行政院國家科學委員會補助專題研究計畫 ■成果報告
□期中進度報告
前發炎物質在硫代乙醯胺引發之肝毒性中所扮演的角色
計畫類別:■個別型計畫 □整合型計畫 計畫編號:
NSC98-2312-B-006-002-MY3
執行期間:98 年 08 月 01 日至 100 年 12 月 31 日
執行機構及系所:
國立成功大學 工業衛生科暨環境醫學研究所
計畫主持人:許德榮 共同主持人:
計畫參與人員:
成果報告類型(依經費核定清單規定繳交):□精簡報告 ■完整報告
本計畫除繳交成果報告外,另須繳交以下出國心得報告:
□赴國外出差或研習心得報告
□赴大陸地區出差或研習心得報告
■出席國際學術會議心得報告
□國際合作研究計畫國外研究報告
處理方式:除列管計畫及下列情形者外,得立即公開查詢
□涉及專利或其他智慧財產權,□一年□二年後可公開查詢
中 華 民 國 101 年 3 月 9 日
目 錄
項目 頁次
中文摘要 3
英文摘要 4
報告內容
前言與目的 5
研究方法 6-8
結果 9-16
討論 17-18
參考文獻 19-21
致謝 22
中文摘要
研究背景:以乙醯胺為基底的化合物被各種工業製程廣泛使用,而其毒性卻尚未被研 究。硫代乙醯胺(thioacetamide, TAA)是一種被大量使用在工業上的化學物質,且在動物 實驗中發現硫代乙醯胺是一種造成肝臟毒性的物質,然而,硫代乙醯胺所引起急性肝臟 發炎的機制目前並不清楚。
目的:本研究目的為探討硫代乙醯胺所引起的急性肝臟發炎之機制,並找出其可能引起 致毒性之主要代謝物。
方法:本研究利用不同劑量以腹腔注射硫代乙醯胺(0, 10, 30, 100 mg/kg)誘導急性肝臟發 炎;而在硫代乙醯胺注射後 0, 1, 3, 6, 以及 12 小時,以測量發炎激素以及一氧化氮含量 以代表肝臟發炎指標。利用免疫酵素連結反應測定肝臟中之前發炎細胞激素含量;分別 利用兔抗嗜中性球血清以及細胞色素 P450 抑制劑 SKF525A 以評估嗜中性球細胞以及 細胞色素在硫代乙醯胺誘導急性肝臟發炎初始階段中所扮演的角色;此外,TAA-S-oxide 以及乙醯胺等代謝物也都被用來評估是否被包含在硫代乙醯胺所引發之早期急性肝臟 發炎。
結果:研究結果顯示,大鼠注射硫代乙醯胺六小時內會增加腫瘤壞死因子-、介白素 -1與一氧化氮的含量,增加誘導型一氧化氮合成酶的表現以及骨髓過氧化酵素的活 性,而兔抗嗜中性球血清則能明顯抑制這些參數的增加與表現。細胞色素 P450 抑制劑 可明顯抑制硫代乙醯胺所引起之腫瘤壞死因子-、介白素-1與一氧化氮的含量,並能 抑制骨髓過氧化酵素的活性。此外,乙醯胺會增加骨髓過氧化酵素的活性以及增加前發 炎物質的產生,但 TAA-S-oxide 則不會產生任何影響。
結論:乙醯胺相關的嗜中性球活化作用與硫代乙醯胺所誘導的肝臟傷害有關,而暴露乙 醯胺生成的化合物可能是造成急性肝臟傷害的一個危險因子。
關鍵詞:乙醯胺、硫代乙醯胺、肝臟發炎、嗜中性球浸潤、發炎細胞激素、一氧化氮
Abstract
BACKGROUND:Acetamide-based compounds widely used in industries and their toxic effect have not been studied. Thioacetamide (TAA) is widely used in industry and is known to be a hepatotoxicants in experimental animals. However, the mechanism underlying TAA-induced acute inflammation is still unclear.
OBJECTIVES: We investigated the mechanisms and the involvement of main TAA metabo- lites in acute hepatic inflammation induced by TAA in rats.
METHODS:Acute hepatic inflammation was induced by TAA (0, 10, 30, and100 mg/kg, ip), while the inflammatory indicators including cytokines and nitric oxide were determined 0, 1, 3, 6, and 12 h after TAA administration. Hepatic pro-inflammatory cytokines were measured quantitatively using enzyme-linked immunosorbent assay. Rabbit anti-neutrophil serum and SKF525A (cytochrome P450 2E1 [CYP 2E1] inhibitor) were used to examine the roles of neutrophil and cytochrome in the initiation of TAA-induced acute hepatic inflammation, re- spectively. In addition, TAA-S-oxide and acetamide were also used to examine the involve- ment of TAA metabolites in the early stage of TAA-induced hepatic inflammation.
RESULTS:TAA increased, within 6 h, hepatic tumor necrosis factor- production, interleu- kin-1, nitrite levels, inducible nitric oxide synthase expression, and myeloperoxidase activi- ty. Rabbit anti-neutrophil serum markedly inhibited all TAA-increased parameters. CYP 2E1 inhibitors showed significant inhibition of tumor necrosis factor-, interleukin-1, nitrite, and myeloperoxidase activity after TAA treatment. In addition, acetamide, but not TAA-S-oxide, increased myeloperoxidase activity and all tested proinflammatory mediators generation.
CONCLUSION: We conclude that acetamide-associated neutrophil activation is involved, at least partially, in TAA-induced hepatic inflammation. Further, exposure to acetam- ide-generating compounds may be a risk factor of acute hepatic inflammation.
Keywords: Acetamide, thioacetamide, hepatic inflammation, neutrophil infiltration, inflam- matory cytokine, nitric oxide
Introduction
Acetamide's production and use as a solvent, solubilizer, plasticizer, denaturing agent, and in organic synthesis may result in its release to the environment through various waste streams.
