C., Cocquerel, L., Wychowski, C., Penin, F., and
Dubuisson, J. (2002) J Virol 76, 3720-3730
9. Dubuisson, J., Penin, F., and Moradpour, D. (2002) Trends Cell Biol 12, 517-523
10. Egger, D., Wolk, B., Gosert, R., Bianchi, L., Blum, H. E., Moradpour, D., and Bienz, K. (2002)
J Virol 76, 5974-5984
11. Moradpour, D., Gosert, R., Egger, D., Penin, F., Blum, H. E., and Bienz, K. (2003) Antiviral
Res 60, 103-109
12. Dimitrova, M., Imbert, I., Kieny, M. P., and Schuster, C. (2003) J Virol 77, 5401-5414
13. Lohmann, V., Korner, F., Koch, J., Herian, U., Theilmann, L., and Bartenschlager, R. (1999)
Science 285, 110-113
14. Blight, K. J., Kolykhalov, A. A., and Rice, C. M. (2000) Science 290, 1972-1974
15. Welbourn, S., Green, R., Gamache, I., Dandache, S., Lohmann, V., Bartenschlager, R.,
Meerovitch, K., and Pause, A. (2005) J Biol Chem 280, 29604-29611
16. Erdtmann, L., Franck, N., Lerat, H., Le Seyec, J., Gilot, D., Cannie, I., Gripon, P., Hibner, U.,
and Guguen-Guillouzo, C. (2003) J Biol Chem 278, 18256-18264
17. Dumoulin, F. L., von dem Bussche, A., Li, J., Khamzina, L., Wands, J. R., Sauerbruch, T., and
Spengler, U. (2003) Virology 305, 260-266
18. Boucheix, C., and Rubinstein, E. (2001) Cell Mol Life Sci 58, 1189-1205
19. Chen, J. J., Wu, R., Yang, P. C., Huang, J. Y., Sher, Y. P., Han, M. H., Kao, W. C., Lee, P. J.,
Chiu, T. F., Chang, F., Chu, Y. W., Wu, C. W., and Peck, K. (1998) Genomics 51, 313-324
20. Chen, C. C., Shieh, B., Jin, Y. T., Liau, Y. E., Huang, C. H., Liou, J. T., Wu, L. W., Huang, W.,
Young, K. C., Lai, M. D., Liu, H. S., and Li, C. (2001) J Biomed Sci 8, 214-222
21. Guo, Y. L., Chang, H. C., Tsai, J. H., Huang, J. C., Li, C., Young, K. C., Wu, L. W., Lai, M. D.,
Liu, H. S., and Huang, W. (2002) Environ Mol Mutagen 40, 122-128
22. Chang, H. C., Tsai, J., Guo, Y. L., Huang, Y. H., Tsai, H. N., Tsai, P. C., and Huang, W. (2003)
Biochem Biophys Res Commun 305, 1109-1115
23. Tsai, W. C., Tsai, S. T., Ko, J. Y., Jin, Y. T., Li, C., Huang, W., Young, K. C., Lai, M. D., Liu, H.
S., and Wu, L. W. (2004) Oral Oncol 40, 418-426
24. Kim, J. E., Song, W. K., Chung, K. M., Back, S. H., and Jang, S. K. (1999) Arch Virol 144,
329-343
25. Hemler, M. E. (2003) Annu Rev Cell Dev Biol 19, 397-422
26. Raghavachari, N., Krysan, K., Xing, K., and Lou, M. F. (2001) Invest Ophthalmol Vis Sci 42,
1002-1008
27. Krysan, K., and Lou, M. F. (2002) Invest Ophthalmol Vis Sci 43, 1876-1883
28. Koide, S., Yoshida, I., Tsuji, A., and Matsuda, Y. (2003) J Biochem (Tokyo) 134, 433-440
29. Dorner, A. J., Wasley, L. C., Raney, P., Haugejorden, S., Green, M., and Kaufman, R. J. (1990)
J Biol Chem 265, 22029-22034
30. Bush, K. T., Goldberg, A. L., and Nigam, S. K. (1997) J Biol Chem 272, 9086-9092
31. Linden, T., Doutheil, J., and Paschen, W. (1998) Neurosci Lett 247, 103-106
32. Parker, R., Phan, T., Baumeister, P., Roy, B., Cheriyath, V., Roy, A. L., and Lee, A. S. (2001)
Mol Cell Biol 21, 3220-3233
Mori, K., and Alape-Giron, A. (2004) J Biol Chem 279, 21724-21731
34. de Parseval, A., Lerner, D. L., Borrow, P., Willett, B. J., and Elder, J. H. (1997) J Virol 71,
