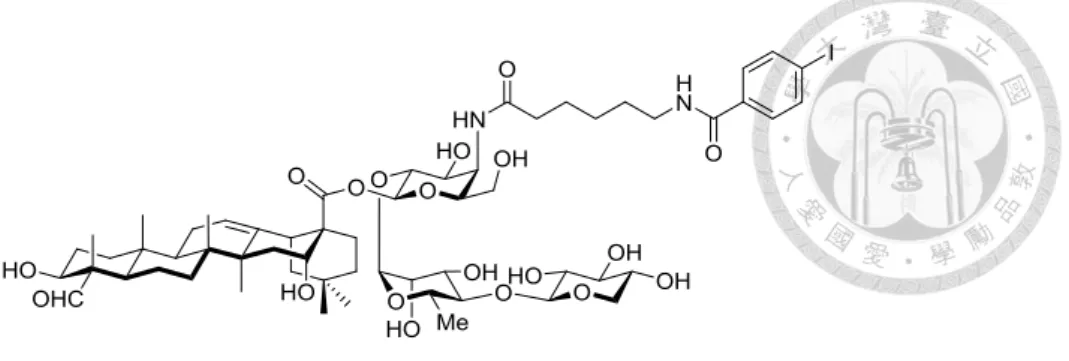
碩士論文
Graduate Institute of Pharmaceutical Sciences College of Medicine
National Taiwan University Master Thesis
半合成 GPI-0100 衍生物暨合成其葡萄醣醛酸類源物作為免疫 活化劑
Semisynthesis of GPI-0100 Analogues and Synthesis of Its Glucuronide-Congeners as Immunostimulators
刁志威 Chee-Wai Chaw
指導教授:梁碧惠 博士 Advisor: Pi-Hui Liang, Ph.D.
中華民國 108 年 7 月
July, 2019
誌謝
在這幾年的研究所生涯裡,投入了好多精神與時間。在這些日子裡,老師們的指 導及同學們的協助,對我的研究及心理歷練是具有非常深刻的幫助的。
首先,感謝梁老師願意收下我並給予循循善誘。在實驗室內也盡可能地提供足夠 的資源及良好的實驗環境讓我們專注於實驗。
接著,實驗室內的各位學長姐及同學們的幫助。除了一起做實驗,由於結構的複
雜度常常無法自己利用200NMR 做分析,所以還要特別感謝幫忙測 400 及 600NMR 和
送測HRMS 的學長們!在 HPLC 的使用上,大家也很願意讓出儀器優先畢業班學生使
用。
當然,這兩年除了實驗以為,在藥學所所學到關於藥學研究的知識,讓我開了眼 界,學到很多以前在化學系從未學會的。這兩年也參加過兩次的研討會都是一些難忘 的回憶。
這是我第一次的論文撰寫,花了不少時間來來回回,從一開始的初稿一路大改好 幾回,才終於得以在期限內完成。在撰寫過程學著更細心謹慎,也感謝口委們的細心 修改及意見。
皂皮樹(Quillaja saponaria)是一種富含皂苷的常綠樹。其樹皮的提取物已被廣泛 研究並證明具有良好的佐劑活性。將其粗樹皮提取物在溫和的鹼性條件下水解化學不穩
定的醯基側鏈,並以醯胺鍵與脂族十二烷基鏈連接,可以得到半合成的GPI-0100。與 QS-
21 相比,它是一種更安全的佐劑,然而,其富含異質成分和潛在的毒性阻礙了其在臨床
中的廣泛應用。由於在合成這種皂苷時,將葡萄醣醛酸基與皂皮酸C-3 的鍵結是困難及
有挑戰性的。為了解決這個問題,我們提出了兩種策略。首先,我們利用6-N-葡萄醣醛
基取代葡萄醣醛酸基來合成 GPI-0100 的葡糖苷酸類源物。在室溫 TBDMSOTf 酸催化
下,6-N-葡萄醣醛基與皂皮酸 C-3 位置的醣基化可以達到 71%的產率。然後將 N-6'位置
的疊氮還原,並以Cbz 保護基做保護,除去 C-28 的烯丙基後得到化合物 39。去保護後
的羧基與三醣進行醣基化得到88% 產率(α/β 比例 = 1/10)的 40。在 40 個別與不同芳
香基烷基酸鍵結後,進行去保護得到產物42a 和 42b。另外,我們也開發了一種能夠從
皂皮樹皮提取物中以 38%分離產率分離出在 C-3 含有三醣的皂皮酸皂苷 44。接著以半
合成的方式在C-28 的羧基接上三醣,以 BF3•OEt2為活化劑得到87%含有六醣的皂苷化
合物。在全部去保護後,與末端含有芳香環的烷基胺以醯胺鍵做鍵結,得到最終產物51a
和51b。這兩系列的化合物將會進一步評估其免疫刺激能力。
關鍵詞: 疫苗佐劑、皂苷、QS-21、GPI-0100
Quillaja saponaria is an evergreen tree and rich in quillaic acid containing saponins.
Extracts from its bark have been extensively studied and demonstrated as promising adjuvant activity. The crude bark extract was processed under mild basic hydrolysis and then conjugated with an aliphatic dodeacyl chain via a hydrolytically stable amide bond to give GPI-0100.
Comparing to QS-21, it is a safer adjuvant. However, its heterogeneous composition and inherent toxicity prevent its wider use in clinic. These saponins bearing a glucuronyl group at C-3 position of quillaic acid made it difficult in synthesis process. To solve this problem, two strategies were conducted. Firstly, we synthesized the glucuronide congeners of GPI-0100 via replacing glucurosyl group by 6-N-glycosyl group. Using TBDMSOTf as a promotor, the glycosylation of 6-N-glycosyl donor with quillaic acid 3 gave 36 in 71% yield. Followed by azide reduction and Cbz formation at N-6′ position, and then removal of the allyl group at C-28 afforded compound 39. 39 was glycosylated with trisaccharide donor to afford 40 in 88% yield (/β ratio = 1/10). Compound 40 was then subjected to hydrogenolysis under 55 psi H2
catalyzed by 10% Pd(OH)2/C, followed by amide formation with two arylalkanoic acids, acid/base hydrolysis to afford final products 42a and 42b. Secondly, we developed an isolation method from Q. saponaria tree bark extract to get 3-O-trisaccharide containing quillaic acid 44 in 38% isolated yield, 44 was also semi-synthesized with trisachharide at C-28 position by using
will be evaluated for immunostimuating ability in the future.
