國立臺灣大學工學院高分子科學與工程學研究所 碩士論文
Institute of Polymer Science and Engineering College of Engineering
National Taiwan University Master Thesis
探討含有三噻吩的共軛側鏈其分子結構以及二維延伸共軛 性質對於聚噻吩高分子之紫外/可見光吸收光譜的影響 Effect of Terthiophene-Based Conjugated Side-Chain Architecture
on the Two-Dimensional Extended Conjugation Properties of the UV Absorption Spectra of Polythiophenes
楊育雯 Yu-Wen Yang
指導教授:黃慶怡 博士 Advisor: Ching-I Huang, Ph.D.
中華民國 103 年 8 月
August, 2014
i
ii
致謝 致謝致謝 致謝
能完成本篇研究,我要特別感謝我的指導教授黃慶怡老師,從老師的身上讓 我學到了做研究時所應抱持的熱忱與積極的態度,也讓我了解到不同於以往在做 合成的實驗室裡偏向注重最後的結果呈現,在理論計算的部分更要細心去觀察每 一步的流程。同時感謝口試委員王立義老師對於本論文的指導,提供寶貴的意見,
在此對於口試委員至上由衷的謝意。
另一方面,我也要感謝實驗室每一位成員對我的支持與鼓勵。喻傑學長總會 在一旁幫我加油打氣,讓我有動力繼續堅持下去;從錦文學長身上讓我了解目前 最新穎的電腦硬體設備以及軟體資源的潮流,幫我解決在研究上所面臨的問題;
而榆臻學姊一直是我的依靠,除了在平日裡時常關心我的近況並與我分享日常生 活中的趣事之外,在研究上更給予我莫大的幫助;軒宏學長總會在我心情低落時,
給予我安慰與鼓勵,特別是學長的笑容總能帶給人溫暖的感受;宜康學長與子中 學長則在研究上給予我相當多的技術指導,使我的研究得以順利的進行;我的好 鄰居易緯總會透過各種方式紓緩我的緊張情緒,而他積極正向的態度是值得我要 向他學習的;柏江常在我心情低靡時,與我分享親身的經歷,常有一語驚醒夢中 人的感覺,而在軟體或電腦使用上遇到困難,他也都不吝惜幫我解決;當然還有 實驗室裡的學弟妹何穎、文皓、于璇、威霆與偉豪,因為有你們常常在一旁陪著 我、逗我笑,讓我很快又能振作起來繼續奮鬥。
最後,我要感謝我的家人,他們總是在背後默默的給予我最大的支持,也常 以話語來開導我、鼓勵我,讓我得到很多心靈上的幫助。我也感謝身邊曾經幫助 過我的老師、學長姐、親朋好友等,因為有你們,讓我的研究以及人生都更加的 完整,真的很感謝大家。
iii
中文摘要 中文摘要中文摘要 中文摘要
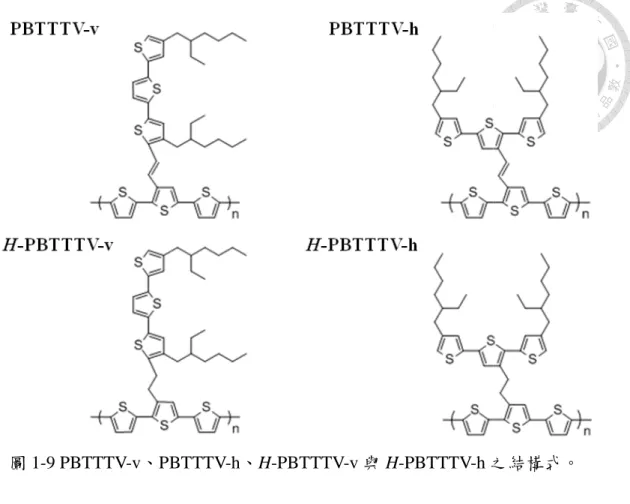
本研究主要可分成兩個部分來進行:首先,我們利用全原子分子動力學(AAMD) 模擬單一條高分子鏈在溶液無序狀態下之分子構形,並探討溶劑效應對於分子構 形之影響;接著,再利用量子力學法來探討聚噻吩系列之共軛高分子其分子構形 與紫外/可見光吸收光譜之關聯性。我們選擇以三噻吩為主軸的四個高分子材料 PBTTTV-v、PBTTTV-h、H-PBTTTV-v 與 H-PBTTTV-h 作為研究對象,藉此來探 討平行或垂直主鏈之側鏈位向以及銜接處為乙烯基(Vinyl)或三個單鍵對於紫外/可 見光吸收光譜的影響。
藉由觀察全原子分子動力學所得到在溶液無序狀態下之高分子構形,可以得 知 PBTTTV-v、PBTTTV-h、H-PBTTTV-v 與 H-PBTTTV-h 四個材料因受到銜接處 上碳原子之立體阻礙的影響,使得其不論是在主鏈上的 τMain chain_A與側鏈上的 τSide
chain_B皆擁有較廣的扭轉角度分布。比較特別的是,在銜接處的扭轉角度分析中,
又以單鍵與單鍵之間的運動較為自由,其幾乎是呈現偏離共平面的 90 度(270 度) 附近,而引入乙烯基(Vinyl)能使其維持良好的共平面性(180 度)。從這些結果中,
能看到溶劑效應對於分子構形之影響。
於紫外/可見光吸收光譜的結果中,觀察 PBTTTV-v 與 PBTTTV-h 之吸收光譜,
可發現到其圖譜變的較為複雜,透過人工調控角度的測試,會得知其中存在著二 維方向的延伸共軛,而於軌域分布中也看到電子雲分布會由主鏈延續到側鏈上。
我們從中擷取片段以不同比例的方式進行組合,並與兩組不同聚合度之實驗結果 相互比較,可以得知與實驗結果相符合。將銜接處改以三個單鍵後,可以發現到 不同側鏈位向的 H-PBTTTV-v(垂直)與 H-PBTTTV-h(平行)皆有很相似的吸收光譜 範圍,且其吸收光譜主要是受到主鏈與側鏈兩種片段結構所提供之貢獻所影響,
而從其軌域分布中,也能得到良好的印證結果。在未來,我們將修正其中的扭轉 角度,改從分子動力學達動態平衡後,擷取最大訊號峰值,再以量子力學法計算 吸收光譜,從中探討並比較兩種不同之分子構形與紫外/可見光吸收光譜的變化。
iv
Abstract
We employed all-atom molecular dynamics (AAMD) simulation and the quantum
mechanical (QM) methods to explore the correlation between the molecular
conformation and the UV absorption behaviors of terthiophene-based conjugated
polymers. The four model systems examined here are PBTTTV-v, PBTTTV-h,
H-PBTTTV-v and H-PBTTTV-h. We focus on the effects of side-chain architecture on
the two-dimensional extended conjugation properties of the UV absorption spectra of
polythiophenes.
First, we observe the molecular conformation displaying disordered state of
polymer chains within dilute solution via AAMD. In the PBTTTV-v, PBTTTV-h,
H-PBTTTV-v and H-PBTTTV-h molecules, due to a large steric hindrance from the
linkage between main chain and side chain, they have broader torsional angle
distribution of main chain (τMain chain_A) and side chain (τSide chain_B). Because of the free
motion of single bonds, the single bond linkages between main chain and side chain
don’t intend to preserve a coplanar conformation as the H-PBTTTV-v and H-PBTTTV-h
molecules. In contrast, as for the PBTTTV-v and PBTTTV-h polymers with vinyl
linkage tend to keep a coplanar conformation.
Then, we explore the UV absorption spectra of PBTTTV-v and PBTTTV-h
molecules via QM. We notice that PBTTTV-v and PBTTTV-h have the two-dimensional
v
extended conjugation properties in the UV absorption spectra, which make the UV
absorption spectra more complicated. On the other hand, we observe the molecular
orbital and discover the electron cloud may scatter along the main chain to side chain.
As a result, we constitute several segments from the molecular orbitals by adjusting
each segment’s fraction. The resultant UV absorption spectra of PBTTTV-v and
PBTTTV-h show good match to the experimental results. When the single bonds are
substituted for the vinyl linkages, the UV absorption spectra of H-PBTTTV-v and
H-PBTTTV-h dimer show two peaks in the same range. The two absorption peaks are
from either the main chain or the side chain. In the future, we will modify the torsional
angle of molecular conformation within AAMD. We will systematically explore the
correlation between molecular conformation and optoelectronic properties of
PBTTTV-v, PBTTTV-h, H-PBTTTV-v and H-PBTTTV-h.
