科技部補助專題研究計畫報告
探討ZNF322A致癌蛋白參與肺癌形成與轉移的分子機制(第3年)
報 告 類 別 : 成果報告 計 畫 類 別 : 個別型計畫 計 畫 編 號 : MOST 106-2320-B-006-071-MY3 執 行 期 間 : 108年08月01日至109年07月31日 執 行 單 位 : 國立成功大學藥理學科暨研究所 計 畫 主 持 人 : 王憶卿 計畫參與人員: 碩士級-專任助理:張欣慈 碩士班研究生-兼任助理:官彣徽 碩士班研究生-兼任助理:余旻樺 碩士班研究生-兼任助理:吳思亭 博士後研究-博士後研究:謝智雄本研究具有政策應用參考價值:■否 □是,建議提供機關
(勾選「是」者,請列舉建議可提供施政參考之業務主管機關)
本研究具影響公共利益之重大發現:□否 □是
中 華 民 國 109 年 10 月 08 日
中 文 摘 要 : 研究背景: ZNF322A為致癌轉錄因子。本實驗室先前發現ZNF322A會 與c-Jun形成異源二具體,正向調控alpha-adducin (ADD1) 和 cyclin D1 (CCND1) 基因的之表現,進一步促進腫瘤的生長及轉移 。然而,對於調控ZNF322A蛋白質功能的轉譯後修飾及訊息傳遞路徑 機制尚未釐清。 研究目的:本計畫欲探討轉譯後修飾如蛋白質磷酸化對於 ZNF322A蛋白質穩定性及轉錄活性之調控機制,並進一步研究其上游 激酶 (kinase) 及其訊息傳遞路徑。 研究結果: 本研究利用蛋白質體學及蛋白質磷酸化資料庫分析 ,發現到一些ZNF322A磷酸化位點,而這些位點透過質譜分析被預測 出可能受到AKT所調控。進一步以試管內激酶反應 (In vitro
kinase assay) 證實了ZNF322A確實為AKT的激酶受質。在細胞實驗 中,透過西方墨點法 (Immunoblot) 及雙螢光酶啟動子試驗 (Dual luciferase promoter assay) 證實AKT磷酸化ZNF322A能促進 ZNF322A的蛋白質穩定性及轉錄活性。進一步分析四個AKT磷酸化 ZNF322A的位點,發現ZNF322A Thr262位點被AKT磷酸化會促使 ZNF322A蛋白質穩定性增加進而促進其對ADD1基因的轉錄活性;而 AKT調控ZNF322A Thr150、Ser224、Thr234位點的磷酸化雖不改變其 蛋白質穩定性,卻也能促使ZNF322A結合到ADD1與CCND1啟動子的區 域上,正向調控ADD1及CCND1轉錄活性及基因表現,進一步促進肺癌 細胞的增生及爬行。此外 epidermal growth factor 的刺激能夠活 化AKT進一步維持ZNF322A的蛋白質穩定性及轉錄活性。在動物實驗 中發現AKT與ZNF322A同時過表達較ZNF322A單獨過表達更能誘導肺癌 細胞肺部轉移。由以上研究結果顯示AKT藉由磷酸化ZNF322A,正向 調節ZNF322A的轉錄活性與致癌轉移功能。此外,AKT所調控的Thr-150,Ser-224,Thr-234和Thr-262磷酸化在試管內和動物實驗皆證 實可促進肺癌細胞生長和轉移。臨床上,收集150位肺癌病人的腫瘤 樣本,分析結果顯示ZNF322A磷酸化的表達與AKT磷酸化活性相關 ,多變項Cox迴歸分析表示結合AKT磷酸化和ZNF322A磷酸化表達資料 可做為預測肺癌病人疾病預後的重要參數。 結論:本篇研究發現AKT磷酸化ZNF322A不僅能促進蛋白質的穩定 性,也能活化下游基因之轉錄調控,進而導致肺癌的形成;本篇研 究揭露AKT訊息傳遞路徑在肺癌細胞,動物模式和臨床病人中證實可 促進ZNF322A蛋白質穩定性,並能正向調控下游基因轉錄活性的新穎 分子致癌機轉。 中 文 關 鍵 詞 : 肺癌、AKT、ZNF322A、磷酸化、轉錄因子。
英 文 摘 要 : Background: ZNF322A is an oncogenic zinc-finger
transcription factor. Our published result shows that ZNF322A positively regulates alpha-adducin (ADD1) and cyclin D1 gene transcription to promote tumorgenicity of lung cancer. However, the upstream regulatory mechanisms of ZNF322A protein function are still unknown. Previously, our cell-based mass spectrometry analysis identified several ZNF322A phosphorylation sites which may be targeted by AKT, suggesting that ZNF322A may be a protein substrate of AKT. Purpose: This study aimed to investigate the role of AKT
in regulation of ZNF322A protein function and to identify the upstream signaling pathway involved in AKT-mediated ZNF322A functions.
Results: Our data demonstrated that AKT could
phosphorylate ZNF322A by in vitro kinase assay. In cell-based studies, AKT indeed interacted with ZNF322A in lung cancer cells. Moreover, we found that overexpression of AKT promoted ZNF322A protein stability and transcriptional activity whereas these effects were inhibited by knocking down of AKT or treating with AKT inhibitor. In addition, ZNF322A phosphorylation at Thr-262 by AKT promoted ZNF322A protein stability thus increased ADD1 promoter activity. Interestingly, ZNF322A Thr-150, Ser-224, and Thr-234 phosphorylation by AKT enhanced ADD1 and CCND1 promoter activity without affecting protein stability. Chromatin immunoprecipitation assay showed that ZNF322A
phosphorylation defective mutants T150A-, S224A-, and
T234A-ZNF322A attenuated binding affinity to ADD1 and CCND1 promoter compared with wild-type (WT)-ZNF322A. Furthermore, AKT-mediated Thr-150, Ser-224, and Thr-234 phosphorylation promoted lung cancer cell proliferation and motility. In addition, we found that AKT-promoted ZNF322A protein stability and transcriptional activity could be regulated by epidermal growth factor receptor signaling.
Consistently, experimental tumor metastasis assay showed that overexpression of WT-ZNF322A significantly increased tumor metastasis in vivo which was further enhanced by co-overexpression of AKT in comparison with control group. Furthermore, AKT-mediated Thr-150, Ser-224, Thr-234 and Thr-262 phosphorylation promoted lung cancer cell growth and metastasis in vitro and in vivo. Clinically, expression of phosphorylated ZNF322A correlated with actively
phosphorylated AKT in tumor specimens from 150 lung cancer patients. Multivariate Cox regression analysis indicated that combined phosphorylated AKT and phosphorylated ZNF322A expression profile was an independent factor to predict the clinical outcome in lung cancer patients.
Conclusions: This study provided the novel mechanism of AKT signaling axis in promoting ZNF322A protein stability and transcriptional activity in lung cancer cell, xenograft and clinical models. We proposed that therapeutic
strategies by targeting ZNF322A oncoprotein for
transcription suppressing or by blocking AKT signaling may provide new treatment for lung cancer patients.
英 文 關 鍵 詞 : Lung cancer, AKT, ZNF322A, phosphorylation, and transcription factor.
科技部補助專題研究計畫成果報告
期中進度報告
探討 ZNF322A 致癌蛋白參與肺癌形成與轉移的分子機制
(3/3)
Investigating the molecular mechanism(s) of ZNF322A oncoprotein in lung
tumorigenesis (3/3)
計畫類別:個別型計畫 (特約計畫)
計畫編號:MOST 106-2320-B-006-071-MY3
全程執行期間: 106 年 08 月 01 日至 109 年 07 月 31 日
執行機構及系所:國立成功大學藥學科暨研究所
計畫主持人:王憶卿
計畫參與人員:劉雅棻、郭懿瑩、謝智雄、廖昇佑、陳玉婷、林哲仲、
黃士宣、張欣慈、官彣徽、余旻樺、吳思亭
期末報告處理方式:
1. 公開方式:
非列管計畫亦不具下列情形,立即公開查詢
□涉及專利或其他智慧財產權,□一年□二年後可公開查詢
2.「本研究」是否已有嚴重損及公共利益之發現:否 □是
3.「本報告」是否建議提供政府單位施政參考 否 □是
中 華 民 國 109 年 10 月 8 日
摘要
中文關鍵詞:肺癌、AKT、ZNF322A、磷酸化、轉錄因子。
研究背景: ZNF322A 為致癌轉錄因子。本實驗室先前發現 ZNF322A 會與 c-Jun 形成異源二具體,正向調控 alpha-adducin (ADD1) 和 cyclin D1 (CCND1) 基 因的之表現,進一步促進腫瘤的生長及轉移。然而,對於調控 ZNF322A 蛋白質 功能的轉譯後修飾及訊息傳遞路徑機制尚未釐清。 研究目的:本計畫欲探討轉譯後修飾如蛋白質磷酸化對於 ZNF322A 蛋白質 穩定性及轉錄活性之調控機制,並進一步研究其上游激酶 (kinase) 及其訊息傳 遞路徑。 研究結果: 本研究利用蛋白質體學及蛋白質磷酸化資料庫分析,發現到一 些 ZNF322A 磷酸化位點,而這些位點透過質譜分析被預測出可能受到 AKT 所 調控。進一步以試管內激酶反應 (In vitro kinase assay) 證實了 ZNF322A 確實為 AKT 的激酶受質。在細胞實驗中,透過西方墨點法 (Immunoblot) 及雙螢光酶啟 動子試驗 (Dual luciferase promoter assay) 證實 AKT 磷酸化 ZNF322A 能促進 ZNF322A 的蛋白質穩定性及轉錄活性。進一步分析四個 AKT 磷酸化 ZNF322A 的位點,發現 ZNF322A Thr262 位點被 AKT 磷酸化會促使 ZNF322A 蛋白質穩定 性增加進而促進其對 ADD1 基因的轉錄活性;而 AKT 調控 ZNF322A Thr150、 Ser224、Thr234 位點的磷酸化雖不改變其蛋白質穩定性,卻也能促使 ZNF322A 結合到 ADD1 與 CCND1 啟動子的區域上,正向調控 ADD1 及 CCND1 轉錄活性 及基因表現,進一步促進肺癌細胞的增生及爬行。此外 epidermal growth factor 的刺激能夠活化 AKT 進一步維持 ZNF322A 的蛋白質穩定性及轉錄活性。在動 物實驗中發現 AKT 與 ZNF322A 同時過表達較 ZNF322A 單獨過表達更能誘導肺 癌細胞肺部轉移。由以上研究結果顯示 AKT 藉由磷酸化 ZNF322A,正向調節 ZNF322A 的轉錄活性與致癌轉移功能。此外,AKT 所調控的 Thr-150,Ser-224, Thr-234 和 Thr-262 磷酸化在試管內和動物實驗皆證實可促進肺癌細胞生長和轉 移。臨床上,收集 150 位肺癌病人的腫瘤樣本,分析結果顯示 ZNF322A 磷酸化 的表達與 AKT 磷酸化活性相關,多變項 Cox 迴歸分析表示結合 AKT 磷酸化和 ZNF322A 磷酸化表達資料可做為預測肺癌病人疾病預後的重要參數。
結論:本篇研究發現 AKT 磷酸化 ZNF322A 不僅能促進蛋白質的穩定性, 也能活化下游基因之轉錄調控,進而導致肺癌的形成;本篇研究揭露 AKT 訊息 傳遞路徑在肺癌細胞,動物模式和臨床病人中證實可促進 ZNF322A 蛋白質穩定 性,並能正向調控下游基因轉錄活性的新穎分子致癌機轉。
Abstract
Keyword:Lung cancer, AKT, ZNF322A, phosphorylation, and transcription factor.
