國立臺灣大學理學院化學所 博士論文
Department of Chemistry College of Science
National Taiwan University Doctoral Dissertation
類黃酮化合物在毛細管電泳法之分離與線上濃縮之研究 Studies of Separation and On-line Concentration of Flavonoids in
Capillary Electrophoresis
郭家銘
Chia-Ming Kuo
指導教授﹕林萬寅 博士, 林敬二 博士 Advisor: Wann-Yin Lin, Ph.D.
Ching-Erh Lin, Ph.D.
中華民國 101 年 7 月
July, 2012
ii
謝誌
首先要感謝林敬二教授及林萬寅教授這幾年來對我的指導,老師們的學者風 範總令我景仰不已,無論做研究或待人處事都使我長了許多視野,也知道我還有 很多不足得好好再學習的地方。並感謝口試委員林金全老師、桂椿雄老師及吳劍 侯老師蒞臨指導,並提醒我論文不足之處,還有很感謝預口試請的劉春櫻老師各 方面的包容及配合。
一路求學過程非常感謝帶我長大的奶奶,她一直希望家裡小孩能唸個博士,
感謝一直各方面支援我的叔公跟叔叔還有伯父伯母,一直督導我好好做個正派有 用的人,外婆、大舅舅、舅媽跟表弟表妹也一直讓我回南部的時候感到家庭的溫 暖,還有一直很疼我的媽媽,感謝妳讓我到這世上體驗人生。
謝謝研究所生活中認識的所有人,旭敦學長引導我來學習毛細管電泳,一開 始教我如何報告的郁庭學姊,實驗上幫我解決非常多問題的宇智大學長、碩班時 照顧過我的怡茹學姊、幫我解決任何 word 處理及懂很多有的沒有的的艾傑學長、
常常一起聊些五四三的育錚學姊、方旬、信凱、33、番仔、小白兔和阿毛、碩班 時的夥伴思親及雯堯、總是有很多有趣新奇事物的巍儒、廷璋、育生、losie、常 一起去看電影的夠用、紅白、很白癡的崇元,因為你們這些系上的朋友來實驗室 才會一直有動力。
也非常感謝高中大學一路相挺同學們,讓我一開始來台北有黑戶可以住的卓 勳跟建安,建安常常給我很多其他的幫助,讓我在學業之餘也可以追求音樂的夢 想、在低潮時可互相吐苦水的小米、總提供很多有用的資訊以前常借我機車的紅 燒魚、超好約的玩樂咖林白爛、常找我吃飯的君豪、鬍子、河馬、愛玉、林彥伯、
哲維。
感謝詞曲創作社認識的朋友一直學業上及音樂上的互相鼓勵,幫我得到政大
iii
金旋獎創作組第二名的壩子跟熱拿鐵,常關心我然後常找我打桌球保齡球的 skippy,還有感謝樂園工作室跟工作室的大夥,冠宇、kelly、貞子、小茵,讓我 周末有可以去放鬆心情的市內桃園,感謝小馬讓我在課業外玩音樂能玩到賣出第 一首歌,完成了來台北後很希望能達到的夢想。
感謝所有教過的家教學生及他們的家長,有家教薪水讓我有更多機會去探索 台北這個複雜的城市,也開了很多眼界並學習到許多事物。
最後感謝我的女朋友小武,一直陪在我身邊陪我,鼓勵我,而且總是能陪我 嘗試各樣的事物或吃吃喝喝,讓我總是保持著開心幸福的感覺。
在台大這麼多年終於要畢業了,碩班畢業的時候忘了寫謝誌,所以這次多寫 了一些,真的很感謝這段時間遇到的每個親朋好友對我的照顧,希望往後的人生 還能有很多機會互相打擾指教,這段時間能遇到你們真是我運氣非常好。
iv
中文摘要
本論文選擇以類黃酮化合物(flavonoids)以毛細管電泳法來探討偵測靈敏度 的增強,注重小分子在毛細管電泳上之線上濃縮技巧。結果分為兩大部分:
第一部份先以毛細管區帶電泳法(CZE)及微胞電動力層析法(MEKC)做分離,
接著測試幾種不同堆積模式,包括電極極性反向堆積模式(large volume sample stacking with reverse electrode polarity, LVSS)及大體積反向電壓微胞掃集模式 (large volume sample stacking-sweeping, LVSS-sweeping)或正向添加 SDS 的掃集 模式,其中 LVSS-Sweeping 及正向掃集的最佳化條件較難求得,而 LVSS 法於 0.4 分鐘時切換電壓方向可得良好的堆積,並測得 hesperetin、naringenin、quercetin 與 kaempferol 的偵測極限分別 14.87、14.71、19.76 與 16.66 ng/mL(S/N = 3),並 可用於真實樣品偵測。
第二部份強調於逆向掃集模式的改良,最佳條件為使用掃集模式(sweeping),
於分離緩衝中及樣品基質中添加適當電解質,有助改善長時間進樣的樣品濃縮。
甚至可讓注入類黃酮化合物時間達 480 秒,最大樣品體積約佔毛細管總長的 96.5
%。樣品基質使用 pH 2.0 的 20 mM 磷酸鹽緩衝液,分離緩衝液為 pH 2.0 的 20 mM 樣品注入時間為 120 秒時,磷酸緩衝液並分別添加 50 mM的 SDS,ACN 則分別添加 10 %。而 120 秒的樣品注入所得hesperetin、naringenin、quercetin及 kaempferol 之偵 測極限分別為 43.53、38.25、57.38 及 48.55 ng/mL(S/N = 3)。而 480 秒的樣品注入所得 hesperetin、naringenin、quercetin及 kaempferol 之偵測極限分別為 21.28、15.37、24.26 及 24.7 ng/mL(S/N = 3)。並能有效應用於真實樣品之檢測。
關鍵字:類黃酮化合物、毛細管電泳、線上濃縮、樣品掃集、大體積堆積。
v
Abstract
In this dissertation, four flavonoid analytes were selected to investigate the enhancement of detection sensitivity by capillary electrophoresis. Several on-line concentration modes studied were divided into two parts:
In the first part, CZE and MEKC modes were studied first, and then the large volume stacking with switching the electrode polarity (LVSS) mode and the LVSS-sweeping mode were used to concentrate the anlaytes. Amoung these modes, the LVSS with switching polarity at 0.4 min was the optimal condition. The limits of detections (S/N = 3) of hesperetin、naringenin、quercetin and kaempferol were determined to be 14.87、14.71、19.76 and 16.66 ng/mL, respectively.
