行政院國家科學委員會專題研究計畫 成果報告
以 RAFT 活自由基聚合法合成奈米級核殼型橡膠及無機/有 機核殼型顆粒添加劑及探討其對乙烯基酯樹脂及環氧樹脂
之聚合固化行為及性質之影響(第 3 年) 研究成果報告(完整版)
計 畫 類 別 : 個別型
計 畫 編 號 : NSC 97-2221-E-011-014-MY3
執 行 期 間 : 99 年 08 月 01 日至 100 年 07 月 31 日 執 行 單 位 : 國立臺灣科技大學化學工程系
計 畫 主 持 人 : 黃延吉
計畫參與人員: 碩士班研究生-兼任助理人員:許毓倫 碩士班研究生-兼任助理人員:劉家豪 碩士班研究生-兼任助理人員:邱淑微 碩士班研究生-兼任助理人員:許勝裕 碩士班研究生-兼任助理人員:陳彥廷 碩士班研究生-兼任助理人員:黃新堯
報 告 附 件 : 出席國際會議研究心得報告及發表論文
公 開 資 訊 : 本計畫涉及專利或其他智慧財產權,2 年後可公開查詢
中 華 民 國 100 年 11 月 08 日
中文摘要: 本計劃為奈米級核殼型橡膠及無機/有機核殼型顆粒之控管合 成,及該添加劑對乙烯基酯樹脂系統(由苯乙烯、乙烯基酯、
添加劑三成份聚合固化而成之熱固性聚摻物)及環氧樹脂系統 (由環氧樹脂、硬化劑、添加劑三成份聚合固化而成之熱固性 聚摻物)之聚合固化行為、体積收縮及增韌性質的影響。這些 添加劑,吾人擬利用可逆加成-斷裂鏈轉移(RAFT)之活自由基 聚合法合成之,因其能夠得到規格優良的高分子,如設定的分 子量、低分子量分佈、及設定的結構。
第一年計劃乃在於(i)奈米級通用核殼型橡膠之合成(以傳統乳 化聚合法)及其對乙烯基酯樹脂及環氧樹脂之聚合固化及性質 的影響研究,及(ii)奈米級特用核殼型橡膠之控管合成(以 RAFT 活自由基聚合法)。第二年計劃乃在於(i)奈米級特用核殼 型橡膠(以 RAFT 活自由基乳化聚合法合成)對乙烯基酯樹脂及 環氧樹脂之聚合固化及性質的影響研究及(ii)無機(二氧化矽 奈米顆粒)/有機核殼型顆粒之控管合成(以 RAFT 活自由基溶液 聚合法合成;其中二氧化矽奈米顆粒乃利用水解矽元素所自行 合成者)。第三年則為無機(二氧化矽奈米顆粒)/有機核殼型顆 粒對乙烯基酯樹脂及環氧樹脂之聚合固化及性質的影響研究。
英文摘要: This project is to deal with the controlled synthesis of special additives, such as nano-scale core-shell rubbers (CSR), and inorganic/organic core-shell particles (CSP), and their effects on the curing behavior, volume shrinkage, and toughening of vinyl ester resins, which are made from styrene (St), vinyl ester (VE), and the special additive, and epoxy
resins, which are made from epoxy, curing agent, and the special additive. These special additives will be synthesized by living free-radical polymerizations by reversible addition -fragmentation chain transfer (RAFT), which can afford well-defined polymers with controlled molecular weight, low polydispersity, and controlled architectures.
The research for the first year has been aimed at (i) the synthesis of general-purpose nano-scale core-shell rubbers (gp-CSR) by conventional emulsion
polymerizations and their effects on the curing behavior and properties of VER and epoxy resins, and (ii) the controlled synthesis of specialized nano- scale core-shell rubbers (s-CSR) by RAFT living free- radical polymerizations. The research for the second
year has been aimed at (i) the effects of s-CSR, which has been synthesized by RAFT living free-radical
emulsion polymerizations, on the curing behavior and properties of VER and epoxy resins, and (ii) the controlled synthesis of inorganic/organic core-shell particles (s-CSP) by RAFT living free-radical solution polymerizations using silica nano-particles,which were synthesized by hydrolysis of elemental silicon, as the core. Our effort for the third year research has been focused on the effects of s-CSP on the curing behavior and properties of VER and epoxy resins.
行政院國家科學委員會補助專題研究計畫成果報告
以RAFT活自由基聚合法合成奈米級核殼型橡膠及無機/有機核殼型顆 粒添加劑及探討其對乙烯基酯樹脂及環氧樹脂之聚合固化行為及性質 之影響
計畫類別:ˇ個別型計畫 □整合型計畫 計畫編號:97-2221-E-011 -014 -MY3
執行期間:97 年 8 月 1 日至 100 年 7 月 31 日 執行機構及系所:台灣科技大學化工系
計畫主持人:黃延吉教授 共同主持人:
計畫參與人員:黃俊翰、謝宇軒、張瀚文、許廷宇、戴孟祥、陳 曉蘭(第一年);戴夢祥、林建辰、陳曉蘭、林優 恩(Hyurin Oktavia)、劉家豪、邱淑微(第二 年);許毓倫、劉家豪、邱淑微、許勝裕、陳彥 廷、黃新堯(第三年)
成果報告類型(依經費核定清單規定繳交):ˇ完整報告
本成果報告包括以下應繳交之附件:
□赴國外出差或研習心得報告一份
□赴大陸地區出差或研習心得報告一份
█出席國際學術會議心得報告
□國際合作研究計畫國外研究報告書一份
處理方式:除列管計畫及下列情形者外,得立即公開查詢
□涉及專利或其他智慧財產權,□一年ˇ二年後 可公開查詢
中華民國 100 年 10 月 31 日
Table of Contents
Page
Chinese Abstract 5
English Abstract 5
1. Introduction 6
2. Experimental 7
2.1 First Year 7
2.1.1 Synthesis of general-purpose core-shell rubber additives by conventional emulsion
polymerizations 7
2.1.2 UP, vinyl ester, and epoxy resins 7
2.1.3 Preparations of sample solutions and cure conditions 7
2.1.4 Characterization and property measurements 8
2.1.5 Synthesis of core-shell rubber (CSR) additives by reversible addition-fragmentation
chain transfer (RAFT) emulsion polymerizations 8
2.2 Second Year 8
2.2.1 Synthesis of specialized nano-scale core-shell rubber (s-CSR) additives by reversible addition-fragmentation chain transfer (RAFT) emulsion polymerizations 8
2.2.2 UP, vinyl ester, and epoxy resins 9
2.2.3 Preparations of sample solutions and cure conditions 9
2.2.4 Characterization and property measurements 9
2.2.5 Synthesis of colloidal silica nanoparticles via hydrolysis of elemental silicon 9 2.2.6 Synthesis of polymer-grafted silica nanoparticles by reversible addition-fragmentation
chain transfer (RAFT) solution polymerizations 10
2.3 Third Year 10
2.3.1 Synthesis of colloidal silica nanoparticles via hydrolysis of elemental silicon 10 2.3.2 Synthesis of polymer-grafted silica nanoparticles by reversible addition-fragmentation
chain transfer (RAFT) solution polymerizations 10
2.3.3 Chain extension polymerization to synthesize diblock copolymer-silica hybrids 10 2.3.4 Synthesis of core-shell rubber additives of MA-Gx type by conventional emulsion
polymerizations 11
2.3.5 UP, vinyl ester, and epoxy resins 11
2.3.6 Preparations of sample solutions and cure conditions 11
2.3.7 Characterization and property measurements 11
3. Results and Discussion 11
3.1 First Year 11
3.1.1 Compatibility of styrene/VER/CSR systems 11
3.1.2 Relationship between morphologies and mechanical properties – the Takayanagi models 12
3.1.3 Volume shrinkage for St/VER/gp-CSR systems 13
3.1.4 Volume shrinkage for Epoxy/DDM/gp-CSR systems 13
3.1.5 Glass transition temperatures for Epoxy/DDM/CSR and Epoxy/DDS/CSR systems by
DMA 14
3.1.6 Mechanical properties for St/VER/gp-CSR and Epoxy/DDM/gp-CSR systems 14
3.2 Second Year 14
3.2.1 Compatibility of styrene/VER/s-CSR and Epoxy/DDM/s-CSR systems 14 3.2.2 Volume shrinkage for St/VER/s-CSR and Epoxy/DDM/s-CSR systems 15