Background: ZNF322A is an oncogenic zinc-finger transcription factor. Our published result shows that ZNF322A positively regulates alpha-adducin (ADD1) and cyclin D1 gene transcription to promote tumorgenicity of lung cancer. However, the upstream regulatory mechanisms of ZNF322A protein function are still unknown. Previously, our cell-based mass spectrometry analysis identified several ZNF322A phosphorylation sites which may be targeted by AKT, suggesting that ZNF322A may be a protein substrate of AKT.
Purpose: This study aimed to investigate the role of AKT in regulation of ZNF322A protein function and to identify the upstream signaling pathway involved in AKT-mediated ZNF322A functions.
Results: Our data demonstrated that AKT could phosphorylate ZNF322A by in vitro kinase assay. In cell-based studies, AKT indeed interacted with ZNF322A in lung cancer cells. Moreover, we found that overexpression of AKT promoted ZNF322A protein stability and transcriptional activity whereas these effects were inhibited by knocking down of AKT or treating with AKT inhibitor. In addition, ZNF322A phosphorylation at Thr-262 by AKT promoted ZNF322A protein stability thus increased ADD1 promoter activity. Interestingly, ZNF322A Thr-150, Ser-224, and Thr-234 phosphorylation by AKT enhanced ADD1 and CCND1 promoter activity without affecting protein stability. Chromatin immunoprecipitation assay showed that ZNF322A phosphorylation defective mutants T150A-, S224A-, and T234A-ZNF322A attenuated binding affinity to ADD1 and CCND1 promoter compared with wild-type (WT)-ZNF322A. Furthermore, AKT-mediated Thr-150, Ser-224, and Thr-234 phosphorylation promoted lung cancer cell proliferation and motility. In addition, we found that AKT-promoted ZNF322A protein stability and transcriptional activity could be regulated by epidermal growth factor receptor signaling. Consistently, experimental tumor metastasis assay showed that overexpression of WT-ZNF322A significantly increased tumor metastasis in vivo which was further enhanced by co-overexpression of AKT in comparison with control group. Furthermore, AKT-mediated Thr-150, Ser-224, Thr-234 and Thr-262 phosphorylation promoted lung cancer cell growth and metastasis in vitro and in vivo. Clinically, expression of phosphorylated ZNF322A correlated with actively phosphorylated AKT in tumor specimens from 150 lung cancer patients. Multivariate Cox regression analysis indicated that combined phosphorylated AKT and phosphorylated ZNF322A expression profile was an independent factor to predict the clinical outcome in lung cancer patients.
Conclusions: This study provided the novel mechanism of AKT signaling axis in promoting ZNF322A protein stability and transcriptional activity in lung cancer cell, xenograft and clinical models. We proposed that therapeutic strategies by targeting ZNF322A oncoprotein for transcription suppressing or by blocking AKT signaling may provide new treatment for lung cancer patients.
INTRODUCTION
Several studies reported that deregulation of zinc fingers (ZNFs) transcription factors would lead to tumorigenesis in different cancer type. For example, ZNF139 increases expression of survivin, x-IAP and Bcl-2 to promote gastric cancer proliferation and inhibits apoptosis (Fan et al., 2015). In addition, ZNF139 has been shown to promote gastric cancer cell migration and invasion through up-regulating MMP-2, MMP-9 and ICAM-1 expression, but suppressing TIMP-1 expression (Li et al., 2014). ZEB1facilitates breast cancer angiogenesis by recruiting Sp1 to VEGFA promoter region to enhance VEGFA expression and secretion (Liu et al., 2016). Furthermore, ZNF926 could transcriptionally up-regulate c-Myc expression to induce glioma stem cell maintenance (Fang et al., 2014). Conversely, other ZNF proteins are defined as tumor suppressor gene such as ZNF24, ZNF668 and ZHX1. ZNF24 suppresses breast cancer angiogenesis through inhibiting VEGF expression (Harper et al., 2007; Jia et al., 2013). Moreover, ZNF668 promotes p53 protein stability by disrupting MDM2-mediated degradation thus inhibiting cell proliferation in breast cancer (Hu et al., 2011). ZHX1 decreases cyclin D1, cyclin E and Bcl-2 expression to induce cell cycle arrest and apoptosis in gastric cancer (Ma et al., 2016). These findings indicate that dysregulation of ZNF may lead to induce tumorigenesis. Therefore, it is crucial to elucidate the regulatory mechanism of ZNF protein in tumorigenesis.
Aberrant expression of transcription factors implicates in many diseases including cancer, therefore, the protein abundance of transcription factor must be tightly controlled. Post-translational modifications (PTMs) such as phosphorylation are reported to regulate the protein stability control of transcription factors. For instance, Twist1 phosphorylation by MAPKs at Ser-68 promotes Twist1 protein stability to facilitate breast cancer invasiveness (Hong et al., 2011). IKK-mediated Gli1 phosphorylation stabilizes its protein stability and transcriptional activity in diffuse large B-cell lymphoma (Agarwal et al., 2016). In contrast, Slug has been showed to inhibit lung cancer cell metastasis through phosphorylating by GSK3 and reducing Slug protein stability by CHIP through proteasome degradation (Kao et al., 2013). Moreover, PDK1-mediated Snail phosphorylation promotes FBXO11-induced proteasome degradation, which suppresses Snail-mediated epithelial-to-mesenchymal transition and metastasis in breast cancer (Zheng et al., 2014). These results implicate that protein stability control of zinc finger transcription factor is associated with cancer progression.
It has been showed that activation signaling enhances ZNF activity. For example, activation RAS/RAF/MEK/ERK signaling enhances KLF4 promoter activity to promote melanoma cell growth (Riverso et al., 2017). ZEB1 interacts with YAP1 to activate ZEB1 transcriptional activity in aggressive cancer types including breast cancer (Lehmann et al., 2016). Furthermore, AKT-mediated SP-1 phosphorylation at S42, T679 and S698 promotes CCR7 expression upon COX-2 stimulation (Chuang et al., 2013). Notably, Gli1 promotes lung cancer cell proliferation and stemness by activating MAPK/ERK signaling pathway, instead of canonical Hedgehog pathway (Po et al., 2017). ZFP91 facilitates proliferation by interacting with NF-B/p65 and cooperatively bind to HIF-1 promoter thus up-regulating expression of HIF-1 in colon cancer (Ma et al., 2016). These studies demonstrate that activation of transcriptional activity of zinc finger protein by kinase signaling is relative to cancer development.
ZNF322A, also known as ZNF388 or ZNF489, is a zinc-finger transcription factor, which belongs to the C2H2 type kruppel-like zinc-finger protein family.
ZNF322A consists of 11 tandem repeats of C2H2 zinc finger motif. Li and associates
found that ZNF322A is mostly expressed in the nucleus and ZNF322A could activate SRE and AP-1 transcriptional activity through MAPK signaling pathway (Li et al., 2004). Our previous study showed that ZNF322A overexpression correlates with poor prognosis in lung cancer patients both in Asian and Caucasian. In addition, ZNF322A promotes lung tumor growth and metastasis partially because ZNF322A forms complex with c-Jun and cooperatively activates promoter activity of alpha-adducin (ADD1) and cyclin D1 (CCND1), but suppresses promoter activity of p53 (Jen et al., 2016). In addition, we found that CK1 and GSK3 phosphorylate ZNF322A at Ser396 and Ser391 to facilitate FBW7α-induce protein degradation (Liao et al., 2017). Taken together, ZNF322A is an oncoprotein that contributes to lung tumorigenesis.
Therefore, this study aims to investigate the post-translational modifications on ZNF322A protein stability and transcriptional activity associated with its oncogenic effects in lung cancer. We identified several ZNF322A phosphorylation sites in vivo including Ser-224 with high LC-MS/MS experimental confidence. Other ZNF322A phosphorylation sites, such as Thr-234 and Thr-262 were discovered from PhosphoSitePlus online database. Moreover, Thr-150 phosphorylation site was found by in vitro kinase assay using LC-MS/MS. Interestingly, these phosphorylation sites of ZNF322A were predicted to be regulated by AKT using several phosphorylation site prediction software. Therefore, we proposed to investigate the effects of AKT in regulation of ZNF322A protein function in this study.
MATERIALS AND METHODS
Cell Lines and culture conditions
Human lung cancer cell lines H1299 and H460 cells were purchased from the American Type Culture Collection (Rockville, MD, USA), where they were characterized by DNA-fingerprinting and isozyme detection. H1299 was cultured in Dulbecco's Modified Eagle Medium (DMEM; Gibco, Grand Island, NY, USA); H460 in Roswell Park Memorial Institute medium (RPMI) 1640 (Gibco). All media were supplemented with 10% fetal bovine serum (FBS, Gibco) and 1% penicillin/streptomycin (Gibco). Cells were incubated at 37°C in a humidified incubator containing 5% CO2.