If released to air, acetamide will exist solely as a vapor in the ambient atmosphere. Va- por-phase acetamide will be degraded in the atmosphere by reaction with photochemical- ly-produced hydroxyl radicals. If released to soil, acetamide is expected to have very high mobility and volatilization from moist soil surfaces is not expected to be an important fate process. If released into water, acetamide is not expected to adsorb to suspended solids and sediment (IARC, 1984). Chemical hydrolysis studies demonstrate that acetamide will not hy- drolyze under environmental conditions (HSDB, 1993). Occupational exposure to acetamide may occur through inhalation and dermal contact with this compound at workplaces where acetamide is produced or used. It causes mild skin irritation from acute (short-term) exposure.
No information is available on the chronic (long-term), reproductive/developmental, or car- cinogenic effects of acetamide in humans. Tests involving acute exposure of rats and mice have shown acetamide to have low to moderate acute toxicity from oral exposure (HSDB, 1993). Further, acetamide is currently available directly as a product or end-metabolite of some toxic substances. One such toxic substance is thioacetamide and its end-product is ac- etamide after metabolized in animals.
Thioacetamide (TAA) is a chemical originally widely used as a fungicide, a curing in- gredient, a chemical reagent, a raw medicine, a textile dye, and a finishing auxiliary (Chen et al. 2006). TAA is also used to stabilize motor fuels, purify hydrochloric and sulfuric acids, process leather, and to substitute for hydrogen sulfide in laboratories (IARC 1974; HSDB 2009). Although its hepatotoxicity is rarely reported in the workers exposed to TAA, it is a potent hepatotoxicant and has being used to study chronic hepatic diseases such as fibrosis in experimental animals (Fitzhugh and Nelson 1948).
TAA contains a thiono-sulfur group which undergoes an extensive metabolism to acet- amide and TAA-S-oxide (TASO) by the mixed function oxidase system (Chieli and Malvaldi 1983). Cytochrome P450 (CYP), one of the important components in mixed function oxidase system, plays an important role in the biotransformation of TAA (Ramaiah et al. 2001). In- ducing CYP 2E1 deteriorates TAA-induced hepatic injury (Ramaiah et al. 2001). TASO is further metabolized to TAA-S-dioxide, a very highly reactive compound (Hunter et al. 1977).
Its binding to tissue macromolecules might induce hepatic necrosis and oxidative stress (Tunez et al. 2005). In contrast, some reports indicate that acetamide does not participate in TAA-associated hepatic lesions (Chilakapati et al. 2007).
Most chronic liver diseases follow a common course, they progress from mild inflam- mation, to more severe inflammation, to fibrosis, and finally to cirrhosis (Brenner 2009).
Therefore, hepatic acute inflammation may play a crucial role in the pathogenesis and the development of TAA-induced chronic fibrosis (Ide et al. 2003; Kornek et al. 2006). During inflammatory response, activated neutrophils infiltrate target organs and release proinflam- matory mediators, such as tumor necrosis factor (TNF)-, interleukin (IL)-1, and nitric ox- ide (NO) (Cassatella et al. 1997). In addition, nitric oxide is generated by various isoforms of nitric oxide synthases (NOS), and inducible NOS (iNOS) are dominated in inflamma- tion-associated nitric oxide production (Nussler and Billiar 1993; Wong and Billiar 1995).
Furthermore, the proinflammatory mediators are the important biomarkers for evaluating in- flammatory response. This study was conducted to address two unanswered questions: (1) Is TAA-metabolites are the cause for hepatic inflammation: (2) Do acetamide plays any im- portant role during TAA-induced acute hepatic inflammation?
Materials and methods
Chemicals. TAA was purchased from Fluka (Tokyo, Japan). Specific CYP 2E1 inhibitor SKF525A and acetamide were purchased from Enzo Life Sciences Itd. (Plymouth Meeting, PA) and Sigma (St Louis, MO), respectively.
Animals. Male Wistar rats (weighing 200-300 g) and New Zealand White rabbits (weighing 1.8-2.0 kg) were obtained from and housed in our institution’s laboratory animal center. They were housed individually in a room with a 12/12-h light/dark cycle and with central air conditioning (25C, 70% humidity). The animal care and experimental protocols were in accordance with institutional guidelines, which are approved by the National Labor- atory Animal Breeding and Research Center in Taiwan.
Experimental design. Experiment 1: Dose-response of TAA-induced inflammatory me- diators. Rats were intraperitoneally (ip) injected with TAA (0, 10, 30, and 100 mg/kg) (n = 6 per group). Six hours later, hepatic TNF-, IL-1, nitrite, and iNOS expression were as- sessed.
Experiment 2: Time-course of TAA-induced inflammatory mediators. Rats were inject- ed with 100 mg/kg of TAA (ip). Hepatic TNF-, IL-1, nitrite, and iNOS expression were assessed 0, 1, 3, 6, and 12 h after TAA treatment (n = 6 per time point).
Experiment 3: Effects of TAA on hepatic inflammatory cell infiltration. In the dose-response study, rats were injected with TAA (0, 10, 30, and 100 mg/kg; ip) (n = 6 per group). In the time-course study, rats were injected with 100 mg/kg of TAA (ip). Hepatic Myeloperoxidase (MPO, a specific marker for neutrophil infiltration) activity was determined 0, 1, 3, 6, and 12 h after TAA treatment (n = 6 per time point). Hematoxylin and eosin (H &
E) stain and CD68 immunostain (a specific marker for monocyte/macrophage) also were done 6 h after TAA treatment. In addition, macrophage inactivator gadolinium chloride (GdCl3) (ranging form 0 to 30 mg/kg, intravenously (iv)) were given 24 before TAA (100 mg/kg, ip) administration, while hepatic nitrite was determined 6 h after TAA.