Key words: Vaccine adjuvant, Saponins, QS-21, GPI-0100
Ac Acetyl
ACN Acetonitrile
All Allyl
Bn Benzyl
Bz Benzoyl
Cbz Benzyloxycarbonyl
CbzCl Benzyl chloroformate COSY Correlation spectroscopy CpG Cytosine-phosphate-guanine CTL Cytotoxic T lymphocyte
DBU 1,8-Diazabicyclo[5.4.0]undec-7-ene
DC Dendritic cell
DC-SIGN Dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin
DCC Dicyclohexylcarbodiimide
DDQ 2,3-Dichloro-5,6-dicyano-1,4-benzoquinone DIPEA N,N-Diisopropylethylamine
DMA Dimethyl acetamide DMAP 4-Dimethyl aminopyridine EtOAc Ethyl acetate
FDA Food and Drug Administration
GD3 Ganglioside GD3 (disialoganglioside)
HBV Hepatitis B vaccine
HPLC High performance liquid chromatography HRMS High resolution mass spectrometry
HSQC Heteronuclear single quantum coherence spectroscopy ISCOM Immune stimulating complexes
KLH Keyhole limpet hemocyanin
MeOH Methanol
MHC-I Major histocompatibility complex-1 MPL Monophosphoryl lipid A
MS Molecular sieves
MUC1 Mucin 1 (also called CA 15-3, KL-6 and BM7)
NBS N-Bromosuccinimide
NHS N-Hydroxysuccinimide
NIS N-Iodosuccinimide
NMR Nuclear magnetic resonance ODN Oligodeoxynucleotide
OVA Ovalbumin
QS-21 Quillaja saponaria Molina, fraction 21 Q. saponaria Quillaja saponaria
rt Room temperature
SAR Structure-activity relationship STol p-Methylphenylthiol
TBAI Tetrabutylammonium iodide
TFA Trifluoroacetic acid
THF Tetrahydrofuran
TLC Thin layer chromatography TLR Toll-like receptor
TES Triethylsilyl
TESOTf Trimethylsilyl trifluoromethanesulfonate TsOH p-Toluenesulfonic acid
Table of Contents
誌謝 ... i
中文摘要 ... ii
Abstract ... iii
Abbreviations ... v
1. Introduction ... 1
1.1. Quillaja saponaria ... 1
1.1.1. Phytochemicals from Quillaja saponaria ... 2
1.1.2. Biological activity ... 3
1.1.3. Quillaic acid ... 4
1.1.4. Production of Q. Saponaria saponins ... 5
1.1.5. Quil-A®... 6
1.1.6. QS-21 and its purified analogues ... 7
1.2. Vaccine adjuvant ... 11
1.2.1. Approved vaccine adjuvants ... 12
1.2.2. QS-21 adjuvanted vaccine ... 13
1.2.3. Plausible mechanism of QS-21 ... 15
1.3. Development of QS-21 ... 16
1.3.1. Challenges of using QS-21 ... 16
1.3.2. Synthesis of QS-21 ... 17
1.3.3. GPI-0100 ... 18
1.3.4. Synthesis of QS-21 analogues inspired by GPI-0100 ... 19
1.3.5. Truncation of glucuronide and 28-O-linked tetrasaccharide ... 21
1.3.6. Development of GPI-0100 analogues in our lab ... 23
2. Motivation ... 24
3. Results and discussion ... 26
3.1. Retrosynthetic analysis ... 26
3.2. Part 1: Synthesis of building blocks and trisaccharide ... 27
3.3. Part 2: Synthesis of azido-glucose donor... 29
3.4. 3-O-Glycosylation of Quillaic ester... 31
3.5. Synthesis of tetrasaccharide saponin ... 31
3.6. Part 3: Semisynthesis of hexasaccharide saponins ... 35
3.7. Global deprotection and amide formation of hexasaccharide saponins ... 37
4. Conclusion ... 41
5. Experimental section ... 43
5.1. General Procedures ... 43
5.2. Chemical reagents ... 43
5.4. Synthetic Procedures ... 46
6. References ... 84
Appendixes ... 97
List of figures
Figure 1. Quillaja saponaria soap-bark tree. ... 1Figure 2. General structure of Q. saponaria saponins... 3
Figure 3. Structure of quillaic acid (1). ... 4
Figure 4. Production process of saponin extracts from Q. saponaria. ... 6
Figure 5. HPLC chromatogram of an aqueous bark extract. ... 8
Figure 6. Deacylated saponin (DS-1) and reacylated saponin (RDS-1). .... 20
Figure 7. Synthetic GPI-0100 analogues. ... 21
Figure 8. Structure of 10. ... 22
Figure 9. Truncation of 28-O-linkage tetrasaccharides.56 ... 22
Figure 10. Our preliminary work on the modification of GPI-0100 analogues. 23 Figure 11. Change of %concentration of saponin starting material and product. 37
List of tables
Table 1. Reported triterpene aglycones in Quillaja saponaria. ... 2Table 2. List of commercial saponin products from Q. saponaria. ... 5
Table 3. Lethality of saponins to CD-1 mice. ... 8
Table 4. Natural saponins from of Quillaja saponaria bark. ... 10
Table 5. Licensed vaccine adjuvants, their class, components and registered vaccine. ... 13
Table 6. QS-21 adjuvanted vaccines. ... 14
Table 7. Modification of our targets. ... 25
Table 8. Global deprotection of 40 and conditions of hydrogenation. ... 33
Table 9. HPLC gradient profile to monitor deacylation of QA saponin. .... 36
Table 10. Global deprotection and amide formation steps. ... 38
List of schemes
Scheme 4. Synthesis of four building blocks. ... 28
Scheme 5. Synthesis of trisaccharide donor 21. ... 29
Scheme 6. Synthesis of azido-glucose donor 20. ... 30
Scheme 7. 3-O-Glycosylation of quillaic ester 36. ... 31
Scheme 8. Synthesis of fully-protected tetrasaccharide saponin 40. ... 32
Scheme 9. Amide formation then global deprotection. ... 34
Scheme 10. Semisynthesis of fully-protected hexasaccharide saponin 46. 36 Scheme 11. Global deprotection then amide formation. ... 40
1.1. Quillaja saponaria
Quillaja saponaria Molina (from Quillajaceae family, Figure 1), also known as soap bark or soap bark tree, is commonly found in Chile.1-2 It is an evergreen tree growing up to 18 m (59 ft.) by 6 m (19 ft.) at a slow rate. The tree has thick, rough and dark bark, while its evergreen leaf is smooth, leathery and shiny with 3-5 cm long. The flowers of the tree are white and 15 mm diameter 5-parted borne in dense corymbs. Each dry fruit from this tree is star shape and containing 10-20 seeds.
Figure 1. Quillaja saponaria soap-bark tree. (cited fromGera-Untermhaus 1887)3
1.1.1. Phytochemicals from Quillaja saponaria
The word sapo is a Latin word which means “soap”, since saponin forms foam when it mixes with water due to its amphiphilic chemical structure which contains lipophilic aglycons and hydrophilic glycon. Nearly 60 saponins were isolated from the extracts of Q. saponaria. Most of them contain several triterpenoid structural motifs, as listed in
Table 1.4-5
Table 1. Reported triterpene aglycones in Quillaja saponaria.
Triterpene Ra Rb Rc Rd
Quillaic acid (1) CHO OH H CH3
Quillaic acid, 22β-OH CHO OH OH CH3
Gypsogenin CHO H H CH3
Phytolaccinic acid CH2OH H H CO2CH3
Phytolaccinic acid, 23-O-Ac CH2OAc H H CO2CH3
Echynocystic acid CH3 OH H CH3
The general structure of the saponins in Q. saponaria was glycosylated at the C-3 and C-28 positions (Figure 2). In most cases, the C-3 of triterpene was O-linked with
further O-glycosylated by either a β-D-Xylp or a α-L-Rhap residue. On the other hand, C- 28 position normally glysosylated with complex sugar moieties conserved with a disaccharide α-L-Rhap-(1→2)-β-D-Fucp.6
Figure 2. General structure of Q. saponaria saponins.
1.1.2. Biological activity
Historically, saponins from Q. saponaria have been attributed to a wide range of applications in cosmetics, such as antidandruff, cleansing, emulsifying, masking, skin conditioning, and surfactant.4 Nowadays, they are approved for use as food additives in European Union, the United Kingdom, the United States, China, Japan, and so on.7-8
Since the aglycons of these saponins might have the affinity with cell membrane cholesterol leading to antimicrobial activities, some pharmacological activities, such as antiviral, antifungal, antibacterial, and antiparasitic ones, have been reported for Quillaja saponins.9-11
Interestingly, Quillaja saponins even showed hepatoprotective activity in Wistar mice assay. In the studies of Abdel-Reheim et al., these saponins significantly reduced oxidative stress and suppressed NOS production.12
1.1.3. Quillaic acid
The most well-known structure of the phytochemicals from the bark and root extracts of Q. saponaria is the aglycon part of the saponins, quillaic acid, which is characterized by an aldehyde group and hydroxyl group substituted at C-4 and C-16α respectively.13-14
Figure 3. Structure of quillaic acid (1).
Besides bark of Q. Saponaria, quillaic acid can also be isolated from leaves of Quillaja brasiliensis, roots of Saponaria officinalis, or whole plants of Silene armeria.15-
17 According to studies, most quillaic acid containing saponins had been reported to
possess immunoadjuvant activity.18
1.1.4. Production of Q. Saponaria saponins
The extract from Q. saponaria is very useful in multiple applications. Table 2 shows the list of commercial products of saponin extract from Q. saponaria.
Table 2. List of commercial saponin products from Q. saponaria.
Producer Commercial name Composition declared by producer
Desert King International, USA
QY-150 Liquid, non-refined QE
Natural Response, Chile QP-1000 Spray-dried powder, non-refined QE
Producer 1, USA BF 3399 Liquid, non-refined QE
Quest, Ireland Saponin 5012 Spray-dried powder, non-refined QE
Maruzen Pharmaceuticals, Japan
Quillajanin C-100 Liquid, partially refined QE
QE: Quillaja extracts
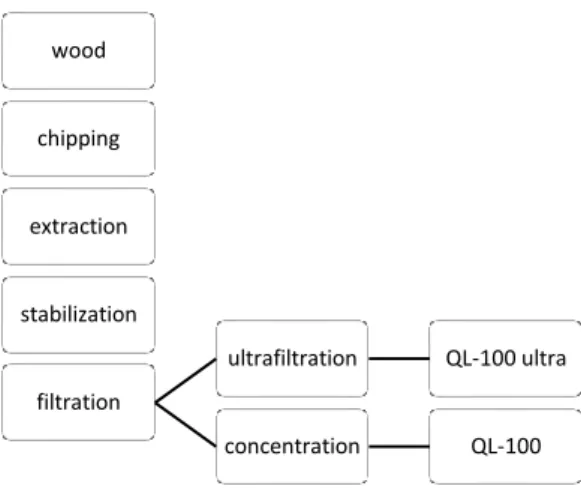
To satisfy the demand of Quillaja saponin, there has been a great ecological impact by felling and debarking about 60,000 old trees every year by the year 1998. To reduce the number of felling trees and augment the efficacy of extractions, a new production process (Figure 4) was introduced in 1996 which allowed to extract desired saponins from not only the bark but also the whole biomass of Q. saponaria. As a result, 25,000 trees were survived every year and less organic solvent was used through this ultrafiltration technique.19 Even through this method was efficient, it remained a need to provide greener
method to obtain Quillaja saponin.