vi
目 目目 目 錄錄錄 錄
口試委員會審定書...i
致謝...ii
中文摘要...iii
Abstract...iv
目錄...vi
圖目錄...viii
表目錄...xi
第一章 前言...1
第二章 模擬方法...16
2-1 全原子分子動力學(All-atom Molecular Dynamics)模擬...17
2-1-1 力場選擇...17
2-1-2 模擬系統介紹與初始架構...18
2-1-3 模擬流程...18
2-2 量子力學計算...19
2-2-1 密度泛函理論(Density Functional Theory, DFT)與時間依賴-密度泛 函理論(Time Dependent-Density Functional Theory, TD-DFT)...19
2-2-2 相關條件測試與計算流程...19
第三章 結果與討論...21
3-1 利用分子動力學與量子力學法模擬探討 PBTTTV-v 之分子構形及其對於紫 外/可見光吸收光譜的影響...21
3-2 利用分子動力學與量子力學法模擬探討 PBTTTV-h 之分子構形及其對於紫 外/可見光吸收光譜的影響...24
3-3 利用分子動力學與量子力學法模擬探討 H-PBTTTV-v 之分子構形及其對於 紫外/可見光吸收光譜的影響...27
vii
3-4 利用分子動力學與量子力學法模擬探討 H-PBTTTV-h 之分子構形及其對於 紫外/可見光吸收光譜的影響...29 3-5 探討以分子動力學與量子力學模擬之分子構形及其對於紫外/可見光吸收 光譜的影響...31 第四章 結論…...57 參考文獻...59
viii
圖目錄 圖目錄圖目錄 圖目錄
圖 1-1 於不同位置規整度下(a)HT 達 97%與(b)HT 達 64% P3HT 在氯仿溶液下之紫 外/可見光吸收光譜[19]。...7 圖 1-2 (a)烷基取代型之單體結構與聚合後之高分子材料於(b)氯仿溶液中及(c)成膜
後之紫外/可見光吸收光譜[31]。...8 圖 1-3 原子取代型之高分子結構與其在成膜後之紫外/可見光吸收光譜[32]。...9 圖 1-4 PBDTTT-S 與 PBDTDTTT-S-T 於薄膜下之紫外/可見光吸收光譜[43]。...10 圖 1-5 含有純聚噻吩系統之衍生物(a)其結構式及其在(b)氯仿溶液中和(c)薄膜之紫 外/可見光吸收光譜[40]。...11 圖 1-6 含有苯乙烯基三苯胺官能基共軛側鏈之 PT5TPA 二維共軛高分子結構與其
紫外/可見光吸收光譜[49]。...12 圖 1-7 (a)不同共軛側鏈接枝位向之二維共軛高分子結構其(b)在成膜升溫後與氯仿
溶液下之紫外/可見光吸收光譜結果,並經由模擬進行其在(c)堆疊狀態和單 一條鏈於真空下之主鏈扭轉角度分布分析[51]。...13 圖 1-8 聚噻吩系統之衍生物(a)其結構式與 P3HT、PDBT 於良溶劑中(b)高分子鏈之 蜷曲構形及其(c)主鏈上之扭轉角度分布[52]。...14 圖 1-9 PBTTTV-v、PBTTTV-h、H-PBTTTV-v 與 H-PBTTTV-h 之結構式。...15 圖 2-1 (a)為單體至三聚體之有/無甲基取代示意圖與其(b)所對應之吸收光譜。...20 圖 3-1 在氯苯中無序狀態下(a) PBTTTV-v、(b) PBTTTV-h、(c) H-PBTTTV-v 與
(d) H-PBTTTV-h 於 300K 經 300ps 動態平衡後之分子鏈構形。...40 圖 3-2 (a) PBTTTV-v、(b) PBTTTV-h、(c) H-PBTTTV-v 與(d) H-PBTTTV-h 之相關
扭轉角度對應示意圖。...41 圖 3-3 PBTTTV-v 在主鏈上之(a)A 處(b)B 處與(c)C 處、側鏈上之(d)A 處與(e)B 處
與(f)銜接處的扭轉角度分布圖。...42 圖 3-4 人為調控 PBTTTV-v 主鏈上之右側扭轉角度(τMain chain_B)其分子構形示意圖
ix
與其紫外/可見光吸收光譜。...43 圖 3-5 人為調控 PBTTTV-v 主鏈上之左側扭轉角度(τMain chain_A)其分子構形示意圖
與其紫外/可見光吸收光譜。...44 圖 3-6 PBTTTV-v 二聚體中各個拆解片段之(a)分子構形與其計算之(b)紫外/可見光
吸收光譜。...45 圖 3-7 (a)PBTTTV-v 與(b)PBTTTV-h 在氯苯溶劑下其不同聚合度之紫外/可見光吸
收光譜。...46 圖 3-8 不同比例下之 PBTTTV-v 各片段於組合後之紫外/可見光吸收光譜並與實驗 之低分子量結果比較。...47 圖 3-9 不同比例下之 PBTTTV-v 各片段於組合後之紫外/可見光吸收光譜並與實驗 之高分子量結果比較。...47 圖 3-10 擷取分子動力學之(a) PBTTTV-v 二聚體、(b) PBTTTV-h 三聚體、
(c) H-PBTTTV-v 二聚體與(d) H-PBTTTV-h 二聚體之分子構形。...48 圖 3-11 PBTTTV-h 在主鏈上之(a)A 處(b)B 處與(c)C 處、側鏈上之(d)A 處與(e)B 處 與(f)銜接處的扭轉角度分布圖。...49 圖 3-12 人為調控 PBTTTV-h 之扭轉角度其分子構形示意圖與其紫外/可見光吸收
光譜。...50 圖 3-13 PBTTTV-h 三聚體中各個拆解片段之(a)分子構形與其計算之(b)紫外/可見光 吸收光譜。……...51 圖 3-14 不同比例下之 PBTTTV-h 各片段於組合後之紫外/可見光吸收光譜並與實
驗之低分子量結果比較。...52 圖 3-15 不同比例下之 PBTTTV-h 各片段於組合後之紫外/可見光吸收光譜並與實
驗之高分子量結果比較。...52 圖 3-16 H-PBTTTV-v 在主鏈上之(a)A 處(b)B 處與(c)C 處、側鏈上之(d)A 處與(e)B
處與(f)銜接處的扭轉角度分布圖。...53
x
圖 3-17 人為調控 H-PBTTTV-v 主鏈與側鏈間銜接處之扭轉角度(τsingle bond)並計算 其紫外/可見光吸收光譜。...54 圖 3-18 H-PBTTTV-h 在主鏈上之(a)A 處(b)B 處與(c)C 處、側鏈上之(d)A 處與(e)B
處與(f)銜接處的扭轉角度分布圖。...55 圖 3-19 人為調控 H-PBTTTV-h 主鏈與側鏈間銜接處之扭轉角度(τsingle bond)並計算
其紫外/可見光吸收光譜。...56
xi
表目錄 表目錄表目錄 表目錄
表 3-1 PBTTTV-v、PBTTTV-h、H-PBTTTV-v 與 H-PBTTTV-h 之主鏈、側鏈與銜接
處之扭轉角度分布。...33
表 3-2 PBTTTV-v 二聚體之相關軌域分布數據。...34
表 3-3 PBTTTV-v 與 PBTTTV-h 之實驗相關數據。...35
表 3-4 PBTTTV-v 在低分子量時之各片段於不同比例下之組合數據。...35
表 3-5 PBTTTV-v 在高分子量時之各片段於不同比例下之組合數據。...35
表 3-6 PBTTTV-h 三聚體之相關軌域分布數據。...36
表 3-7 PBTTTV-h 在低分子量時之各片段於不同比例下之組合數據。...37
表 3-8 PBTTTV-h 在高分子量時之各片段於不同比例下之組合數據。...37
表 3-9 H-PBTTTV-v 二聚體之相關軌域分布數據。...38
表 3-10 H-PBTTTV-h 二聚體之相關軌域分布數據。...39
1
第一章 第一章 第一章第一章 前言前言前言前言
於現今太陽能電池領域中,依據材料主要可分成三大類,分別為矽類(Silicon)、
化合物(Compound)與有機材料(Organic materials)。其中矽類太陽能電池雖為目前研 究之主流,但由於其製備過程較為複雜且合成環境之要求也較為嚴苛,促使製備 成本也隨之提高;而在化合物半導體中,則是利用 III-V 族與 II-VI 族之稀有元素 與其他過渡金屬,因而有高成本與環境汙染的問題產生。但在有機半導體中幾乎 不會遇到上面所提到之困難,其中又以高分子太陽能電池最受矚目,由於其材料 擁有較佳之可塑性、柔軟性、低成本及製備簡單等優點,使得有機高分子太陽電 池成為外界研究與開發之新一代再生能源主流[1-7]。