Experiment 4: Effects of anti-neutrophil serum on TAA-induced hepatic MPO activity, proinflammatory mediators, and hepatic injury. Rats were divided into five groups of six. Sa- line-group rats were given saline (1 ml/kg, ip) only; TAA-group rats were given TAA (100 mg/kg, ip) only; NS-group rats were given normal serum (1 ml/rat) and then, 1 h later, TAA treatment; AS-group rats were given anti-neutrophil serum (1 ml/rat) and then, 1 h later, TAA treatment. Hepatic MPO activity and TNF- levels were determined 1 h after TAA treatment.
IL-1, and nitrite levels were determined 6 h after TAA treatment.
Experiment 5: Effects of CYP2E1 inhibitor SKF525A on TAA-induced hepatic MPO activity and proinflammatory mediator releases. Rats were divided into five groups of six.
The rats in group I were given their first dose of saline (1 ml/kg, ip) and then, 1 h later, a se- cond dose of saline (1 ml/kg, ip); the rats in groups II-V were given SKF525A (0, 10, 20, or 50 mg/kg, ip) and then, 1 h later, TAA treatment (100 mg/kg, ip). Hepatic MPO activity and TNF- were determined 1 h after TAA treatment. IL-1, and nitrite levels were determined 6 h after TAA treatment.
Experiment 6: Effects of TASO on hepatic MPO and proinflammatory mediator releases.
Rats were divided into two groups. Group I, rats were injected with saline (1 ml/kg, ip).
Group II, rats were injected with 100 mg/kg of TASO (ip). Hepatic MPO, TNF-, IL-1, and nitrite levels were assessed 0, 1, 3, and 6 h after saline or TASO treatment (n = 6 per time point).
Experiment 7: Effects of acetamide on hepatic MPO and proinflammatory mediator re- leases. Rats were divided into two groups. Group I, rats were injected with saline (1 ml/kg, ip). Group II, rats were injected with 100 mg/kg of acetamide (ip). Hepatic MPO, TNF-,
IL-1, and nitrite levels were assessed 0, 1, 3, and 6 h after saline or acetamide treatment (n = 6 per time point).
Measuring cytokines. Cytokine levels in tissue homogenates were measured quantita- tively using enzyme-linked immunosorbent assay (ELISA) kits (DuoSet; R&D Systems Inc., Minneapolis, MN) with some modifications. Briefly, 100 µ l of test sample and serial stand- ards dilutedin sample buffer (PBS containing 1% (w/v) BSA [pH 7.6]) were incubated at room temperature for 2 h. After the samples had been incubated with 100 µ l of biotinylated rabbit anti-rat TNF- or IL-1 antibody, streptavidin-conjugated horseradish peroxidase was added at room temperature for 20 min. The peroxidase reaction was initiatedby adding 100 µl of TMB (Sigma-Aldrich Co., St. Louis, MO) for 30 min, and then stopped by adding 50 µ l of 0.5 M H2SO4. The absorbance was measured at 450 nm with an ELISA reader (Hsu et al.
2007).
Measuring nitrite concentration. Briefly, the amounts of nitrite in tissue were measured after the Griess reaction by incubating 100 l of tissue homogenate with 100 l of Griess so- lution at room temperature for 20 min. We used the spectrophotometer to measure the ab- sorbance at 550 nm. Nitrite concentration was calculated by comparing it with a standard so- lution of known sodium nitrite concentration (Hsu et al. 2006).
Western Blotting. The expression of specific proteins was determined using Western blotting. Liver tissue was homogenized in 200 l of ice-cold protein lysis buffer and then al- lowed to stand on ice for 30 min. After the tissue had been centrifuged (12000 rpm, 30 min), a protein assay dye (Bio-Rad Laboratories, Hercules, CA) with bovine serum albumin as the standard was used to determine protein concentration in the supernatant. We used electro- phoresis to separate between 20 and 50 g of protein in 8-12% sodium dodecyl sulfate-PAGE gel, and then transferred the product to nitrocellulose sheets (NEN Life Science Products, Inc, Boston, MA) with a Western blot apparatus (Bio-Rad Laboratories, Hercules, CA) run at 1.2 A for 3 h. After they had been blocked in 5% non-fat skim milk in TBST, the immunoblots were incubated with primary IgG polyclonal antibody against iNOS (dilution 1:1500) (BD Biosciences, San Diego, CA) in 5% non-fat skim milk. After they had been washed three times with TBST, the immunoblots were incubated with secondary antibody conjugated with alkaline phosphatase (dilution 1:3000) (Jackson ImmunoResearch Laboratories, Inc., Phila- delphia, PA). The immunoblots were developed using bromo-chloro-indolyl phos- phate/nitroblue tetrazolium solution (Kirkegaard & Perry Laboratories, Inc., Baltimore, MD).
Measuring hepatic MPO activity. Hepatic tissue was homogenized in 20 mM phosphate buffer (pH 7.4) and then centrifuged (13,000 rpm for 10 min at 4C). The pellet was resus- pended in 1 ml of 50 mM phosphate buffer containing 0.5% hexadecyltrimethylammonium bromide. The suspension was subjected to four cycles of freezing and thawing, and then cen- trifuged (13,000 rpm for 5 min at 4C). Supernatant (0.5 ml) was mixed with tetramethylben- zidine (0.5 ml) and incubated for exactly 1 min. The reaction was stopped by adding 0.5 ml of 2N H2SO4. The spectrophotometer was then used to measure the absorbance at 405 nm.
MPO activity is shown as absorbance at 405 nm/min/mg protein (Liu et al. 2008).
Histological examination. A histological examination was used to further assess hepatic neutrophil infiltration. Briefly, organ tissues were fixed in 4% formaldehyde buffered with a phosphate solution (0.1 M [pH 7.4]) at room temperature. Organ fragments were washed in phosphate buffer, dehydrated in graded concentrations of ethanol, and then embedded in par- affin. From each tissue, 4-m-thick sections were created and stained with hematoxylin and eosin (Sewerynek et al. 1996).