3.3 Third Year 16
3.3.1 Synthesis of Si-PMA from commercial silica particles and their effects on properties of
UP and VER resins 16
3.3.1.1 Si-PMA synthesis and properties of Si-PMA 16
3.3.1.2 Compatibility of St/UP/Si-PMA and St/VER/Si-PMA systems 17
Page 3.3.1.3 Volume shrinkage for St/UP/Si-PMA and St/VER/Si-PMA systems 17 3.3.2 Synthesis of Si-PMA and Si-PBA-b-PMA from self-synthesized colloidal silica using
elemental silicon by hydrolysis and their effects on properties of VER resins 18 3.3.2.1 Si-PMA and Si-PBA-b-PMA synthesis and their properties 18 3.3.2.2 Compatibility of uncured St/VER/Si-PMA and Epoxy/DDM/Si-PMA systems
3.3.2.3 Cured sample morphology for St/VER/Si-PMA and Epoxy/DDM/Si-PMA systems by
SEM and TEM 19
3.3.2.4 Glass transition temperature as measured by DMA 19
3.3.2.5 Mechanical properties for St/VER/Si-PMA cured systems 20 3.3.2.6 Factors to improve toughening properties for nano-scale CSR or CSP toughened St/
VER and Epoxy/DDM cured systems 20
3.3.2.7 Mechanical properties for St/VER/Si-PBA-b-PMA cured systems 21 3.3.3 Synthesis of MA-Gx type of general-purpose core-shell rubber (gp-CSR) and their
effects on properties of epoxy/DDM resin systems 21
3.3.3.1 Compatibility of Epoxy/DDM/CSR and St/VER/CSR systems 22 3.3.3.2 Cured sample morphology for Epoxy/DDM/gp-CSR(MA-Gx-30) systems by SEM
and TEM 22
3.3.3.3 DSC cure kinetics for Epoxy/DDM/5% MA-Gx-30 systems 23 3.3.3.4 Glass transition temperature for Epoxy/DDM/gp-CSR(MA-Gx-30) systems as
measured by DMA 23
3.3.3.5 Mechanical properties for Epoxy/DDM/gp-CSR(MA-Gx-30) cured systems 24 3.3.4 Curing behavior and properties for neat St/VER and Epoxy/DDM systems 25 3.3.4.1 Cured sample morphology for neat St/VER and Epoxy/DDM systems by SEM 25 3.3.4.2 DSC cure kinetics for neat St/VER and Epoxy/DDM systems 26 3.3.4.3 Glass transition temperature for neat St/VER and Epoxy/DDM systems as measured by
DMA 27
3.3.4.4 Mechanical properties for neat St/VER and Epoxy/DDM cured systems 27 3.3.5 Effects of particle size of nano-scale Si-PMA on the volume shrinkage in the cure of
vinyl ester resins 28
3.3.5.1 Cured sample morphology for St/VER/Si-PMA systems by SEM 28
3.3.5.2 Volume shrinkage 28
4. Conclusions 29
4.1 First Year 29
4.1.1 Volume shrinkage for St/VER/gp-CSR systems 29
4.1.2 Volume shrinkage for Epoxy/DDM/gp-CSR systems 29
4.1.3 Mechanical properties for St/VER/gp-CSR and epoxy/DDM/gp-CSR systems 29
4.2 Second Year 30
4.2.1 Synthesis of nano-scale living core-shell rubber 30 4.2.2 Volume shrinkage for St/VER/s-CSR and Epoxy/DDM/s-CSR systems 30
4.3 Third Year 30
4.3.1 Effects of micron and nano-scale inorganic/organic core-shell particle on the volume shrinkage in the cure of unsaturated polyester and vinyl ester resins 30 4.3.2 Synthesis of colloidal silica nanoparticles via hydrolysis of elemental silicon and
synthesis of Si-PMA and Si-PBA-b-PMA by RAFT solution polymerizations 31 4.3.3 Mechanical properties for St/VER/Si-PMA and St/VER/Si-PBA-b-PMA cured systems 31
4.3.4 Volume shrinkage for St/VER/Si-PMA cured systems 32
4.3.5 Synthesis of core-shell rubber additives of MA-Gx type by conventional emulsion
polymerizations 32
4.3.6 Mechanical properties for Epoxy/DDM/gp-CSR(MA-Gx-30) cured systems 32 4.3.7 Curing behavior and properties for neat St/VER and Epoxy/DDM systems 33
Page 4.3.7.1 Cured sample morphology for neat St/VER and Epoxy/DDM systems by SEM 33 4.3.7.2 DSC cure kinetics for neat St/VER and Epoxy/DDM systems 33 4.3.7.3 Glass transition temperature for neat St/VER and Epoxy/DDM systems as measured
by DMA 34
4.3.7.4 Mechanical properties for neat St/VER and Epoxy/DDM cured systems 34
5. Self Evaluation of the Project 34
6. References 35
7. Tables and Figures 36
計劃名稱:以RAFT活自由基聚合法合成奈米級核殼型橡膠及無機/有機核殼型顆粒添加劑及 探討其對乙烯基酯樹脂及環氧樹脂之聚合固化行為及性質之影響
Synthesis of Nano-Scale Core-Shell Rubber and Inorganic/Organic Core-Shell Particle as Additives by RAFT Living Free Radical Polymerizations, and Their Effects on Curing Behavior and Properties of Vinyl Ester and Epoxy Resins
計劃編號:97-2221-E-011 -014 -MY3 執行期限:97/8/1 – 100/7/31
計劃主持人:黃延吉教授
執行機構:台灣科技大學化工系
中文摘要
本計劃為奈米級核殼型橡膠及無機/有機核殼型顆粒之控管合成,及該添加劑對乙烯基酯 樹脂系統(由苯乙烯、乙烯基酯、添加劑三成份聚合固化而成之熱固性聚摻物)及環氧樹脂系統 (由環氧樹脂、硬化劑、添加劑三成份聚合固化而成之熱固性聚摻物)之聚合固化行為、体積收 縮及增韌性質的影響。這些添加劑,吾人擬利用可逆加成-斷裂鏈轉移(RAFT)之活自由基聚合 法合成之,因其能夠得到規格優良的高分子,如設定的分子量、低分子量分佈、及設定的結 構。
第一年計劃乃在於(i)奈米級通用核殼型橡膠之合成(以傳統乳化聚合法)及其對乙烯基酯 樹脂及環氧樹脂之聚合固化及性質的影響研究,及(ii)奈米級特用核殼型橡膠之控管合成(以 RAFT 活自由基聚合法)。第二年計劃乃在於(i)奈米級特用核殼型橡膠(以 RAFT 活自由基乳化 聚合法合成)對乙烯基酯樹脂及環氧樹脂之聚合固化及性質的影響研究及(ii)無機(二氧化矽奈 米顆粒)/有機核殼型顆粒之控管合成(以 RAFT 活自由基溶液聚合法合成;其中二氧化矽奈米 顆粒乃利用水解矽元素所自行合成者)。第三年則為無機(二氧化矽奈米顆粒)/有機核殼型顆粒 對乙烯基酯樹脂及環氧樹脂之聚合固化及性質的影響研究。
關鍵詞:乙烯基酯樹脂;環氧樹脂;以可逆加成-斷裂鏈轉移之活自由基聚合;乳化聚合;奈 米級核殼型橡膠;無機/有機核殼型顆粒;二氧化矽奈米顆粒;体積收縮控制;增韌;聚合固 化
ABSTRACT
This project is to deal with the controlled synthesis of special additives, such as nano-scale core-shell rubbers (CSR), and inorganic/organic core-shell particles (CSP), and their effects on the curing behavior, volume shrinkage, and toughening of vinyl ester resins, which are made from styrene (St), vinyl ester (VE), and the special additive, and epoxy resins, which are made from epoxy, curing agent, and the special additive. These special additives will be synthesized by living free-radical polymerizations by reversible addition -fragmentation chain transfer (RAFT), which can afford well-defined polymers with controlled molecular weight, low polydispersity, and
controlled architectures.
