Potential Energy Surface (PES)
It is essential to construct a potential energy surface (PES) in order to perform a classical trajectory simulation. The PES describes the potential energy (for ground electronic state Kr+Az) as a function of the coordinates of all atoms in the system. Thus, in principle, the PES is a function of 57 degrees of freedom. The most rigorous method to obtain a PES is to perform ab initio quantum chemistry calculations at a large set of nuclear geometries and then fit the energies to an analytical function. This approach been applied successfully to a number of three and four atom systems. For the present problem, such a first principles calculation is impractical. The large number of degrees of freedom make it impossible to compute energies at enough geometries (at any level of quantum chemistry) to construct the potential function. Thus, we adopt a more approximate method.
We decompose the PES into the sum of two parts: the intramolecular potential of Az, and the intermolecular potential between Kr and Az.
The intramolecular potential is built from semi-empirical terms based on local bond coordinates. The intermolecular potential is constructed based on ab initio calculations where the Az molecule is held fixed at its equilibrium geometry and thus only the relative position of the Kr-atom is modeled.
A. Intramolecular Azulene Potential
The intramolecular azulene potential23 is a sum terms representing bond stretching (r), bending (θ), torsion (φ), and wagging contributions (ω) with the respective force constants kr, kθ, kφ, and kω and equilibrium values r0, θ0, and ω0.
∑ −
∑ +
∑ +
∑ +
=
Φ 2
0) ( k 2 ] 1 ) cos(n -k
| k [|
2 2 1 ) 0 - ( k 2 2 1 ) 0 r - r(r k 2 1 Azu
ω ω ω φ φ
θ φ θ θ
(All contributions are harmonic in internal coordinates except for those describing dihedral angles.) Although most terms in this expression appear harmonic, it should be noted that this expression is anharmonic in the normal mode coordinates and so intramolecular energy transfer occurs readily using this potential.
The intramolecular potential is based on local contribution in bond coordinates. It is built from optimized contribution from the bond stretching, bending, wagging, and torsion motion within the molecule.
Each component has its own parameters, such as natural bond lengths, natural bond angles, and force constants. These parameters were taken from Heidelbach and Schroeder23 and can be seen in Table2. Each component is described as follows:
Contributions from bond stretches:
)2 0 r - r(r k 2 1∑
= U(r)
kr : the force constant in kcal-mol-1/A0 2
r0 : the natural bond length inA0
Contributions from bendings:
)2 0 - ( k 2 ) 1
( θ θ
θ = ∑ θ U
kθ : the force constant in kcal-mol-1/deg2 θ0 : the natural bond angle in degree
Contributions from torsions:
] ) cos(n -k
| k [|
2 ) 1
( φ
φ φ = ∑ φ
U
kψ : the force constant in kcal/mol n : periodicity
Contributions from wags:
∑ −
= 2
0) ( k 2 ) 1
( ω ω
ω ω U
kω : the force constant in kcal-mol-1/deg2 ω - ω0 : the out of plane angle in degree
A reasonable test of the intramolecular potential is provided by a normal mode analysis. Using the PES, the normal mode frequencies are computed. These frequencies can then be compared to experimental spectroscopic measurements of the transition energies of the fundamentals. As seen in Table 3, the PES provides a reasonably accurate model of the observed transition frequencies.
We are thus encouraged to believe that this model may be adequate to describe the intramolecular dynamics.
Table2. Force constants of the intramolecular azulene potential
a. Equilibrium bond lengths are given in Figure 2.
b. The phase and periodicity are 180.0 and 2, respectively.
Figure2. C2v structure of azulene. Black numbers are the respective bond lengths. Blue numbers are the respective width and length of azulene. Brown numbers are the distances form the top hydrogen and the lowest hydrogen, respectively, to the center of mass (COM) of azulene. The distance unit is A0 .
1 189 116 17 900 981 33 1378 1398
2 240 133 18 911 1016 34 1396 1419
3 304 225 19 941 1029 35 1443 1479
4 323 275 20 952 1041 36 1448 1496
5 331 330 21 965 1060 37 1457 1527
6 406 395 22 971 1067 38 1480 1578
7 486 407 23 987 1128 39 1536 1598
8 542 434 24 1012 1129 40 1579 1641
9 562 515 25 1049 1174 41 2968 3027
10 680 563 26 1058 1181 42 3018 3028
11 712 582 27 1117 1224 43 3037 3030
12 731 745 28 1160 1233 44 3037 3034
13 762 759 29 1210 1296 45 3042 3035
14 795 785 30 1216 1296 46 3072 3035
15 813 866 31 1268 1301 47 3077 3035
16 825 967 32 1300 1340 48 3098 3038
Table3. Comparison of vibrational frequencies a normal-mode analysis with experimental frequencies24 in cm-1.