Plasmids, RNAi and transfection
Plasmid and siRNA transfection was carried out using TurboFect (Thermo Fisher Scientific, MA, USA) and Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) reagent according to manufacturer’s protocol. siRNAs against AKT were purchased from Invitrogen.
Site-directed mutagenesis
ZNF322A phosphorylation-defective and -mimetic mutants, including T150A/E, S224A/E, T234A/E and T262A/E were generated by site-directed mutagenesis. Wild-type (WT) ZNF322A plasmid DNA was used as the PCR template and ZNF322A mutant cDNAs were amplified using indicated primers and High Fidelity PCR enzyme system (Thermo Fisher Scientific, Waltham, MA, USA). The PCR condition comprised at 98oC for 10 sec followed by 30 cycles at 98oC for 1 sec, 60oC for 5 sec and 72oC for 75 sec and finally at 72oC for 1 min. To digest template plasmid DNA, 1l of DpnI restriction enzyme (New England Biolabs, Ipswich, MA, USA) was added to the PCR product and then incubated at 37oC for 1 hr. The product mix was eluted by gel extraction kit (Favorgen biotech corp, Pingtung, Taiwan) to purify ZNF322A mutant plasmid DNA.
Cloning, expression and purification of the human ZNF322A recombinant protein
Human ZNF322A cDNA was PCR-amplified and cloned into the SacI and SalI sites of pGEX4T-1 expression vector containing glutathione S-transferase (GST) to generate GST-tagged ZNF322A expression vector. GST-tagged ZNF322A expression vector were transformed into BL21 E.coli strain, and single-colony transformant was grown in Luria-Bertani (LB) broth containing 50 µg/ml ampicillin at 37oC. When an OD600nm of 0.4-0.6 was reached, protein expression was induced with 0.5 mM
isopropyl-β-D-thiogalactopyranoside (IPTG) (Milipore, Bedford, MA, USA) for 16 hr at 16oC. The E.coli cells were harvested by centrifugation, resuspended in 50 ml 1× Phosphate-buffered saline (PBS), and immediately sonicated with cooling. The bacteria lysate containing GST-tagged ZNF322A recombinant protein was clarified by centrifugation, the supernatant was applied to columns pre-packed with Glutathione sepharose (GE Healthcare, Chicago, IL, USA). The column was washed three times with PBS, and the protein was eluted with the elution buffer (50mM Tris-HCl, pH 8.0) supplemented 10 mM glutathione. The purity of the GST-tagged ZNF322A recombinant proteins was determined by SDS-PAGE.
In vitro kinase assay
For AKT in vitro kinase assay, 1 g human recombinant GST-tagged ZNF322A protein was incubated with the increasing amount of active full-length recombinant AKT protein in kinase buffer [25 mM Tris-HCl (pH 7.5), 5 mM -glycerophosphate (pH 7.3), 2 mM DTT, 0.1mM Na3VO4, and 10 mM MgCl2] containing 25 M
unlabeled ATP and 2.5 Ci of -[32P]-ATP. The phosphorylation reactions were incubated at 30°C for 30 min. The reactions were stopped by adding an equal volume of 2X SDS loadingbuffer and heated to 95°C for 5 min. Samples were then separated by 8% SDS-PAGE and transferred onto nitrocellulose membranes and detected by autoradiography.
RNA extraction and quantitative reverse-transcriptase polymerase chain reaction (RT-qPCR) assays
Total RNA was extracted using Trizol reagent (Invitrogen). A total of 4 g RNA was used to synthesize cDNA using SuperScriptTM reverse transcriptase (Invitrogen) according protocols provided by manufacturer. mRNA expression of ZNF322A, AKT, ADD1 and GAPDH genes was analyzed by RT-qPCR using a LightCycler® 480 SYBR Green I Master (Roche Diagnostics, Indianapolis, IN, USA), and GAPDH gene served as an internal control. Quantitative real time PCR was performed on the StepOnePlus™ Real-Time PCR System machine (Applied Biosystems, Foster city, CA, USA). Relative quantification using the comparative Ct method with the data from Roter-Gene 3000 was performed according to manufacturer’s protocol.
Dual luciferase promoter assay
Cells were plated in 12-well plates the day before transfection. The pGL3-Basic constructs and pGL4-Renilla plasmid were included as internal controls. Cells were transfected with or without AKT for 16 hr prior to co-transfected with empty vector, wild-type (WT)-ZNF322A, ZNF322A phospho-mutants and gene promoter vector or pGL3-Basic for 16 hr. The dual luciferase reporter assay kit (Promega, Madison, WI,
USA) was used to determine gene promoter activity according to the protocols provided by the manufacturer. The data were represented as the means of ratio of firefly luciferase to Renilla luciferase activity by triplicate experiments.
Immunoblotting
Cells were lysed on ice using RIPA buffer in the presence of protease inhibitor cocktail (Sigma-Aldrich). Cell lysate was centrifuged at 13,200 rpm for 15 min at 4°C. Protein extracts were solubilized in loadingbuffer (60 mM Tris-base, 2% SDS, 10% glycerol, and 5% -mercaptoethanol). Samples containing equal amounts of protein (75 µg) were separated on an 8% sodium dodecyl sulfate/ polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto PVDF membranes (Millipore). Proteins of interest were immunoblotted using indicated antibodies. The signal was visualized by reacting with Western blot chemiluminescence reagent (Millipore). Chromatin immunoprecipitation (ChIP)-qPCR assay
Cell lysates were cross-linked with 1% formaldehyde and sonicated on ice to shear DNA to lengths between 300 and 500 bp. Subsequent steps were performed with the Magna ChIP G assay kit (Millipore) according to the manufacturer's instructions. Chromatin was immunoprecipitated for 16 hr at 4°C using anti-HA and normal IgG listed in Table 3. The DNA samples recovered from ChIP were analyzed by quantitative real time PCR using Fast SYBR Green Master Mix and StepOnePlus™ System (Applied Biosystems).
Transwell migration assay
For transwell migration assay, H1299 cells (1.5 × 105) ectopically expressed with WT- or phospho-mutant of T150A, S224A or T234A-ZNF322A in the presence or absence of AKT in serum free medium were seeded onto the upper chamber of transwell (Falcon, Lincoln Park, NJ, USA). Medium containing of 20% FBS (Gibco) was added to the lower chamber as chemoattracttants and cells were incubated at 37°C for 24 hr. Cells attached on the reverse side of the membrane were fixed and then stained with crystal violet. Six random selected views were photographed and quantified.
Statistical analysis
Quantification of the immunoblotting was analyzed using ImageJ software. Three independent experiments for cell studies and six mice per group for animal studies were analyzed unless indicated otherwise. Two-tailed Student’s t-test was used in cell and animal studies. Data represent mean + SEM. The levels of statistical significance were expressed as p-values, *p 0.05; ** p 0.01; *** p 0.001.
RESULTS
ZNF322A was a protein substrate of AKT
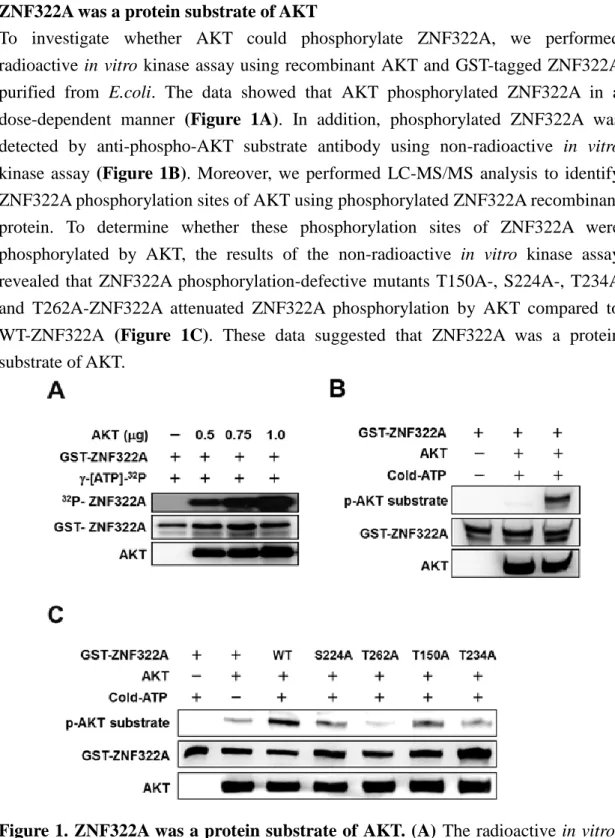
To investigate whether AKT could phosphorylate ZNF322A, we performed radioactive in vitro kinase assay using recombinant AKT and GST-tagged ZNF322A purified from E.coli. The data showed that AKT phosphorylated ZNF322A in a dose-dependent manner (Figure 1A). In addition, phosphorylated ZNF322A was detected by anti-phospho-AKT substrate antibody using non-radioactive in vitro kinase assay (Figure 1B). Moreover, we performed LC-MS/MS analysis to identify ZNF322A phosphorylation sites of AKT using phosphorylated ZNF322A recombinant protein. To determine whether these phosphorylation sites of ZNF322A were phosphorylated by AKT, the results of the non-radioactive in vitro kinase assay revealed that ZNF322A phosphorylation-defective mutants T150A-, S224A-, T234A and T262A-ZNF322A attenuated ZNF322A phosphorylation by AKT compared to WT-ZNF322A (Figure 1C). These data suggested that ZNF322A was a protein substrate of AKT.
Figure 1. ZNF322A was a protein substrate of AKT. (A) The radioactive in vitro kinase assay demonstrated that AKT phosphorylated ZNF322A in a dose dependent. (B) The nonradioactive in vitro kinase assay confirmed that ZNF322A was phosphorylated by AKT. (C) The non-radioactive in vitro kinase assay revealed that ZNF322A phosphorylation-defective mutants T150A-, S224A-, T234A and T262A-ZNF322A attenuated ZNF322A phosphorylation by AKT compared to WT-ZNF322A.