5742-5749
35. Willett, B., Hosie, M., Shaw, A., and Neil, J. (1997) J Gen Virol 78 ( Pt 3), 611-618
36. Loffler, S., Lottspeich, F., Lanza, F., Azorsa, D. O., ter Meulen, V., and Schneider-Schaulies, J.
(1997) J Virol 71, 42-49
37. Ida-Hosonuma, M., Iwasaki, T., Yoshikawa, T., Nagata, N., Sato, Y., Sata, T., Yoneyama, M.,
Fujita, T., Taya, C., Yonekawa, H., and Koike, S. (2005) J Virol 79, 4460-4469
38. Le Naour, F., Prenant, M., Francastel, C., Rubinstein, E., Uzan, G., and Boucheix, C. (1996)
Oncogene 13, 481-486
39. Muramatsu, S., Ishido, S., Fujita, T., Itoh, M., and Hotta, H. (1997) J Virol 71, 4954-4961
40. Hemler, M. E. (2001) J Cell Biol 155, 1103-1107
41. Ikeyama, S., Koyama, M., Yamaoko, M., Sasada, R., and Miyake, M. (1993) J Exp Med 177,
1231-1237
42. Funakoshi, T., Tachibana, I., Hoshida, Y., Kimura, H., Takeda, Y., Kijima, T., Nishino, K., Goto,
H., Yoneda, T., Kumagai, T., Osaki, T., Hayashi, S., Aozasa, K., and Kawase, I. (2003)
Oncogene 22, 674-687
43. Miyado, K., Yamada, G., Yamada, S., Hasuwa, H., Nakamura, Y., Ryu, F., Suzuki, K., Kosai, K.,
Inoue, K., Ogura, A., Okabe, M., and Mekada, E. (2000) Science 287, 321-324
44. Tachibana, I., and Hemler, M. E. (1999) J Cell Biol 146, 893-904
45. Tai, X. G., Yashiro, Y., Abe, R., Toyooka, K., Wood, C. R., Morris, J., Long, A., Ono, S.,
Kobayashi, M., Hamaoka, T., Neben, S., and Fujiwara, H. (1996) J Exp Med 184, 753-758
46. Wack, A., Soldaini, E., Tseng, C., Nuti, S., Klimpel, G., and Abrignani, S. (2001) Eur J
Immunol 31, 166-175
47. Mittelbrunn, M., Yanez-Mo, M., Sancho, D., Ursa, A., and Sanchez-Madrid, F. (2002) J
Immunol 169, 6691-6695
48. Pileri, P., Uematsu, Y., Campagnoli, S., Galli, G., Falugi, F., Petracca, R., Weiner, A. J.,
Houghton, M., Rosa, D., Grandi, G., and Abrignani, S. (1998) Science 282, 938-941
49. Silvie, O., Rubinstein, E., Franetich, J. F., Prenant, M., Belnoue, E., Renia, L., Hannoun, L.,
Eling, W., Levy, S., Boucheix, C., and Mazier, D. (2003) Nat Med 9, 93-96
50. Charrin, S., Manie, S., Oualid, M., Billard, M., Boucheix, C., and Rubinstein, E. (2002) FEBS
Lett 516, 139-144
51. Schmid, E., Zurbriggen, A., Gassen, U., Rima, B., ter Meulen, V., and Schneider-Schaulies, J.
(2000) J Virol 74, 7554-7561
52. Holmgren, A. (1989) J Biol Chem 264, 13963-13966
53. Smeekens, S. P. (1993) Biotechnology (N Y) 11, 182-186
54. Taylor, N. A., Van De Ven, W. J., and Creemers, J. W. (2003) Faseb J 17, 1215-1227
55. Kuznetsov, G., Chen, L. B., and Nigam, S. K. (1994) J Biol Chem 269, 22990-22995
56. Kuznetsov, G., Chen, L. B., and Nigam, S. K. (1997) J Biol Chem 272, 3057-3063
57. Schaiff, W. T., Hruska, K. A., Jr., McCourt, D. W., Green, M., and Schwartz, B. D. (1992) J
Exp Med 176, 657-666
58. Feng, W., Bedows, E., Norton, S. E., and Ruddon, R. W. (1996) J Biol Chem 271, 18543-18548
Footnotes
*This work was supported by research grants NSC 92-2314-B-006-093, NSC 93-2314-B-006-119 and NSC 94-2320-B-006-081 from the National Science Council, Taiwan, R.O.C. The authors are grateful to the members of the laboratories of Drs. Ching Li, Li-Wha Wu, Wenya Huang, Ming-Derg Lai and Hsiao-Sheng Liu for their collaboration in setting up the microarray core facility at National Cheng Kung University.