The research for the first year has been aimed at (i) the synthesis of general-purpose
nano-scale core-shell rubbers (gp-CSR) by conventional emulsion polymerizations and their effects on the curing behavior and properties of VER and epoxy resins, and (ii) the controlled synthesis of specialized nano-scale core-shell rubbers (s-CSR) by RAFT living free-radical polymerizations. The research for the second year has been aimed at (i) the effects of s-CSR, which has been synthesized by RAFT living free-radical emulsion polymerizations, on the curing behavior and properties of VER and epoxy resins, and (ii) the controlled synthesis of inorganic/organic core-shell particles (s-CSP) by RAFT living free-radical solution polymerizations using silica nano-particles,which were synthesized by hydrolysis of elemental silicon, as the core. Our effort for the third year research has been focused on the effects of s-CSP on the curing behavior and properties of VER and epoxy resins.
Key words: vinyl ester resin (VER); epoxy resin; living free-radical polymerization by reversible addition-fragmentation chain transfer (RAFT); emulsion polymerization; nano-scale core-shell rubbers (CSR); inorganic/organic core-shell particles (CSP); silica nanoparticles (SNP); volume shrinkage control; toughening; curing
1. Introduction
The large volume shrinkage of unsaturated polyester resins (UP) during the cure, typically ranging from 7-10%1, is due to the extensive intramolecular or cyclization reactions of UP molecules and the formation of compact microgel structures2 during the cure. Adding specific thermoplastic polymers as low profile additives (LPA) in the UP can lead to a reduction or even elimination of the polymerization shrinkage during the cure3. However, low-profile polyester molding compounds when formulated with pigments may usually exhibit an unacceptable hazing of the pigment's color. For the past 15 years, unique non-reactive4 and reactive5 LPAs for high temperature applications have been developed that give significantly improved deep color pigmentation while maintaining a smooth surface and zero shrinkage, yet the fundamental principle has not been treated until lately6-7. By employing non-reactive LPA6, microvoids8 can be generated during the cure, which can then compensate for the volume shrinkage. In contrast, by employing reactive LPA7, the intrinsic polymerization shrinkage may be reduced, which is due to the reduction in cyclization reaction of UP resin during the cure caused by the favorable intermolecular crosslinking reaction between UP and reactive LPA.
For special additives of unsaturated polyester resins (UP), such as core-shell rubbers (CSR)9, their outer polymer shells are usually synthesized by conventional polymerization methods, where well-defined polymer chains would not be easily prepared to better control the physical and mechanical properties of the cured resins. In recent years, controlled radical polymerization10, such as reversible addition-fragmentation chain transfer (RAFT) polymerization11-12, have been widely applied to synthesize well-defined polymers with controlled molecular weight, low polydispersity, and controlled chain-end functionality due to its tolerance to a wide range of reaction conditions, the straightforward setup to result in a block copolymer, and its versatility toward the range of monomers with variable functionality13-14.
The objective of the first-year project is to investigate the effects of general-purpose nano-scale core-shell rubber (gp-CSR) as an LPA, which was synthesized by conventional emulsion polymerizations, on the volume shrinkage characteristics and internal pigmentability for styrene
(ST)/vinyl ester resin (VER)/CSR, epoxy (EP)/4,4-diaminodiphenymethane (DDM)/CSR, and EP/diamino diphenylsulfone (DDS)/CSR ternary systems. Also, in order to pave the way for the second-year project, the controlled synthesis of specialized nano-scale core-shell rubbers (s-CSR) by RAFT living free-radical polymerizations has also been carried out. The objective of the second-year project is to investigate the effects of specialized nano-scale core-shell rubber (s-CSR) as an LPA on the curing behavior, volume shrinkage characteristics, internal pigmentability, and mechanical properties for styrene (ST)/vinyl ester resin (VER)/CSR, epoxy (EP)/4,4-diaminodiphenymethane (DDM)/CSR, and EP/diamino diphenylsulfone (DDS)/CSR ternary systems. Also, in order to pave the way for the third-year project, the controlled synthesis of inorganic/organic core-shell particles (s-CSP) by RAFT living free-radical solution polymerizations using silica nano-particles as the core has also been carried out. Our effort for the third year research has been focused on the effects of s-CSP on the curing behavior and properties of VER and epoxy resins.
2. Experimental 2.1 First Year
2.1.1 Synthesis of general-purpose core-shell rubber additives by conventional emulsion polymerizations
The nano-scale general-purpose core-shell rubbers (gp-CSR), with poly(butyl acrylate) (PBA) as the core and poly(methyl methacrylate) (PMMA) as the shell, were synthesized by two-stage emulsion polymerizations15-17. The shells of the gp-CSR were also modified by introducing ethylene glycol dimethacrylate (EGDMA) as a crosslinking agent with or without glycidyl methacrylate (GMA) as a comonomer. The properties of the twelve gp-CSRs synthesized in this study, including BA/MMA-EGDMA (i.e. G0 type), BA/MMA-EGDMA-GMA(5) (i.e. G1 type), BA/MMA-EGDMA-GMA(10) (i.e. G2 type), and BA/MMA-EGDMA-GMA(16) (i.e. G3 type), are summarized in Table 1.
2.1.2 UP, vinyl ester, and epoxy resins
Two UP resins18, one vinyl ester resin (VER)19-20 , and one epoxy resin (EPR), were synthesized. One of the UP resins was made from maleic anhydride (MA), 1,2-propylene glycol (PG), and phthalic anhydride (PA), and the other was made from MA and PG by polycondensation reactions in the bulk phase. In the synthesis of VER, bisphenol-A (BPA) epoxy was made from BPA and epichlorohydrin at a molar ratio of 1:10 by solution polymerization at 70oC first. The epoxy then reacted with methacrylic acid (MAA) at a molar ratio of 1:2.14 by bulk polymerization at 115-120oC to obtain VER. The molecular characteristics of the UP, VER, and EPR are summarized in Table2.
2.1.3 Preparations of sample solutions and cure conditions
For all of the sample solution, 0%, 5% or 10% by wt. of CSR was added. For St/UP/CSR and St/VER/CSR systems, the molar ratio of styrene to polyester or VER C=C bonds was fixed at MR = 2/1, and the reaction was initiated by 1 wt. % of tert-butyl perbenzoate (TBPB). For the sample solution with pigments, 10% by weight of Bordeaux Red was added as pigments. All the cure reactions of UP and VER systems were carried out at 110oC isothermally for 1 hr.
For the epoxy (EP)/4,4-diaminodiphenylmethane (DDM)/CSR and EP/diamino diphenylsulfone (DDS)/CSR ternary systems, the molar ratio of epoxy group in EP and active hydrogen in DDM or DDS was kept at MR = 1/1. For DDM systems, the mixing step was maintained at 80oC for 30 min, while the cure reaction was conducted at 100oC for 2 hr, followed by a post-cure at 180oC by another 2 hr. In contrast, for DDS systems, the mixing step was maintained at 115oC for 10 min, while the cure reaction was conducted at 140oC for 3 hr, followed by a post-cure at 200oC by another 3 hr.
2.1.4 Characterization and property measurements
The compatibility of St/UP/CSR, St/VER/CSR, EP/DDM/CSR, EP/DDS/CSR systems at 25oC (for UP and VER systems), 80oC (for EP/DDM systems), and 95oC (for EP/DDS systems) prior to reaction21, cure kinetics of the ternary systems by DSC22, cured sample morphology for the fractured surface by SEM23, cured sample morphology by TEM9, and glass transition temperatures by DMA have been investigated. Volume shrinkage and color depth4 of the cured sample were measured by density methods23 and by using a chromameter (Minolta, CR-300) respectively.
2.1.5 Synthesis of core-shell rubber (CSR) additives by reversible addition-fragmentation chain transfer (RAFT) emulsion polymerizations
The CSR, with poly(butyl acrylate) (PBA) as the core and poly(styrene-co-acrylic acid) (SAA) as the shell, was synthesized by emulsifier-free controlled free-radical emulsion polymerization of styrene via RAFT using dibenzyltrithiocarbonate (DBTTC) as a chain transfer agent and acrylic acid as an ionogenic comonomer and involving both batch and spontaneous phase inversion processs. 24
First, DBTTC was synthesized according to literature24. In the spontaneous phase inversion process, the polymerizations took place in two different steps. The first step was a bulk copolymerization at 60oC of styrene and acrylic acid with DBTTC as a RAFT agent and AIBN (2,2‟-azobisisobutyronitrile) as an initiator, where 75.0 g of styrene (0.721 mol) and 25.0 g of acrylic acid (0.347 mol; 0.325 molar fraction in the comonomer feed) with 0.16 g (9.8 x 10-4 mol) of AIBN initiator were placed in the five-neck glass vessel reactor with 2.9 g of the dibenzyltrithiocarbonate RAFT agent (0.010 mol; target Mn = 10300 g mol-1). The whole system was heated to 60oC for 6h, to reach 54% conversion.