Immunostaining and quantification of Macrophage using CD 68 marker. Tissue sec- tions were deparaffinized, rehydrated, and then incubated with CD 68 (Macrophage marker) Ab-3 (clone KP1) antibody (1:200) (Thermo Fisher Scientific Inc., Fremont, CA) for 30 min at room temprature and developed (Ultra Vision Detection System Anti-Polyvalent,
Alk-Phos/BCIP/NBT Kit; Thermo Fisher Scientific Inc.,). The sections were counterstained with Nuclear Fast red, cleared, and mounted using 3H-diethylphenylxanthine (DPX). CD 68 positive cells were counted in 10 high-power fields (100×).
Isolating neutrophils from rats. Rats were injected (i.p.) with 20 ml of 0.1% glycogen.
After 4 h, peritoneal cells were harvested by washing the abdominal cavity with 40 mL of PBS (containing 1 U/ml of heparin [pH 7.4]). The cells were centrifuged twice (500 × g for 7 min), suspended again in RBC lysis buffer (0.15 M NH4Cl, 0.01 M NaHCO3, and 0.001 M EDTA-Na), and then centrifuged again (320 × g for 10 min). The pellet was then resuspended in Hank’s balanced salt solution (pH 7.35). Smears of the cell suspension were stained using Liu-stain. The percentage of neutrophils in the cell suspension was > 95%.
Preparing rabbit anti-neutrophil serum. Two rabbits were each intravenously injected with 2 ml of neutrophils (1 × 107/ml). Whole blood was collected from the rabbits’ ear veins and allowed to clot for 2 h at 37C to create the anti-neutrophil serum. Before it was used, the rabbit anti-neutrophil serum was heated at 56C for 45 min to inactivate the complement.
Synthesis of TASO. TAA (40 g, 0.53 mol) was dissolved in dimethylformamide (120 mol) and cooled to -20C. Then 30% H2O2 (60 ml) was added dropwise at 1 ml/min with vigorous stirring. The mixture was stirred for 20 h at -20C. The product was recovered by filtration, levigated with CS2 (100 ml), and then recrystallized from acetone to get the long white nee- dles. The infrared spectrum was consistent with that reported for stable for 6 months if stored at -70C (Porter and Neal 1987).
Statistical analysis. Data are means standard deviation (SD). Group comparisons were done using SPSS statistical software (SPSS Institute, Chicago, IL). Student’s t-test was used for pairwise comparisons between treatments. For the antiserum and CYP 2E1 inhibitor analyses, one-way ANOVA and then Tukey’s honestly significant difference post-hoc tests were used. Statistical significance was set at P < 0.05.
Results
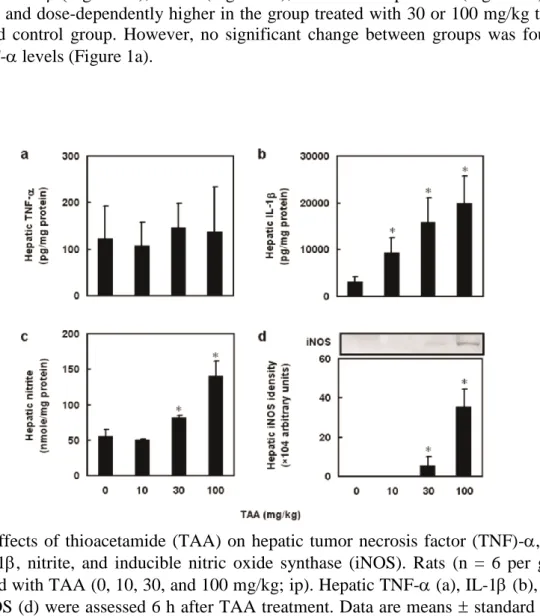
To examine the effect of TAA on acute hepatic function, dose response of TAA on he- patic TNF-, IL-1, nitrite and iNOS expression were assessed. Six hours after TAA treat- ment, hepatic IL-1Figure 1b), nitrite (Figure 1c), and iNOS expression (Figure 1d) were significantly and dose-dependently higher in the group treated with 30 or 100 mg/kg than in the untreated control group. However, no significant change between groups was found in hepatic TNF- levels (Figure 1a).
Figure 1. Effects of thioacetamide (TAA) on hepatic tumor necrosis factor (TNF)-, inter- leukin (IL)-1, nitrite, and inducible nitric oxide synthase (iNOS). Rats (n = 6 per group) were injected with TAA (0, 10, 30, and 100 mg/kg; ip). Hepatic TNF- (a), IL-1 (b), nitrite (c), and iNOS (d) were assessed 6 h after TAA treatment. Data are means standard devia- tion (SD). *P < 0.05 compared with the control group.
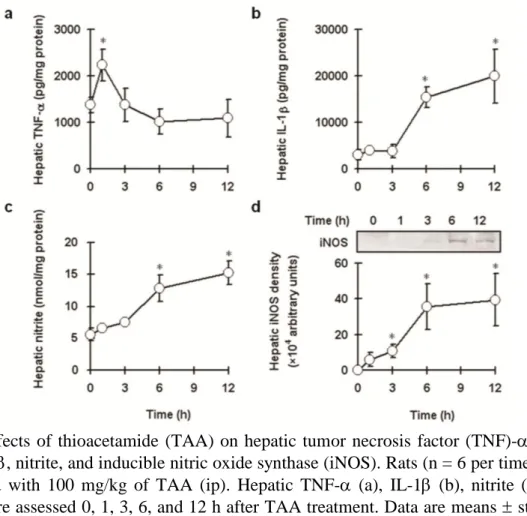
To examine the effect of TAA on proinflammatory mediator releases, hepatic TNF-, IL-1, nitrite and iNOS expression levels were determined. The expression of hepatic proin- flammatory mediators was significantly higher in TAA-treated rats than in control rats. He- patic TNF- production was significantly higher 1 h post-treatment (Figure 2a); hepatic IL-1 and nitrite were significantly higher 6 and 12 h post-treatment (Figures 2b&2c), and iNOS expression was significantly higher 3, 6, and 12 h post-treatment after (Figure 2d).