1Abbreviations used are: HCV, hepatitis C virus; E, envelope; NS, non-structural; ER, endoplasmic
enhanced green fluorescent protein; PCR, polymerase chain reaction; N-terminal, amino-terminal; C-terminal, carboxy-terminal; G6PDH, glucose-6-phosphate dehydrogenase; HSV-TK, herpes simplex virus thymidine kinase; MFI, mean fluorescence intensity.
Figure Legends
Fig. 1. Subcellular localization of HCV NS2-EGFP fusion proteins. Huh7 cells transiently transfected with plasmids expressing EGFP or various NS2-EGFP fusion proteins for 48 h were trypsinized and reseeded on coverslips overnight. Cell nuclei were stained with DAPI for 30 min. The cells were then visualized with a Leica confocal fluorescence microscope with a fluorescent isothiocyanate filter (A, C, E, G) and UV light source (B, D, F, H). Cells that expressed EGFP (A and B), NS2(827-1026)-EGFP (C and D), NS2(827-926)-EGFP (E and F) and NS2(926-1026)-EGFP (G
and H) are shown. Scalebar= 20 μm.
Fig. 2. CDNA microarray analysis and quantification of CD9- and CD81-RNA levels by real-time PCR. (A) CDNA microarray analysis with mRNA isolated from stable Huh7 cells expressing EGFP or NS2(827-1026)-EGFP. Rectangular boxes indicate enlarged images, each containing 6 local gene spots shown in their relative positions, from a representative cDNA microarray chip. The 6 internal control
genes (1 = bacteriophage lamda Pst I-digested 1.2- kb fragment; 2 = plant rbcL; 3 = plant GA4; 4 =
human GAPDH; 5 = bacteriophage lamda Pst I-digested 1.1- kb fragment; 6 = plant rca), CD9
(Hs.114286), glutaredoxin (Hs.28988), PACE4 (Hs.498494) and ERp72 (Hs.93659) are marked in black in the corresponding box. The right-most column shows the induction folds of gene expression in the NS2(827-1026)-EGFP cells as compared to the EGFP control cells. (B) Total RNAs were extracted from the EGFP cells (open) or the NS2(827-1026)-EGFP cells (filled), and reverse-transcribed to cDNAs with specific primer pairs encompassing nucleotide sequences of CD9, CD81 and G6PDH, respectively. Real-time PCR reactions with the LightCycler instrument were performed to quantify the specific RNA levels. The quantities of CD9 and CD81 RNAs were normalized to the level of G6PDH RNA present in the same sample, as expressed by mean ± SD from duplicate determinations.
Fig. 3. Dual-color flow cytometric analysis and quantification of surface CD9 and CD81 proteins. (A) Huh7 cells were transiently transfected with pEGFP-N1 or pNS2(827-1026)-EGFP DNA for 48 h, stained with R-PE-conjugated MoAbs recognizing CD9 or CD81, and the quantities of fluorescent proteins and fluorochromes were then measured by dual-color flow cytometric analysis. A representative histogram for green fluorescent signals showed that 56.4%, 24.8% and 0.1% were EGFP-positive in cells transfected with pEGFP-N1 (solid curve), pNS2(827-1026)-EGFP (dotted curve) and mock transfection control (shaded region), respectively. The EGFP-positive subpopulations were recognized as transfectants expressing either EGFP or NS2-EGFP fusion proteins. (B) Quantitative results for surface CD9 and CD81 proteins in the EGFP-positive transfectants. The MFI values from the cells expressing NS2(827-1026)-EGFP protein were divided by the counterpart cells expressing EGFP and were indicated as relative level by considering the corresponding control as 1 fold (open). The tested cells included 293A (gray), HeLa (stripped), Huh7 (black), PLC (dotted) and HepG2 (oblique). Results are shown as mean ± SD from triplicate determinations.
Fig. 4. Immunostaining of cell surface CD9 and CD81 proteins and nuclei. Huh7 cells stably transfected with EGFP (A, B, E, F) or NS2(827-1026)-EGFP (C, D, G, H) were seeded on coverslips and stained with R-PE-labeled MoAbs against CD9 (A, C) or CD81 (E, G) and DAPI (B, D, F, H) for
nuclei. Images were photographed with a Leica confocal fluorescence microscope. Scalebar= 20 μm.
Fig. 5. Induction of surface CD9 and CD81 levels with different HCV NS2 constructs. The levels of surface CD9 and CD81 were quantified by dual-color flow cytometric analysis in Huh7 transfected
cells expressing NS2(827-1026)-EGFP (black), NS2(827-926)-EGFP (dotted), or
NS2(926-1026)-EGFP (oblique). Data are expressed as fold changes of MFI compared to the control cells expressing EGFP alone (open).