於高分子太陽能電池中,共軛高分子材料常作為電子予體(Electron donor),因 而必須擁有較佳的吸光能力並產生激子(Excitons),以進行後續的電荷轉移及分離 的過程[8]。除了透過基本的光電性質來讓非定域化(Delocalized)的 π 電子可以在高 分子鏈上移動外,重要的是還必須擁有低能隙及較寬廣之吸收光譜範圍的兩個特 性。當共軛高分子材料具備有低能隙之特性時,由於其電荷被激發所需的能量較 低,電荷容易從最高佔據分子軌域(Highest occupied molecular orbital, HOMO)躍遷 至最低未佔據分子軌域(Lowest unoccupied molecular orbital, LUMO),導致吸收峰 大多呈現於長波長區,而在短波長區的吸收略顯不足,進而影響其整體吸收光譜 範圍不夠廣泛[9],限制了可利用之太陽光的範圍。因此,如何增加共軛高分子對 短波長區的太陽光吸收與提升整體之吸收光譜範圍成為非常重要的課題。
在諸多的共軛高分子材料中,以聚噻吩類(Polythiophenes, PTs)因具有高載子遷 移率及較低之能隙[10-14],而成為目前研究主流。此外,聚噻吩類的衍生物 (Poly(3-alkylthiophenes), P3ATs)又因溶解度佳、環境之穩定性及良好的光電性質,
而被廣泛的研究與探討。在實驗上有許多研究指出此一類之聚噻吩衍生物(P3ATs) 在吸收光譜上的吸光能力深受分子之位置選擇性(Regioregularities)[15-19]、分子量 (Molecular weight)[20-24]與加工條件(Processing conditions)[25-26]的影響。其中,
2
Bout 等人[19]探討 P3HT 在不同的頭接尾(Head-to-tail, HT)與頭接頭(Head-to-head, HH)位置下對於紫外/可見光吸收光譜的影響中,如圖 1-1(a)與(b)所示,指出當 P3HT 在氯仿(Chloroform, CHCl3)稀薄溶液狀態下,其 HT 達 64 %下所呈現之最大吸收峰 值為 420 nm,吸收範圍為 300 nm~550 nm;而當 HT 達 97%時,最大吸收峰值略 為增加至 454 nm,吸收範圍亦些微增加至 300 nm~570 nm。從這些實驗結果中,
我們可以觀察到當分子鏈趨近於 HT 的位置時,不僅能使最大吸收峰值呈現紅位移 現象,也有效提升了整體吸收光譜之吸收範圍。但不可忽略的是,在溶液無序狀 態下,由於分子鏈間缺少 π-π 作用力,使得分子鏈不易維持共平面性。我們從 Inganäs 等人[25]的研究中,也提到了溶劑效應對於高分子構形的影響,說明在氯 仿(Chloroform, CHCl3)良溶劑(Good solvent)中,高分子鏈運動較為自由,因而能夠 膨潤開來,使高分子鏈易形成較為蜷曲的樣貌,整體的共平面性不易被維持住,
縮短了有效共軛長度,進而減少吸收光譜的吸收範圍。由此可見,溶劑效應對於 高分子鏈之構形及其所呈現之紫外/可見光吸收光譜是有一定的影響程度。如何有 效增加可見光的吸收範圍,是許多研究學者正在設法改良的課題,因而致力投入 於開發設計新穎共軛高分子材料。
誠如上述所述,為了能有效增加吸收可見光的範圍,有學者開始致力於改良 側鏈官能基,大致上可分為烷基取代型[27-32]與原子取代型[32-33]兩大類。以 Segalman 等 人 [31] 為 例 , 他 們 於 純 聚 噻 吩 系 統 之 3 號 位 置 分 別 以 乙 基 己 基 (Ethylhexyl, EH)、己基(Hexyl, H)與十二烷基(Dodecyl, DD)進行取代後,其單體結 構如圖 1-2(a)所示,再將之聚合成 P3EHT、P3HT 與 P3DDT,從吸收光譜的結果 中可觀察到,經由烷基官能基取代之高分子對於吸收光譜確實有一定程度之影響。
在氯仿(CHCl3)溶液中,含有直鏈型烷基取代之 P3HT 與 P3DDT 皆有較紅位移的現 象產生,其最大吸收峰值為 450 nm,吸收範圍約在 350 nm~550 nm,而於分支型 的烷基 P3EHT 中,其最大吸收峰值則些微藍位移至 440 nm;而在成膜下,可以明 顯看到三者皆有紅位移的現象產生,且其不再僅有一個吸收峰,於最大吸收峰旁
3
有鄰近的小峰產生,而整體之吸收範圍也明顯增加至 350 nm~650 nm,如圖 1-2(b) 與(c)所示;而在 Jo 等人[32]的研究中,以含有拉電子性質之氟原子取代噻吩上之 氫原子,可以發現到於成膜下以氟原子取代之 F12 與 FEH 與原氫原子之 H12 與 HEH 相比,其最大吸收峰增加至 598nm,且整體吸收範圍為 350 nm~650 nm,如 圖 1-3 所示。由此可知,利用烷基型或原子型官能基取代之方式來修飾可見光的吸 收範圍是可行的,但是對於吸收短波長區域的可見光仍然相當有限。
因此,開始有學者嘗試將含有共軛性質之側鏈引入聚噻吩中,形成二維共軛 高分子,期望能夠延長有效共軛長度,並增加在吸收光譜上的吸收[34-45]。其中 如圖 1-4 所示,Hou 等人[43]的研究中合成一維方向共軛之 PBDTTT-S 與二維方向 延伸之 PBDTDTTT-S-T,在吸收光譜中僅呈現單一且較寬廣之吸收峰,其整體吸 收範圍可達到 300 nm~800nm。而從 Li 等人[40]合成純聚噻吩系統之衍生物,其結 構式如圖 1-5(a)所示。藉由吸收光譜的結果呈現可以看到,不論是在成膜後的有序 排 列 下 或 是 在 無 序 溶 液 中 , 都 可 以 看 到 在 一 維 共 軛 高 分 子 之 Poly(3-hexyl-2,2’-bithiophene) (P1)僅呈現單一的吸收峰,相較於在側鏈上引入乙烯 基之二維共軛高分子 Poly(3-[2-(5-hexyl-2-thienyl)ethenyl]-2,2’-bithiophene) (P2)所 呈現出的兩個吸收峰,說明在接枝共軛側鏈後,能明顯看到在短波長區域對於可 見光的吸收幅度大為增強,如圖 1-5(b)與(c)所示。除此之外,Li 等人亦於含有乙 烯基連接之共軛側鏈上做了許多官能基的改良與修飾,像是噻吩乙烯(Thienylene vinylene)[46-47] 、 吩 噻 乙 烯 (Phenothiazine vinylene)[48] 與 苯 乙 烯 基 三 苯 胺 (Styryl-triphenylamine)[49]等。如圖 1-6 所示,從 Li 等人[49]合成含有苯乙烯基三 苯胺官能基共軛側鏈之 PT5TPA,從其紫外/可見光吸收光譜可以看到,不論是在 短波長區或長波長區,整體的吸光強度明顯有增加的趨勢,其最大吸收峰值為 555 nm,而整體吸收範圍為 300 nm~650 nm。由這些實驗的結果中,我們可以說明乙 烯基(Vinyl)連接在二維共軛高分子主鏈與共軛側鏈間,能夠增加高分子主鏈上之共 平面性[50],以延續共軛的延伸性,也因此導致擁有較寬廣的吸收範圍。另一方面,
4
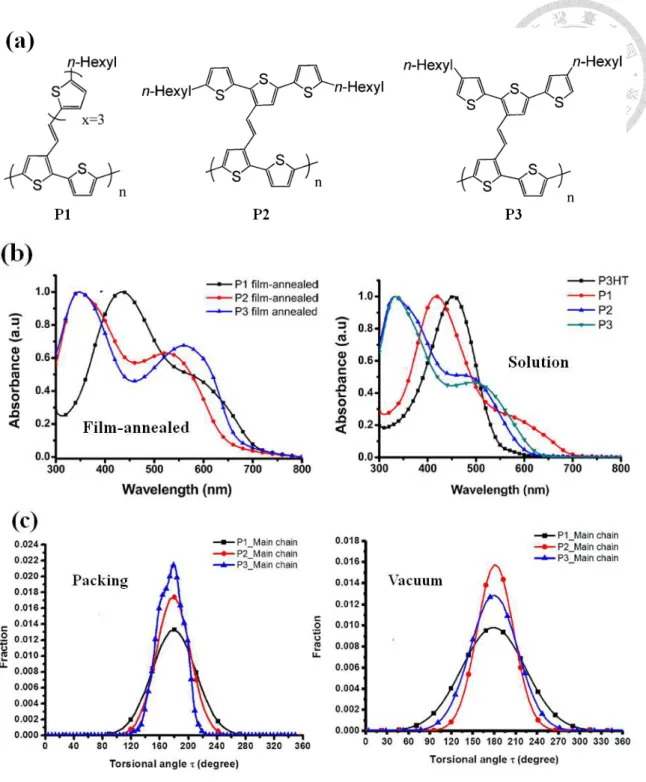
比較特別的是,在 Wang 等人[51]合成一系列不同共軛側鏈接枝位向分別為垂直或 平 行 於 主 鏈 上 之 二 維 共 軛 高 分 子 , 其 結 構 式 如 圖 1-7(a) 所 示 。 可 以 發 現 Poly{3-(5”-hexyl-2,2’:5’,2”-terthiophenyl-5-vinyl)thiophene-alt-thiophene} (P1) 、 Poly{3-(5,5”-dihexyl-2,2’:5’,2”-terthiophenyl-3’-vinyl)thiophene-alt-thiophene} (P2)及 Poly{3-(4,4”-dihexyl-2,2:5’,2”-terthiophene-3’-vinyl)thiophene-alt-thiophene} (P3) 其 在成膜升溫狀態下,最大吸收峰值分別為 431 nm、346 nm 及 344 nm,三者之吸收 範圍皆約為 300 nm~700 nm;但在溶液無序狀態下則有些微藍位移的現象產生,P1、
P2 及 P3 最大吸收峰值分別降至 417 nm、332 nm 及 333 nm,整體的吸收範圍依序 為 300 nm~700 nm、300 nm~600 nm 與 300 nm~650 nm,其紫外/吸收光譜結果如圖 1-7(b)所示。從實驗的結果中,可以觀察到不同接枝位向之共軛側鏈會使紫外/可見 光吸收光譜呈現不一樣的樣貌,由此現象也更加說明了紫外/可見光吸收光譜將不 再是僅由單純的主鏈與側鏈兩者所貢獻,其主鏈與側鏈之間的共軛延續性與否,
將是值得我們深入去探討的重點之一。