Figure 4. Production process of saponin extracts from Q. saponaria.
1.1.5. Quil-A®
Quil-A®, a purified Q. saponaria extracts, is a saponin vaccine adjuvant product of Brenntag Biosector. In 1974, it was first prepared from the aqueous extract from Q.
saponaria by using gel exclusion, dialysis, and ion exchange chromatography.20 It was demonstrated to have antitumor potential. Although the results revealed that the survival rate of mice with leukemia was improved, high toxicity was presented in high dosage in the clinical practice.21-22 Nowadays, Quil-A® was mainly applied as adjuvant in veterinary vaccines, active component of ISCOM, nanoparticle adjuvants, raw material for QS-21 and other saponin fractions.23
Q. saponaria was ever considered as an ideal substance for vaccine development
due to its strong immunoadjuvant activity in the reports for Quil-A®.24-25 However, the
wood
chipping
extraction
stabilization
filtration
ultrafiltration QL-100 ultra
concentration QL-100
heterogeneous mixture of saponins contributed to its toxicological effects.24 Therefore, efforts have been taken to find purified fractions of Q. saponaria extract, or even a single pure analogues from Quil-A® .
1.1.6. QS-21 and its purified analogues
Quillaja saponins have been extensively used alone and mixed with other adjuvants
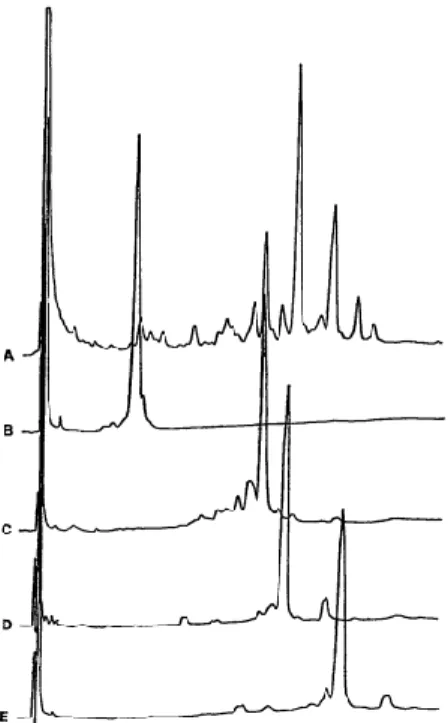
since that saponins were known to enhance antibody responses, helper and cytotoxic T- cell responses.26 Using reverse-phase chromatography (RP-HPLC), a series of fractions from Q. saponaria bark were purified by Kensil et al.(Figure 5), with QS-7 (B in Table 3), QS-17 (C in Table 3), Q-18 (D in Table 3) and QS-21 (E in Table 3). Among these component, QS-7 and QS-21 were found to be less toxic (Table 3).27 Between, QS-21 has been studied extensively and evaluated in over 100 clinical trials of vaccines against cancers and infectious diseases saponin adjuvant due to its higher abundance compared to QS-17.28
Figure 5. HPLC chromatogram of an aqueous bark extract. (cited from J. Immunol 1991, 146, 433.)27
Table 3. Lethality of saponins to CD-1 micea Dose (μg) Quil-A QS-7 QS-18 QS-21
125 1/5 0/5 4/5 0/5
250 2/5 0/5 5/5 0/5
500 4/5 0/5 5/5 1/5
aNumber of deaths per group of mice within 72 hours after intradermal injection of saponins.
QS-21 was a particular fraction from HPLC separation, and it contains two major isomeric molecular. Both of these saponins incorporate quillaic acid as a central triterpene aglycon core, with a branched trisaccharide residue is linked to the C-3 hydroxyl group, and a linear tetrasaccharide attached to the C-28 through an ester bond. The fourth component is a glycosylated pseudo-dimeric acyl chain linked to the fucose moiety via a
labile ester conjugation. The major and minor components differ in the constitution of the terminal sugar residue of the tetrasaccharide, which incorporate either an β-D- apiofuranose (Api) (65%) or a β-D-xylopyranose (Xyl) (35%), respectively.24, 29
Although QS-21 has been proven to be an exceedingly powerful adjuvant in immunotherapy, its tolerated dose in patients was limited typically to below 100 µg. To overcome its drawback, QS-7 was found not only negligible toxicity in mice, but also significant stand-alone adjuvant activity.30-31 QS-7 differs from QS-21 at the 4-O position of the fucopyranosyl unit that connects to the C-28 carboxyl group of quillaic acid (Table 4), that is, a small acetyl instead of characteristic long chiral acyl side chain. Furthermore, compared with QS-21, it has two extra monosaccharide units in the C-28 oligosaccharide domain. It was reported that the 3- and/or 4-O acetyl groups of the fucosyl unit might play an important role in tuning the adjuvanticity of the QS-7 analogs.31
Besides the most extensively studied QS-21, other fraction of the extracts has drawn the interest of research which pursued a reduced toxicity QS saponin-based immunostimulants, still retained or advanced adjuvant activities. QS-17 and the most abundant saponin in the extract, QS-18, were found to be the valuable leads. Comparing to QS-21, QS-17/18 has one additional β-D-glucopyranosyl (Glc) unit linked to the α-L- rhamnopyranosyl (Rha) unit at its 3-O position. QS-17 and QS-18 has a different sugar moiety at the end position of the acyl side chain, that is, disaccharide unit and
monosaccharide unit, respectively (Table 4).32
Conclusively, the RP-HPLC fractions of the Quil-A® mixture was studied by Kensil et al in 1991, who found the fractions QS-7, QS-17, QS-18 and QS-21 to be particularly
potent. QS-17 and QS-18 were the major component of Q. saponaria but suffered highly toxic in mice. On the other hand, QS-7 and QS-21 demonstrated far less toxicity and gained more extensive studies. To pursue the adjuvant with favorable immune responses, these compounds and their further developed analogues have been extensively studied.
Table 4. Natural saponins from of Quillaja saponaria bark.
Compound R1 R2 R3 R4 R5
QS-21api H Acyl H H β-D-Apif
QS-21xyl H Acyl H H β-D-Xylp
QS-7 α-L-Rhap Acetyl N/A β-D-Glcp β-D-Apif QS-17 H Acyl α-L-Rhap β-D-Glcp β-D-Apif
QS-18 H Acyl H β-D-Glcp β-D-Apif
1.2. Vaccine adjuvant
Vaccine immunology has been well developed since last century. To combat against a specific disease by vaccine, an innocuous form of disease agent such as killed or weakened bacteria or viruses was usually employed to stimulate antibody production and activate cellular immunology. Many vaccines such as smallpox, measles, and polio in most cases have been proven very effective in human protection.33-34 For example, reported cases of wild polio virus was reduced to less than 100 cases globally in 2015.35 However, vaccines often come with risks of adverse events and large-scale production of vaccine is very challenging.36 Therefore, there is a growing need for a potentiator, so called adjuvants, which incorporate in therapeutic vaccine formation to enhance, modulate and prolong the immune response.
An adjuvant is a substance that added to vaccines to increase the immunogenicity and protection against infection. The word “adjuvant” means “to aid” which comes from the Latin word adjuvare. Adjuvants help activating the immune system by regulating the humoral or cellular immunity according to the objective of vaccination. Combining with adjuvants, human vaccines based on weakened or inactivated pathogens could elicit robust protective immune responses. For example, adjuvanted H5N1 pandemic influenza vaccines showed improved immune responses compared to un-adjuvanted vaccines in animal models.37 Therefore, adjuvants have several important benefits in vaccine
immunology, especially reducing the amount of required antigen and reducing the number of vaccine doses.
1.2.1.