AKT upregulated ZNF322A protein expression in a transcription-independent manner
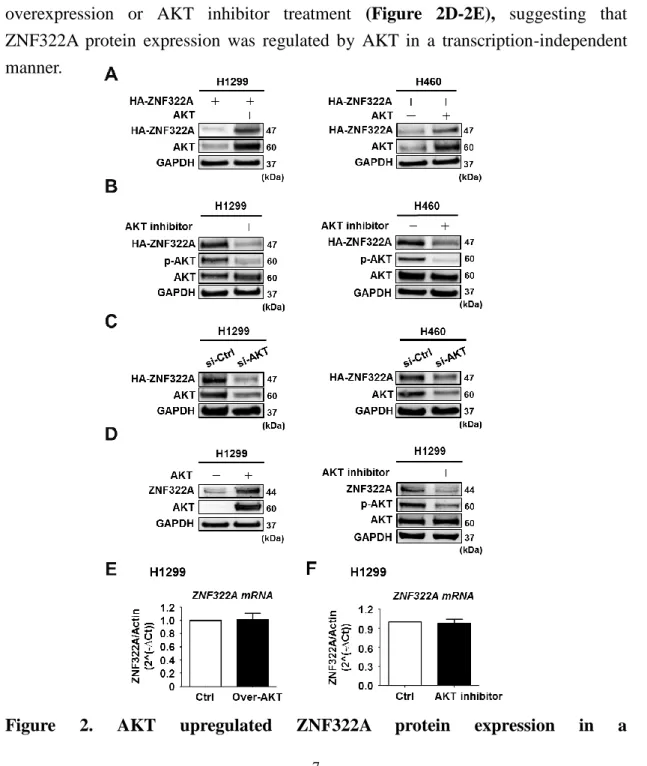
AKT has been reported to regulate protein expression through phosphorylation-dependent manner. We further examined whether ZNF322A protein expression was regulated by AKT. We transiently expressed HA-tagged ZNF322A (HA-ZNF322A) with or without AKT in H1299 cells and H460 cells and examined the ZNF322A protein level by immunoblotting. The data revealed that ZNF322A protein level was upregulated upon AKT overexpression, whereas the protein level was inhibited by knocking down of AKT or by AKT inhibitor treatment (Figure 2A-2C). Moreover, we performed RT-qPCR assay to examine ZNF322A mRNA expression. We observed that ZNF322A mRNA levels were not affected by AKT overexpression or AKT inhibitor treatment (Figure 2D-2E), suggesting that ZNF322A protein expression was regulated by AKT in a transcription-independent manner.
transcription-independent manner. (A) H1299 (left) and H460 (right) cells ectopically expressed AKT and HA-tagged ZNF322A (HA-ZNF322A) were subjected to Western blot analysis as indicated. (B) Western blot analysis of ectopically expressed HA-ZNF322A protein expression in H1299 (left) and H460 (right) cells treated with or without AKT inhibitor MK2206. (C) H1299 (left) and H460 (right) cells transfected with si-control (si-Ctrl) or si-AKT oligo were detected for ZNF322A protein level. (D) Western blot analysis of endogenous ZNF322A protein expression in H1299 cells treated with AKT inhibitor MK2206 (right) or ectopically expressed of AKT (left). (E, F) RT-qPCR analysis of mRNA level of ZNF322A in H1299 cells (E) overexpressed with AKT or (F) treated with AKT inhibitor
AKT enhanced ZNF322A transcriptional activity
Our laboratory has previously reported that ZNF322A binds to ADD1 promoter, which contains AP-1 element. To determine whether AKT could promote ZNF322A transcriptional activity, luciferase promoter activity assay of ADD1 promoter was performed on cells expressing HA-tagged ZNF322A with or without AKT inhibitor. Importantly, overexpression ZNF322A enhanced ADD1 promoter activity, whereas ADD1 promoter activity was inhibited by AKT inhibitor treatment (Figure 3A). In addition, cells with knockdown of AKT blocked ADD1 promoter activity (Figure 3B). Furthermore, overexpression of AKT enhanced ADD1 promoter activity whereas the activity was abolished by knocking down of ZNF322A (Figure 3C). These results demonstrated that AKT promoted ZNF322A transcriptional activity.
activity assay was performed using ADD1 promoter in H1299 cells expressing WT-ZNF322A and treated with or without AKT inhibitor MK2206 for 3hr. (B) Dual luciferase activity assay was performed using ADD1 promoter in H1299 si-Ctrl or si-AKT cells expressing WT-ZNF322A. (C) Dual luciferase activity assay was performed using ADD1 promoter in AKT overexpressed H1299 cells transfected with si-Ctrl or ZNF322A siRNA. Data are presented as mean + SEM. P-values determined using two-tailed Student’s t-test. *p 0.05; ** p 0.01; *** p 0.001.
Recruitment of ZNF322A transcriptional complex to promoter region of ADD1 and CCND1
Transcription factor activity has been reported by regulating DNA binding affinity (Filtz et al., 2014). This prompted us to further investigate whether T150-ZNF322A, S224-ZNF322A and T234-ZNF322A would enrich at ADD1 and CCND1 promoter region. We transiently expressed EV, WT-, T150A-, T150E-, S224A-, S224E-, T234A or T234E-ZNF322A vector in H460 cells and examined the chromatin binding affinity of ZNF322A to ADD1 and CCND1 promoter by ChIP-qPCR. The results revealed that cells expressing WT-ZNF322A enhanced ZNF322A binding compared to control group. Interestingly, we observed that T150A-, S224A- and T234A-ZNF322A significantly decreased the binding efficiency compared to WT-ZNF322A (Figure 4A-4C). To further investigate whether T150-, S224- and T234-ZNF322A phosphorylation mediated by AKT facilitated the recruitment of ZNF322A transcriptional complex to ADD1 and CCND1 promoter region, H460 cells were transiently expressed WT-, T150A-, S224A- or T234A-ZNF322A with or without AKT. ChIP-qPCR assay data showed that the attenuation of T150A-, S224A- or T234A-ZNF322A binding to ADD1 promoter could not be further promoted by AKT overexpression (Figure 4D-4F). In addition, we observed that the impairment of T150A-, S224A- or T234A-ZNF322A binding to CCND1 promoter could not be further promoted by AKT overexpression (Figure 4G-4I). These results indicated that AKT-mediated T150, S224 and T234 phosphorylation enhanced the recruitment of ZNF322A transcriptional complex to ADD1 and CCND1 promoter region.
Figure 4.Recruitment of ZNF322A transcriptional complex to promoter region of ADD1 and CCND1. (A-C) ChIP-qPCR assay confirmed the ZNF322A occupancy at the ADD1 promoter. ZNF322A-bound chromatin was immunoprecipitated using anti-HA antibody in H460 cells expressing T150A/E- (A), S224A/E- (B) or T234A/E- (C) HA-ZNF322A mutants. WT-HA-ZNF322A was included for comparison. (D-F) ChIP-qPCR assay confirmed that the ZNF322A occupancy at the ADD1 promoter was further enhanced by AKT overexpression. ZNF322A-bound chromatin was immunoprecipitated using anti-HA antibody in H460 cells expressing T150A- (D), S224A- (E) or T234A- (F) HA-ZNF322A mutants with or without AKT. (G-I) ChIP-qPCR assay confirmed that the ZNF322A occupancy at the CCND1 promoter was further enhanced by AKT overexpression. ZNF322A-bound chromatin was immunoprecipitated using anti-HA antibody in H460 cells expressing T150A- (G), S224A- (H) or T234A- (I) HA-ZNF322A mutants with or without AKT.WT-HA-ZNF322A was included for comparison. Data are presented as mean + SEM. P-values determined using two-tailed Student’s t-test. *p 0.05; ** p 0.01; *** p 0.001.
AKT-mediated Thr-150, Ser-224, Thr-234 phosphorylation induced lung cancer cell migration
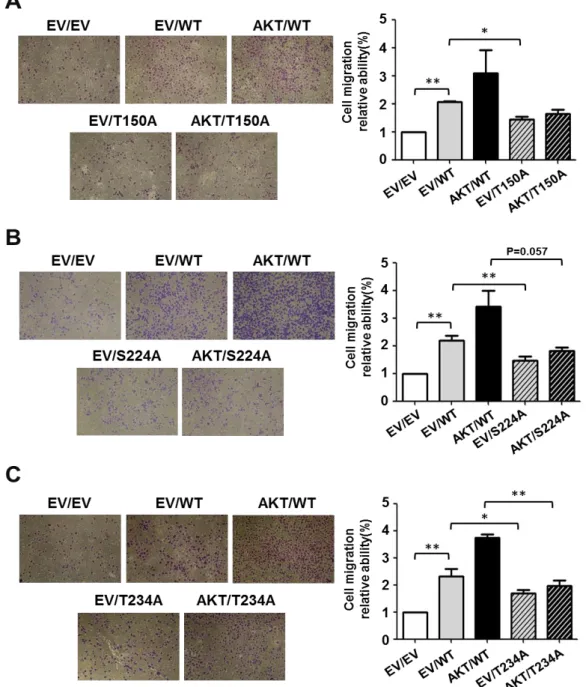
Previous study showed that ZNF322A activates ADD1 expression to enhance lung cancer cell migration (Jen et al., 2016). Therefore, we evaluated whether ZNF322A phosphorylation sites regulated lung cancer cell migration. We found that lung cancer cells migration ability were accelerated when cells expressing WT-ZNF322A and overexpression of AKT compared to WT-ZNF322A alone. Notably, transwell assay results showed that T150A-, S224A- and T234A-ZNF322A could not induce lung cancer cell migration by AKT overexpression (Figure 5A-5C). These results suggested that ZNF322A phosphorylation at T150, S224 and T234 by AKT promoted lung cancer cell migration ability.
lung cancer cell migration. (A-C, left) Representative images and (A-C, right) quantification of transwell migration of H1299 cells expressing WT-, T150A- (A), S224A- (B) or T234A-ZNF322A (C) with or without AKT expression. Data are presented as mean + SEM. P-values determined using two-tailed Student’s t-test. *p 0.05; ** p 0.01; *** p 0.001.