誠如先前所述,二維共軛高分子在無序溶液狀態下之分子鏈構形深受共軛側 鏈在二維方向上的效應所影響,且與紫外/可見光吸收光譜有著複雜且密不可分的 關係。但由於在實驗上僅能觀察到高分子在巨觀下的行為表現,其細部分子鏈的 構形卻往往不得而知,因而凸顯了理論模擬研究的重要性。然而,值得注意的是,
目前文獻上對於共軛高分子在無序溶液狀態下之構形與光電性質之理論模擬相關 研究上,尚未有完整的探討。於 Wang[51]等人的研究成果中,初步模擬單一條高 分子鏈在真空的狀態下其分子鏈構形之變化,間接說明實驗上高分子鏈在溶劑中 的效應,並模擬堆疊狀態下之分子鏈構形變化進行比較與探討。經由主鏈上的硫
環 扭 轉 角 度 分 析 來 看 , 可 以 發 現 到
Poly{3-(5”-hexyl-2,2’:5’,2”-terthiophenyl-5-vinyl)thiophene-alt-thiophene} (P1) 、 Poly{3-(5,5”-dihexyl-2,2’:5’,2”-terthiophenyl-3’-vinyl)thiophene-alt-thiophene} (P2)與 Poly{3-(4,4”-dihexyl-2,2:5’,2”-terthiophene-3’-vinyl)thiophene-alt-thiophene} (P3) 以
5
單一條高分子鏈的形式在真空狀態下時,因主鏈上缺乏堆疊的作用導致其擁有較 廣的扭轉角度分布,表示其高分子鏈運動較為自由,也間接暗指著其高分子鏈是 處於較為無序的環境下,進而影響了實驗的吸收光譜;而透過模擬高分子鏈堆疊 之分子鏈構形,因主鏈上受到 π-π 作用力的吸引,使其高分子鏈擁有較為有序之堆 疊狀況,因而其主鏈上的扭轉角度分布範圍較窄,其結構式與扭轉角度分布如圖 1-7(c)所示。而於 Huang 等人[52]針對一系列聚噻吩衍生物 Poly(3-hexylthiophene) (P3HT) 、 Poly(4,4’-dihexyl-2,2’-bithiophene) (PDBT) 、 Poly(5,5’’’-dihexyl-[2,3’;5’,2’’;3’’,2’’’]quarter thiophene) (PD3QT) 與 Poly(5,5’’’-dihexyl-[2,3’;5’,2’’;4’’,2’’’]quarter thiophene) (PD4QT)進行理論計算,其 結構式如圖 1-8(a)所示。為了符合真實的實驗狀況,初步將溶劑效應考量進去,模 擬單一條高分子鏈於良溶劑之稀薄溶液中其分子鏈構形變化。經由主鏈上硫環之 扭轉角度分布分析結果,可以發現到 P3HT 與 PDBT 皆擁有較廣且較為凌亂之扭 轉角度分布,主要是受到了溶劑添加的效應影響,使高分子鏈運動較為自由,因 而呈現較為蜷曲的樣貌,如圖 1-8(b)與(c)所示。這些結果都隱含著,共軛高分子在 溶液無序狀態下之分子構形對於吸收光譜確實是相當重要且不可忽視的因素。
因此,本研究將有系統的探討含有不同共軛側鏈接枝位向之共軛高分子在溶 液無序狀態下其分子鏈構形與光電性質之間的關聯性。我們延續先前與 Wang 等人 [51]所合成之噻吩乙烯(Thienylene vinylene)系統的二維共軛高分子材料,並在主鏈 上改良以三噻吩做為主軸,以減少因龐大側鏈之立體障礙所造成主鏈上硫環之扭 轉角度偏轉過大,並且因實驗上合成製備所需具備較佳的溶解度特性,將原本的 己基(Hexyl)側鏈基團統一改良為分支的乙基己基(Ethylhexyl)側鏈基團。我們選用 在主鏈及側鏈間以乙烯基(Vinyl)銜接,其側鏈以垂直於主鏈(PBTTTV-v)或平行於 主鏈(PBTTTV-h)的方式接枝於主鏈上之兩種共軛高分子,更加深入了解共軛效應 在二維方向上的延伸性,且此兩個材料已投稿於 Physical Chemistry Chemical Physics[53]。接著,我們在主鏈及側鏈間改以三個連續單鍵銜接,再將側鏈分別以
6
垂直於主鏈(H-PBTTTV-v)與平行於主鏈(H-PBTTTV-h)的方式接枝於主鏈上,以探 討不同的側鏈位向對於分子鏈構形及其光電性質之間的關聯性,其結構式如圖 1-9 所示。首先,我們利用分子動力學(Molecular dynamics)模擬單一條分子鏈於良溶劑 (Good solvent)之稀薄溶液中,並且從其分子鏈之構形與相關分析中,我們也初步 得到一些結論。接著,利用量子力學法(Quantum mechanical calculation)計算其紫外 /可見光吸收光譜,從其分子鏈的構形中探討其中二維延伸共軛性質對於紫外/可見 光吸收光譜之貢獻程度。
7
圖 1-1 於不同位置規整度下(a)HT 達 97%與(b)HT 達 64% P3HT 在氯仿溶液下之紫 外/可見光吸收光譜[19]。
8
圖 1-2 (a)烷基取代型之單體結構與聚合後之高分子材料於(b)氯仿溶液中及(c)成膜 後之紫外/可見光吸收光譜[31]。
9
圖 1-3 原子取代型之高分子結構與其在成膜後之紫外/可見光吸收光譜[32]。
10
圖 1-4 PBDTTT-S 與 PBDTDTTT-S-T 於薄膜下之紫外/可見光吸收光譜[43]。
11
圖 1-5 含有純聚噻吩系統之衍生物(a)其結構式及其在(b)氯仿溶液中和(c)薄膜之紫 外/可見光吸收光譜[40]。
12
圖 1-6 含有苯乙烯基三苯胺官能基共軛側鏈之 PT5TPA 二維共軛高分子結構與其 紫外/可見光吸收光譜[49]。
13
圖 1-7 (a)不同共軛側鏈接枝位向之二維共軛高分子結構其(b)在成膜升溫後與氯仿 溶液下之紫外/可見光吸收光譜結果,並經由模擬進行其在(c)堆疊狀態和單一條鏈
於真空下之主鏈扭轉角度分布分析[51]。
14
圖 1-8 聚噻吩系統之衍生物(a)其結構式與 P3HT、PDBT 於良溶劑中(b)高分子鏈之 蜷曲構形及其(c)主鏈上之扭轉角度分布[52]。
圖 1-9 PBTTTV-v、PBTTTV
15
BTTTV-h、H-PBTTTV-v 與 H-PBTTTV-h之結構式之結構式。
16
第二章 第二章 第二章
第二章 模擬方法模擬方法模擬方法模擬方法
在本研究中我們採用全原子分子動力學與量子力學二種模擬方法來探討含有 三噻吩共軛側鏈之共軛高分子其分子結構與二維延伸共軛性質對於紫外/可見光吸 收光譜之影響。我們選擇 PBTTTV-v、PBTTTV-h、H-PBTTTV-v 與 H-PBTTTV-h 作為研究的對象,來協助釐清二維延伸共軛性質於不同側鏈分子結構之共軛高分 子中如何影響吸收圖譜。其模擬流程可分為兩部分進行,首先,於 2-1 節我們將描 述如何利用全原子分子動力學模擬來得到分子鏈於溶液無序狀態下之穩定構形;
接著在 2-2 節,將會介紹關於量子力學的模擬條件與方法。
17
2-1 全原子分子動力學全原子分子動力學全原子分子動力學全原子分子動力學(All-atom Molecular Dynamics)模擬模擬模擬模擬 2-1-1 力場選擇力場選擇力場選擇力場選擇
在本研究中,我們選用 Material Studio (MS)軟體中 Forcite 模組裡的 Dreiding 力場,來計算全原子模型中原子間之交互作用力參數[54]。此力場能夠描述大量的 有機分子、生物分子以及週期表中所有主族無機分子的結構與動力學性質,且對 於分子的幾何結構、能量與晶體的堆疊皆有相當高的準確性,所有位能函數項將 表示於式(1)至式(10)。但要特別注意的是,在 Dreiding 力場並無給定電荷值,因此 我們必須使用電荷平衡法(QEq)來給定電荷,其為一種實驗性的近似法[55],經由 此法所得到的電荷分布,能夠反映出分子周圍環境的變化,且原子之電荷分佈會 隨著分子構形而有所改變,所需要的參數為電子親和力、原子半徑、原子游離能 以及原子的相對位置。
nb
val U
U
U = + (1)
inversion torsion
bend stretch
val U U U U
U = + + + (2)
( )
[ ]
2 12, 2
1
=
−
= − −
e R e
R e stretch
D e k
D
U αn e α (3)
[ ]
2 00 2
, sin cos
2 cos 1
J IJK IJK
J IJK
IJK bend
C K C
U = θ − θ = θ (4)
( )
[ ]
{
1 cos 0}
2 1
JK JK
JK
torsion V n
U = − ϕ−ϕ (5)
(
0)
22
1 φ−φ
= K
Uinversion (6)
hb els vdw
nb U U U
U = + + (7)
−
=
6
0 12
0
0 2
R R R
D R
Uvdw (8)
( )
ij j i
els R
Q U Q
0637 ε .