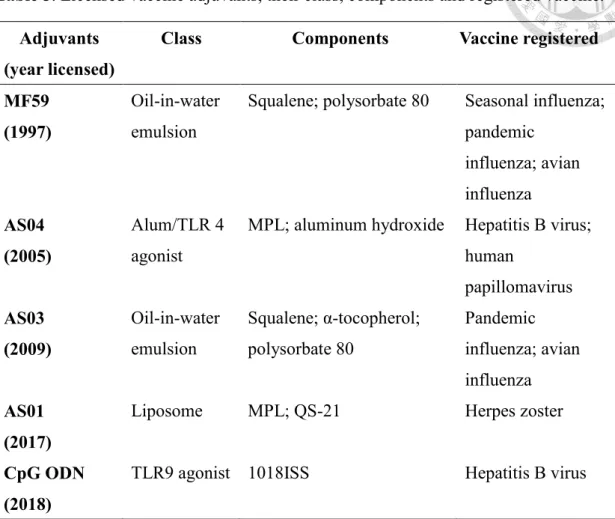
Approved vaccine adjuvantsTable 5 lists series of adjuvants that have been licensed in use with human vaccines to date.
Various of adjuvants have been used in human vaccines, such as mineral salts, oil in water, liposome, and TLR agonist. The development of adjuvants has been very slow.
Until the early 1990’s, aluminum salts were the only licensed adjuvants used in human vaccines in the United States.38 It is also known as Alum, generally considered as a stimulator of Th2 type immune response, and is the most widely used adjuvants in practical human vaccination.39-40 After decades, oil-in-water MF59 and AS03 adjuvants were used in pandemic influenza and avian influenza vaccines to improve the humoral and cell-mediated immunity.41 Both MF59 and AS03 contain squalene, but they have different compositions. AS03 has a component of α-tocopherol to modulate innate immune response.42 HBV vaccines were registered with TLR agonist class adjuvants AS04 and CpG ODN which were licensed in 2005 and 2018, respectively. Recently, liposome class adjuvant AS01 with the components of MPL and QS-21 were approved to be used in Herpes zoster vaccines. It is worthy mentioning that QS-21 is the second non-
endogenous substrate that was approved by FDA as an adjuvant.
Table 5. Licensed vaccine adjuvants, their class, components and registered vaccine.
Adjuvants (year licensed)
Class Components Vaccine registered
MF59 (1997)
Oil-in-water emulsion
Squalene; polysorbate 80 Seasonal influenza;
pandemic influenza; avian influenza AS04
(2005)
Alum/TLR 4 agonist
MPL; aluminum hydroxide Hepatitis B virus;
human
papillomavirus AS03
(2009)
Oil-in-water emulsion
Squalene; α-tocopherol;
polysorbate 80
Pandemic influenza; avian influenza AS01
(2017)
Liposome MPL; QS-21 Herpes zoster
CpG ODN (2018)
TLR9 agonist 1018ISS Hepatitis B virus
1.2.2. QS-21 adjuvanted vaccine
QS-21 is one of the most potential adjuvant with favorable immune responses by promoting high antigen-specific antibody responses.43-44 Unlike aluminum hydroxide, QS-21 promotes a balanced of both IgG1 and IgG2 production.27, 45-46 Further studies showed that QS-21 induces IL-2, IFN-γ, and Th1 bias immunity in vaccine responses.43,
47-48
Besides of being used solely as an adjuvant , the amphiphilic property of QS-21 allowed it to be formulated into a liposome form (AS01) or an oil-in-water emulsion (AS02) to induce humoral and cellular immunity against cancers and pathogens (Table 6).49 Some of them had been proceeded to clinical trial, SHIGRIX®, a combination of varicella-zoster virus glycoprotein gE and AS01, is the only approved vaccine to prevent herpes in 2017. Both Maria vaccine Mosqurix® and OBI-822/QS-21 are being evaluated in phase III clinical trial for the intervention of malaria and triple negative breast cancer (NCT03562637, NCT03608878) respectively.50-51 The phase III trial of GM2/QS-21 was terminated due to low efficacy.52 In addition, QS-21 has also been applied with ACC-001 in phase II trial of early Alzheimer’s disease.53-54
Table 6. QS-21 adjuvanted vaccines.
Combination (brand name) Intervention Status
VSV gE/AS01 (SHIGRIX®) Herpes zoster Approved (Oct. 2017) RTS, S/AS01 (Mosqurix®) Malaria Phase III
GM2-KLH/QS-21 Stage II melanoma Phase III (terminated) OBI-822/QS-21 Triple negative breast cancer Phase III
ACC-001/QS-21 Early Alzheimer’s disease Phase II (completed)
1.2.3. Plausible mechanism of QS-21
Although the potency of QS-21 had been investigated in over 100 clinical trials, the
exact mechanism of action of QS-21 in vaccination still remains unknown. It is generally agreed that QS-21 does not directly interact with Toll-like receptors (TLR), and does not operate by depot effect.55
Since QS-21 has the potential ability to increase T-cell response, it was proposed that the C4-aldehyde moiety could form a Schiff base with amino groups on T-cell surface receptors to activate T-cell.56 To investigate its importance in activating immune responses Fernández-Tejada et al. synthesized QS-21 analogues by removing the C4- aldehyde group. However, the results revealed that either reducing the C4-aldehyde to alcohol or alkyl group, saponins demonstrated the similar immunoresponses in mouse vaccination with GD3-KLH, MUC1-KLH, and OVA.57 Thus, the hypothesis of the Schiff base mechanistic hypothesis remains as question.
DC-SIGN, a receptor on DC, usually binds to fucopyranosyl residues and biases its response toward Th2 immunity.58 Since QS-21 induce both Th1 and Th2 immunity, The fucose residue of QS-21 was proposed to take responsibility to be an internal moiety binding to DC-SIGN to induce both Th2 immunity.59
The amphiphilic property of QS-21 has been postulated that is important since the deacylated saponin (DS-1, Figure 6) lost the capacity to stimulate Th1 immunity.60 The
lipophilic acyl chain might facilitate the delivery of exogenous antigens into the APC and further inducing immunity in T-cells.61 In addition, triterpene’s high affinity for cholesterol could make QS-21 facilitates the endosomal escape of antigens, leading to DC activation and antigen cross-presentation by collecting the endosomes-lysosomes and destabilizing them.62-63
1.3. Development of QS-21
1.3.1.
Challenges of using QS-21Despite the success of QS-21, there are several limitations while employing QS-21 in cancer vaccine or adjuvant. Firstly, it was limited to only 50 μg of dosage for the usage in healthy human due to its inherent toxicity.64 Secondly, QS-21 is chemically unstable, it was easily decomposed in either high temperature or pH ≥ 7.4 environment, which made it more difficult to store.65 Finally, the isolated amount of QS-21 from the soap bark extract was limited (below 0.5%) and it greatly impacts on the sustainable cultivation of Q. saponaria.66-67 Together, these disadvantages might prevent QS-21 for a wider use in clinic. Thus, to overcome these obstacles developing of synthetic approaches toward QS- 21 and its analogues would allow us to understand the structure activity relationship and might also help to explore their mechanism.
1.3.2. Synthesis of QS-21
QS-21api and its analogues were firstly synthesis by D. Gin’s group in 2005 (Scheme 1).68-69 B(PhF5)3 was applied to build the most challenging linkage between glucuronate 2 and quillaic ester 3. After a series of modifications on glucuronide 4, the acylated tetrasaccharide 6 was attached to C-28 position under the catalysis of BF3·OEt2 at -78 °C to obtain beta-selective glycosidic bond. The similar procedures were also applied on the synthesis of other analogues such as QS-21xyl and QS-7 by the same group.31, 70 Briefly, they achieved total synthesis of QS-21 in 70 steps. Although this synthetic methods was still lacked of efficiency, their pioneer studies provided valuable synthetic methods for constructing complex triterpenoid glucosides.
Scheme 1. Synthesis of QS-21.68
1.3.3. GPI-0100
To overcome the drawbacks of QS-21 and QS-7, a novel semisynthetic saponin, GPI-0100, has been developed from QS-7. By deletion of the unstable acyl linker of QS- 21 and installation of a dodecyl long chain at the glucuronic acid domain through a more stable amide bond (Scheme 2), GPI-0100 demonstrated higher stability in aqueous solution and lower toxicity in mice. It was 10 times less toxic than QS-21 with a lethal
dose of 5 mg in mice.71-72 However, clinical trial showed that only 20 times dosage of GPI-0100 could achieve the same efficacy of QS-21, which under such dose, it led to hepatoxicity.73
Scheme 2. Semisythesis of GPI-0100.72
1.3.4. Synthesis of QS-21 analogues inspired by GPI-0100
The chemically unstable acyl side of QS-21 had been hydrolyzed to give DS-1 (Figure 6) by Lui et al..74. Besides, DS-1 was further conjugated with aliphatic chain to give RDS-1, which also known as an isolated GPI-0100. These derivatives demonstrated
significant reduction of hemolytic affect than QS-21. However, RDS-1 needed higher dose to induce comparable IgG1 titer and the IgG2a antibody secretion and DS-1 lost adjuvantivity. These results revealed that the deacylated saponin was a poor immunostimulator, but the aliphatic chain-bearing saponin restored the adjuvant activity.