B. Intermolecular Potential between Kr and Azulene
The intermolecular potential is a fit of an analytical function to ab initio energies computed at many geometries. Ab Initio calculations provide the Born-Oppenheimer potential from a quantum mechanical calculation of the ground state electronic energy without inclusion of experimental data. Usually, significant approximations are required to make the quantum calculations practical.
The ab initio calculation method used in this study is MP2,25-27 which is the Møller-Plesset perturbation theory to second order. The basis set used is 6-31G**.28-37 This method takes into account the electron correlation, by Rayleigh-Schrodinger perturbation theory.38
The ab initio calculations were done keeping azulene in its equilibrium planar structure and varying the position of the krypton atom. The ab initio calculations were performed for 1074 geometries (830 geometries where Kr is above or under the plane of azulene. 244 geometries where Kr and azulene lie on the same plane.). The ab initio calculations were done using Gaussian03 program.39 After getting the data points, I represent the PES by fitting the data points to a “model function” that depends on adjustable parameters. The model function used for the intramolecular potential is a sum of pair-wise Kr-C and Kr-H potentials.
The pair-wise potential has the following form:
exp 8
r (-Br) C A
V(r)= +
where A, B, C are adjustable parameters. The first term describes a repulsive potential and the second describes an attractive one. The Kr-C potential is:
exp 8
r Cc cr)
c (-B A KrC(r)
V = +
and the Kr-H potential is:
exp 8
r r) C (-B A
KrH(r)
V = H H + H
The intramolecular azulene potential is a multidimensional function and consists of eighteen variables: ten different Kr-C distances (r KrC 1, r KrC 2 …r KrC 10) and eight different Kr-H distances (r KrH 1, r KrH 2 …r KrH 8).
The intramolecular azulene potential is :
∑=
∑ +
= = ⎟
⎠
⎜ ⎞
⎝
⎟ ⎛
⎠
⎜ ⎞
⎝
⎟ ⎛
⎠
⎜ ⎞
⎝
⎛ 8
1 10
8 1 , 1
, 10
1 i
iKrH KrH r
i V
iKrC KrC r
KrH V KrH r
KrC r KrC r
r
V K K
The intermolecular potential between Kr and azulene depends linearly on the parameters (AC, CC, AH, and CH) and nonlinearly on the parameters (BC and BH). The parameters were determined using least squares fitting procedure. The usual Linear Least Squares method can only be used for functions which depend linearly on the parameter to be fitted. However, the present intermolecular potential has non-linear parameters. On other hand Levenberg-Marquardt Method, for nonlinear fitting, needs a good initial guess for each parameter;
unfortunately, it is not easy to come up with a good initial guess.
Therefore, the combination of the two kinds of methods is necessary.
First, using General Linear Least Squares method with given BC and BH we obtain the four linear parameters: AC, CC, AH, and CH, then using AC, CC, AH, and CH fitted by using General Linear Least Squares method and the given BC and BH as an initial guess we perform Levenberg-Marquardt Method. Different sets of given BC and BH, were done to get the best fit parameters.
The best fits are as follows:
r . -
r) .
e(- . KrC(r)
V 8
2611 40359 26984330
895 3
142011 +
=
8 87564 1893
77108368 9364 3
33998
r. - r) .
e(- .
KrH(r)
V = +
AC: 142011.895 (kcal/mol) BC: 3.26984330 (A0 -1)
CC: -40359.2611 (kcal/mol-A0 -8) AH: 33998.9364 (kcal/mol) BH: 3.77108368 (A0 -1)
CH: -1893.87564 (kcal/mol-A0 -8)
The intermolecular potential between Kr and azulene is then obtained by summing over all the Kr-H and Kr-C pairs in the system:
( ) ( ) ( )
) ri
. i
ri e .
. (
) ri
. i
ri e .