= 332 (9)
DHA DA
hb DA
hb hb
hb R
R R
D R
U 4θ
6 12
cos 6
5
−
= (10)
18
2-1-2 模擬系統介紹與初始架構模擬系統介紹與初始架構模擬系統介紹與初始架構模擬系統介紹與初始架構
於高分子鏈在稀薄溶液之無序狀態下的模擬,考慮到軟體本身之總原子數目 上限值為十萬個,因而我們先將系統中所有的溶劑分子與單體分子之氫原子除去,
並與其原本相接之碳原子建立聯合原子(United atom)系統,以減少總原子數。且為 了之後擴展成高分子鏈,我們保留住主鏈上末端的兩個氫原子,並修正其電荷。
我們選擇於每組系統中建立單一條且聚合度為 10 之高分子鏈,再將高分子鏈進行 最佳化,其末端距離(End-to-end distance)約為 113 Å。為了確保不受到鄰近盒子內 高分子鏈的相互影響,我們取高分子鏈之末端距離的兩倍作為盒子的對角線長度,
經由換算後,可將盒子的邊長設定為 131 Å,再於盒子中填入符合實際狀況之氯苯 (C6H5Cl)溶劑分子數目,以符合其密度為 1.11 g/cm3。
2-1-3 模擬流程模擬流程模擬流程模擬流程
整體初始架構完成後,考慮到軟性高分子中 C-H 鍵之震動週期最低,約為 10 fs,因此設定 1 fs(粒子運動最小週期的 1/10)作為時間步階。在進行模擬前,必 須先將初始架構進行幾何最佳化,釋放過高的能量以取得穩定的構形,再進行升 溫退火程序,考慮在恆溫-恆體積下(NVT ensemble),以 10 K/ps 之速率從 300 K 升溫至 500 K,再由相同速率降溫至 300 K,整個循環過程進行 3 次,以確保系統 能夠達到熱力學平衡。再將升溫退火後之構形於 300K 下進行長時間 300 ps 的等溫 動態平衡,我們發現在 250 ps 之後溫度與系統能量之誤差均小於 1 %。因此,選 擇取最後之 50 ps 的等溫動態平衡後之高分子構形,並對其來進行分析與統計。
19
2-2 量子力學計算量子力學計算量子力學計算量子力學計算
2-2-1 密度泛函理論密度泛函理論密度泛函理論密度泛函理論(Density Functional Theory, DFT)與時間依賴與時間依賴與時間依賴與時間依賴---密度泛函理論-密度泛函理論密度泛函理論密度泛函理論 (Time Dependent-Density Functional Theory, TD-DFT)
我們選用量子力學軟件 Gaussain 03 來進行計算[55],並以最常運用於分子結 構計算之密度泛函理論(Density Functional Theory, DFT) [56],搭配交換-相關泛函 為 B3LYP (Becke, three-parameter, Lee-Yang-Parr)[57]以及 6-31G*基組,來對其分子 結構進行幾何最佳化(Geometry Optimization)。但 DFT 並無法描述電子的激發態,
讓我們用來計算紫外/可見光吸收光譜。因而需要經由時間依賴-密度泛函理論 (Time Dependent-Density Functional Theory, TD-DFT)[58-61],迅速且準確描述分子 於激發態時之性質,且目前於文獻上使用此方法對於分子結構之紫外/可見光吸收 光譜的預測有很高的準確性。也因此,在後續我們先利用 DFT/B3LYP/6-31G*來對 分子結構進行幾何最佳化,再結合 TD-DFT/B3LYP/6-31G*來進行激發態的能量計 算,並模擬其分子結構之紫外/可見光吸收光譜。
2-2-2 相關條件測試與計算流程相關條件測試與計算流程相關條件測試與計算流程相關條件測試與計算流程
為了瞭解不同的烷基側鏈對於紫外/可見光吸收光譜的影響幅度,我們初步將 H-PBTTTV-v 的 單體 至三聚 體之分 子構 形 進行幾 何最佳 化, 並 且將乙 基己基
(Ethylhexyl)側鏈改以甲基(Methyl)側鏈進行取代,結構式如圖 2-1(a)所示,再利用 TD-DFT 計算各自的紫外/可見光吸收光譜,如圖 2-1(b)所示。我們可以發現到不論 是在單體、二聚體或三聚體上,烷基側鏈若改以甲基側鏈進行取代,兩者所呈現 的吸收光譜幾乎是相似的,也因此在往後與量子力學相關的計算中,我們將一律 進行此代換。
20
圖 2-1 (a)為單體至三聚體之有/無甲基取代示意圖與其(b)所對應之吸收光譜。
21
第三章第三章
第三章第三章 結果與討論結果與討論結果與討論結果與討論
本研究為了探討二維延伸共軛性質對於聚噻吩高分子系統之紫外/可見光吸收 光譜的影響,我們有系統地選擇以三噻吩為主軸的四個高分子材料 PBTTTV-v、
PBTTTV-h、H-PBTTTV-v 與 H-PBTTTV-h 作為研究對象。本研究主要可分成兩大 主軸進行探討:首先,第一個主軸是為了能符合並反映出真實溶劑狀態下之結果,
因而我們利用分子動力學模擬來得到四個高分子材料於溶液無序狀態(Disordered state)下的構形,並對此提出初步的結論與探討;而在第二個主軸上,則是利用量 子力學法計算來得到其中的紫外/可見光吸收光譜,並探討其分子構形與光電性質 之間的關聯性。且為了釐清不同側鏈接枝位向之共軛高分子對於紫外/可見光吸收 光譜的影響,我們將逐一針對四個材料來進行探討,並於最後的 3-5 節進行整合及 相互比較。藉由此兩個主軸可以有系統的探討側鏈垂直接枝於主鏈上或平行接枝 於主鏈上之共軛高分子材料,其二維共軛高分子材料中之二維延伸共軛性質對於 紫外/可見光吸收光譜的影響。
3-1 利用分子動力學與量子力學模擬利用分子動力學與量子力學模擬利用分子動力學與量子力學模擬利用分子動力學與量子力學模擬探討探討探討 PBTTTV-v 之分子構形探討 之分子構形之分子構形及其對於之分子構形及其對於及其對於紫外及其對於紫外紫外/可紫外可可可 見光吸收光譜的影響
見光吸收光譜的影響 見光吸收光譜的影響 見光吸收光譜的影響
圖 3-1(a)為單一條 PBTTTV-v 分子鏈於良溶劑之稀薄溶液中 300 K 下經過 300ps 動態平衡後之分子鏈構形圖,可看到 PBTTTV-v 於氯苯溶劑中,分子鏈會明 顯地伸展開來,且整條分子鏈也有類似 S 型曲線之樣貌。我們針對其分子鏈進行 定量分析,且為了可以細部來探討 PBTTTV-v 的構形,將主鏈上硫環與硫環之間 的扭轉角度細分為 τMain chain_A、τMain chain_B與 τMain chain_C三部分,側鏈上的硫環與硫 環間的扭轉角度則分為 τSide chain_A與 τSide chain_B兩部分,另外,也針對其主鏈與側鏈 之間銜接處的扭轉角度 τVinyl進行分析,相關扭轉角度標示如圖 3-2(a)的示意圖所 示,而其扭轉角度分布將統一整理並列於表 3-1。
由圖 3-3(a)、(b)與(c)中,我們可以發現到在 A 位置其最大訊號峰位置約為 140 度,偏離共平面將近 40 度,其主要原因是銜接於主鏈與側鏈間的乙烯基(Vinyl),
22
其碳原子距離 A 處硫環上的硫原子較近,受限於立體障礙,兩者呈現排斥的作用,
使得 A 處的扭轉角度不但不會維持在共平面,甚至還有三種不同的扭轉角度分布;
而於主鏈上之 B 處和 C 處,則因為本身較不受到乙烯基(Vinyl)或側鏈之立體空間 上的影響,因而使兩者之最大訊號峰位置幾乎集中接近於共平面的角度 180°或於 0° (360°)附近。接著,如圖 3-3 (d)與(e)所示,經由側鏈上的扭轉角度分析,我們可 以發現到由於其側鏈結構為垂直接枝於主鏈上,其 A 與 B 位置較不受限於立體空 間的影響,因而皆擁有相似的扭轉角度分布,且其最大訊號峰位置幾乎維持在共 平面 180 度。再延伸探討其主鏈與側鏈之銜接處扭轉角度分布 τVinyl,如圖 3-3(f) 所示。經由乙烯基(Vinyl)的扭轉角度分析,可以發現到其擁有良好的共平面性,我 們推測這是因為乙烯基(Vinyl)本身結構中含有雙鍵,在立體空間上,雙鍵不易因自 由的運動而產生扭轉,即便加入了良溶劑,乙烯基(Vinyl)依舊能幫助主鏈與側鏈之 間維持良好的共平面性,因而使其呈現了較窄的扭轉角度分布狀況。
但在這裡我們將先排除溶劑效應所帶來的影響,初步以量子力學法來探討真 空下之分子構形及其對於紫外/可見光吸收光譜的影響。為了要找到影響紫外/可見 光吸收光譜之因素,我們必須先回歸到材料結構本身,了解分子構形的變化如何 影響整體的吸收光譜。因而我們選擇先從高分子材料之最小單元結構-單體著手,
保留其經最佳化後所得的原始構形,以人為調控角度的方式來觀察紫外/可見光吸 收光譜之變化。我們以每 30 度為間隔,選擇只改變主鏈上其中一個扭轉角度(τmain
chain)再計算其吸收光譜。從圖 3-4 與 3-5 中,我們能夠明顯觀察到,不論我們怎麼
去改變主鏈右側硫環與硫環間的扭轉角度(τmain chain_B),其最大吸收峰的位置幾乎都 不會改變(最大吸收峰值約為 490 nm),僅有在短波長區的部分有藍、紅位移之變 化,而整體吸收範圍也都維持在 300 nm~600 nm 左右;但當我們改變主鏈左側硫 環與硫環之間的扭轉角度(τmain chain_A)時,可以清楚的觀察到最大吸收峰值開始伴隨 著角度偏轉越來越大而有明顯的藍位移變化,而整體的吸收範圍亦跟著逐漸變窄。