Figure 6. Deacylated saponin (DS-1) and reacylated saponin (RDS-1).
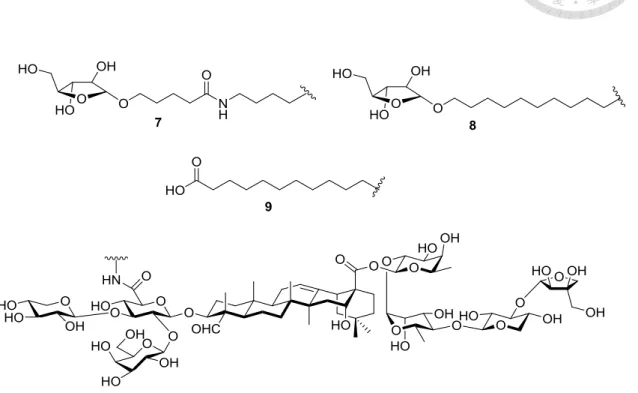
Another series of GPI-0100 inspired synthetic analogues was developed by Wang et al.75-76 In their studies the acyl side chain of QS-21 was completely removed, instead a long alkyl chain was attached on the glucuronide domain through a stable amide bond (Figure 7). Evaluation of the adjuvant activity of the four analogues with different terminal-functionalized long chains have been demonstrated. RDS-1 had poor water solubility while the glycosylated 7 and 8, which were designed to improve the solubility,
were poor in inducing Th1 immunity. Among these, only the carboxylated 9 demonstrated comparable IgG titer and IgG2a sub-type switching comparable to that of GPI-0100.
Figure 7. Synthetic GPI-0100 analogues.
1.3.5. Truncation of glucuronide and 28-O-linked tetrasaccharide
The influence of adjuvant activity by modifying glucuronic acid site of QS-21 have been investigated by Gin’s group. The results showed that conjugation of glycine,
ethylamine or ethylene diamine on glucuronic site retained similar effect on humoral and cellular immunity induction but no effect on the physical properties.56 Furthermore, compound 10 (Figure 8), structure of removal of C-3 glucuronide trisaccharide and modification on acyl side chain, surprisingly demonstrated comparable immunological response to natural QS-21.57
Figure 8. Structure of 10.
To investigate the function of C-28 linked moieties, the 28-O-linked tetrasaccharide
was then truncated by Gin’s group to trisaccharide, disaccharide, and monosaccharide
(Figure 9).56 As a result, the adjuvant activity was declined along with the decrease of the sugar number.56 Thus, trisaccharide in compound 12 was concluded to be the simplest moiety at C-28 to retain comparable immune stimulating effect to QS-21.
Figure 9. Truncation of 28-O-linkage tetrasaccharides.56
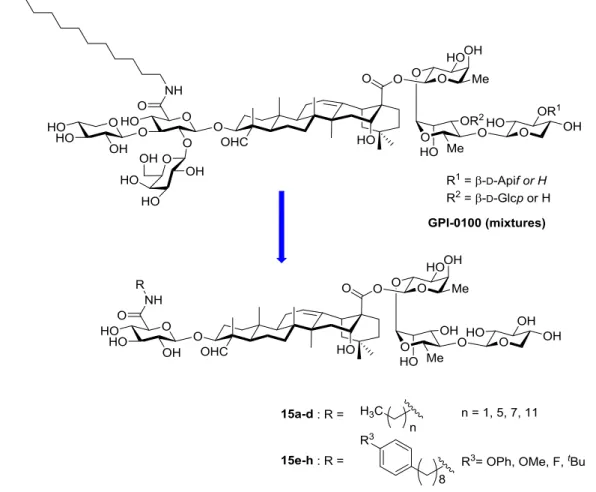
1.3.6. Development of GPI-0100 analogues in our lab
We have developed a concise chemical approach towards a variety of GPI-0100 analogues. These analogues consisted of truncated sugar moieties at C-28 domain and a series of diversified alkyl and p-substituted phenyloctyl chains were attached to glucuronic acid via an amide linkage (Figure 10). Murine immunology studies demonstrated that phenyloctyl chain-bearing saponin variants 15e-h possessed promising activities in antigen specific CD8 T-cell immunity.
Figure 10. Our preliminary work on the modification of GPI-0100 analogues.
2. Motivation
According to the development of QS-21 as immunostimulators previously, a blueprint to make a potent and safe saponin-based immunostimulator was proposed. We have developed a series of immunity-favorable GPI-0100 analogues by chemical synthesis. In this study, we still faced some obstacles, 1) quillaic acid was expensive (USD $4000 for 5 g) making the synthesis of final compounds not cost-effective; 2) glucuronidation with quillaic acid was low yield; 3) our compounds 15a-h were a bit poor water solubility, analogue with higher solubility is demanded.
To address these obstacles, two objectives were proposed (Table 7). First objective:
6-N-glucosyl group at C-3 position was introduced, it could form a “reverse” amide moiety of our targets 15a-h. So far no such compounds have been proposed, according to the best of our attention in the literature. Second objective: Since most of quillaic acids
were obtained from the Q. saponaria bark extract through several hydrolysis process, we proposed to extract 2′-D-galactosyl-3′-D-xylosyl glucuronyl 3-quillaic acid directly from
Ultra Q-100. This would allow us to skip the glucuronidation step and also would have intact trisaccharide at C-3 position which might help to improve water solubility. Actually, such strategy was disclosed in the patent for the preparation of GPI-0100,77 however, the extraction process from Ultra Q-100 remained to be fine-tuned .
Upon obtaining the desired compounds from both objectives, they would be further
conjugated the trisaccharide at C-28 position and then attached the arylalkyl moiety at 6-N-glucosyl or 6-position of glucuronyl through “reversed amide” bond formation or
amide to give final compounds A and B, respectively.
Table 7. Modification of our targets.
Modifications R1 R2 R3
Target A Target B
H H Reversed amide linkage
β-D-Galp β-D-Xylp Amide linkage
3. Results and discussion
3.1. Retrosynthetic analysis
The target A & B were divided into three parts, including lipophilic long chain domain, trissaccharide region, and aglycon (Scheme 3). For the convenience of diverting various long carbon chain, the first disconnection was the amide bond to achieve late stage modifications. Secondly, the preferred protecting groups on the saponins were benzyl, carbamoyl, acetyl, and triethylsilyl groups, which could be removed under mild conditions. Thirdly, construction of 21 would be achieved through (2+1) pathway, which was glycosylation of 24 and 25 to give disaccharide 22 and then conjugated it with 23.
Tricloroacetimidate 21 would be conjugated with nature extracted trisaccharide-saponin or 6-N-glucosyl bearing quillaic acid and then global deprotection to give our target saponins A and B.
Scheme 3. Retrosynthesis analysis of targets A and B
3.2. Part 1: Synthesis of building blocks and trisaccharide
The starting material L-rhamnose was proceeded under acetylation and thio- glycosylation to afford thio-rhamnoside 26 (Scheme 4). The acetyl groups on 26 were then removed by the treatment of 26 with sodium methoxide. Then, the 2,3-cis-diol was protected by isopropylidene to afford acetonide 24.
D-Xylosyl imidate donor 25 was prepared by the starting material D-xylose underwent
acetylation and then selectively removal of 1-O-acetyl group by ethylene diamine/AcOH to give hemiacetal 27. Followed by trichloroacetimidate formation, D-xylosyl donor 25 was readily available to conjugate with rhamnose building block.
D-Fucose was subjected to Fisher glycosylation with benzyl alcohol and its 3,4-cis- diol was protected by isopropylidene to afford protected 23.
The allylic group was introduced to the C-28 carboxylic acid to afford quillaic ester 3 by allylic bromide in 84% yield (Scheme 4).
Scheme 4. Synthesis of four building blocks.
The coupling of rhamnose 24 with xylose 25 under catalytic amount of BF3·OEt2
afford disaccharide 28 in 90% yield (Scheme 5). The disaccharide 28 was then proceeded under removal of isopropylidene group by 80% AcOH(aq), followed by acetylation to obtain thio-disaccharide 22 in 33% yield over two steps. Thio-disaccharide 22 was subsequently coupled with fucose 23 to furnish desired trisaccharide 23 in 74%. Finally, after hydrogenation and trichloroacetimidate formation of trisaccharide 29, penta- acetylated trisaccharide donor 21 was readily available.