In the second step, the polymerization medium was cooled to room temperature and a concentrated aqueous solution containing the sodium hydroxide in equimolar amount with respect to the acid groups was slowly introduced. The system was left under stirring for a while to ensure its homogenization. Then, the remaining part of water was added progressively (755 g, i.e. 12.8 wt% targeted solids content). Initially the viscosity of the system built up and a gel was obtained.
The rate of water addition was reduced to dilute the gel without too much heterogeneity in the flow, at a stirring speed of 30 rpm. In the end a transparent, yellow colored liquid was obtained. The system was heated again for 12h at 60oC until complete conversion (94%). Such final dispersion can be used as a seed for further chain extension by n-butyl acrylate leading to 100% conversion within 1 h, where the size of the final product would be 180 nm according to the literature24.
As of July 31, 2009, the synthesis of final dispersion of the submicron-scale and nano-scale SAA latex had been completed. The further chain extension by styrene and n-butyl acrylate to obtain the SAA and PBA-SAA type of CSR, respectively, had also been performed, the size of which, however, ranged from 175-210 nm with a submicron size in diameter, which is the same as that reported in the literature24. Table 3 summarizes the experimental conditions for the surfactant-free, batch emulsion polymerizations of styrene in the presence of acrylic acid at 60oC.
Table 4 shows the experimental conditions for the synthesis of poly(styrene-co-acrylic acid) dispersion as the seed for subsequent preparation of the SAA and PBA-SAA type of CSR. Table 5 displays emulsifier-free seeded emulsion polymerizations using a spontaneous phase inversion process: chain extension with styrene and n-butyl acrylate of the poly(styrene-co-acrylic acid) dispersion E5 at 60oC and alkaline pH. Table 6 shows characteristics of the copolymers and latexes. The results in Table 3 to 6 had been obtained as planned in the first-year project25.
2.2 Second Year
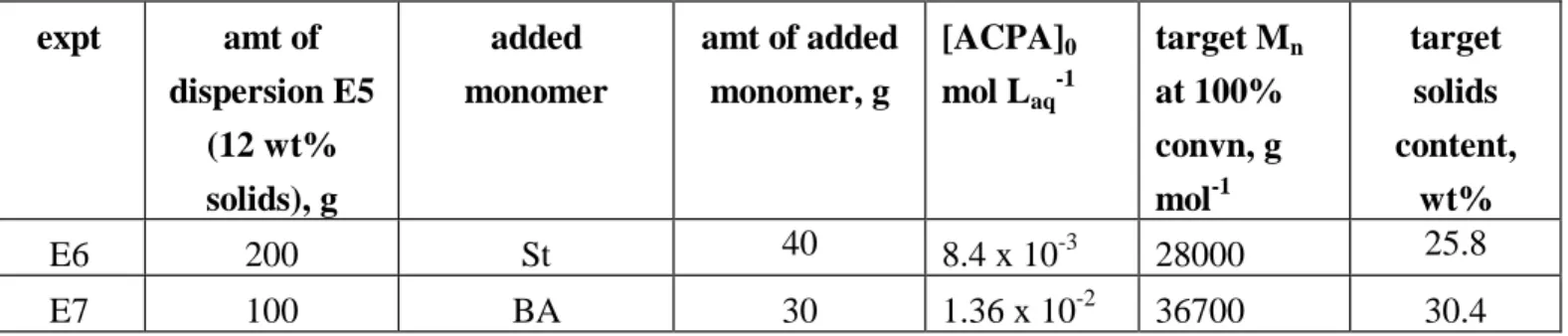
2.2.1 Synthesis of specialized nano-scale core-shell rubber (s-CSR) additives by reversible addition-fragmentation chain transfer (RAFT) emulsion polymerizations
The synthesis procedures have been described in section 2.1.1, where the nano-scale s-CSR ranging from 30 to 100 nm in diameter was not synthesized successfully in the first year of the three-year research project.
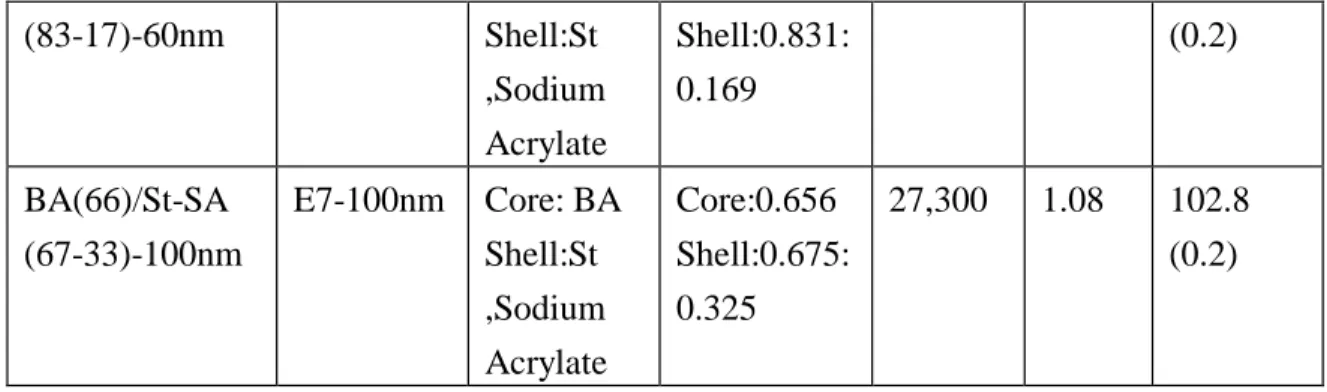
It has been realized in the second-year research project that in the synthesis of final dispersion of the nano-scale SAA latex, the size of the SAA latex can be controlled by adjusting the pH value prior to the reaction, while that of the BA-SAA latex, which is made by the further chain extension by n-butyl acrylate (BA) to obtain the BA-SAA type of CSR, can also be controlled by pH prior to the synthesis of BA-SAA latex. As of July 31, 2010, 15 nm, 60 nm and 100 nm of nano-scale s-CSR (i.e BA-SAA latex) have been synthesized by RAFT emulsion polymerizations.
The corresponding data similar to those in Tables 4-6 are summarized in Table 7-926. 2.2.2 UP, vinyl ester, and epoxy resins
The synthesis procedures are the same as those in the first year of the three-year research project, which have been described in section 2.1.2. The molecular characteristics of the UP, VER, and EPR are summarized in Table2.
2.2.3 Preparations of sample solutions and cure conditions
The preparation procedures for sample solutions and cure conditions are essentially the same as those in the first year of the three-year research project (except the epoxy/DDS/CSR systems), which have been described in section 2.1.3. For epoxy/DDS/CSR systems, the mixing step was maintained at 115oC for 10 min, while the cure reaction was conducted at 180oC for 3 hr, followed by a post-cure at 200oC by another 3 hr.
2.2.4 Characterization and property measurements
The characterization and property measurements including static phase characteristics, cure kinetics, cure sample morphologies by SEM and TEM, glass transition temperature by DMA, volume shrinkage and color depth are the same as those in the first year of the three-year research project, which have been described in section 2.1.4. In addition, mechanical properties9 for St/VER/CSR and Epoxy/DDM/CSR cured systems, including impact strength, tensile strength, and Young‟s modulus, have also been studied.