Antibody specificity tests of homemade p-S224 antibody in cell and clinical samples
To further confirm the role of AKT in promoting ZNF322A protein function in the context of phosphorylation at Ser-224, we generated anti-phospho-ZNF322A specific antibodies which could recognize phosphorylation of ZNF322A at Ser-224. We have preliminarily tested the specificity of this homemade p-S224 antibody in cells expressing WT- or S224A-ZNF322A. Immunoprecipitation (IP) results showed that signal of two individual clones of p-S224 antibody significantly decreased in cells expressing S224A-ZNF322A (Figure 6A). Immunoblotting results showed that p-S224 antibody signal was attenuated in cells expressing S224A-ZNF322A (Figure 6B). Using this p-S224 antibody, we performed immunohistochemistry (IHC) staining in tumor specimens from three lung cancer patients whose IHC staining of total-ZNF322A antibody was positive. The IHC results illustrated that p-S224 antibody also stained positively in these two lung cancer patients (Figure 6C).
cell and clinical samples. The specificity of phosphorylated homemade p-S224 antibody was examined by immunoprecipitation (IP)-Western blot in cells expressing GFP-tagged WT- or S224-ZNF322A (A), immunoblotting (B) in cells expressing HA-tagged EV, WT- or S224-ZNF322A, and immunohistochemistry in lung cancer patients (C).
DISCUSSION
In this study, we provided the novel regulatory mechanism of post-translational modifications in ZNF322A protein stability control and transcriptional activity. We identified that AKT served as the positive regulator of ZNF322A via promoting protein stability and transcriptional activity of ZNF322A. ZNF322A T262 phosphorylation by AKT enhanced ZNF322A protein stability thereby increased its transcriptional activity. Moreover, AKT-mediated ZNF322A phosphorylation at T150, S224 and T234 promoted chromatin binding of ZNF322A transcriptional complex thereby up-regulated the expression of ZNF322A downstream genes such as ADD1 and CCND1. In addition, EGF stimulation also induced ZNF322A protein stability and transcriptional activity through AKT activation. Furthermore, we observed that AKT-mediated ZNF322A activation could promote lung cancer cell proliferation and migration abilities. Our findings delineated previously undefined regulatory mechanisms of ZNF322A oncoprotein by AKT signaling. Deregulation of the EGFR-AKT signaling axis resulted in ZNF322A overexpression and activation thus promoted cancer progression.
Transcription factor activity has been reported to be regulated by protein stability, DNA binding affinity and transcription complex formation (Filtz et al., 2014). Here, we found that Thr150A-, Ser224A- and Thr234A-ZNF322A attenuated ZNF322A binding to ADD1 promoter region thus suppressed promoter activity and mRNA expression of ADD1. Studies have demonstrated that two to three successive C2H2-type zinc finger motifs are the most suitable unit to interact with DNA (Wolfe et al., 2000). In addition, we found conserved linker regions right behind ZNF322A zinc finger motifs 1, 4, 5, 7 and 8. We therefore generated recombinant protein, which consisted of ZNF322A zinc finger motifs 4-8, designated as ZNF127-266. Using electrophoretic mobility shift assay (EMSA), ZNF127-266 was found to interact with ADD1 AP1-2 oligo in a dose-dependent manner, suggesting zinc finger motifs 4-8 as the DNA binding domain of ZNF322A. We will next performed EMSA to confirm whether ZNF322A phosphorylation-defective mutant may abrogate the DNA binding affinity to ADD1 promoter DNA sequences. Importantly, we have conducted molecule modeling analysis to elucidate the possible effect of ZNF322A phosphorylation on DNA binding affinity. The modeling data revealed that Ser224 residue could directly contact with DNA and phosphorylation on this site may increase the non-covalent bonding between ZNF22A and DNA. These data supported our finding that ZNF322A Ser-224 affected the DNA binding affinity of ZNF322A.
Of note, Thr-150 and Thr-234 residues did not contact with DNA, but located on the end of helix of zinc finger motif illustrated by molecule modeling results,
suggesting that phosphorylation on these sites may cause the instabilities of ZNF tandem motif structure (data not shown). It has been reported that C2H2-type ZNF motifs are dedicated to homo-dimerization and hetero-dimerization with closely related family members to mediate the transcriptional regulation (Razin et al., 2012; McCarty et al., 2003). Our preliminary immunoprecipitation assay results showed that ZNF322A could form homodimers. Therefore, it is possible that ZNF322A Thr-150 and Thr-234 site may affect the formation of ZNF322A dimer thus attenuate ZNF322A transcriptional activity. In addition, our previous studies demonstrate that ZNF322A forms complex with c-Jun and cooperatively binds to promoter containing AP-1 site thus activates promoter activity of ADD1 (Jen et al., 2016). Therefore, we speculated that ZNF322A phosphorylation site(s) may interact with c-Jun to regulate ADD1 promoter activity. We will perform immunoprecipitation and dual luciferase promoter assay to determine whether c-Jun could not interact with ZNF322A phosphorylation-defective mutant therefore ADD1 promoter activity was not inhibited by the c-Jun/ZNF322A complex. Furthermore, we still could not exclude that ZNF322A may form transcriptional complex with others transcription factor. To validate whether ZNF322A would interact with other transcription factors, we will perform GST pull down assay using GST-tagged phosphorylation-defective mutant of ZNF322A purified from E.coli and then identify ZNF322A-interacting transcription factor-like proteins by LC-MS/MS. Further studies will be needed to elucidate the mechanism of ZNF322A phosphorylation by AKT on ZNF322A transcriptional activity.
It is well known that EGFR-PI3K-AKT signaling pathway contributes to tumorigenesis. In this study, we demonstrated that AKT facilitated ZNF322A protein stability and transcriptional activity upon EGF stimulation (data not shown). It has been reported that expression of p-AKT positively correlates with the EGFR or TGF-β level in NSCLC patients (Mukohara et al., 2004). In addition, there are positive correlation between EGFR and p-AKT in metastatic breast cancer patients (Kallergi et al., 2008). This prompted us to further examine whether EGFR overexpression and p-AKT had positive correlation in cancer patients with p-S224 ZNF322A expression pattern. Therefore, EGFR and p-AKT or p-ZNF322A can be tested as potential prognosis biomarkers in the future. We proposed that AKT inhibitor in combination with TKI could further suppress ZNF322A expression thereby promote cell cycle arrest and apoptosis. To confirm if ZNF322A expression was blocked, proliferation assay and apoptosis assay will be examined upon TKI synergizing with AKT inhibitor. Clinically, cancer patients who highly expressed both ZNF322A and AKT may have a better outcome to such combination treatment.
In conclusion, we had uncovered the effects of AKT in regulation of ZNF322A protein function. We found that T262-ZNF322A phosphorylation by AKT stabilized ZNF322A protein stability. In addition, AKT-mediated ZNF322A phosphorylation at T150, S224 and T234 promoted ZNF322A recruitment to ADD1 promoter region and perhaps CCND1 promoter thereby enhancing lung cancer cell proliferation and migration. We also demonstrated that EGFR-AKT axis facilitated protein stability and transcriptional activity of ZNF322A. This study collectively revealed that AKT serve as the positive role in regulating ZNF322A protein function.
REFERENCES
Agarwal NK, Kim CH, Kunkalla K, Konno H, Tjendra Y, Kwon D, Blonska M, Kozloski GA, Moy VT, Verdun RE, Barber GN, Lossos IS, Vega F. (2016). Active IKKβ promotes the stability of GLI1 oncogene in diffuse large B-cell lymphoma. Blood. 127(5):605-15.
Chuang CW, Pan MR, Hou MF, Hung WC. (2013). Cyclooxygenase-2 up-regulates CCR7 expression via AKT-mediated phosphorylation and activation of Sp1 in breast cancer cells. J Cell Physiol. 228(2):341-8.
Fan L, Tan B, Li Y, Zhao Q, Liu Y, Wang D, Zhang Z. (2015). Silencing of ZNF139-siRNA induces apoptosis in human gastric cancer cell line BGC823. Int J Clin Exp Pathol. 8(10):12428-36.
Fang L, Zhang L, Wei W, Jin X, Wang P, Tong Y, Li J, Du JX, Wong J. (2014). A methylation-phosphorylation switch determines Sox2 stability and function in ESC maintenance or differentiation. Mol Cell. 55(4):537-51.
Filtz TM, Vogel WK, Leid M. (2014). Regulation of transcription factor activity by interconnected post-translational modifications. Trends Pharmacol Sci. 35(2):76-85.
Harper J, Yan L, Loureiro RM, Wu I, Fang J, D’Amore PA, Moses MA. (2007). Repression of vascular endothelial growth factor expression by the zinc finger transcription factor ZNF24. Cancer Res. 67:8736–41.
Hong J, Zhou J, Fu J, He T, Qin J, Wang L, Liao L, Xu J. (2011). Phosphorylation of serine 68 of Twist1 by MAPKs stabilizes Twist1 protein and promotes breast cancer cell invasiveness. Cancer Res. 71(11):3980-90.
Hu R, Peng G, Dai H, Breuer EK, Stemke-Hale K, Li K, Gonzalez-Angulo AM, Mills GB, Lin SY. (2011). ZNF668 functions as a tumor suppressor by regulating p53 stability and function in breast cancer. Cancer Res. 71(20):6524–34.
Jen J, Lin LL, Chen HT, Liao SY, Lo FY, Tang YA, Su WC, Salgia R, Hsu CL, Huang HC, Juan HF, Wang YC. (2016). Oncoprotein ZNF322A transcriptionally deregulates alpha-adducin, cyclin D1 and p53 to promote tumor growth and metastasis in lung cancer. Oncogene. 35(18):2357-69.
Jia D, Hasso SM, Chan J, Filingeri D, D’Amore PA, Rice L, Pampo C, Siemann DW, Zurakowski D, Rodig SJ, Moses MA. (2013). Transcriptional repression of VEGF by ZNF24: mechanistic studies and vascular consequences in vivo. Blood. 121(4):707-15.
Kallergi G, Agelaki S, Kalykaki A, Stournaras C, Mavroudis D, Georgoulias V. (2008). Phosphorylated EGFR and PI3K/Akt signaling kinases are expressed in circulating tumor cells of breast cancer patients. Breast Cancer Res. 10(5):R80.
Kao SH, Wang WL, Chen CY, Chang YL, Wu YY, Wang YT, Wang SP, Nesvizhskii AI, Chen YJ, Hong TM, Yang PC. (2013). GSK3beta controls epithelial-mesenchymal transition and tumor metastasis by CHIP-mediated degradation of Slug. Oncogene. 33(24):3172-82.
Lehmann W, Mossmann D, Kleemann J, Mock K, Meisinger C, Brummer T, Herr R, Brabletz S, Stemmler MP, Brabletz T. (2016). ZEB1 turns into a transcriptional activator by interacting with YAP1 in aggressive cancer types. Nat Commun. 7:10498.