Figure 2. Effects of thioacetamide (TAA) on hepatic tumor necrosis factor (TNF)-, inter- leukin (IL)-1, nitrite, and inducible nitric oxide synthase (iNOS). Rats (n = 6 per time point) were injected with 100 mg/kg of TAA (ip). Hepatic TNF- (a), IL-1 (b), nitrite (c), and iNOS (d) were assessed 0, 1, 3, 6, and 12 h after TAA treatment. Data are means standard deviation (SD). *P < 0.05 compared with control group.
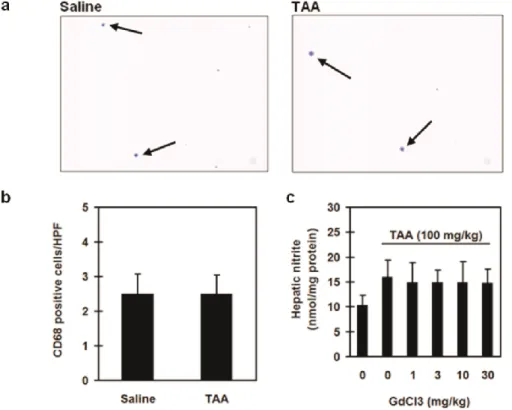
To examine the involvement of macrophage and neutrophil in TAA-induced hepatic in- flammation, we determined hepatic CD68 staining, histological examination, and MPO activ- ity. We found no significant alterations in the recruitment of CD 68 positive cells in all our experimental groups (Figures 3a & 3b). Furthermore, GdCl3 failed to affect hepatic nitrite production compared with TAA alone groups (Figure 3c).
Figure 3. Effect of thioacetamide (TAA) on hepatic macrophages infiltration. In CD68 im- munostain, rats were divided into two groups. Group I rats were treated with saline only and Group II rats were treated with TAA (100 mg/kg). Hepatic histological changes were as- sessed 6 h after treatment (CD68 immunostain; magnification = ×100). The arrows indicated the hepatic CD68 positive cells (a). CD 68 positive cells were counted in 10 high-power fields (100×) (b). In GdCl3 study, rats were divided into 6 groups (n = 6 per group). Group I, rats were given saline alone; Group II, rats were given TAA (100 mg/kg) alone; and Group III-VI, rats were given TAA plus CdCl3 (1, 3, 10, or 30 mg/kg, respectively) (c). Hepatic ni- trite was determined 6 h after saline or TAA administration. Data are means standard devi- ation (SD).
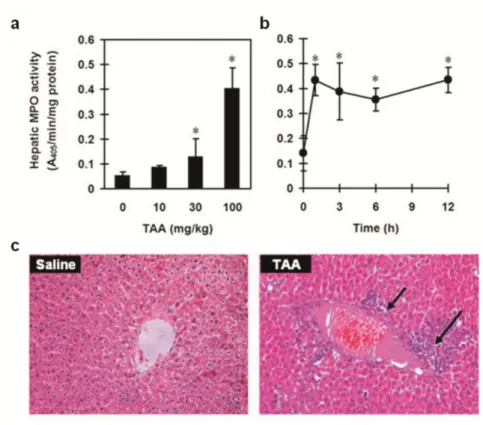
However, hepatic MPO activity was significantly higher in the groups treated with 30- and 100-mg/kg doses of TAA than in the control group (Figure 4a) and in all TAA-treated groups 1, 3, 6, and 12 h post-treatment (Figure 4b). In addition, we found significant hepatic neutrophil infiltration in 6 h after 100 mg/kg of TAA challenge (Figure 4c).
Figure 4. Effect of thioacetamide (TAA) on hepatic neutrophil infiltration. In dose-response study, rats (n = 6 per group) were injected with TAA (0, 10, 30, and 100 mg/kg; ip). Myelop- eroxidase (MPO) activity was detected 6 h after TAA administration (a). In time-course study, rats (n = 6 per time point) were injected with 100 mg/kg of TAA (ip). Hepatic MPO activity was assessed 0, 1, 3, 6, and 12 h after TAA treatment (b). In the histological examination, rats were given saline (Saline group) or TAA (100 mg/kg, ip) (TAA group). Hepatic histological changes were assessed 6 h after treatment. The arrows indicated neutrophil infiltration and a necrotic area (c) (Hematoxylin and eosin stain magnification = ×100). Data are means standard deviation (SD). *P < 0.05 compared with control group.
To examine the role of neutrophil infiltration in TAA-induced acute hepatic inflammation, rabbit anti-neutrophil serum was used. There were no significant differences between the groups treated with rabbit normal serum (NS) and TAA. Hepatic MPO (Figure 5a), TNF-
(Figure 5b), IL-1 (Figure 5c), and nitrite (Figure 5d) levels were significantly lower in rats treated with rabbit anti-neutrophil serum (AS) than in those treated with TAA and NS, but not significantly different from those treated with saline only.
Figure 5. Effects of rabbit anti-neutrophil serum on myeloperoxidase (MPO) activity and proinflammatory mediator production in thioacetamide (TAA)-induced hepatic acute in- flammation. Rats were divided into four groups of six. Saline-group rats were given saline (1 ml/kg, ip) only; TAA-group rats were given TAA (100 mg/kg, ip) treatment; NS-group rats were given normal serum (1 ml/rat) 1 h before TAA treatment; AS-group rats were given an- ti-neutrophil serum (1 ml/rat) 1 h before TAA treatment. Hepatic MPO (a) activity and TNF- (b) were detected 1 h after TAA treatment. Hepatic IL-1 (c), and nitrite (d) were as- sessed 6 h after TAA treatment. Data are means standard deviation (SD). Differences be- tween treatments with different letters are statistically significant (P < 0.05).