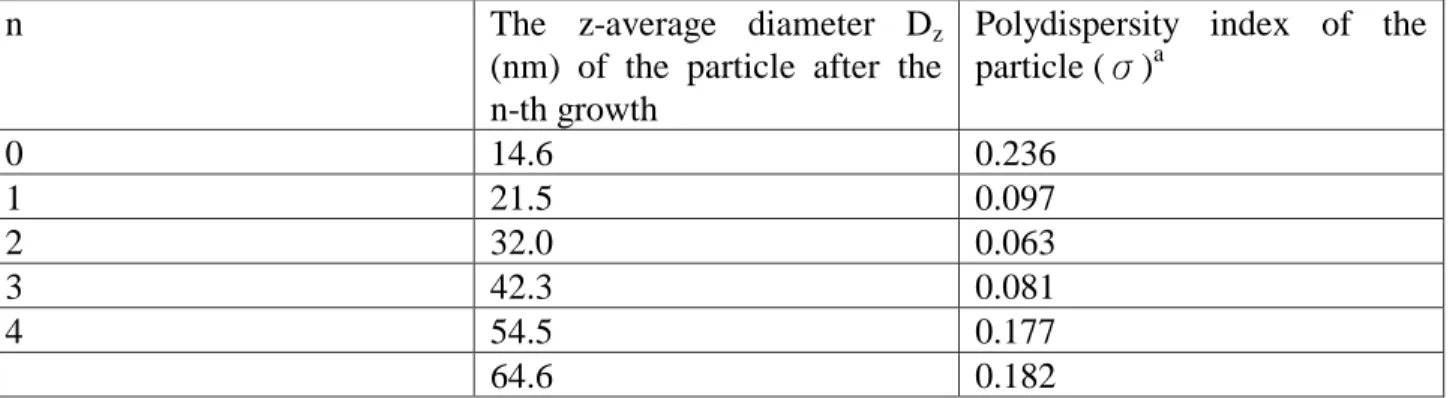
2.2.5 Synthesis of colloidal silica nanoparticles via hydrolysis of elemental silicon27
A typical synthesis of the silica particles of the test series was performed in a 500 mL reaction vessel and involved addition of NaOH and NH4OH to 200 mL of water while thoroughly mixing the solution (pH = 12). The mole ratio of NaOH and NH4OH was 10:1. Then the temperature was heated to 70oC and 5 g of silicon powders (+99.9% purity(i.e. grade 3N), 100 mesh (i.e. ~150 μm) in particle size, Sigma-Aldrich), which was mixed with 5 mL of ultra-purification water to form a slurry first, were added to the solution. The mixture was vigorously stirred with a stirring rate of 250 rpm to mix the aqueous solution well. After 20 min, 20 g of silicon powder, which was again mixed with 20 mL of ultra-purification water to form a slurry first, was added to the reaction vessel at once. 100 mL of 0.5 M NaOH was added with a rate of 25 mL/h subsequently. Then the reaction progressed during 2 h at the same temperature. After completion of the reaction, the remaining silicon powder was removed by filtration with 0.8 μm filtration membrane. The obtained colloidal suspensions were then mixed with acetone (a volume ratio of 1:2) to facilitate the precipitation of silica particles. After subsequent filtration and drying at room temperature, the silica nanoparticles can be obtained.
To regrow silica seeds to a desired size, an appropriate portion of the seeds was taken and diluted with water so that the final silica concentration was in the range of 2 wt% to prevent flocculation. The mixture was then brought to 70oC under constant stirring of ca. 250 rpm, and 5 g of silicon powder and 50 mL of 0.5 M NaOH per 100 mL of the mixture were added as described above.
The size and size distribution of colloidal silica after the nucleation and the subsequent four growths are displayed in Table 10.
2.2.6 Synthesis of polymer-grafted silica nanoparticles by reversible addition-fragmentation chain transfer (RAFT) solution polymerizations13-14
Self-synthesized silica nanoparticles were boiled in 6M HCl at 90oC for 24hr, washed with distilled water and acetone, and dried under vacuum at 60oC overnight13. 3-(benzylsulfanylthiocarbonylsulfanyl) propionic acid (BSPA)28 were synthesized and purified according to literature methods.
The synthesis of polymer-grafted silica particles, namely, inorganic/organic core-shell particle (CSP) employed as low-profile additives (LPA) for low-shrink unsaturated polyester (UP), vinyl ester resins (VER), and epoxy (EP), were synthesized by Z-supported RAFT polymerizations13-14 . In this work, the following key steps13-14,29-30
were involved: (i) synthesis of BSPA-grafted silica particles (Si-BSPA), where silica was reacted with 4-(chloromethyl)phenyltrimethoxysilane to produce benzyl chloride functionalized silica (Si-Cl) first, followed by reacting with BSPA to make the Si-BSPA , (ii) RFAT solution polymerization of methyl acrylate mediated by Si-BSPA at 60oC for 18-21 hr, with or without adding free BSPA in the reacting mixtures, and (iii) aminolysis to cleave the grafted polymer chains on the silica gel for the characterization of molecular weight and molecular weight distribution by GPC, and (iv) structure characterization of BSPA, Si-Cl, Si-BSPA, Si-poly(methyl acrylate) (i.e. Si-PMA) by FTIR, H1 NMR, C13 NMR, GPC, TGA, EA, and DSC.
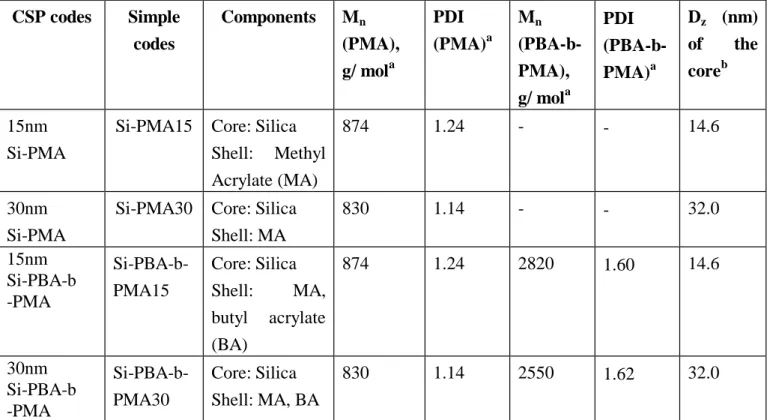
The synthesis conditions and properties of Si-PMA, employed as CSP additive for UP, VE, and EP resins for a similar work29-30 , where commercial fumed silica particle with a size of 5-15 nm was employed, to this study, are summarized in Table 11. The loading and grafting properties of Si-Cl, Si-BSPA, and Si-PMA on solid surface of silica gel particles are listed in Table 12, revealing a loading in active sites of 0.40 mmole of CTA/g of solid (GSi-BSPA), corresponding to a grafting density of 0.38 molecule of CTA/nm2 (Gd). The weight grafting ratio of PMA grafted silica particles, Gr(%), determined by TGA was 7.82 g PMA/100 g of silica.
As of July 31, 2010, the synthesis of 15 nm Si-PMA (i.e. Si-PMA15nm) and 30 nm Si-PMA (i.e. Si-PMA30nm) using the self-synthesized of silica nanoparticle in our laboratory had been completed31. Table 13 shows TGA results for the 15 nm and 30 nm size of silica particles grafted with benzyl chloride, BSPA, and poly(methyl acrylate) (PMA). Table 14 displays the loading and grafting efficiency of Si-Cl and Si-BSPA for the 15 nm and 30 nm size of silica particles. Table 15 shows polymerization results for RAFT graft polymerization mediated by Si-BSPA in the presence of free CTA (BSPA).
2.3 Third Year
2.3.1 Synthesis of colloidal silica nanoparticles via hydrolysis of elemental silicon
The synthesis procedures have been described in section 2.2.5. For the third-year research project, the size and size distribution of colloidal silica synthesized after the nucleation and the subsequent five growths are displayed in Table 1632.
2.3.2 Synthesis of polymer-grafted silica nanoparticles by reversible addition-fragmentation chain transfer (RAFT) solution polymerizations
The synthesis procedures for polymer-grafted silica particles, Si-PMA, have been described in section 2.2.6. For the third-year research project, the synthesis of 15 nm Si-PMA (i.e.
Si-PMA15nm) and 30 nm Si-PMA (i.e. Si-PMA30nm) using the self-synthesized of silica nanoparticle in our laboratory has also been completed32. Table 17 shows TGA results for the 15 nm and 30 nm size of silica particles grafted with benzyl chloride, BSPA, and poly(methyl acrylate) (PMA). Table 18 displays the loading and grafting efficiency of Si-Cl and Si-BSPA for the 15 nm and 30 nm size of silica particles. Table 19 shows polymerization results for RAFT graft polymerization mediated by Si-BSPA in the presence of free CTA (BSPA).