Li Y, Tan BB, Zhao Q, Fan LQ, Wang D, Liu Y. (2014). ZNF139 promotes tumor metastasis by increasing migration and invasion in human gastric cancer cells. Neoplasma. 61(3):291-8.
Li Y, Wang Y, Zhang C, Yuan W, Wang J, Zhu C, Chen L, Huang W, Zeng W, Wu X, Liu M. (2004). ZNF322, a novel human C2H2 Kruppel-like zinc-finger protein, regulates transcriptional activation in MAPK signaling pathways. Biochem Biophys Res Commun. 325(4):1383-92.
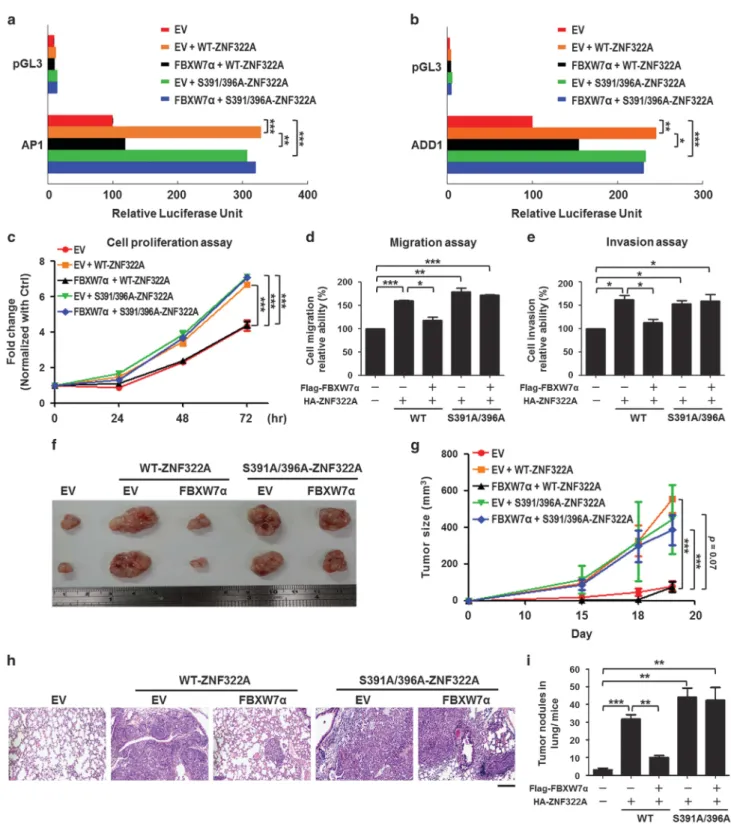
Liao SY, Chiang CW, Hsu CH, Chen YT, Jen J, Juan HF, Lai WW, Wang YC. (2017). CK1δ/GSK3β/FBXW7α axis promotes degradation of the ZNF322A oncoprotein to suppress lung cancer progression. Oncogene. 36(41):5722-5733
Liu L, Tong Q, Liu S, Cui J, Zhang Q, Sun W, Yang S. (2016). ZEB1 upregulates VEGF expression and stimulates angiogenesis in breast cancer. PLoS One. 11(2):e0148774.
Ma Juan, Mi Chunliu, Wang Ke Si, Lee Jung Joon, Jin Xuejun. (2016). Zinc finger protein 91 (ZFP91) activates HIF-1α via NF-κB/p65 to promote proliferation and tumorigenesis of colon cancer. Oncotarget. 7(24):36551–36562.
McCarty AS, Kleiger G, Eisenberg D, Smale ST. (2003). Selective dimerization of a C2H2 zinc finger subfamily. Mol Cell. 11(2):459-70.
Mukohara T, Kudoh S, Matsuura K, Yamauchi S, Kimura T, Yoshimura N, Kanazawa H, Hirata K, Inoue K, Wanibuchi H, Fukushima S, Yoshikawa J. (2004). Activated Akt expression has significant correlation with EGFR and TGF-alpha expressions in stage I NSCLC. Anticancer Res. 24(1):11-7.
Po A, Silvano M, Miele E, Capalbo C, Eramo A, Salvati V, Todaro M, Besharat ZM, Catanzaro G, Cucchi D, Coni S, Di Marcotullio L, Canettieri G, Vacca A, Stassi G, De Smaele E, Tartaglia M, Screpanti I, De Maria R, Ferretti E. (2017). Noncanonical GLI1 signaling promotes stemness features and in vivo growth in lung adenocarcinoma. Oncogene. 36(32):4641-4652.
Razin SV, Borunova VV, Maksimenko OG, Kantidze OL. (2012). Cys2His2 zinc finger protein family: classification, functions, and major members. Biochemistry (Mosc). 77(3):217-26.
Riverso M, Montagnani V, Stecca B. (2017). KLF4 is regulated by RAS/RAF/MEK/ERK signaling through E2F1 and promotes melanoma cell growth. Oncogene. 36(23):3322-3333.
Wolfe SA, Nekludova L, Pabo CO. (2000). DNA recognition by Cys2His2 zinc finger proteins. Annu Rev Biophys Biomol Struct. 29: 183-212.
Zheng H, Shen M, Zha YL, Li W, Wei Y, Blanco MA, Ren G, Zhou T, Storz P, Wang HY, Kang Y. (2014). PKD1 phosphorylation-dependent degradation of SNAIL by SCF-FBXO11 regulates epithelial-mesenchymal transition and metastasis. Cancer Cell. 26(3):358-73.
本計畫產出之 SCI 論文:
1. Seng-You Liao, Chi-Wu Chiang, Chun-Hseng Hsu, Yu-Ting Chen, Jayu Jen,
Hsuen-Feng Juan, Wu-Wei Lai, Yi-Ching Wang. 2017.
CK1δ/GSK3/FBXW7 axis promotes degradation of the ZNF322A oncoprotein to suppress lung cancer progression. Oncogene 36(41):5722-5733.
2. Sheng-You Liao, I-Ying Kuo, Yu-Ting Chen, Pao-Chi Liao, Ya-Fen Liu, Hsin-Yi Wu, Wu-Wei Lai, Yi-Ching Wang. 2019. AKT-mediated phosphorylation enhances protein stability and transcription activity of ZNF322A to promote lung cancer progression. Oncogene. 38(41):6723-6736.
ORIGINAL ARTICLE
CK1
δ/GSK3β/FBXW7α axis promotes degradation of the
ZNF322A oncoprotein to suppress lung cancer progression
S-Y Liao1, C-W Chiang1,2, C-H Hsu3, Y-T Chen4, J Jen4, H-F Juan5, W-W Lai6and Y-C Wang1,4
Overexpression of Cys2His2 zinc-finger 322A (ZNF322A) oncogenic transcription factor is associated with lung tumorigenesis. However, the mechanism of ZNF322A overexpression remains poorly understood. Here, we discover that protein stability of ZNF322A is regulated by coordinated phosphorylation and ubiquitination through the CK1δ/GSK3β/FBXW7α axis. CK1δ and GSK3β kinases sequentially phosphorylate ZNF322A at serine-396 and then serine-391. Moreover, the doubly phosphorylated ZNF322A protein creates a destruction motif for the ubiquitin ligase FBXW7α leading to ZNF322A protein destruction. Overexpression of FBXW7α induces ZNF322A protein degradation, thereby blocks ZNF322A transcription activity and suppresses ZNF322A-induced tumor growth and metastasis in vitro and in vivo. Clinically, overexpression of ZNF322A correlates with low FBXW7α or defective CK1δ/GSK3β-mediated phosphorylation in lung cancer patients. Multivariate Cox regression analysis indicates that patients with ZNF322A high/FBXW7 low expression profile can be used as an independent factor to predict the clinical outcome in lung cancer patients. Our results reveal a new mechanism of ZNF322A oncoprotein destruction regulated by the CK1δ/GSK3β/FBXW7α axis. Deregulation of this signaling axis results in ZNF322A overexpression and promotes cancer progression.
Oncogene (2017)36, 5722–5733; doi:10.1038/onc.2017.168; published online 5 June 2017
INTRODUCTION
Cys2His2 zinc-finger (ZNF) proteins are the largest class of
DNA-binding protein in eukaryotic transcription factors.1 Malfunction of ZNF transcription factors is involved in tumorigenesis and the regulatory mechanism varies among different ZNF proteins. For instance, ZNF24 represses transcription of vascular endothelial growth factor to inhibit angiogenesis and tumor growth of breast cancer.2 ZNF331 acts as a tumor suppressor and downregulates the gene expression associated with cell proliferation and invasion in gastric cancer.3In contrast, other ZNF proteins are defined as oncogenes, like ZNF217 and zinc-finger 322A (ZNF322A). Overexpression of ZNF217 promotes epithelial–mesenchymal transition (EMT) and chemoresistance in breast cancer.4,5Using lung cancer cell and xenograft models, we demonstrate that ZNF322A promotes tumor growth through positive transcriptional regulation of cyclin D1 and negative regulation of p53 expression, whereas upregulation of adducin 1 (ADD1) expression is required for ZNF322A-induced tumor metastases. Clinically, ZNF322A overexpression is found in Asian and Caucasian lung cancer patients and correlates with poor prognosis.6However, the mechanism of ZNF322A overexpression
remains unclear.
It has been reported that the expression of many ZNF transcription factors are regulated by phosphorylation and ubiquitin–proteasome degradation. Glycogen synthase kinase 3 beta (GSK3β), the serine (Ser)/threonine (Thr) kinase, has been reported to regulate protein stability of many ZNF transcription factors like Slug, KLF2 and Snail through
phosphorylation-dependent proteasome degradation.7–9 Some protein substrates of GSK3β require priming phosphorylation on Ser/Thr amino residue located downstream of the GSK3β phosphorylation site. CK1 is known to primely phosphorylate substrates includingβ-catenin, Snail and pVHL, which is required for subsequent GSK3β-mediated phosphorylation.10–12 For example, phosphorylation of Snail at Ser-104 and Ser-107 by casein kinase 1ε (CK1ε) is required for subsequent GSK3β-mediated phosphorylation targeting it for proteasomal degradation.10 Of note, the F-box and WD40 repeat domain-containing 7 (FBXW7), a SCF (complex of SKP1, CUL1 and F-box protein) type E3 ubiquitin ligase, has been reported to involve in GSK3β-mediated protein degradation. The FBXW7 consensus-binding motif is called Cdc4 phosphodegron (CPD) (Ser/Thr)-PXX-(S/T/E), its Ser/Thr residues can be phosphorylated by GSK3β or other kinases and further recognized by FBXW7 E3 ubiquitin ligase.13,14FBXW7 regulates cell cycle progression and cell survival by promoting degradation of many oncogenic transcription factors such as c-Jun, MCL1, p100 and KLF5.15–18 These studies indicated that control of protein stability of transcription factors is associated with cancer progression. However, whether ZNF322A can be regulated by phosphorylation and ubiquitin–proteasome degradation remains elusive.