藉由這些人為調控單體主鏈上的扭轉角度測試,我們發現到在此單體中由於單鍵
23
與雙鍵的方向延續,因而有 L 型共軛的結構產生。由此我們推測 PBTTTV-v 因擁 有乙烯基(Vinyl)銜接於主鏈與側鏈之間,電子會由主鏈傳遞到側鏈上,得以使共軛 性延續下去,也正因為如此,其紫外/可見光吸收光譜將不再僅是由單純的主鏈或 側鏈所貢獻,還多了二維方向的 L 型共軛。也就是說在短波長區的部分主要是由 主鏈及側鏈所貢獻,而長波長區的部分則是由共軛長度較長之二維延伸性的 L 型 共軛所貢獻。另一方面,為了更加瞭解二維延伸共軛性質對於吸收光譜的影響,
我們延伸至二聚體來進行探討並與王立義老師實驗室團隊之實驗結果進行比較。
從其軌域分布出發,我們觀察並挑選出幾個振子強度(Oscillator strength)較強的激 發態作為代表性,如表 3-2 所示,能很清楚的看到電子雲主要的分布狀況變的較為 複雜,且有同時存在於主鏈以及側鏈上之可能,我們將這些電子雲分布狀況做為 參考依據,大致上可將其分成四種可能的片段,除了出現在主鏈上,還包括三種 L 型共軛的片段上,如圖 3-6(a)所示。我們保留其原始片段之構形,並計算個別的紫 外/可見光吸收光譜,如圖 3-6(b)所示。從這些片段所計算出的吸收圖譜中,我們 依然能看到在短長區為主鏈與側鏈所提供的貢獻,而長波長區則是整體之 L 型共 軛的效應。比較特別的是,主鏈上含有六個硫環之 6T 與含有五個硫環再加上乙烯 基(Vinyl)之 L 型 5L 兩者的最大吸收峰位置極為相近,且整體吸收範圍亦幾乎相同,
唯有在吸收強度上有些許的差異性,這主要歸因於乙烯基(Vinyl)與硫環所提供之貢 獻程度不盡相同,才會造成此差異現象出現。接著,我們調控不同比例將四種可 能片段進行組合,再分別與兩組不同分子量之實驗圖譜來相互比較,其實驗之相 關條件如表 3-3 所示,而其於氯苯溶劑下之吸收光譜結果則如圖 3-7(a)所示。從表 3-4 中,我們可以看到經由組合後的曲線,與低分子量的結果相比,5L、6L、8L 與 6T 四個片段比例分別為 0.19、0.19、0.43 與 0.19,能清楚的觀察到二聚體中共 軛長度最長之 L 型共軛(8L)是影響整個紫外/可見光吸收光譜最大的關鍵來源,如 圖 3-8 所示。而伴隨著分子量的增加,從實驗的圖譜上來看,僅有在長波長區的部 分逐漸有明顯的訊號峰產生,但其整體吸收範圍仍與低分子量之實驗結果相近。
24
有鑑於此,我們分別讓四個片段的吸收曲線做消長變化,其 5L、6L、8L 與 6T 四 個片段比例分別微調成 0.08、0.24、0.46 與 0.22,如表 3-5 所示,可以很明顯的看 到伴隨著分子量的提升,其 L 型共軛的效應也越來越強,如圖 3-9 所示。
從上述的扭轉角度分布分析中,我們能得知在溶液無序狀態下,分子鏈構形 容易因溶劑效應的影響呈現較為雜亂的扭轉角度分布,與真空下經幾何最佳化後 之結果相比較,兩者有不同的分子構形,使我們不能忽略了溶劑效應所帶來的影 響。也因此,在之後我們將修正其中的扭轉角度,如圖 3-10(a)所示,改從分子動 力學達動態平衡後,擷取其中較具代表性的扭轉角度,即最大訊號峰值,再分別 以量子力學法計算各片段之吸收光譜並進行組合後,與在真空下組合之結果相比 較,從中探討兩種不同之分子構形,其紫外/可見光吸收光譜會有怎樣的變化。
3-2 利用分子動力學與量子力學模擬利用分子動力學與量子力學模擬利用分子動力學與量子力學模擬利用分子動力學與量子力學模擬探討探討探討探討 PBTTTV-h 之分子構形之分子構形之分子構形及其對於之分子構形及其對於及其對於紫外及其對於紫外紫外/可紫外可可可 見光吸收光譜的影響
見光吸收光譜的影響 見光吸收光譜的影響 見光吸收光譜的影響
圖 3-1(b)為單一條 PBTTTV-h 分子鏈於良溶劑之稀薄溶液中 300 K 下經過 300ps 動態平衡後之分子鏈構形圖,可看到 PBTTTV-h 於氯苯溶劑中,分子鏈會明 顯地伸展開來,且整條分子鏈也有類似 S 型曲線之樣貌。我們針對其分子鏈進行 定量分析,且將其構形細分為三大部分來做探討,其主鏈上硫環與硫環之間的扭 轉角度細分為 τMain chain_A、τMain chain_B與 τMain chain_C,側鏈上的硫環與硫環間的扭轉 角度則分為 τSide chain_A與 τSide chain_B以及主鏈與側鏈之間銜接處的扭轉角度 τVinyl, 相關扭轉角度示意圖標示於圖 3-2(b),其扭轉角度分布將整理於表 3-1。
經由主鏈上之扭轉角度分析,由於其受限於銜接於主鏈與側鏈間的乙烯基 (Vinyl)之立體障礙,使其 A 處擁有較廣之扭轉角度分布,且其最大訊號峰位置偏 離共平面將近 40 度;而於主鏈上之 B 處和 C 處,則因為本身較不受到乙烯基(Vinyl) 或側鏈之立體空間上的影響,因而使兩者較容易維持於共平面的 180 度附近,如 圖 3-11(a)、(b)與(c)所示。接著,由圖 3-11(d)與(e)中,因其側鏈結構與主鏈相互平 行,使其容易受到乙烯基(Vinyl)所影響,特別是在 B 處,因為 B 處硫環上的硫原
25
子與乙烯基(Vinyl)上的碳原子距離較近,為了避免兩原子重疊,使得 B 處有三種 不同的扭轉角度分布,且幾乎不可能維持在共平面。經由乙烯基(Vinyl)的扭轉角度 分析,我們一樣能發現到其與 PBTTTV-v 有相近之結果,乙烯基(Vinyl)能幫助主鏈 與側鏈之間維持良好的共平面性,其最大訊號峰值幾乎集中於 180 度,如圖 3-11(f) 所示。
在這裡我們一樣先不考量溶劑效應所帶來的影響,初步以量子力學法來探討 真空下經幾何最佳化後之分子構形及其對於紫外/可見光吸收光譜的影響。從圖 3-12 所示,我們採用相似的人工調控測試方式及相同的設定條件,從高分子材料 之最小單元結構-單體著手,保留其經最佳化後所得的原始構形,人為調控兩對 角硫環與硫環間的扭轉角度為 90 度,並觀察計算後之紫外/可見光吸收光譜變化。
從其吸收光譜中,我們能很清楚的觀察到當同時改變示意圖中的 1 和 4 位置為 90 度時,其會有兩個較明顯的吸收峰產生;但當我們同時改變示意圖中的 2 和 3 位 置為 90 度時,卻僅有一個特徵峰的訊號出現。同樣都是改變對角的兩個扭轉角度,
但計算出來的吸收光譜卻有很大的不同,這也暗指出其中有二維延伸共軛性的可 能,才會使圖譜呈現不同的樣貌。而我們藉由仔細觀察結構中的單鍵和雙鍵之方 向延續性,發現到其中有 Z 型共軛的存在,且其方向會通過示意圖中 2 和 3 兩處,
這也能說明先前將 1 和 4 位置偏轉 90 度時,其吸收光譜會有兩個吸收峰產生,其 一是來自於主鏈與側鏈上之單一硫環,而另一個則是來自於共軛長度較長之 Z 型 共軛所貢獻;相反的,當同時改變示意圖中的 2 和 3 位置為 90 度時,則會因為中 斷其中二維方向的共軛延續性,使電子不易被傳遞下去,因而會在兩吸收峰之間 形成一個主要吸收峰,則是來自於兩兩硫環間的貢獻。從這些測試的結果中,我 們都能發現到添加乙烯基(Vinyl)於主鏈與側鏈之間,能有效的幫助整體結構多了另 一個維度的共軛延續性,使得紫外/可見光吸收光譜的吸收範圍將變得更為寬廣。
另一方面,為了更加瞭解二維延伸共軛性質對於吸收光譜的影響,我們延伸至三 聚體來進行探討。首先,我們觀察其軌域分布,並從中挑選出幾個振子強度
26
(Oscillator strength)較強的激發態作為代表性,如表 3-6 所示,能很清楚的看到電子 雲主要的分布狀況同時並存於主鏈以及側鏈上,因而將這些電子雲分布狀況大致 上分成七種可能的片段,除了最常見的出現在主鏈與側鏈上,還包括五種 Z 型共 軛的片段上,如圖 3-13(a)所示,並計算各片段之紫外/可見光吸收光譜,如圖 3-13(b) 所示。從這些片段所計算出的吸收圖譜中,我們能看到在短長區的部分主要為主 鏈或側鏈所提供的貢獻,長波長區則是整體之 Z 型共軛的效應,而隨著硫環數目 的增加,其吸收強度也有略為提升的效果。比較特別的是,主鏈上含有九個硫環 之 9T 與含有十個硫環再加上乙烯基(Vinyl)之 Z 型 10Z 兩者的最大吸收峰位置極為 相近,且整體吸收範圍亦幾乎相同,唯有在吸收強度上有些許的差異性,也因此 我們推測當共軛長度達三個單體時,其紫外/可見光吸收光譜相較於四聚體幾乎不 會有太大的變化。也正因如此,我們選擇調控七種可能片段之不同比例再將之進 行組合,並分別與兩組不同分子量之實驗圖譜來相互比較,其實驗之相關條件已 列於表 3-3,其於氯苯溶劑下之吸收光譜結果則如圖 3-7(b)所示。我們可以看到經 由組合後的曲線,與低分子量的結果相比,如表 3-7 所示,3T、4Z、5Z、7Z、8Z、
9T 與 10Z 七個片段比例分別為 0.24、0.23、0.23、0.17、0.02、0.05 與 0.06,能清 楚的觀察到二維延伸共軛性質之存在是影響整個紫外/可見光吸收光譜的重要因素,
如圖 3-14 所示。當伴隨著分子量的增加,從實驗的圖譜上來看,在長波長區的部 分其吸收峰強度變強,而我們從模擬中也看到在共軛長度較長之片段比例也有略 為提升的現象,如表 3-8 所示,正好與實驗相互呼應,其 3T、4Z、5Z、7Z、8Z、