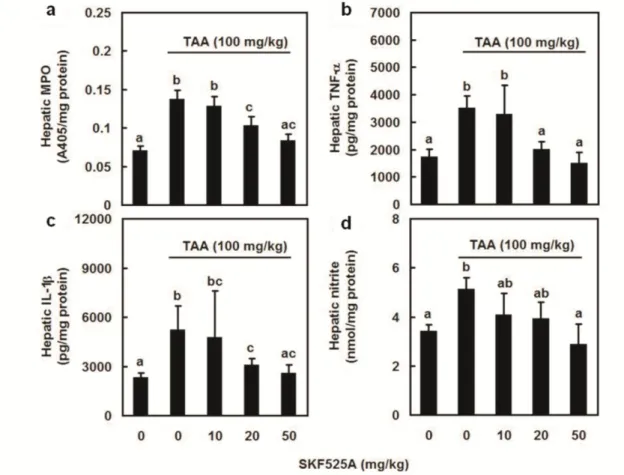
To examine the involvement of CYP 2E1 in neutrophil activation and proinflammatory mediator release, hepatic MPO (Figure 6a), TNF- (Figure 6b), IL-1 (Figure 6c), and nitrite (Figure 6d) levels were assessed. Hepatic MPO activity, TNF-, IL-1, and nitrite levels were significantly lower in all groups treated with the indicated doses of SKF525A than in the group treated with TAA alone.
Figure 6. Effects of CYP 2E1 inhibitor SKF525A on myeloperoxidase (MPO) activity and proinflammatory mediator production after thioacetamide (TAA) administration. Rats were divided into five groups of six. Group-I rats were given a first dose of saline (1 ml/kg, ip) and then, 1 h later, a second dose of saline (1 ml/kg, ip); rats in groups II-V were given methima- zole (0, 10, 20, or 50 mg/kg, respectively; ip) 1 h before TAA (100 mg/kg, ip) treatment. He- patic MPO activity (a) and TNF- (b) were detected 1 h after TAA treatment. Hepatic IL-1
(c) and nitrite (d) levels were assessed 6 h after TAA treatment. Data are means standard deviation (SD) (n = 6). The differences between treatments with different letters are statisti- cally significant (P < 0.05).
To examine the involvement of TAA metabolites in TAA-associated neutrophil activation and hepatic inflammation, TASO (Figure 7) and acetamide (Figure 8) were used. TASO did not affect hepatic MPO activity (Figure 7a) and TNF- (Figure 7b) levels within 6 h com- pared with Saline group. TASO increased hepatic IL-1 at 3 and 6 h after administration (Figure 7c). Further, TASO increased hepatic nitrite levels at 6 h compared to Saline group (Figure 7d).
Figure 7. Effects of TASO on hepatic myeloperoxidase (MPO) activity and proinflam- matory mediator production. Rats were divided into two groups. Group I, rats were injected with saline (1 ml/kg, ip). Group II, rats were injected with 100 mg/kg of TASO (ip). Hepatic MPO (a), TNF- (b), IL-1 (c), and nitrite (d) levels were assessed 0, 1, 3, and 6 h after sa- line or TASO treatment. Data are means standard deviation (SD) (n = 6). *P < 0.05 com- pared with control group.
On the other hand, acetamide significantly increased hepatic MPO activity compared with Saline group at 1, 3, and 6 h after treatment (Figure 8a). Acetamide significantly increased hepatic TNF- at 1 h (Figure 8b), and increased hepatic IL-1 at 3 and 6 h (Figure 8c) after treatment compared to Saline group. Further, acetamide significantly increased hepatic nitrite at 6 h compared to Saline group (Figure 8d).
Figure 8. Effects of acetamide on hepatic myeloperoxidase (MPO) activity and proinflam- matory mediator production. Rats were divided into two groups. Group I, rats were injected with saline (1 ml/kg, ip). Group II, rats were injected with 100 mg/kg of acetamide (ip). He- patic MPO (a), TNF- (b), IL-1 (c), and nitrite (d) levels were assessed 0, 1, 3, and 6 h after saline or acetamide treatment. Data are means standard deviation (SD) (n = 6). *P < 0.05 compared with control group.
Discussion
We have presented strong evidence that hepatic neutrophil infiltration is a key factor in initi- ating TAA-induced acute hepatic inflammation. Inhibiting neutrophil activation blocked the release of proinflammatory mediators. Specific CYP2E1 inhibitor SKF525A decreased TAA-associated neutrophil activation and the release of proinflammatory mediators. Fur- thermore, acetamide, similar to TAA, caused hepatic neutrophil infiltration and inflammation.
Therefore, we hypothesize that acetamide-associated neutrophil activation is important in the initiating TAA-induced acute hepatic inflammation.
TNF- is a pleiotropic cytokine that induces cellular responses such as proliferation, production of inflammatory mediators, and cell death (Allenbach et al. 2006; Hatano 2007).
TNF- is involved in the pathophysiology of viral hepatitis, alcoholic liver disease, nonalco- holic fatty liver disease, and ischemia-reperfusion injury in the liver (Somi et al. 2006; Hata- no 2007). TNF- regulates the release of proinflammatory mediators, such as IL-1 and ni- tric oxide, which complete the inflammatory response (Ksontini et al. 1998; Strieter et al.
1993; Cerami 1993; Jana et al. 2005). We found that TNF- levels increased as early as 1 h after TAA treatment and that IL-1 and iNOS expressions did not increase until 3 h after.
Therefore, we hypothesize that TNF--associated inflammatory response is crucial in initiat- ing TAA-induced acute hepatic inflammation.
Neutrophil, but not macrophage, activation and infiltration into the liver may be involved in the production of TNF- in TAA-induced acute hepatic inflammation. Although TNF- is produced primarily by macrophages and neutrophils (Welbourn and Young 1992; Tucker- mann et al. 2007), inhibiting macrophages does not attenuate TAA-induced early hepatic cy- tokine production and inflammation (IARC 1974). In the present study, TAA did not induce hepatic macrophage infiltration. Further, macrophage inactivator GdCl3 failed to affect TAA-enhanced hepatic nitrite production. It is likely that macrophages are not involved in initiating TAA-induced acute hepatic inflammation. In contrast, inhibiting neutrophil activa- tion significantly inhibited TAA-induced TNF- production and subsequent inflammatory mediator generation. Therefore, we hypothesize that neutrophil activation is also crucial in initiating TAA-induced acute hepatic inflammation.