Fig. 6. HCV NS2 increased plaque size and the number of plaques formed by poliovirus infection of monolayer cells. Plaque assay by poliovirus infection was performed with monolayer Huh7 cells stably expressing the various NS2-EGFP fusion proteins. (A) The numbers of plaques were counted and represented as mean ± SD from at least four measurements. Results included monolayer cells from 2 independent clones for each of NS2(827-1026)-EGFP and NS2(926-1026)-EGFP, and a single clone for each of EGFP and NS2(827-926)-EGFP. (B) Plaques on the monolayer cells were photographed and are shown from a representative experiment. (C) After poliovirus infection, total RNA was extracted from the EGFP cells () and the NS2(827-1026)-EGFP cells () at the indicated time points for quantification of poliovirus RNA by real-time PCR. The amount of intracellular poliovirus RNA was normalized to the quantity of G6PDH, which served as the internal control for RNA sample preparation. Results are shown as mean ± SD from triplicate determinations.
Fig. 7. Plaque reduction assay. The plaque reduction assay was conducted in poliovirus-infected Huh7 monolayer cells stably expressing NS2(827-1026)-EGFP. Cells were treated with medium, mouse IgG Ab, anti-CD9 MoAb, anti-CD81 MoAb or combined anti-CD9 and anti-CD81 MoAbs. (A) The numbers of plaques were counted and represented as mean ± SD from duplicate determinants. (B) Plaques on the tested monolayer cells were photographed and are shown from a representative experiment.
Fig. 8. Effects of the HCV NS2 protein on CD9 and CD81 promoters. (A) Illustration of putative
motifs on CD9 and CD81 promoters. The TATA box (□), the Sp1 binding motif () and the NFkappaB
binding motif (▲) are indicated by the number from the translation start site. CD9 promoter has neither
a TATA box nor a CAAT box (38). (B) The firefly luciferase reporterscontaining a5’-flanking CD9
promoter or CD81 promoter were co-transfected into Huh7 cells with plasmids expressing EGFP or NS2(827-1026)-EGFP at increasing concentrations. Reporters pGL3-Control and pGL3-Basic were used as the positive and the negative control, respectively. In each assay, phRL-TK, a constitutive renilla luciferase reporter served as an internal control to normalize the transfection efficiency. The values for firefly luciferase activity are presented relative to the renilla luciferase activity, as shown by the averages from six independent measurements.
Figure 1
C
D
A
B
E
F
Internal control
genes
CD9
Glutaredoxin
PACE4
ERp72
Figure 2
(A)
Genes
Position
EGFP
NS2(827-1026)
Induction folds
-EGFP
(B)
0 40 80 120 160 CD9 CD81 R N A le ve ls (c op ie s/ G 6P D H )
_
4.0 ± 0.1
3.4 ± 0.1
3.1 ± 0.5
2.9 ± 0.6
Figure 3
(A)
(B)
0 1 2 3 4 5 6 7 CD9 CD81 Fo ld (s )Figure 4
A
C
E
G
B
D
F
H
Figure 5
0
0.5
1
1.5
2
2.5
3
3.5
4
4.5
CD9
CD81
F
ol
d(
s)
Figure 6
(A)
0
40
80
120
160
200
EGFP NS2(827-1026)-EGFP-1 NS2(827-1026)-EGFP-2 NS2(827-926)-EGFP NS2(926-1026)-EGFP-1 NS2(926-1026)-EGFP-2Pl
aq
ue
s
pe
r
w
el
l
-Figure 6
(B)
(C)
0
40
80
120
0
2
4
6
8
10
12
14
Post infection (hr)
In
tr
ac
el
lu
la
r
po
li
ov
ir
us
R
N
A
(c
op
ie
s/
G
6P
D
H
)
EGFP
NS2(827-
NS2(827-1026)-EGFP-1
1026)-EGFP-2
NS2(827-
NS2(926-
Figure 7
(A)
(B)
0
40
80
120
160
200
240
mock IgG CD9 CD81 CD9+CD81 Pl aq ue s pe r w el l-Mock
IgG
CD9 MoAb
CD81 MoAb
CD9 MoAb +
Figure 8
(A)
(B)
-334
-327
0
1
2
3
4
F
ir
ef
ly
/r
en
ill
a
lu
ci
fe
ra
se
ac
tiv
ity
EGFP NS2(827 EGFP NS2(827 EGFP NS2(827- EGFP NS2(827
-1026)-EGFP -1026)-EGFP -1026)-EGFP -1026)-EGFP