. (
i
iKrH KrH r
i V
iKrC KrC r KrH V
KrH r KrC ,r
r KrC , r V
8 87564 8 1893
1
77108368 9364 3
33998
8 2611 40359 10
1
26984330 895 3
142011
8 1 10
8 1 1
10 1
∑ −
= + −
∑ −
=
= −
∑=
∑ +
= = K
K
Ab initio calculations can be very time-consuming, especially when done using a high level theory with a large basis. The present MP2 calculation is not a particularly high level of ab initio calculation, but it is believed that the results are sufficiently accurate to obtain meaningful results for this system. In particular, we believe that qualitative aspect of the collision dynamics will depend on the gross features of the PES and will not be overly sensitive to the fine points of the potential. Thus, the intermolecular potential was obtained by fitting 1074 geometries of an MP2/6-31G** calculation to a multidimensional function.
C. Checking the Fit
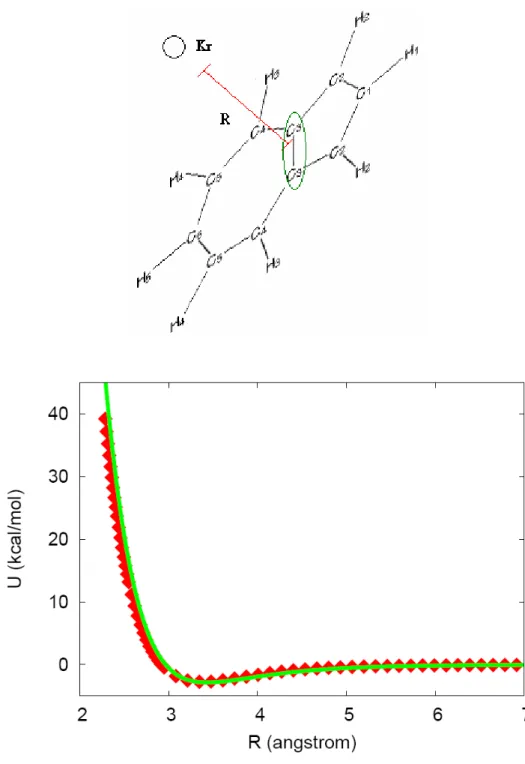
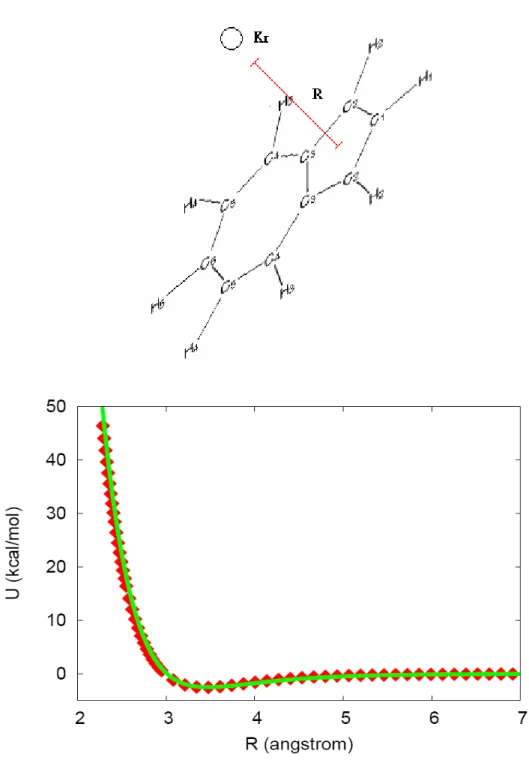
A plot of our fitted potential and the ab initio points are given in Figures 3-8. In the first three figures, we give those in which the Kr is in the plane of Ar, and in the latter three we present those in which the Kr is out of the plane of Ar. We see that the out of plane potential values are fitted with high accuracy, however, those for the in plane potential are somewhat poorer. However, in the trajectory calculations, the chance of Kr hitting azulene from a direction normal to the molecular plane is much higher than that of Kr hitting the azulene with in-plane.
Thus, the fit is good in the most important regions.
The qualitative character of the potential consists of a fairly deep van der Waals well centered above the molecular plane near the carbon rings. The well depth is approximately 4 kcal/mol. The potential for geometries with the Kr close to the Az molecule is dominated by a steep repulsive wall when the Kr-atom closely approach any atom in the Az-molecule. Dynamically, the vdW-well will steer the long range motion of an approaching Kr-atom and tend to align the Kr in the normal plane to Az.