Here we demonstrate that ZNF322A is a short half-life protein regulated by coordinated phosphorylation and ubiquitination through CK1δ/GSK3β/FBXW7α signaling axis in normal physiological condition. Deregulation of this signaling axis results in ZNF322A protein overexpression and contributes to lung tumorigenesis.
1
Institute of Basic Medical Sciences, College of Medicine, National Cheng Kung University, Tainan, Taiwan;2Institute of Molecular Medicine, College of Medicine, National Cheng
Kung University, Tainan, Taiwan;3
Department of Agricultural Chemistry, National Taiwan University, Taipei, Taiwan;4
Department of Pharmacology, College of Medicine, National
Cheng Kung University, Tainan, Taiwan;5
Department of Life Science, Institute of Molecular and Cellular Biology, Graduate Institute of Biomedical Electronics and Bioinformatics,
National Taiwan University, Taipei, Taiwan and6
Department of Surgery, National Cheng Kung University Hospital, College of Medicine, National Cheng Kung University, Tainan, Taiwan. Correspondence: Professor Y-C Wang, Department of Pharmacology and Institute of Basic Medical Sciences, College of Medicine, National Cheng Kung University, No.1, University Road, Tainan 70101, Taiwan.
E-mail: [email protected]
Oncogene (2017)36, 5722–5733 © 2017 Macmillan Publishers Limited, part of Springer Nature. All rights reserved 0950-9232/17 www.nature.com/onc
RESULTS
ZNF322A is a short half-life protein regulated by ubiquitin– proteasome system
Many transcription factors are short-lived and regulated by the ubiquitin–proteasome system. Therefore, we used cycloheximide (CHX) chase assay to analyze the protein half-life of ZNF322A in human lung cell lines. Endogenous ZNF322A protein was found to have a short half-life of about 15 min (Supplementary Figure S1A) and ectopically expressed HA-tagged ZNF322A protein was similarly rapidly degraded with a half-life of 15 min in cancer cells and even as short as 6 min in BEAS-2B bronchial epithelial cells (Supplementary Figures S1B–D). ZNF322A protein level increased upon treatment with the proteasome inhibitor MG132 (Supplementary Figures S1E–I), suggesting that ZNF322A protein is regulated by the ubiquitin–proteasome system.
Phosphorylation of ZNF322A by CK1δ triggers the subsequent phosphorylation by GSK3β
Protein phosphorylation is often required for ubiquitination/protea-some-dependent degradation. Our liquid chromatography-tandem mass spectrometry (LC-MS/MS) experiment identified several ZNF322A phosphorylation sites in vivo including Ser-224 and Ser-391 with high LC-MS/MS experimental confidence, and Ser-83, Tyr-95, Ser-396 and Ser-399 with low LC-MS/MS experimental confidence. We focused on Ser-391 phosphorylation site with high LC-MS/MS experimental confidence (Supplementary Figure S1J) because Ser-391 was a putative GSK3β phosphorylation site predicted by several phosphorylation software. Priming phosphor-ylation of protein substrates by other kinases such as CK1 are required for subsequent GSK3β-mediated phosphorylation.10–12We
observed that the putative CK1 phosphorylation sites Ser-396 and Ser-399 were close to Ser-391 site (Figure 1a, upper panel) and a highly conserved region between amino acids 390 and 402 was identified by sequence analysis across multiple species (Figure 1a, lower panel). Our pilot studies in cells showed that overexpression of CK1δ, but not CK1α or CK1ε, reduced the protein expression level of ZNF322A (Supplementary Figures S1K and L). Indeed, in vitro kinase assay showed that CK1δ phosphorylated bacterial expressed GST-tagged ZNF322A recombinant protein in a dose-dependent manner, and the phosphorylation signal was decreased upon treatment with CK1δ inhibitor IC261 (Figure 1b). In addition, the phosphorylation signal of phosphorylation-defective mutant of S396A (S396A-ZNF322A) or S399A (S399A-ZNF322A) was reduced compared with wild-type ZNF322A (WT-ZNF322A) (Figure 1c and Supplementary Figure S1M). However, CHX chase data showed that S396A-ZNF322A mutant maintained a longer protein half-life, whereas the half-life of S399A mutant was similar to that of WT-ZNF322A in cell-based experiments (Supplementary Figure S1N). These data suggested that Ser-396 was the main phosphorylation site of CK1δ in regulating ZNF322A protein stability.
Next, we examined whether ZNF322A phosphorylation by CK1δ may create the priming phosphorylation site, which could facilitate GSK3β-mediated phosphorylation. Indeed, the results of the in vitro kinase assay showed that CK1δ-mediated phosphor-ylation further enhanced ZNF322A phosphorphosphor-ylation by GSK3β (lanes 5 and 6 vs lanes 2 and 3, Figure 1d), and the phosphorylation signal was attenuated upon treatment of GSK3β inhibitor SB216763 (lanes 8 and 9 vs lanes 5 and 6, Figure 1d). In addition, S391A phosphorylation-defective mutant (391A-ZNF322A) obviously blocked ZNF322A phosphorylation by GSK3β compared with WT-ZNF322A (lanes 4 vs 3; lanes 8 vs 7, Figure 1e), indicating that Ser-391 is the phosphorylation site of GSK3β. Notably, CK1δ phosphorylation-defective mutant S396A-ZNF322A protein greatly decreased GSK3β-mediated phosphorylation level (lanes 4 vs 3, Figure 1f) and attenuated CK1δ/GSK3β-mediated phosphorylation level (lanes 8 vs 7, Figure 1f). Together, these data suggested that CK1δ-mediated
ZNF322A phosphorylation at Ser-396 is a prerequisite for effective GSK3β-mediated phosphorylation at Ser-391 of ZNF322A. CK1δ and GSK3β promote ZNF322A protein degradation Given that ZNF322A is phosphorylated in vitro by CK1 and GSK3β, both of which have been reported to regulate protein stability through phosphorylation-dependent proteolysis, we next determined if ZNF322A could interact with CK1δ and GSK3β in cells. The immunoprecipitation (IP) results showed that ZNF322A interacted with CK1δ and GSK3β (Supplementary Figures S2A and B). Of note, co-IP data showed that CK1δ, GSK3β and ZNF322A existed in the same protein complex (Figure 2a). As expected, overexpression of CK1δ or GSK3β reduced ZNF322A protein level in both H1299 and H460 cells, this decrease in ZNF322A protein was blocked by MG132 treatment (Figures 2b and c; Supplementary Figures S2C and D). Similarly, constitutively active GSK3β (GSK3β-CA), but not the kinase-dead GSK3β (GSK3β-KD), reduced the ZNF322A protein level (Supplementary Figure S2E). Conversely, ZNF322A protein expression level was increased in cells treated with CK1 inhibitors IC261 and D4476 or GSK3β inhibitor GSK3β XI (Figures 2d and e; Supplementary Figures S2F and G). In addition, depletion of CK1δ and GSK3β by short hairpin RNAs increased ZNF322A protein level (Supplementary Figure S2H), but did not affect ZNF322A mRNA levels (Supplementary Figures S2I and J), indicating that CK1δ and GSK3β regulated ZNF322A protein stability via post-translational modification.
We further analyzed the protein half-life of ZNF322A by CHX chase assay upon depletion of CK1δ or GSK3β. Our results confirmed that the half-life of ZNF322A protein was extended upon depletion of CK1δ or GSK3β (Figures 2f and g; Supplementary Figures S2K and L). Importantly, our cell-based ubiquitination assay revealed that knockdown of CK1δ or GSK3β reduced ZNF322A protein ubiquitination (Figures 2h and i), whereas ectopic expression of CK1δ or GSK3β-CA increased ZNF322A protein ubiquitination (Figure 2j; Supplementary Figures S2M and N). Importantly, the ubiquitination level of the S391A/S396A-ZNF322A mutant protein was decreased upon GSK3β overexpression as compared with WT-ZNF322A (Figure 2k). Collectively, these results indicated that both CK1δ and GSK3β promoted ZNF322A protein degradation via phosphorylation at Ser-391 and Ser-396.
The CK1δ–GSK3β axis phosphorylates ZNF322A at Ser-396 and Ser-391, and thus promotes ZNF322A turnover
We have shown that phosphorylation of ZNF322A by CK1δ at Ser-396 triggered the subsequent phosphorylation of Ser-391 by GSK3β in vitro. In addition, both CK1δ and GSK3β promoted ZNF322A protein degradation in cells. It is conceivable that Ser-391 and Ser-396 double phosphorylation mediated by GSK3β and CK1δ has a role in the regulation of ZNF322A protein degradation. As expected, phosphorylation-defective S391A-, S396A- and S391A/396A-ZNF322A proteins all exhibited longer protein half-life than WT-ZNF322A protein (Figures 3a–c).
To further characterize the interplay between GSK3β and CK1δ in promoting ZNF322A protein destruction in the context of phosphorylation at Ser-391 and Ser-396, we generated anti-phospho-ZNF322A-specific antibodies, which could recognize phosphorylation of ZNF322A at Ser-391 or at Ser-396 (Supplementary Figures S3A-D). Using these anti-phospho-ZNF322A antibodies, we found that overexpression of GSK3β or CK1δ promoted endogenous ZNF322A phosphorylation level at Ser-391 and Ser-396, respectively (Figures 3d and e). Conversely, phosphorylation levels of Ser-391 and Ser-396 were attenuated by GSK3β inhibitor and CK1δ inhibitor treatments, respectively, accompanied with an increase of total ZNF322A protein level in WT-ZNF322A-expressing cells (Figures 3f and g; Supplementary
Accumulated ZNF322A oncoprotein in lung cancer S-Y Liao et al
Figures S3E–G). Importantly, overexpression of GSK3β could not promote degradation of phosphorylation-defective S391A-ZNF322A mutant protein compared with WT-ZNF322A protein. Similarly, S396A-ZNF322A mutant was not degraded by overexpression of CK1δ (Figures 3h and i). These data suggested that GSK3β and CK1δ phosphorylate ZNF322A at Ser-391 and Ser-396, respectively, thereby resulting in ZNF322A protein turnover.