2.3.3 Chain extension polymerization to synthesize diblock copolymer-silica hybrids13,29,32
In a typical experiment (run 1 of Table 20), Si-PMA (33.79 mol), toluene (17.2 ml), PMA (137 mol, used as a macro-RAFT agent), butyl acrylate (BA, 51.5 mmol), and AIBN (29 mol) were added to a reaction vessel. The reaction vessel was degassed by three freeze-pump-thaw cycles and then put into an oil bath preset to 60oC for 8h. After polymerization, the samples were treated according to a method similar to the previously described RAFT polymerization of MA13,29,32. For the 15 nm Si-PBA-b-PMA, GPC analyses (Table 20) showed that the degrafted PMA-b-PBA diblock copolymer had number-average molecular weight of 2820 and polydispersity of 1.60, and free PMA-b-PBA produced in solution had number-average molecular weight of 91520 and polydispersity of 1.15. In contrast, for the 30 nm Si-PBA-b-PMA, GPC analyses (Table 20) showed that the degrafted PMA-b-PBA diblock copolymer had number-average molecular weight of 2550 and polydispersity of 1.62, and free PMA-b-PBA produced in solution had number-average molecular weight of 73610 and polydispersity of 1.14.
2.3.4 Synthesis of core-shell rubber additives of MA-Gx type by conventional emulsion polymerizations33
The nano-scale core-shell rubbers (CSR), with poly(butyl acrylate) (PBA) as the core and poly(methyl acrylate) (PMA) as the shell, were synthesized by two-stage emulsion polymerizations15-17,33. The shells of the CSR were also modified by introducing ethylene glycol dimethacrylate (EGDMA) as a crosslinking agent with or without glycidyl methacrylate (GMA) as a comonomer. The properties of the six CSRs synthesized in this study, including BA/MA-EGDMA (i.e. G0 type), BA/MA-EGDMA-GMA(5) (i.e. G1 type), and BA/MA-EGDMA-GMA(10) (i.e. G2 type) are summarized in Table 21.
2.3.5 UP, vinyl ester, and epoxy resins
The synthesis procedures are the same as those in the first year of the three-year research project, which have been described in section 2.1.2. The molecular characteristics of the UP, VER, and EPR are summarized in Table2.
2.3.6 Preparations of sample solutions and cure conditions
The preparation procedures for sample solutions and cure conditions are essentially the same as those in the first year of the three-year research project, which have been described in section 2.1.3.
2.3.7 Characterization and property measurements
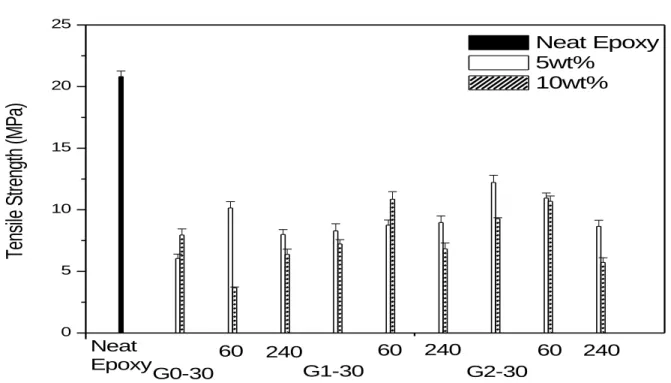
The characterization and property measurements including static phase characteristics, cure kinetics, cure sample morphologies by SEM and TEM, glass transition temperature by DMA, volume shrinkage and color depth are the same as those in the first year of the three-year research project, which have been described in section 2.1.4. In addition, mechanical properties9 for St/VER/Si-PMA-15 and Epoxy/DDM/CSR(MA-Gx-30) cured systems, including impact strength, tensile strength, Young‟s modulus, fracture toughness, Poisson ratio, and fracture energy, have also been studied34.
3. Results and Discussion 3.1 First Year
3.1.1 Compatibility of styrene/VER/CSR systems
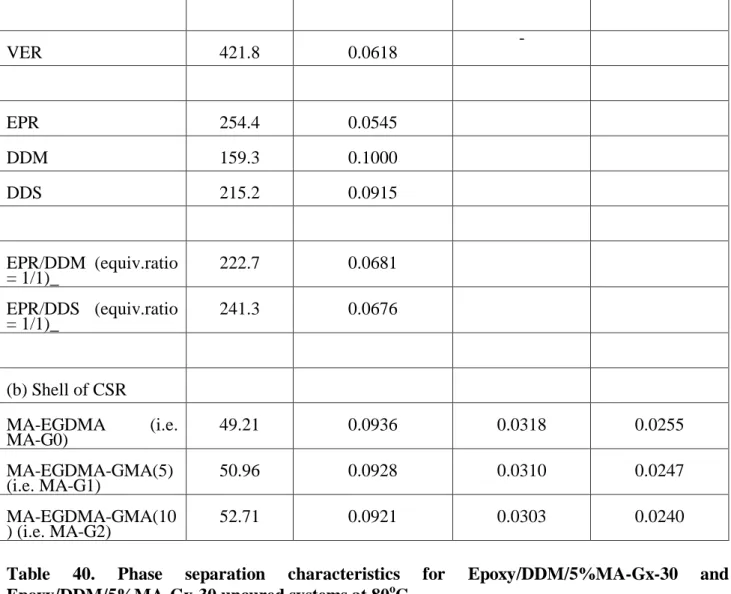
The molecular polarity of vinyl ester (VE) and the shell of gp-CSR was evaluated in terms of the calculated dipole moment per unit volume18,25, /V1/2 (Table 22). It was lower for VE than that for the shell of CSR (0.0618 vs. 0.0723-0.0737).
In general, the higher the polarity difference per unit volume between VER and the shell of CSR, the less compatibility for the St/VER/CSR system at 25oC and at 110oC prior to reaction.
Data in Table 22 reveal that the St/VER/CSR system containing G2 type of CSR would theoretically be the least compatible (|‟VER - ‟Shell of CSR| = 0.0119), followed by the G1-CSR system (|‟VER - ‟Shell of CSR| = 0.0112), and the G0-CSR system would be the most compatible (|‟VER - ‟Shell of CSR| = 0.0105). This is generally in agreement with the static phase characteristics data for the uncured St/VER/gp-CSR systems at 110oC (Table 23), where the phase separation time for G2-30 CSR system was the shortest, followed by the G1-30 CSR and G0-30 CSR systems.
During the cure at 110oC for the eighteen St/VER/CSR systems, SEM micrographs (Figures 1-3) show that a two phase microstructure, consisting of a major continuous phase of styrene-crosslinked vinyl ester and a CSR-rich dispersed phase, would result.
For the St/VER/G0-CSR ternary systems (Fig. 1), at a fixed particle size of G0-CSR at either 30 nm (i.e. G0-30, Fig. 1(a)-(b)) or 240 nm (i.e. G0-240, Fig. 1(e)-(f)), the system containing 10 wt% CSR would be more incompatible than that of 5 wt% CSR, whereas the trend was reversed for the G0-60 system (Fig. 1(c)-(d)). At a fixed CSR content at either 5 wt% or 10 wt%, the compatibility of the St/VER/G0-CSR system was decreased as the CSR size added was increased from 30nm, to 60 nm, and to 240 nm. (cf. Fig. 1(a), (c), and (e); cf. Fig. 1(b), (d), and (f))
For the St/VER/G1-CSR ternary systems (Fig. 2), irrespective of the CSR size, the system containing 10 wt% CSR would be more incompatible than that of 5 wt% CSR. At a fixed CSR content at either 5 wt% or 10 wt%, the compatibility of the St/VER/G1-CSR system was decreased as the CSR size added was increased from 30nm to 60 nm. Further increasing the CSR size to 240 nm would lead to an increase in the compatibility of the ternary system during the cure, which is due to the increase of viscosity of the ternary system and the concomitant slower rate of phase separation during the cure. (cf. Fig. 2(a), (c), and (e); cf. Fig. 2(b), (d), and (f))
For the St/VER/G2-CSR ternary systems (Fig. 3), irrespective of the CSR size, the system containing 10 wt% CSR would be more compatible than that of 5 wt% CSR, which is in contrast to the G1-CSR systems in Fig. 2. At a fixed CSR content at either 5 wt% or 10 wt%, the compatibility of the St/VER/G2-CSR system was increased as the CSR size added was increased from 30nm to 60 nm, which is due to the increase of viscosity of the ternary system and the concomitant slower rate of phase separation during the cure. Further increasing the CSR size to 240 nm would lead to a decrease in the compatibility of the ternary system during the cure. (cf. Fig.