Scheme 5. Synthesis of trisaccharide donor 21
3.3. Part 2: Synthesis of azido-glucose donor
The starting material diacetone D-glucose was proceeded to benzylation with benzyl
alcohol, followed by deacetonization then acetylation to afford 30 (Scheme 6). After the 1-O-Acetyl group was selectively replaced by thiotoluene group, the remaining acetyl groups was then hydrolyzed by the treatment of sodium methoxide. Then, 4-O and 6-O position were temporary masked by benzylidene and followed by benzylchloride (BzCl) installation on 2-O position to afford 33. By the treatment of anhydrous Borane in THF under the catalysis of trimethylsilyl trifluoromethanesulfonate (TMSOTf), the benzylidene group was hydrolyzed and selectively free 6-O while 4-O was still under protection of benzyl group.
To prepare the 6-azido deoxy-glucose 35, glucose 34 was first treated with methanesulfonyl chloride, then the mesylate group was replaced by azide group under the suspension with sodium azide in DMF at 80 °C. Finally, followed by trichloroacetimidate formation, glucosyl donor 20 was available to conjugate with quillaic ester 3.
Scheme 6. Synthesis of azido-glucose donor 20.
3.4. 3-O-Glycosylation of Quillaic ester
The conjugation of quillaic ester 3 with azido-glucose 20 was successfully resulted in 71% yield of product 36 (Scheme 7). Interestingly, this result revealed the selectivity of 3-O glycosylation over 16-O position.
Scheme 7. 3-O-Glycosylation of quillaic ester 36.
3.5. Synthesis of tetrasaccharide saponin
With the glycoside 36 in hand, further modifications had been conducted to unmask the C-28 carboxylic acid (Scheme 8). First, the benzoyl group was hydrolyzed under basic condition at elevated temperature. Surprisingly, 28-O-allyl ester was not affected under this harsh environment. The resulting azido-glycoside was then proceeded under triethylsilylation, reduction of azide group to amine and protection of amine group by benzyl carbamate formation to afford fully-protected quillaic ester 37.
The O-allyl ester 38 was hydrolyzed by the catalysis of Pd(OAc)2 under mild acidic
environment to give glycoside acceptor 39. Under the catalysis of Lewis acid at -78 °C, the monoacid 39 was conjugated with trisaccharide 21 to obtain 40 in 88% yield (α/β = 1/10).
Scheme 8. Synthesis of fully-protected tetrasaccharide saponin 40.
40 was proceeded to global deprotection prior to amide bond formation. The fully protected saponin was suspended with Pd/C in THF under H2 atmosphere to hydrolyze the amino protecting carbamoyl group of 6-N and the benzyl groups on 3-O and 4-O of glucose. However, there was no reaction under this mild condition. After attempting with a series of reagent and conditions (Table 8), the hydrogenation was furnished by suspending with Pd(OH)2 in THF/MeOH under 55 psi of H2 for 36 hours (Entry 3), but the TES and acetyl protecting group were hydrolyzed as well under this condition. Upon complement of acidic hydrolysis and basic methanolysis, 41 was obtained in 11% yield
after HPLC purification
Table 8. Global deprotection of 40 and conditions of hydrogenation.
Entry Catalysis reagent Solvent Concentration of H2 Results
1 10% Pd/C THF 1 atm No reaction in 24 h
2 10% Pd(OH)2/C THF/MeOH 1 atm Only few product in
24 h
3 10% Pd(OH)2/C THF/MeOH 55 Psi Successfully
removed aryl
protections in 36 h.
Subjection of fully deprotected 41 with amide formation condion might appear side reactions, such as forming ester linkage coupling with 10-(4-(4- fluorophenoxy)phenyl)decanoic acid 17 or (E)-10-(4-(4-fluorophenoxy)phenyl)dec-9- enoic acid 16, the procedures were reversed by arranging the aidic and basic deprotections to the last step. After debenzylation of 40 which was confirmed mass spectrometry, the
amide bond formation was successively carried out with HBTU/DIPEA coupling system to afford conjugate amides (Scheme 9). After that, the amides were proceeded acidic hydrolysis and methanolysis to remove all the remaining protections to give compounds 42a and 42b.
Scheme 9. Amide formation then global deprotection
3.6. Part 3: Semisynthesis of hexasaccharide saponins
With the available mixture of Ultra Q-100, the semisynthesis of quillaic acid saponin was carried out with the hydrolyzation and protecting installation (Scheme 9). First, the ester bond conjugation at C-28 was hydrolyzed under basic condition at elevated temperature. To control the deacylation process in this harsh environment, the reaction mixture was monitored by HPLC chromatography following the gradient conditions as shown in Table 9 to monitor the consumption of starting materials and the yield of deacylated products. The results (Figure 11) showed that the starting material was completely hydrolyzed after 7 hours of reaction time in this basic condition The crude resulting mixture was then proceeded to triethylsilylation and selectively hydrolysis under mild condition to obtain diacid 44.
To selectively protect the carboxylic acid group on glucuronide with benzyl group, diacid 44 was treated with benzylchloroformate and the bulky tri-t-butylpyridine as a base to avoid to protect the carboxylic acid group at C-28. The monoacid 45 with 65% of yield was available to conjugate with trisaccharide. Finally, coupling of trisaccharide-imidate 21 with monoacid 45 using BF3•OEt2 brought 46 in 87%.
Scheme 10. Semisynthesis of fully-protected hexasaccharide saponin 46.
Table 9. HPLC gradient profile to monitor deacylation of QA saponin.
Time (min) % H2O %ACN Flow (mL/min)
0 90.0 10.0 1.0
30.0 60.0 40.0 1.0
40.0 30.0 70.0 1.0
42.0 90.0 10.0 1.0
50.0 90.0 10.0 1.0
Column: Vydac 214TP C4 5μ UV Detector: Wavelength = 210 nm
Figure 11. Change of %concentration of saponin starting material and product.
3.7. Global deprotection and amide formation of hexasaccharide saponins
With compound 46 in hands, our initial strategy to finish saponin target B was to conjugate with amines at final stage before removing all the protections (Table 10). The fully protected 46 was proceeded under hydrogenolysis of benzyl group, then followed by amide coupling. However, the resulting residue was a mixture of saponins with different numbers of deprotection of TES groups. Furthermore, the mixture became even more messy after conjugating with amine on TLC analysis (Table 10, Entry 2).
-20 0 20 40 60 80 100 120
0 2 4 6 8 10
product starting material
%Concentration
reaction time (h)
Table 10. Global deprotection and amide formation steps.
Entry Step 1 Step 2 Step 3 Results
1 THF, 1 atm H2, 18 h - - Debenzylation not
completed 2 THF, 50 Psi H2, 18 h Amide
formation
Acid & basic hydrolysis
Messy in HPLC
3 THF/MeOH, 1 atm H2, 18 h
Acid & basic hydrolysis
Amide formation
30-70% yield
With the experience of entry 2, the deprotection step was arranged prior to the amide
bond formation step (Table 10, Entry 3). After the debenzylation, the resulting residue was proceeded under acidic hydrolysis the isopropylidene, triethylsilyl groups, and then methanolysis the remaining acetyl groups. Because C-28 ester bond was easily to be hydrolyzed, the concentration of TFA was adjusted from 80% to 75%, and the yield was improved from 13% to 28% for three steps. Followed by HPLC purification and concentration, compound 50 was obtained in 28% ready for late stage amine conjugation with 4-phenoxyphenyl-octanoic acid and 4-methoxyphenyl-octanoic acid by using HBTU/DIPEA in DMA to give final compound 51a (80%) and 51b (30%), respectively (Scheme 11).
Scheme 11. Global deprotection then amide formation.
4. Conclusion
In order to get various GPI-0100 analogues for SAR study, as well as development of efficient strategy to get these compounds, two series of GPI-0100 analogues which contained the same trisaccharide moieties at C-28 of quillaic acid, but different modifications on 3-O-linkage were synthesized.
Target A was designed to replace 3-glucuronic acid of GPI-0100 analogues by 6-N- glucosyl moiety. Synthesizing target A was concise and efficient, two glycosylation steps were catalyzed by different promotors, TBDMSOTf was applied to build the linkage between 6-N3-glycoside 20 and quillaic ester 3 which resulted in β-selective compound 36 in 71% yield. BF3•OEt was employed to promote glycosylation of 39 with trisaccharide 21 to obtain tetrasaccharide sapnin 40 in 88% yield (α/β = 1/10). Amide bond formation and deprotection steps to give final A was tedious, first removal of benzyl of 40 was achieved under 55 psi H2 and 10% Pd(OH)2/C. Amide bond formation of 41 with arylalkyl acids by using HBTU/DIPEA as coupling reagents, and then global deprotection of the remaining protecting groups, the final 42a and 42b were obtained by purification of using HPLC.