Next, we verified that CK1δ-mediated ZNF322A phosphorylation at Ser-396 may be critical for ZNF322A protein degradation targeted by GSK3β. To this end, we first examined the protein interaction between S396A-ZNF322A mutant and GSK3β. The IP assay results showed that the protein interaction between S396A-ZNF322A and GSK3β was attenuated compared with WT-ZNF322A (Supplementary Figure S4A). Moreover, knocking down of CK1δ indeed abolished the protein interaction between WT-ZNF322A and GSK3β (Supplementary Figure S4B). These data suggested that CK1δ-mediated ZNF322A phosphorylation at Ser-396 was required for targeting of ZNF322A protein by GSK3β. Moreover, we ectopically expressed WT- or S396A-ZNF322A in GSK3β-overexpressing cells. As expected, high level of S396A-ZNF322A was expressed in cells even when GSK3β was overexpressed (Figure 3j), supporting the notion that GSK3β phosphorylates ZNF322A in a CK1δ phosphorylation-dependent manner to regulate ZNF322A protein level. In addition, phosphorylation-mimetic mutant S396E-ZNF322A showed lower expression level than WT-ZNF322A. The expression level of phosphorylation-mimetic mutant S396E-ZNF322A could be restored to the level similar to that of WT-ZNF322A upon GSK3β depletion, suggesting the importance of subsequent S391 phosphorylation by GSK3β in regulation of ZNF322A protein expression (Figure 3k). In addition, GSK3β-induced WT-ZNF322A
protein degradation could be blocked by depletion of CK1δ (Figure 3l; Supplementary Figure S4C). Consistently, overexpres-sion of CK1δ promoted WT-ZNF322A protein degradation, whereas GSK3β depletion blocked CK1δ-mediated WT-ZNF322A protein turnover (Figure 3m; Supplementary Figure S4D). Collectively, these data suggested that the sequential order of phosphorylation of ZNF322A at Ser-396 by CK1δ and then Ser-391 by GSK3β is crucial for ZNF322A protein degradation.
FBXW7α is an E3 ligase targeting ZNF322A protein for ubiquitination and degradation
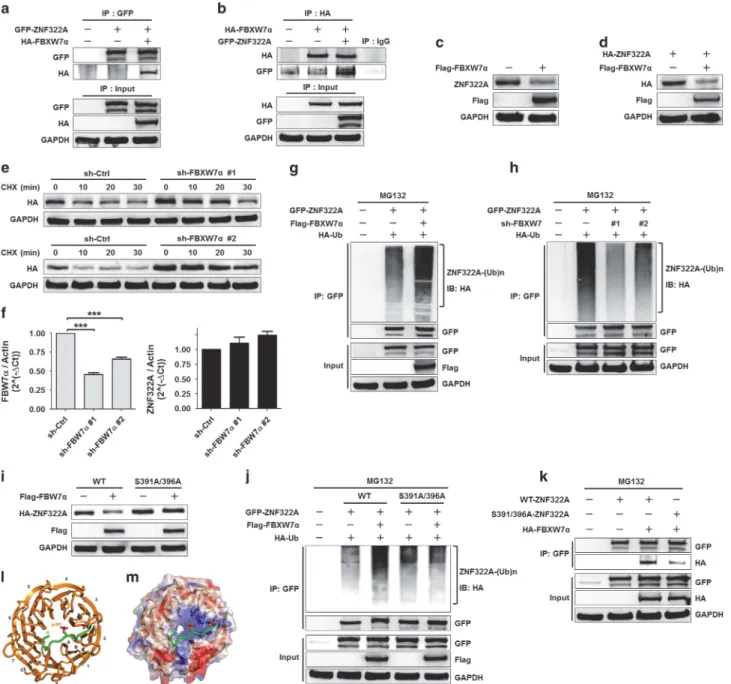
We next addressed the question of which E3 ubiquitin ligase is required for CK1δ/GSK3β-mediated ZNF322A protein degradation. FBXW7, a SCF type E3 ubiquitin ligase, binds to the CPD motif in its protein substrates and targets them for degradation. Sequence analysis revealed that ZNF322A protein contains CPD-like sequences from amino acids 391 to 396, which were highly conserved in ZNF322A orthologs (Supplementary Figure S5A). Therefore, we investigated whether ZNF322A was the target of FBXW7 E3 ligase. The IP results showed that ZNF322A interacted with FBXW7α in both H460 and H1299 cells (Figures 4a and b; Supplementary Figure S5B). In addition, overexpression of FBXW7α decreased endogenous or ectopically expressed ZNF322A protein level (Figures 4c and d; Supplementary Figure S5C). Furthermore, knockdown of FBXW7α increased ZNF322A protein half-life (Figure 4e), but did not affect ZNF322A mRNA level (Figure 4f), suggesting that FBXW7α regulates ZNF322A protein expression at the post-translational modification level.
As MG132 treatment blocked FBXW7α-induced ZNF322A protein degradation (Supplementary Figure S5D), we therefore
Figure 1. CK1δ-primed phosphorylation of ZNF322A at Ser-396 is required for GSK3β-mediated phosphorylation of ZNF322A at Ser-391 in vitro. (a) Sequence alignment of GSK3β and CK1 consensus motifs in ZNF322A (upper) and across multiple species (lower). The targeted phosphorylation residue of GSK3β is shown in red and CK1 in blue. (b) In vitro kinase assay using recombinant CK1δ and GST-ZNF322A upon treatment with or without CK1 inhibitor IC261 in the presence ofγ-[ATP]-32P. (c) In vitro kinase assay in the presence of recombinant CK1δ, wild-type (WT) or S396A mutant (S396A) GST-ZNF322A recombinant protein. (d) In vitro kinase assay using GST-ZNF322A pre-incubated with or without recombinant CK1δ before incubation with recombinant GSK3β upon treatment with or without GSK3β inhibitor (GSK3βI, SB216763). (e) In vitro kinase assay using CK1δ-primed GST-ZNF322A (WT or S391A) and recombinant GSK3β upon treatment with or without GSK3βi. (f) In vitro kinase assay using CK1δ-primed GST-ZNF322A (WT or S396A) and recombinant GSK3β upon treatment with or without GSK3βi.
Accumulated ZNF322A oncoprotein in lung cancer S-Y Liao et al
determined the ZNF322A protein ubiquitination level upon FBXW7α manipulation. Cell-based ubiquitination assay showed that overexpression of FBXW7α increased ZNF322A protein ubiquitination level (Figure 4g; Supplementary Figure S5E), whereas depletion of FBXW7α decreased ZNF322A protein ubiquitination (Figure 4h). Moreover, FBXW7α-promoted ubiquitination of ZNF322A was blocked by lysine (K) 48R mutant, but not by K63R mutant, indicating the formation of K48-linked ubiquitination by FBXW7α (Supplementary Figure S5F). Collectively, these results supported a novelfinding that FBXW7α acted as an E3 ligase for ZNF322A protein degradation.
Phosphorylation of Ser-391 and Ser-396 facilitates the recognition and degradation of ZNF322A by FBXW7α
Our data so far lead us to speculate that CK1δ/GSK3β-mediated ZNF322A phosphorylation is required for FBXW7α-promoted ZNF322A protein destruction. Indeed, CK1δ or GSK3β depletion blocked FBXW7α-mediated ZNF322A protein degradation (Supplementary Figures S5G and H). Notably, FBXW7α degraded WT-ZNF322A protein but not phosphorylation-defective S391A/396A-ZNF322A mutant (Figure 4i). Cell-based ubiquitination assay showed decrease in S391A/396A-ZNF322A mutant protein ubiquitination by FBXW7α compared with WT-ZNF322A (Figure 4j).
Figure 2. CK1δ and GSK3β promote ZNF322A protein degradation. (a) H1299 cells were co-transfected with wild-type GFP-tagged ZNF322A (GFP-WT-ZNF322A), Flag-tagged CK1δ (Flag-CK1δ) and Myc-tagged constitutively active GSK3β (Myc-GSK3β) for 24 h. Cells were treated with the proteasome inhibitor MG132 before being harvested. Co-IP experiment was performed using anti-GFP to pull down GFP-ZNF322A interacting proteins. (b, c) IB analysis of endogenous ZNF322A protein level in H1299 (upper) and H460 (lower) cells expressing with or without Flag-CK1δ (b) or in cells expressing with or without Myc-GSK3β (c). (d, e) IB analysis of endogenous ZNF322A protein level in H1299 (upper) and H460 (lower) cells treated with or without CK1 inhibitor IC261 (d) or GSK3β inhibitor GSK3β XI (e). (f, g) IB analysis of cell lysates from H460 sh-Ctrl and sh-CK1δ #1 (f) or from H460 sh-Ctrl and sh-GSK3β #1 (g) cells expressing HA-ZNF322A for 24 h before CHX (20 μg/ml) treatment at the indicated times. Quantification of ZNF322A band intensities was normalized to GAPDH, and then normalized to the time-point: 0 min. Data are presented as mean± s.e.m. (h, i) IB analysis of the anti-GFP IP products from H460 Ctrl and CK1δ cells (#1 and #2 clones) (h) or from H460 Ctrl and sh-GSK3β cells (#1 and #2 clones) (i). Where indicated, HA-Ub or GFP-WT-ZNF322A was included in the transfection. Cells were treated with proteasome inhibitor MG132 (10μM) for 6 h before being harvested. (j, k) IB analysis of the anti-GFP IP products from H460 cells expressing Flag-CK1δ (j) or cells expressing Myc-GSK3β (k). Where indicated, HA-Ub, GFP-WT-ZNF322A or GFP-S391A/396A-ZNF322A was included in the transfection. Cells were treated with the proteasome inhibitor MG132 before being harvested. IB, Immunoblotting.
Accumulated ZNF322A oncoprotein in lung cancer S-Y Liao et al