9T 與 10Z 七個片段比例分別調整成 0.25、0.23、0.20、0.17、0.01、0.07 與 0.07,
如圖 3-15 所示。從這些測試的結果,都在在告訴了我們二維共軛高分子材料對於 紫外/可見光吸收光譜的影響是較為複雜的,他包含的不僅有主鏈與側鏈而已,甚 至還有從主鏈延伸至側鏈的共軛性存在,由兩個維度的不同貢獻程度影響了最後 圖譜的呈現。而從這些比例上的調整與組合,我們確實能看到與實驗中有類似的 結果產生,且經由組合後的曲線也與實驗結果上有著相符合的印證。
27
在之後我們會加入溶劑效應所帶來的影響,並修正分子鏈其中的扭轉角度,
如圖 3-10(b)所示,改從分子動力學達動態平衡後,擷取其中較具代表性的扭轉角 度,即最大訊號峰值,再分別以量子力學法計算各片段之吸收光譜並進行組合後,
與在真空下組合之結果相比較,從中探討兩種不同之分子構形,其紫外/可見光吸 收光譜的變化。
3-3 利用分子動力學與量子力學模擬利用分子動力學與量子力學模擬利用分子動力學與量子力學模擬探討利用分子動力學與量子力學模擬探討探討 H-PBTTTV-v 之分子構形探討 之分子構形之分子構形及其對於之分子構形及其對於及其對於紫外及其對於紫外紫外/紫外 可見光吸收光譜的影響
可見光吸收光譜的影響 可見光吸收光譜的影響 可見光吸收光譜的影響
圖 3-1(c)為單一條 H-PBTTTV-v 分子鏈在氯苯溶劑中 300 K 下經過 300ps 動態 平衡後之分子鏈構形圖,可看到 H-PBTTTV-v 在氯苯溶劑中,分子鏈會明顯地伸 展開來,且整條分子鏈有類似 S 型曲線之樣貌。我們針對其分子鏈進行定量分析,
且為了可以細部來探討 H-PBTTTV-v 的構形,將主鏈上硫環與硫環之間的扭轉角 度細分為 τMain chain_A、τMain chain_B與 τMain chain_C三部分,側鏈上的硫環與硫環間的扭 轉角度則分為 τSide chain_A與 τSide chain_B兩部分,另外,也針對其主鏈與側鏈之間銜接 處的扭轉角度 τSingle bond進行分析,相關扭轉角度標示如圖 3-2(c)的示意圖所示,而 其扭轉角度分布將統一整理並列於表 3-1。
經由主鏈上的扭轉角度分析,如圖 3-16(a)、(b)與(c)所示,我們可以發現到在 A 位置擁有兩個相近區域的扭轉角度分布,且觀察其最大訊號峰位置偏離共平面 約 40 度,推測是因為銜接於主鏈與側鏈間的三個單鍵上的碳原子距離硫環上的硫 原子較近,兩者產生排斥,使得 A 處的扭轉角度幾乎不會維持在共平面;而相較 於 B 處和 C 處,則因為本身較不受立體空間的影響,因而使兩者之最大訊號峰位 置幾乎集中接近於共平面的角度 180°與 0° (360°)。接著,我們針對側鏈上的硫環 與硫環間的扭轉角度進行分析,如圖 3-16(d)與(e)所示,可以發現到由於其側鏈結 構為垂直接枝於主鏈上,其 A 與 B 位置不受限於立體空間的影響,因而皆擁有相 似的扭轉角度分布,且其最大訊號峰位置幾乎維持在共平面 180 度。由圖 3-16(f) 中,經由主鏈與側鏈間銜接處的扭轉角度分析,我們可以發現到其幾乎是呈現偏
28
離共平面的 90 度附近,幾乎不可能維持在共平面,推測是因為其結構主要是由三 個單鍵所組成,而單鍵與單鍵之間的運動較為自由,為了減低側鏈對於主鏈所造 成的立體效應,會產生較大的扭轉。
在這裡我們先排除溶劑效應所帶來的影響,初步以量子力學法來探討真空下 之分子構形及其對於紫外/可見光吸收光譜的影響。基於想了解紫外/可見光吸收光 譜之影響因素,促使我們必須從結構本身著手,了解分子構形的變化如何影響整 體的吸收光譜。我們先從高分子材料之最小單元結構-單體著手,保留其經最佳 化後所得的原始構形,初步以人為的方式調控主鏈與側鏈間銜接處的扭轉角度
(τsingle bond)並且計算其吸收光譜。從 H-PBTTTV-v 的測試結果,從圖 3-17 中能夠清
楚的觀察到,伴隨著τsingle bond每隔 30 度扭轉一次角度,當由 90 度偏轉至 270 度時,
圖譜中都僅呈現一個較為明顯的特徵峰,其吸收範圍皆落在 310 nm~450 nm 附近,
且最大吸收峰值約為 379 nm,可以得知不論是主要吸收峰的位置或是整體的吸收 範圍幾乎都沒有明顯的差異。由此可知,我們推測主要是因為當主鏈和側鏈間以 三個單鍵相銜接時,中斷了二維方向共軛的延續性,使得電子幾乎不會從主鏈傳 遞到側鏈上,進而縮短了整體的共軛長度,也因此才會在短波長區僅有單一訊號 峰的產生,而此訊號峰則是來自於主鏈以及側鏈上三個硫環所貢獻,也就是說 H-PBTTTV-v 並不具有二維延伸共軛的性質。另一方面,為了更加確保二維延伸共
軛性質並不存在於此材料中,我們延伸至二聚體來進行探討,從其軌域分布來說 明,我們觀察並挑選出幾個振子強度(Oscillator strength)較強的激發態作為代表性,
如表 3-9 所示。我們能很清楚的看到電子雲主要的分布狀況僅有兩種可能性,一是 只會單獨出現在主鏈上,二則是在兩側鏈上。從這個測試的結果,再次驗證了此 材料中並沒有二維延伸共軛的情形發生。
從上述的扭轉角度分布分析中,我們能得知在溶液無序狀態下,分子鏈構形 容易因溶劑效應的影響呈現較為雜亂的扭轉角度分布,與真空下經幾何最佳化後 之結果相比較,兩者有不同的分子構形,使我們不能忽略了溶劑效應所帶來的影
29
響。也因此,在之後我們將修正其中的扭轉角度,改從分子動力學達動態平衡後,
擷取其中較具代表性的扭轉角度,即最大訊號峰值,如 3-10(c)所示,再分別以量 子力學法計算其主鏈與側鏈之吸收光譜,透過組合的方式,從單體延伸探討至二 聚體、三聚體、…,並預測其高分子材料達到一定的高分子量時,其紫外/可見光 吸收光譜會有怎樣的變化。
3-4 利用分子動力學與量子力學模擬利用分子動力學與量子力學模擬利用分子動力學與量子力學模擬利用分子動力學與量子力學模擬探討探討探討 H-PBTTTV-h 之分子構形探討 之分子構形之分子構形及其對於之分子構形及其對於及其對於紫外及其對於紫外紫外/紫外 可見光吸收光譜的影響
可見光吸收光譜的影響 可見光吸收光譜的影響 可見光吸收光譜的影響
圖 3-1(d)為單一條 H-PBTTTV-h 分子鏈於良溶劑之稀薄溶液中 300 K 下經過 300ps 動態平衡後之分子鏈構形圖,可看到 H-PBTTTV-h 於氯苯溶劑中,分子鏈會 明顯地伸展開來,且整條分子鏈也有類似 S 型曲線之樣貌。我們針對其分子鏈進 行定量分析,且將其構形細分為三大部分來做探討,其主鏈上硫環與硫環之間的 扭轉角度細分為 τMain chain_A、τMain chain_B與 τMain chain_C,側鏈上的硫環與硫環間的扭 轉角度則分為 τSide chain_A與 τSide chain_B以及主鏈與側鏈之間銜接處的扭轉角度 τSingle
bond,相關扭轉角度示意圖標示於圖 3-2(d),其扭轉角度分布將整理於表 3-1。
經由主鏈上之扭轉角度分析,由於其受限於銜接於主鏈與側鏈間的三個單鍵 之立體障礙,使其於 A 處擁有較廣之扭轉角度分布,且其最大訊號峰位置偏離共 平面將近 40 度;而於主鏈上之 B 處和 C 處,則因為本身較不受到銜接處或側鏈之 立體空間上的影響,因而使兩者較容易維持於共平面的 180 度附近,如圖 3-18(a)、
(b)與(c)所示。經由側鏈之扭轉角度分析中,由於其側鏈結構與主鏈相互平行,使 其容易受到銜接於主鏈與側鏈間的三個單鍵所束縛,特別是在 B 處,因 B 處硫環 上之硫原子與三個單鍵上的碳原子距離較近,受限於立體障礙的影響,使得 B 處 的扭轉角度不會維持在共平面,甚至出現四個不同區域上的扭轉角度分布,如圖 3-18(d)與(e)所示。從上述主鏈與側鏈的扭轉角度分析中,我們可以得知硫環與硫 環間會有較大的角度偏轉,皆是受到了主鏈與側鏈之間銜接處的影響所致,因而 促使我們必須往下針對這部分來做進一步的分析與探討。如圖 3-18(f)所示,由於
30
其側鏈與主鏈因相互平行,再加上溶劑效應的影響,使其呈現了兩種不同的扭轉 角度分布狀況,且幾乎不可能維持在共平面。這些分析結果都顯示了在稀薄溶液 中,分子鏈間因缺少 π-π 作用力,使分子鏈的運動較為自由,分子鏈因而有 S 型的 樣貌出現,也因此使得扭轉角度分布變的較為雜亂。
在這裡我們一樣先不考量溶劑效應所帶來的影響,初步以量子力學法來探討 真空下經幾何最佳化後之分子構形及其對於紫外/可見光吸收光譜的影響。我們採 用相同的人為測試方式及設定條件,持續探討 H-PBTTTV-h 之分子構形對於吸收 光譜的影響。如圖 3-19 所示,我們一樣能夠清楚的觀察到,伴隨著τsingle bond=90°
偏轉至τsingle bond=270°時,都僅呈現一個吸收峰,且其吸收峰的位置及整體吸收範
圍也都沒有太大的差異,其吸收範圍約在 300 nm~420 nm,最大吸收峰值為 353 nm。
再觀察其二聚體之軌域分布狀況,如表 3-10 所示,我們亦能看到電子雲分布只會 單獨出現於主鏈或是分別於兩側鏈上,皆得到了相同的印證。經由這部分的結果,