Target B was designed to take advantage of Q. Saponaria extract to give 3-O- trisaccharirde containing quillaic acid which avoided notorious glucuronidation step. To
our grateful, conditions of isolation of the desired monoacid prosapogenin from ultra Q- 100 was fine-tuned to have 38% yield. Again, glycosylation of monoacid 45 with
trisaccharide 21 was achieved by using BF3•OEt2 as a promotor to give hexasaccharide saponin 46 in 87% yield. The hexasaccharide saponin finals 51a and 51b were carried out by global deprotection prior to amide formation with arylalkyl amines by HBTU/DMA system. This procedure allowed us to achieve late stage modification in synthesizing derivatives. However, the global deprotection steps could be further improved by adjusting the concentration of acid or the reaction temperature.
The adjuvant capability of these compounds will be evaluated afterward.
5. Experimental section
5.1. General Procedures
All reagents and solvents were reagents grade and used without further purification.
For those reagents which were stored in fridge, were opened and used after materials were recovered to rt. Molecular sieves were activated at 200 °C and cooled down to rt prior to use. Reaction progress was monitored by RP-HPLC or analytical TLC on 0.25 mm Merck Milipore silica gel 60 F254 using p-anisaldehyde and ceriumammonium molybdate as staining agents. Flash column chromatography was performed using 230-400 mesh silica gel.
5.2. Chemical reagents
Acros Organics:
Acetic acid, acetonitrile (ACN), acetone, amberlite IR-120 resin, benzyl chloroformate (CbzCl), boron trifluoride diethyl etherate (BF3·OEt2), N-bromosuccinimide (NBS), 1,8- Diazabicyclo[5.4.0]undec-7-ene (DBU), dichloromethane (CH2Cl2), 4-dimethylamino pyridine (DMAP), dimethylforamide (DMF), DIPEA, N-iodosuccinimide (NIS), Na2S2O3, NH4Cl, pyridine, Pd(OH)2/C, t-Butyldimethylsilyl trifluoromethane (TBDMSOTf), tetrahydrofuran (THF), TFA, p-thiocresol, Triethylsilyl
trifluoroethanesulfonate (TESOTf), Trimethylsilyl trifluoromethanesulfonate (TMSOTf), triethylamine, L-arabinose, D-fucose, D-xylose, L-rhamnose
Alfa Aesar:
Trichloroacetonitrile.
Carbosynth Limited:
HBTU
Desert King:
Ultra Q-100
Fisher Scientific:
2,2-dimethoxypropane, NaHCO3, K2CO3,Pd/C
RDH:
Molecular sieves 4 Å , celite
Merck KGaA:
CDCl3, Kieselgrl 60 silica gel 40-63 μm (230-400 mesh)
Nanjing Spring & Autumn Biological Engineering Co., Ltd. (南京春秋生物工程有限 公司):
Quillaic acid
Sigma-Aldrich:
Ac2O, CD3OD, 2,4,6-tri-tert-butylpyridine (TBP)
Uni-onward corp. (友和貿易股份有限公司):
ACS-CHCl3
5.3. Instruments
NMR spectra were acquired by employing Bruker-AV-400 (400 MHz) and Bruker- AV-600 (600 MHz). Chemical shifts (δ) are presented in ppm relative to 1H: 7.26 ppm,
13C: 77.0 ppm for CDCl3; 1H: 3.31 ppm, 13C: 49.0 ppm for methanol-d4. Splitting patterns
are reported as s (singlet), brs (broad singlet), d (double), t (triplet), q (quartet), dd (double doublet), m (multiplet). Coupling constant (J) are given in Hertz (Hz).
Reversed phase HPLC purification and analyses were performed on HITCHI D-2000 Elite HPLC system equipped with auto-sampler L-2200, UV detector L-2420 and pump L-2130.
Exact mass measurements were carried out on VG platform electrospray ESI/MS or BioTOF II.
5.4. Synthetic Procedures
Trichloroacetimidoyl 2,3,4-tri-O-acetyl-α/β-D-xylopyranoside (25).
Imidate 25 was prepared by known procedures and confirmed by 1H NMR spectrum.78 25α:1H NMR (400 MHz, CDCl3) δ 8.66 (s, 1H), 6.47 (d, J = 3.4 Hz, 1H, H- 1), 5.56 (t, J = 9.9 Hz, 1H, H-3), 5.15–4.97 (m, 2H, H-2, H-4), 3.98 (dd, J = 11.0, 5.9 Hz, 1H, H-5a), 3.80 (t, J = 11.0 Hz, 1H, H-5b), 2.05 (s, 6H, CH3), 2.01 (s, 3H, CH3).
Benzyl 3,4-O-isopropylidene-α/β-D-fucopyranoside (23α/β).
To a stirred suspension of D-fucose (3.0 g, 18mmol) in BnOH (35 mL) was added camphorsulfonic acid (0.42 g, 1.8 mmol) and heated to 50 °C for 3 h. After cooling down to rt, the reaction mixture was diluted with EtOAc, washed by H2O and brine, dried over MgSO4, and then concentrated under reduced pressure. The residue was filtered by flash column (silica gel, MeOH/CH2Cl2 = 1/5) to obtain α/β-benzylated mixture as a white solid. To a stirred solution of α/β-benzylated mixture in ACN was added 2,2- dimethoxypropane (2.2 mL, 17 mmol) and TsOH·H2O (0.21 g, 1.1 mmol) under N2
atmosphere at rt. The reaction mixture was stirred for 1.5 h, quenched by Et3N and then concentrated under reduced pressure. The residue was diluted with CH2Cl2, washed by saturated NaHCO3, brine, dried over MgSO4, and then concentrated under reduced pressure. The residue was purified by column (silica gel, EtOAc/hexanes = 1/3 to 1/2) to afford 23α/β (2.0 g, 58%) as yellow syrups. 23α: Rf 0.28 (EtOAc/hexanes = 1/2) ; 1H NMR (400 MHz, CDCl3) δ 7.40–7.29 (m, 5H), 4.94 (d, J = 3.6 Hz, 1H, H-1), 4.78 (d, J
= 11.8 Hz, 1H, Ph-CH2), 4.57 (d, J = 11.8 Hz, 1H, Ph-CH2), 4.22 (t, J = 6.2 Hz, 1H), 4.19–4.11 (m, 1H), 4.09–4.02 (m, 1H), 3.81 (d, J = 3.8 Hz, 1H), 1.51 (s, 3H), 1.35 (s, 3H), 1.31 (d, J = 6.6 Hz, 3H, H-6); BBD 13C NMR (100 MHz, CDCl3) δ 137.2, 128.5, 128.0, 109.2, 96.9 (C-1), 76.1, 75.6, 69.7, 69.4, 64.1, 27.7, 25.9, 16.2 ppm; HRMS+ (ESI-TOF) calcd. for C16H22O5Na [M+Na]+ 317.1359, found 317.1371. 23β: Rf 0.23 (EtOAc/hexanes
= 1/2); 1H NMR (400 MHz, CDCl3) δ 7.37–7.28 (m, 5H), 4.94 (d, J = 11.6 Hz, 1H, Ph-
CH2), 4.58 (d, J = 11.6 Hz, 1H, Ph-CH2), 4.23 (d, J = 8.3 Hz, 1H, H-1), 4.06–3.97 (m, 2H), 3.85 (d, J = 5.3 Hz, 1H), 3.60 (t, J = 7.6 Hz, 1H), 1.53 (s, 3H), 1.45 (d, J = 6.5 Hz, 3H, H-6), 1.35 (s, 3H); BBD 13C NMR (100 MHz, CDCl3) δ 136.9, 128.5, 128.2, 128.0, 109.9, 100.9 (C-1), 78.7, 76.3, 73.6, 70.7, 69.2, 28.2, 26.3, 16.5 ppm; HRMS+ (ESI-TOF) calcd. for C16H22O5Na [M+Na]+ m/z 317.1359, found 317.1359.
p-Methylphenyl 2,3,4-tri-O-acetyl-β-D-xylopyranosyl-(1→4)-2,3-O-isopropylidene- 1-thio-α-L-rhamnopyranoside (28).