3(a), (c), and (e); cf. Fig. 3(b), (d), and (f))
For the three 5% G-60 CSR systems (Figs. 1(c), 2(c), and 3(c)) the compatibility showed an increase and then a decrease as the CSR-containing ternary system was changed from G0-CSR, G1-CSR, and G2-CSR. For all the other fifteen systems, at fixed concentration and size of CSR added, the higher concentration of GMA introduced in the shell of CSR would lead to the less compatibility of St/VER/CSR system during the cure. (i.e. the G2-CSR system < the G1-CSR system < the G0-CSR system with an increasing order of compatibility)
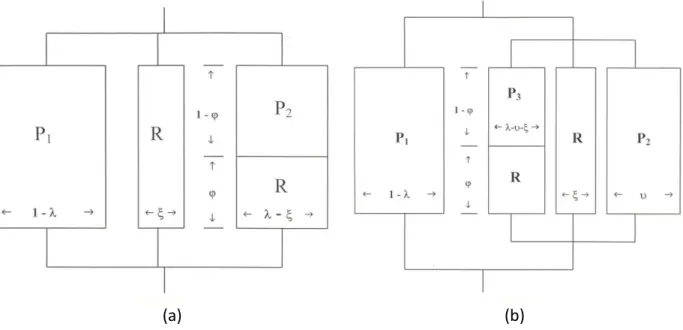
3.1.2 Relationship between morphologies and mechanical properties – the Takayanagi models For the cured CSR-containing VER systems with their characteristic morphologies as shown in Figs. 1-3, their mechanical behavior can be approximately represented by the Takayanagi models36-38, where arrays of weak LPA (R) and stiff styrene-crosslinked vinyl ester (P) phases are indicated (see Figure 4). The subscripts 1, 2, and 3 for P phases are employed due to the distinction of styrene and VER compositions as a result of phase separation during cure, and the quantities λ, φ, ξ, and or their indicated multiplications indicate volume fractions of each phase.
For all of the eighteen St/VER/CSR systems, the two-phase microstructure (Figs. 1-3) consisted of a flake-like continuous phase of styrene-crosslinked vinyl ester (phase P1) and a globule CSR-dispersed phase (i.e., the hole left behind due to the solvent extraction, the globule morphology of which prior to the solvent extraction can be represented by a P-P-S model). For the CSR-dispersed phase, the microgel particles (phase P2) would be segregated by CSR (Phase R).
Between the CSR-segregated microgel particles, there would be some other microgel particles
(Phase P3), with different compositions of styrene and VER from those in phase P2, dispersed in the CSR phase (phase R). Hence, the globule CSR-dispersed phase can be represented by the P-P-S model, which is a parallel combination of the three elements, i.e., P2, R, and P3-R in series.
The upper bound of mechanical behavior for the overall morphology can thus be represented by a P-(P-P-S) model as shown in Figure 4(b), which is simply a parallel combination of the continuous phase P1 and the dispersed phase denoted by a (P-P-S) model.
In contrast, for the St/UP/Low Profile Additive (LPA) system containing relatively polar thermoplastic polyurethane (PU) or poly(vinyl acetate) (PVAc) as an LPA, a homogeneous globule morphology may arise6,18, which can be represented by a parallel-parallel-series (P-P-S) model (Fig.
4(a)).
The mechanical properties of cured samples would change with not only the morphology but also the crosslinking density of styrene-crosslinked polyester in the P1, P2, and P3 phases, with the major continuous phase P1 being the dominant one. The latter information would not be easily obtained, but can be inferred from the static phase characteristics of St/VER/CSR systems at 25oC and 110oC before curing22 (Table 23).
3.1.3 Volume shrinkage for St/VER/gp-CSR systems
The volume shrinkage of the neat VER was about 6.8%, while adding 5 or 10 wt % different gp-CSRs, such as G-30, G-60, and G-240, can reduce the volume shrinkage to the range of -0.9%
(volume expansion) to 5.8% (Table 24). Not enough or too much phase separation during the cure of St/VER/CSR systems would be unfavorable for the volume shrinkage control. (cf Figs. 1-3 and Table 24)
With 5 or 10 wt% of nano-scale CSR (30 nm or 60 nm) added in the St/VER/CSR systems, 10 wt% CSR system would generally result in a better volume shrinkage control than that of 5 wt%
CSR system except for the G0-60, the G0-240, the G1-240, and the G2-30 CSR systems (Table 24). In contrast, with 5 or 10 wt% of submicron CSR (240 nm) added in the St/VER/CSR systems, the trend was reversed except for the G2-240 CSR systems, where the addition of 10 wt% CSR led to a volume expansion (-ΔV/V0 = -0.87 %).
With 5 wt% of G0-CSR, 10 wt% of G0-CSR, or 10wt% of G1-CSR added in the St/VER/CSR systems, as the size of CSR was increased from 30 nm to 60 nm to 240 nm, the fractional volume shrinkage was decreased, followed by an increase, and reached the smallest volume shrinkage at a CSR size of 60 nm (Table 24). However, the effects of CSR size on the fractional volume shrinkage for 5 wt% of G1-CSR systems followed the opposite trend, where the largest volume shrinkage for the G1-60 CSR system (not enough phase separation during the cure) was observed (-ΔV/V0 = 5.7 %).
With 5 or 10 wt% of G2-CSR added in the St/VER/CSR systems, as the size of G2-CSR was increased from 30 nm to 60 nm to 240 nm, the fractional volume shrinkage was increased for the 5 wt% G2-CSR systems, whereas it was decreased for the 10 wt% G2-CSR systems (Table 24). The best volume shrinkage control has been achieved for the 10 wt% G2-60 CSR system with a fractional volume shrinkage of 0.2 %.
The performance of volume shrinkage control for St/VER/CSR system by using G1-CSR. or G2-CSR was generally superior to that by using G0-CSR (Table 24). Due to the larger difference in molecular polarity between the VER and the shell of CSR, the G1-CSR or G2-CSR containing St/VER/CSR system would be more incompatible, as compared with the G0-CSR systems. This would be favorable for the reduction of cyclization reaction for VER during the cure, and the microgel structure during the cure would be less compact due to the more segregating effects of CSR on microgel structures (see Figure 5), and, in turn, be favorable for the decrease of intrinsic polymerization shrinkage after the cure. In addition, the relief of the polymerization shrinkage force caused by the rubbery core of the CSR, which was evidenced by the formation of microvoids as observed by OM and TEM micrographs39 (not shown), may lead to the much less compact
microgel structures, and even an expansion in volume change after the cure could be observed for 10 wt% G2-240 system.
3.1.4 Volume shrinkage for Epoxy/DDM/gp-CSR systems
The volume shrinkage of the neat Epoxy/DDM was about 2.4%, while adding 5 or 10 wt % different CSRs, such as G-30, G-60, and G-240, can reduce the volume shrinkage to the range of -3.8% (volume expansion) to 0.85% (Table 25). Data in Table 25 show that a higher concentration of CSR would lead to a lower volume shrinkage. Also, adding 10wt% of G0-30 or 10 wt% of G0-60 could result in a volume expansion as high as 3.8%. SEM (Figures 6-7) and TEM (Figure 8-9) micrographs for the 10 wt% G0-30 and the 10 wt% G0-60 systems revealed good segregrating effects of CSR on microgel structures, as depicted in Fig. 5, which would be favorable in the reduction of polymerization volume shrinkage during the cure and could even lead to a volume expension. The white spots in the TEM micrographs (Figs. 8-9) were due to the microvoids generated during the cure, which could verify the relief of the polymerization shrinkage force caused by the rubbery core of the CSR as mentioned earlier.