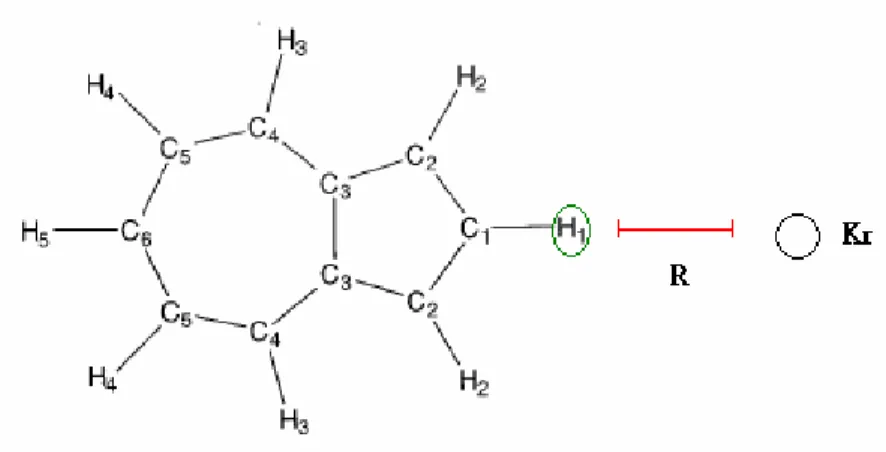
Figure 3. Comparison of the analytical PES to the ab initio data points.
R is the distance from Kr to the circled H-atom. The squares and the line: ab initio points and the fitted intermolecular potential function, respectively.
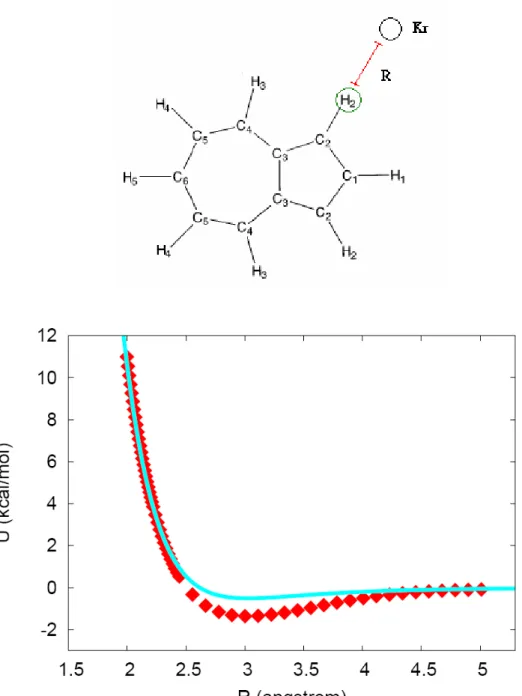
Figure 4. Comparison of the analytical PES to the ab initio data points.
R is the distance from Kr to the circled H-atom. The squares and the line: ab initio points and the fitted intermolecular potential function, respectively.
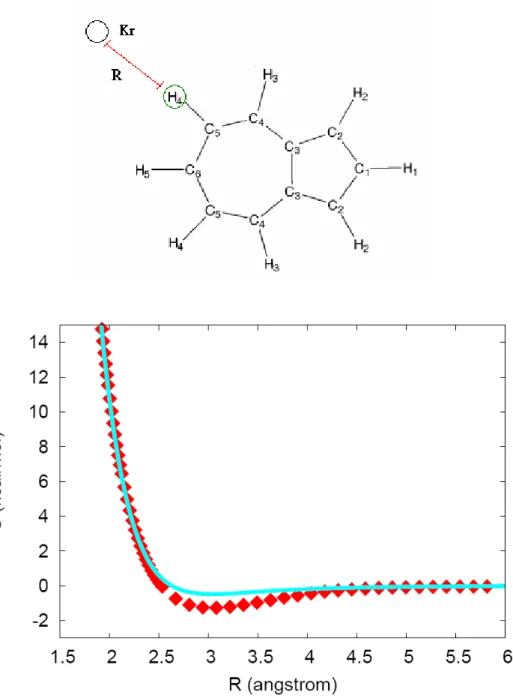
Figure 5. Comparison of the analytical PES to the ab initio data points.
R is the distance from Kr to the circled H-atom. The squares and the line: ab initio points and the fitted intermolecular potential function, respectively.
Figure 6. Comparison of the analytical PES to the ab initio data points.
R is the distance from Kr to the middle of 7 member ring. The squares and the line: ab initio points and the fitted intermolecular potential
Figure 7. Comparison of the analytical PES to the ab initio data points.
R is the distance from Kr to the center of C-C bond circled. The squares and the line: ab initio points and the fitted intermolecular
Figure 8. Comparison of the analytical PES to the ab initio data points.
R is the distance from Kr to the middle of 5 member ring. The squares and the line: ab initio points and the fitted intermolecular potential function, respectively.