我們再次證實了不論是側鏈垂直接枝於主鏈上之 H-PBTTTV-v 或平行接枝於主鏈 上之 H-PBTTTV-h 高分子,兩者因主、側鏈間以三個單鍵相銜接之結構的設計,
使其並不具備有二維延伸的共軛特性,也告訴了我們其紫外/可見光吸收光譜主要 是受到了來自於主鏈及側鏈兩種可能片段結構所提供之貢獻。
從上述的扭轉角度分布分析中,我們能得知在溶液無序狀態下,分子鏈構形 容易因溶劑效應的影響而有較為雜亂的扭轉角度分布,而與真空下經幾何最佳化 後之結果相比較,兩者有不同的分子構形,使我們不能忽略了溶劑效應所帶來的 影響。也因此,在之後我們將修正其中的扭轉角度,改從分子動力學達動態平衡 後,擷取其中較具代表性的扭轉角度,即最大訊號峰值,如圖 3-10(d)所示,再分 別以量子力學法計算其主鏈與側鏈之吸收光譜,透過組合的方式,從單體延伸探 討至二聚體、三聚體、…,並預測其高分子材料達到一定的高分子量時,其紫外/
可見光吸收光譜會有怎樣的變化。
31
3-5 探討探討探討探討以以以以分子動力學與量子力學模擬分子動力學與量子力學模擬分子動力學與量子力學模擬之分子構形分子動力學與量子力學模擬之分子構形之分子構形及之分子構形及及其對於及其對於其對於其對於紫外紫外紫外紫外/可見光吸收光譜可見光吸收光譜可見光吸收光譜可見光吸收光譜 的影響
的影響 的影響的影響
於分子動力學中,經由分子鏈構形之扭轉角度細部分析,我們可以看到 PBTTTV-v 與 PBTTTV-h 因受到立體空間上的影響,主鏈與側鏈銜接處上的碳原子 與 A 處硫環上之硫原子過於接近,彼此產生排斥,使得兩者皆擁有較廣的主鏈扭 轉角度分布(τMain chain_A)。而於側鏈的部份,可以發現到側鏈接枝於主鏈的位向為平 行或垂直會影響其扭轉角度分布。由於 PBTTTV-h 之側鏈與主鏈相互平行,受限 於與銜接處之立體障礙,使其 τSide chain_B擁有三個分布;而 PBTTTV-v 則因其側鏈 與主鏈垂直,不受限於立體空間上的影響,使其幾乎維持在共平面的 180 度。而 於銜接處的扭轉角度分析中,可以發現到引入乙烯基(Vinyl)因雙鍵不易自由運動,
PBTTTV-v 與 PBTTTV-h 之 τVinyl能維持良好的共平面性。在量子力學中,我們人 工調控 PBTTTV-v 單體之主鏈扭轉角度 τMain chain由 0 度到 180 度,可以清楚的觀察 到不論我們如何去改變主鏈右側硫環與硫環間的扭轉角度(τmain chain_B),其最大吸收 峰的位置幾乎都在一固定值,而整體吸收範圍也幾乎沒有變化;但當我們改變主 鏈左側硫環與硫環之間的扭轉角度(τmain chain_A)時,其最大吸收峰值開始伴隨著角度 偏轉越大而有明顯的藍位移變化,整體的吸收範圍亦跟著變窄。由此,我們會得 知 PBTTTV-v 材料中存在著二維方向的 L 型共軛,而從二聚體之軌域分布也看到 電子雲分布會由主鏈延續到側鏈上。我們從中擷取出片段來進行組合並與實驗結 果相互比較,於實驗兩組不同聚合度之比對結果,可以得知透過各片段不同比例 之組合,能與實驗結果有著良好的印證。而我們也採用相似的人工調控單體角度 測試,可以發現到 PBTTTV-h 材料中也存在著二維延伸共軛的特性,當同時改變 1 和 4 位置為 90 度時,會有兩個較明顯的吸收峰產生;但當改變 2 和 3 位置為 90 度時,卻僅有一個吸收峰出現,由此得知其中有 Z 型共軛的存在,且其方向會通 過 2 和 3 兩處。我們以其三聚體之軌域分布狀況做為參考依據,擷取其中的七個 片段來進行組合並與實驗結果比較,可以發現到隨著高分子聚合度的提升,於共
32
軛長度較長之片段 9T 與 10Z 由原本的 0.05 與 0.06 皆略為升至 0.07,而我們從實 驗之吸收光譜中也能觀察到其在長波長區吸收峰之吸收強度有提升,透過不同比 例之組合方式,能說明和實驗有著相符合之結果。
另外,於 H-PBTTTV-v 與 H-PBTTTV-h 也有著與上述兩材料相似的結果,其 主鏈與側鏈銜接處之立體障礙影響了此兩材料的 τMain chain_A與 H-PBTTTV-h 的 τSide
chain_B有著較寬廣之扭轉角度分布。但比較特別的是,在主鏈與側鏈間銜接處的扭
轉角度分析中,由於單鍵與單鍵之間的運動較為自由,其幾乎是呈現偏離共平面 的 90 度(270 度)附近。接著,我們人工調控 H-PBTTTV-v 與 H-PBTTTV-h 單體之 主鏈與側鏈銜接處的扭轉角度 τSsingle bond由 90 度到 270 度,從其計算出的紫外/可 見光吸收光譜中,觀察到在不同扭轉角度下其吸收光譜皆僅有一個吸收峰產生,
即使是在共平面的 180 度仍僅有一個吸收峰,因而推測主鏈與側鏈間並沒有共軛 的延續性,使得其吸收光譜是由主鏈與側鏈兩者所組合而成。而從兩者之二聚體 軌域分布中,也能明顯的看到電子雲僅有於主鏈或側鏈上之兩種分布,印證了我 們的推測是合理的。
綜合以上所述,對於溶液無序狀態下之分子動力學模擬中之分子構形與真空 下以量子力學法對寡聚體進行結構幾何最佳化之分子構形來說,當我們去比較兩 種不同模擬方法下所得到之分子構形,可以觀察到其溶劑效應的存在對於分子構 形是有一定的影響程度,兩者於相對應位置之扭轉角度幾乎呈現偏離共平面 180 度或 90 度(270 度)反向的趨勢,但其兩者之偏差值幾乎是相似的。因此,在未來考 量到溶劑效應對於分子構形之影響,我們將修正其中的扭轉角度,並與先前之結 果進行相互比較。
33
表 3-1 PBTTTV-v、PBTTTV-h、H-PBTTTV-v 與 H-PBTTTV-h 之主鏈、側鏈與銜接 處之扭轉角度分布。
PBTTTV-v PBTTTV-h H-PBTTTV-v H-PBTTTV-h
τ
Main chain_A
55°±18°
140°±30°
225°±55°
110°±19°
156°±27°
219°±55°
146°±18°
207°±43°
146°±49°
219°±55°
τ
Main chain_B 183°±60° 183°±55° 189°±30° 176°±55°
τ
Main chain_C 183°±49° 183°±43° 176°±43°
340°±20° 170°±55°
τ
Side chain_A 176°±60° 176°±48° 183°±55° 183°±48°
τ
Side chain_B 176°±54°
122°±37°
219°±61°
335°±25°
183°±60°
67°±30°
134°±37°
219°±48°
304°±37°
τ
Vinyl / τ
Single bond 176°±49° 183°±43° 97°±60° 116°±55°
249°±43°

![圖 1-1 於不同位置規整度下(a)HT 達 97%與(b)HT 達 64% P3HT 在氯仿溶液下之紫 外/可見光吸收光譜[19]。](https://thumb-ap.123doks.com/thumbv2/9libinfo/9606753.632702/19.892.197.788.110.958/圖於不同位置規整度達達P在氯仿溶液下之紫外可見光吸收光譜.webp)
![圖 1-3 原子取代型之高分子結構與其在成膜後之紫外/可見光吸收光譜[32]。](https://thumb-ap.123doks.com/thumbv2/9libinfo/9606753.632702/21.892.151.721.116.851/圖13原子取代型之高分子結構與其在成膜後之紫外可見光吸收光譜.webp)
![圖 1-4 PBDTTT-S 與 PBDTDTTT-S-T 於薄膜下之紫外/可見光吸收光譜[43]。](https://thumb-ap.123doks.com/thumbv2/9libinfo/9606753.632702/22.892.141.789.124.759/圖14PBDTTTS與PBDTDTTTST於薄膜下之紫外可見光吸收光譜43.webp)
![圖 1-5 含有純聚噻吩系統之衍生物(a)其結構式及其在(b)氯仿溶液中和(c)薄膜之紫 外/可見光吸收光譜[40]。](https://thumb-ap.123doks.com/thumbv2/9libinfo/9606753.632702/23.892.222.796.116.1116/含有純聚噻吩系統之衍生其結構式及其氯仿溶液中和薄膜之紫外可.webp)
![圖 1-6 含有苯乙烯基三苯胺官能基共軛側鏈之 PT5TPA 二維共軛高分子結構與其 紫外/可見光吸收光譜[49]。](https://thumb-ap.123doks.com/thumbv2/9libinfo/9606753.632702/24.892.238.780.125.517/含有苯乙烯基三苯胺官能基共軛側鏈二維共軛高分子結構與其紫外.webp)
![圖 1-8 聚噻吩系統之衍生物(a)其結構式與 P3HT、PDBT 於良溶劑中(b)高分子鏈之 蜷曲構形及其(c)主鏈上之扭轉角度分布[52]。](https://thumb-ap.123doks.com/thumbv2/9libinfo/9606753.632702/26.892.202.791.111.1162/聚噻吩系統之衍生其結構式於良溶劑高分子鏈之蜷曲構形及其主鏈.webp)