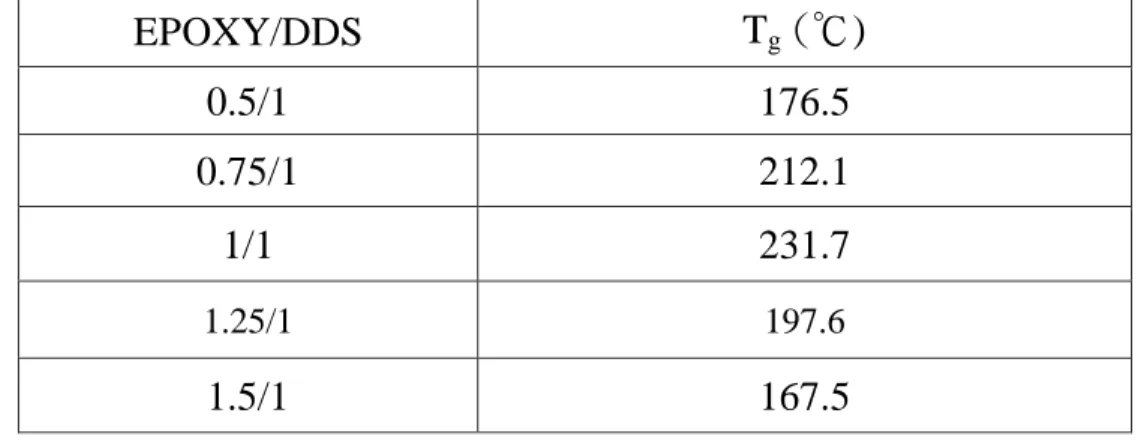
3.1.5 Glass transition temperatures for Epoxy/DDM/CSR and Epoxy/DDS/CSR systems by DMA The glass transition temperature (Tg) for the cured epoxy systems would reach the maximum as the equivalent ratio of epoxy and curing agent (DDM or DDS in this work) was equal to 1.
Either positive or negative deviation of the equivalent ratio from 1 could result in a decrease of Tg. (Tables 26 and 29) For Epoxy/DDM/CSR ternary systems, adding CSR would generally decrease the Tg of the cured matrix. (Tables 27 and 28)
3.1.6 Mechanical properties for St/VER/gp-CSR and Epoxy/DDM/gp-CSR systems
The mechanical properties40 for St/VER/gp-CSR systems are shown in Figures 10-12, and those for Epoxy/DDM/gp-CSR systems are displayed in Figures 13-15. In general, the addition of nano-scale gp-CSR (30 nm and 60 nm) and the submicron (240 nm) gp-CSR in this work would not be able to enhance the impact strength, Young‟s modulus, and tensile strength for the St/VER and Epoxy/DDM systems although most of the gp-CSR used in this work may effectively reduce the volume shrinkage during the cure. Further research in this aspect has been devoted to in the second-year and third-year of the research project.
3.2 Second Year
3.2.1 Compatibility of styrene/VER/s-CSR and Epoxy/DDM/s-CSR systems
The nano-scale living CSR (i.e. s-CSR) used in this work is displayed in Table 30. The molecular polarity of vinyl ester (VE), epoxy/DDM resin (at ER = 1/1) and the shell of s-CSR was evaluated in terms of the calculated dipole moment per unit volume18,26, /V1/2 (Table 31). It was the highest for the epoxy/DDM resin, followed by VE, and the shell polymer of the CSR (i.e.
poly(styrene-co-acrylic acid)) (0.0663 vs. 0.0618 vs. 0.0432).
In general, the higher the polarity difference per unit volume between the resin matrix and the shell of CSR, the less compatibility for the St/VER/CSR and the Epoxy/DDM/CSR systems prior to reaction. Data in Table 31 reveal that the Epoxy/DDM/CSR system containing E7-15nm type of CSR would theoretically be less compatible (|‟EPR/DDM - ‟Shell of CSR| = 0.0231) than the St/VER/CSR system containing E7-15nm (|‟VER - ‟Shell of CSR| = 0.0186). This, however, cannot be verified by the static phase characteristics data for the uncured St/VER/CSR system at 25oC and
the Epoxy/DDM/CSR system at 80oC (Table 32), where the former system remained a single phase at 25oC within 24h, and so did the latter system at 80oC. Adding larger size of s-CSR, such as 60 nm and 100 nm in diameter, could result in a phase separation after the phase equilibrium for Epoxy/DDM/CSR systems at 80oC (Table 32).
During the cure of both St/VER/E7-15nm and Epoxy/DDM/E7-15nm systems, SEM micrographs (Figures 16-17) show that a two phase microstructure, consisting of a major continuous phase of styrene-crosslinked vinyl ester (or DDM-crosslinked epoxy) and a CSR-rich dispersed phase, would result. For the VER systems (Fig. 16), the microgel particles in the continuous phase can be clearly seen especially at SEM micrographs of 5000X magnification, which is due to the segregating effect41 of CSR on the microgel particles. In contrast, for the epoxy/DDM systems (Fig. 17), a flake-like continuous phase can only be observed, which would be due to the overlapping of the microgel particles caused by the lacking of segregating effect of CSR on microgels during the cure. Hence, the St/VER/E7-15nm system would be more compatible than that of the Epoxy/DDM/E7-15nm system during the cure, which exhibited the same trend as that of uncured systems as shown in Table 32.
For the St/VER/E7-15nm ternary systems (Fig. 16), the system containing 10 wt% CSR (Fig.
16(c) and 16(d)) would be more incompatible than that of 5 wt% CSR (Fig. 16(a) and 16(b)) as expected, whereas the trend was reversed for the Epoxy/DDM/E7-15nm ternary systems (Fig. 17).
For the Epoxy/DDM systems (Fig. 17), increasing the CSR concentration from 5wt% to 10wt%
would lead to an increase in viscosity for the ternary system and the concomitant slower rate of phase separation during the cure. As a result, a more compatibility of the 10% CSR system would arise . (cf. Fig. 17(a) and 17(c), or 17(b) and17(d))
TEM micrographs (Figures 18 and 19) are in good agreement with the observations from the SEM pictures. For the St/VER/E7-15nm systems (Fig. 18), judging from the CSR-rich dispersed phase, which was stained as dark color at micrographs of 10000X magnification, the 5% CSR system would be relatively compatible (Fig. 18(a) and (b)) due to the vague boundary between the continuous and dispersed phases, whereas the 10% CSR system was relatively incompatible due to the conspicuous boundary between the continuous and dispersed phases (Fig. 18(c) and (d)).
In contrast, for the Epoxy/DDM/E7-15nm systems (Fig. 19), the TEM micrograph for 5%
CSR system (Fig. 19(b)) somewhat resembled that of St/VER/10%CSR system (Fig. 19(d)), revealing that it was relatively incompatible as well. Increasing the CSR concentration from 5wt% (Fig. 19(a) and 19(b)) to 10wt% (Fig. 19(c) and 19(d)) could lead to a more compatibility of the ternary system, as evidenced by a much smaller domain of the CSR-rich phase . (cf. Fig. 19(b) and 19(d))
It should be noted that the degree of compatibility for the Epoxy/DDM/E7-15nm system during the cure, as revealed by the TEM picture (Fig. 19(d), is moderate, which is unlike those of Fig. 18(b) and Fig. 18(d), the former being somewhat too compatible and the latter somewhat too incompatible in terms of cured sample morphology.
For the cured CSR-containing VER and Epoxy/DDM systems with their characteristic morphologies as shown in Figs. 16-19, their mechanical behavior can be approximately represented by the Takayanagi models36-38, where arrays of weak LPA (R) and stiff styrene-crosslinked vinyl ester (or DDM-crosslinked epoxy) (P) phases are employed (see Fig. 4). For all of the St/VER/CSR and Epoxy/DDM/CSR systems, the two-phase microstructure can be represented by a P-(P-P-S) model (Fig. 4(b)).
3.2.2 Volume shrinkage for St/VER/s-CSR and Epoxy/DDM/s-CSR systems
The volume shrinkage of the neat VER was about 7.5%, while adding 5 or 10 wt % E7-15nm type of CSR is essentially unable to reduce the volume shrinkage (Table 33). Not enough or too much phase separation during the cure of St/VER/CSR systems would be unfavorable for the volume shrinkage control (cf Fig. 16, Fig. 18 and Table 33), where the 5% CSR system was too