To a stirred suspension of 25 (618 mg, 1.5 mmol), 24 (352 mg, 1.1 mmol) and activated 4 Å molecular sieve powder in anhydrous CH2Cl2 (12 mL) was added TMSOTf (0.13 mL, 0.72 mmol) at -50 °C under N2 atmosphere. Upon completion of the reaction after 2 h, the reaction was quenched by addition of Et3N, warmed up to rt, filtered and then concentrated under reduced pressure. The residue was purified by column chromatography (silica gel; EtOAc//hexanes = 1/3) to give 28 (582 mg, 90%) as a yellow syrup: Rf 0.48 (EtOAc/hexanes = 1/1); 1H NMR (400 MHz, CDCl3) δ 7.34 (d, J = 7.9 Hz, 2H), 7.12 (d, J = 7.9 Hz, 2H), 5.64 (s, 1H, H-1), 5.20 (t, J = 8.5 Hz, 1H, H-3′), 5.00 (d, J
= 6.8 Hz, 1H, H-1′), 4.97–4.88 (m, 2H, H-4′, H-2′), 4.29 (d, J = 5.4 Hz, 1H, H-2), 4.17–
4.03 (m, 3H, H-3, H-5, H-5a′), 3.59 (dd, J = 9.6, 7.8 Hz, 1H, H-4), 3.35 (dd, J = 11.6, 8.8 Hz, 1H, H-5b′), 2.33 (s, 3H), 2.10 (s, 3H), 2.04 (s, 6H, Ac × 2), 1.51 (s, 3H), 1.34 (s, 3H), 1.20 (d, J = 6.1 Hz, 3H, H-6); BBD 13C NMR (100 MHz, CDCl3) δ 170.1, 169.9, 169.7, 138.0, 132.6, 129.8, 129.3, 109.5, 99.3 (C-1′), 84.0 (C-1), 79.1 (C-4), 77.9 (C-3), 76.6 (C- 2), 71.4 (C-3′), 71.0 (C-2′), 69.0 (C-4′), 65.4 (C-5), 62.0 (C-5′), 27.9, 26.5, 21.1, 20.8, 20.7, 17.3 ppm; HRMS+ (ESI-TOF) calcd. for C27H36O11SNa [M+Na]+ 591.1871, found 591.1876.
p-Methylphenyl 2,3,4-tri-O-acetyl-β-D-xylopyranosyl-(1→4)-2,3-di-O-acetyl-1-thio- α-L-rhamnopyranoside (22).
To stir a solution of 28 (4.6 g, 8.1 mmol), in 80% AcOH (100 mL) was heated to 60
°C for 12 h. The residue mixture was evaporated and then azeotrope distilled with toluene
(50 mL) twice under reduced pressure. After drying by high vacuum, the crude syrup was treated with Ac2O (2.2 mL, 23 mmol), Et3N (5.2 mL, 38 mmol) and DMAP (9 mg, 0.074 mmol) in CH2Cl2 under N2 atmosphere at rt. Upon completion of the reaction after 2 h, the mixture was diluted with CH2Cl2, washed by H2O, brine, dried over MgSO4, and then
concentrated under reduced pressure, The residue was purified by column chromatography (silica gel; EtOAc/hexanes = 2/3) to give 22 (3.8 g, 76%) as a white solid:
Rf 0.19 (EtOAc/hexanes = 1/2); 1H NMR (400 MHz, CDCl3) δ 7.34 (d, J = 7.9 Hz, 2H), 7.11 (d, J = 7.8 Hz, 2H), 5.39 (brs, 1H, H-2), 5.26 (brs, 1H, H-1), 5.22 (dd, J = 9.6, 3.1 Hz, 1H, H-3), 5.14 (t, J = 9.2 Hz, 1H, H-3′), 5.00–4.93 (m, 1H, H-4′), 4.90 (dd, J = 9.2, 7.6 Hz, 1H, H-2′), 4.66 (d, J = 7.6 Hz, 1H, H-1′), 4.24 (dq, J = 9.6, 6.1 Hz, 1H, H-5), 4.12 (dd, J = 11.6, 5.3 Hz, 1H, H-5a′), 3.72 (t, J = 9.6 Hz, 1H, H-4), 3.39–3.30 (m, 1H, H-5b′), 2.31 (s, 3H), 2.11 (s, 3H), 2.09 (s, 3H), 2.03 (s, 3H), 2.02 (s, 6H, Ac × 2), 1.31 (d, J = 6.1 Hz, 3H, H-6); BBD 13C NMR (100 MHz, CDCl3) δ 170.2, 169.9, 169.8, 169.6, 169.5, 138.1, 132.6, 129.9, 129.3, 101.0 (C-1′), 85.8 (C-1), 76.5 (C-4), 72.1 (C-3′), 71.7 (C-2), 71.6 (C-3), 71.1 (C-2′), 69.2 (C-4′), 68.1 (C-5), 62.5 (C-5′), 21.1, 20.9, 20.7, 20.7, 20.4, 17.5 ppm; HRMS+ (ESI-TOF) calcd. for C28H36O13SNa [M+Na]+ 635.1769, found 635.1774.
Benzyl 2,3,4-tri-O-acetyl-β-D-xylopyranosyl-(1→4)-2,3-di-O-acetyl-α-L- rhamnopyranosyl-(1→2)-3,4-O-isopropylidene-α-D-fucopyranoside (29α).
To a stirred suspension of 23 (111 mg, 0.21 mmol), 22 (214 mg, 0.35 mmol), and activated 4 Å molecular sieves powder in anhydrous CH2Cl2 (1 mL) was added NIS (130 mg, 0.58 mmol) under N2 atmosphere at -50 °C. Upon completion of the reaction after 2 h, the reaction was quenched by addition of saturated NaHCO3 and 10% Na2S2O3 aqueous solution, the reaction mixture was warmed to rt, stirred for 1 h and then filtered. Filtrate was diluted with CH2Cl2, washed by 10% Na2S2O3, saturated NaHCO3, brine, dried over MgSO4, and then concentrated under reduced pressure. The residue was purified by column chromatography (silica gel; EtOAc/hexanes = 1/3 to 2/3) to give 29 (168 mg, 74%) as a white solid: Rf 0.43 (EtOAc/hexanes = 1/1); 1H NMR (600 MHz, CDCl3) δ
7.38–7.27 (m, 5H), 5.32 (dd, J = 3.5, 1.6 Hz, 1H, H-2′), 5.26 (dd, J = 9.3, 3.5 Hz, 1H, H- 3′), 5.13 (t, J = 9.3 Hz, 1H, H-3′′), 5.05 (d, J = 1.5 Hz, 1H, H-1′), 4.96 (td, J = 9.3, 5.4 Hz,
1H, H-4′′), 4.91–4.87 (dd, J = 9.3, 7.6 Hz, 1H, H-2′′), 4.80 (d, J = 3.6 Hz, 1H, H-1), 4.71 (d, J = 12.3 Hz, 1H, Bn CH2), 4.63 (d, J = 7.6 Hz, 1H, H-1′′), 4.54 (d, J = 12.3 Hz, 1H, Bn CH2), 4.34 (dd, J = 8.1, 5.4 Hz, 1H, H-3), 4.14 (qd, J = 6.7, 2.5 Hz, 1H, H-5), 4.10 (dd, J = 11.7, 5.4 Hz, 1H, H-5a′′), 4.02 (dd, J = 5.4, 2.5 Hz, 1H, H-4), 3.74 (dd, J = 8.1, 3.6 Hz, 1H, H-2), 3.62–3.54 (m, 2H, H-4′, H-5′), 3.33 (dd, J = 11.7, 9.5 Hz, 1H, H-5b′′), 2.13 (s, 3H), 2.08 (s, 3H), 2.03 (s, 3H), 2.01 (s, 6H), 1.49 (s, 3H), 1.34 (d, J = 6.7 Hz, 3H,
H-6), 1.32 (s, 3H), 1.14 (d, J = 5.6 Hz, 3H, H-6′); BBD 13C NMR (151 MHz, CDCl3) δ 170.2, 169.8, 169.5, 136.8, 128.6, 127.9, 108.9, 101.1 (C-1′′), 97.9 (C-1′), 96.4 (C-1), 76.5, 76.3, 75.6, 75.5, 72.3, 71.3, 71.1, 70.2, 69.3, 69.1, 67.0, 63.2, 62.5, 28.3, 26.4, 21.0, 21.0, 20.7, 20.7, 20.4, 17.5, 16.3 ppm; HRMS+ (ESI-TOF) calcd. for C37H50O18Na [M+Na]+ 805.2889, found 805.2898.
Trichloroacetimidoyl 2,3,4-Tri-O-acetyl-β-D-xylopyranosyl-(1→4)-2,3-di-O-acetyl- α-L-rhamnopyranosyl-(1→2)-3,4-O-isopropylidene-α/β-D-fucopyranoside (21).
To a suspension of 29 (168 mg, 0.052 mmol) and 10% Pd/C (100 mg) in THF/MeOH/pH 7 PBS buffer = 4/1/1 (6 mL) was stirred at rt under H2 (balloon) atmosphere. After being stirred for 3 days, the mixture was filtered through celite and concentrated under reduced pressure. The residue was purified by column chromatography (silica gel; EtOAc/hexanes = 1/2 to 1/1) to afford hemiacetal (30 mg, 20%) as a white foam solid. To a stirred solution of hemiacetal (110 mg, 0.16 mmol) in anhydrous CH2Cl2 (1 mL) was added Cl3CCN (48 μL, 0.48 mmol), and DBU (9 μL, 0,064 mmol) at rt under N2 atmosphere. After being stirred for 2 h, the reaction was complete