CYP 2E1-associated TAA metabolites have been suggested to be involved in the patho- genesis of TAA-induced liver injury (De Ferreyra et al. 1983; Fall et al. 1997). CYP 2E1 is reported to be primarily involved in the bioactivation of TAA (Ramaiah et al. 2001; Wang et al. 2001). TAA metabolites may be associated with the TAA-induced hepatic inflammatory response and hepatic injury. Metabolites such as TAA-S-oxide and acetamide (Hunter et al.
1977; Porter et al. 1979; Chilakapati et al. 2005) are formed during TAA bioactivation by CYP 2E1. TASO, an earlier and stable intermediate, has been reported to be responsible for TAA-induced hepatotoxicity (Ramaiah et al. 2001; Chilakapati et al. 2005; Wang et al. 2000).
Although TASO increased hepatic IL-1 and nitrite at 3 or 6 h; however, it failed to affect hepatic MPO activity which is significantly enhanced from 1 h after TAA administration. It is likely that TASO did not play a crucial role in initiating TAA-induced acute hepatic inflam- mation, although it has been reported to be associated with TAA-associated chronic hepatic injury (Porter et al.1979).
Acetamide may play an important role in TAA-induced hepatic neutrophil infiltration and acute inflammation. Previous study suggests that acetamide apparently does not produce he- patic lesions similar to those produced by TAA (Chilakapati et al. 2007). However, in the present study, acetamide significantly enhanced hepatic neutrophil infiltration and proin- flammatory cytokine releases at 1 h after administration, as observed in TAA experiments.
Therefore, it seems that acetamide may be crucial in the pathogenesis of the initiation of TAA-induced acute hepatic inflammation.
Occupational exposure to acetamide may occur for those workers in the plastics and chemical industries; however, no information is available on the chronic effects of acetamide in humans or animals (HSDB 1993). In the present study, we demonstrated that acetamide caused acute hepatic inflammation. It is possible that long-term occupational exposure to ac- etamide causes prolonged hepatic inflammation which contributes to chronic hepatic fibrosis (Brenner 2009). Furthermore, acetamide can be generated not only from thioacetamide, but also from various acetamide-generating compounds, such as metronidazole (Chrystal et al.
1980), ammonium acetate (Kohyama et al. 2007), and acetonitrile (DiGeronimo and Antoine 1975). Therefore, we suggest that occupational exposure of acetamide-generating chemicals is a risk factor of hepatic inflammation and dysfunction. However, more investigations will be needed to confirm this. We conclude that acetamide-associated neutrophil activation is involved, at least partially, in the initiation of TAA-induced acute hepatic inflammation.
References
Allenbach C, Zufferey C, Perez C, Launois P, Mueller C, Tacchini-Cottier F. 2006. Macro- phages induce neutrophil apoptosis through membrane TNF, a process amplified by Leishmania major. J Immunol 176:6656-6664.
Brenner DA. 2009. Molecular pathogenesis of liver fibrosis. Trans Am Clin Climatol Assoc 120:361-368.
Cassatella MA, Gasperini S, Russo MP. 1997. Cytokine expression and release by neutro- phils. Ann N Y Acad Sci 832:233-242.
Cerami A. 1993. Tumor necrosis factor as a mediator of shock, cachexia and inflammation.
Blood Purif 11:108-117.
Chen LH, Hsu CY, Weng CF. 2006. Involvement of p53 and Bax/Bad triggering apoptosis in thioacetamide-induced hepatic epithelial cells. World J Gastroenterol 12:5175-5181.
Chieli E, Malvaldi G. 1983. Role of the microsomal FAD-containing monooxygenase in the liver toxicity of thioacetamide S-oxide. Toxicology 31:41-52.
Chilakapati J, Korrapati MC, Hill RA, Warbritton A, Latendresse JR, Mehendale HM. 2007.
Toxicokinetics and toxicity of thioacetamide sulfoxide: a metabolite of thioacetamide.
Toxicology 230:105-116.
Chilakapati J, Shankar K, Korrapati MC, Hill RA, Mehendale HM. 2005. Saturation toxico- kinetics of thioacetamide: role in initiation of liver injury. Drug Metab Dispos 33:1877-1885.
Chrystal EJ, Koch RL, McLafferty MA, Goldman P. 1980. Relationship between metronida- zole metabolism and bactericidal activity. Antimicrob Agents Chemother 18:566-573.
De Ferreyra EC, De Fenos OM, Castro JA. 1983. Prevention of thioacetamide-induced liver necrosis by prior administration of substrates of microsomal flavin-containing monooxygenase. Toxicol Lett 18:127-131.
Digeronimo MJ, Antoine AD. 1975. Metabolism of acetonitrile and propionitrile by Nocardia rhodochrous LL100-21. APPl Environ Microbiol 31:900-906.
Falls JG, Ryu DY, Cao Y, Levi PE, Hodgson E. 1997. Regulation of mouse liver fla- vin-containing monooxygenase 1 and 3 by sex steroids. Arch Biochem Biophys 342:212-223.
Fitzhugh OG, Nelson AA. 1948. Liver tumors in rats fed thiourea or thioacetamide. Science 108:626-628.
Hatano E. 2007. Tumor necrosis factor signaling in hepatocyte apoptosis. J Gastroenterol Hepatol 22 (Suppl. 1):S43-S44.
HSDB (Hazardous Substances Data Bank). 1993. Registry of Toxic Effects of Chemical Substances (RTECS, online database). National Toxicology Information Program, Na- tional Library of Medicine, Bethesda, MD.
HSDB (Hazardous Substances Data Bank). National Library of Medicine. 2009. Available:
http://toxnet.nlm.nih.gov/cgi-bin/sis/search/f?./temp/~MdH4ru:1 [accessed 21 Septem- ber 2009].
Hsu DZ, Chen KT, Chu PY, Li YH, Liu MY. 2007. Sesame oil protects against lead-plus-lipopolysaccharide-induced acute hepatic injury. Shock 27:334-337.
Hsu DZ, Chen KT, Li YH, Chuang, YC, Liu MY. 2006. Sesamol delays mortality and atten- uates hepatic injury after cecal ligation and puncture in rats: role of oxidative stress.
Shock 25:528-532.
Hunter AL, Holscher MA, Neal RA. 1977. Thioacetamide-induced hepatic necrosis. I. In- volvement of the mixed-function oxidase enzyme system. J Pharmacol Exp Ther 200:439-448.
IARC (International Agency for Research on Cancer) .1974. Some Anti-thyroid and Related Substances, Nitrofurans and Industrial Chemicals. IARC Monographs on the Evaluation of Carcinogenic Risk of Chemicals to Humans, vol. 7. Lyon, France. 1974:326.
IARC (International Agency for Research on Cancer). 1987. IARC Monographs on the Evaluation of Carcinogenic Risk to Humans. Supplement 7. World Health Organization, Lyon. 1987.
Ide M, Yamate J, Machida Y, Nakanishi M, Kuwamura M, Kotani T, et al. 2003. Emergence of different macrophage populations in hepatic fibrosis following thioacetam- ide-induced acute hepatocyte injury in rats. J Comp Pathol 128:41-51.
Jana M, Anderson JA, Saha RN, Liu X, Pahan K. 2005. Regulation of inducible nitric oxide synthase in proinflammatory cytokine-stimulated human primary astrocytes. Free Radic Biol Med 38:655-664.
Kohyama E, Dohi M, Yoshimura A, Yoshida T, Nagasawa T. 2007. Remaining acetamide in acetonitrile degradation using nitrile hydratase- and amidase-producing microorganisms.
Appl Microbiol Biotechnol 74:829-835.
Kornek M, Raskopf E, Guetgemann I, Ocker M, Gerceker S, Gonzalez-Carmona MA, et al.
2006. Combination of systemic thioacetamide (TAA) injections and ethanol feeding accelerates hepatic fibrosis in C3H/He mice and is associated with intrahepatic up reg- ulation of MMP-2, VEGF and ICAM-1. J Hepatol 45:370-376.
Ksontini R, MacKay SL, Moldawer LL. 1998. Revisiting the role of tumor necrosis factor alpha and the response to surgical injury and inflammation. Arch Surg 133:558-567.
Liu FC, Day YJ, Liou JT, Lau YT, Yu HP. 2008. Sirtinol attenuates hepatic injury and pro-inflammatory cytokine production following trauma-hemorrhage in male Spra- gue-Dawley rats. Acta Anaesthesiol Scand 52:635-640.
Nussler AK, Billiar TR. 1993. Inflammation, immunoregulation, and inducible nitric oxide synthase. J Leukoc Biol 54:171-178.
Porter WR, Neal R. 1987. Metabolism of thioacetamide and thioacetamide S-oxide by rat liver microsomes. Drug Metab. Dispos 6:379-388.
Proter WR, Gudzinowicz MJ, Neal RA. 1979. Thioacetamide induced hepatic necrosis. II.
Pharmacokinetics of thioacetamide and thioacetamide-S-oxide in the rat. J Pharmacol Exp Ther 208:386-391.
Ramaiah SK, Apte U, Mehendale HM. 2001. Cytochrome P4502E1 induction increases thio- acetamide liver injury in diet-restricted rats. Drug Metab Dispos 29:1088-1095.
Sewerynek E, Melchiorri D, Reiter RJ, Ortiz GG, Lewinski A. 1995. Lipopolysaccha- ride-induced hepatoxicity is inhibited by the antioxidant melatonin. Eur J Pharmacol 293:327-334.
Somi MH, Najafi L, Noori BN, Alizadeh AH, Aghah MR, Shavakhi A, et al. 2006. Tumor necrosis factor-alpha gene promoter polymorphism in Iranian patients with chronic hepatitis B. Indian J Gastroenterol 25:14-15.
Strieter RM, Kunkel SL, Bone RC. 1993. Role of tumor necrosis factor-alpha in disease states and inflammation. Crit Care Med 21:S447-S463.
Tuckermann JP, Kleiman A, Moriggl R, Spanbroek R, Neumann A, Illing A, et al. 2007.
Macrophages and neutrophils are the targets for immune suppression by glucocorticoids in contact allergy. J Clin Invest 117:1381-1390.
Tunez I, Munoz MC, Villavicencio MA, Medina FJ, de Prado EP, Espejo I, et al. 2005.
Hepato- and neurotoxicity induced by thioacetamide: protective effects of melatonin and dimethylsulfoxide. Pharmacol Res 52:223-228.
Wang T, Shankar K, Bucci TJ, Warbritton A, Mehendale HM. 2001. Diallyl sulfide inhibi- tion of CYP2E1 does not rescue diabetic rats from thioacetamide-induced mortality.
Wang T, Shankar K, Ronis MJJ, Mehendale HM. 2000. Potentiation of thioacetamide liver injury in diabetic rats is due to induced CYP2E1. J Pharmacol Exp Ther 294:473-479.
Welbourn CRB, Young Y. 1992. Endotoxin, septic shock and acute lung injury: Neutrophils, macrophages and inflammatory mediators. Br J Surg 10:998-1003.
Wong JM, Billiar TR. 1995. Regulation and function of inducible nitric oxide synthase dur- ing sepsis and acute inflammation. Adv Pharmacol 34:155-170.
Acknowledgment: This study was supported by the National Science Council Taiwan Grant 98-2312-B-006-002-MY3.