In the second part, the sweeping technique was improved to concentrate the analytes. With 120 sec sample injection, the concentration of phosphate buffer at 20 mM was used as the sample matrix, while the separation buffer consisting of 20 mM phosphate electrolyte and 50 mM SDS and 10 % acetonitrile at pH 2.0 was optimized, and the limits of detections (S/N = 3) of hesperetin、naringenin、quercetin and kaempferol were determined to be 43.53、38.25、57.38 and 48.55 ng/mL, respectively.
Sample injection up to 480 sec can also be achieved for baseline separation of four flavonoids in the sweeping mode, and the limits of detections (S/N = 3) of hesperetin、
naringenin、quercetin and kaempferol were determined to be 21.28、15.37、24.26 and 24.7 ng/mL, respectively. The method was successfully applied to determine flavonoids in several real samples.
Key words: flavonoids 、 capillary electrophoresis 、 on-line concentration 、 sweeping、large volume sample stacking.
vi
目錄
審定書... i
謝誌... ii
中文摘要... iv
Abstract ... v
第 1 章 序論... 1
1-1 引言 ... 1
1-2 毛細管電泳發展史 ... 1
1-3 毛細管電泳的分離原理 ... 3
1-3-1 電泳遷移率(Electrophoretic mobility) ... 3
1-3-2 電滲流(Electroosmotic flow, EOF)與流型(Flow profile)... 5
1-3-3 分離效率(Separation efficiency) ... 6
1-4 毛細管電泳的分離模式 ... 8
1-4-1 毛細管區帶電泳法(CZE) ... 8
1-4-2 微胞電動力層析法(MEKC) ... 9
1-4-3 毛細管凝膠電泳法(CGE) ... 9
1-4-4 毛細管等電聚焦電泳法(CIEF)... 10
1-4-5 毛細管等速電泳法(CITP)... 10
1-4-6 毛細管電層析法(CEC) ... 10
1-5 毛細管電泳的注入方式 ... 11
1-5-1 水動力注入法(Hydrodynamic injection) ... 11
1-5-2 水靜力注入法(Hydrostatic injection)... 11
1-5-3 電動力注入法(Electrokinetic injection) ... 12
1-6 毛細管電泳的儀器裝置 ... 13
vii
1-7 線上樣品濃縮的原理與相關計算 ... 13
1-8 毛細管電泳之線上樣品濃縮技術 ... 16
1-8-1 正向堆積模式(NSM)... 17
1-8-2 電極極性反向堆積模式(REPSM) ... 18
1-8-3 反向遷移微胞濃縮(SRMM) ... 18
1-8-4 增強電場樣品進樣(FESI) ... 19
1-8-5 以反向遷移微胞增強電場樣品進樣(FESI-RMM) ... 19
1-8-6 利用反向遷移微胞濃縮與水之堆積(SRW) ... 19
1-8-7 掃集(Sweeping) ... 20
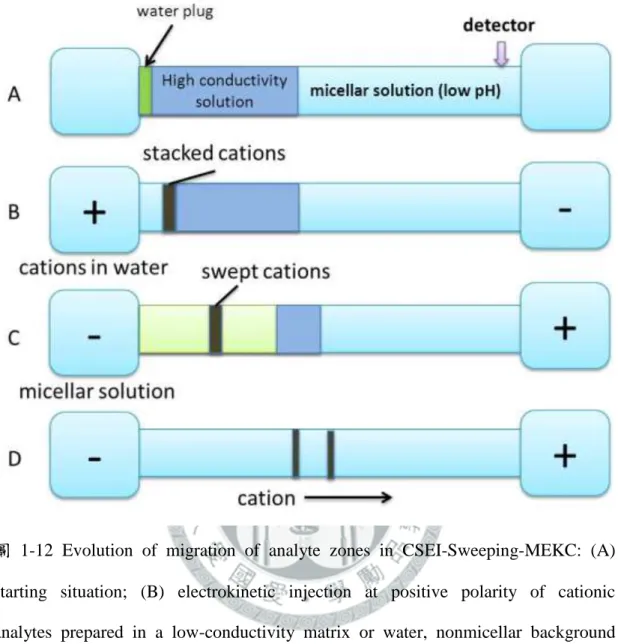
1-8-8 陽離子選擇性注射法與掃集之結合模式(CSEI-Sweeping) .. 21
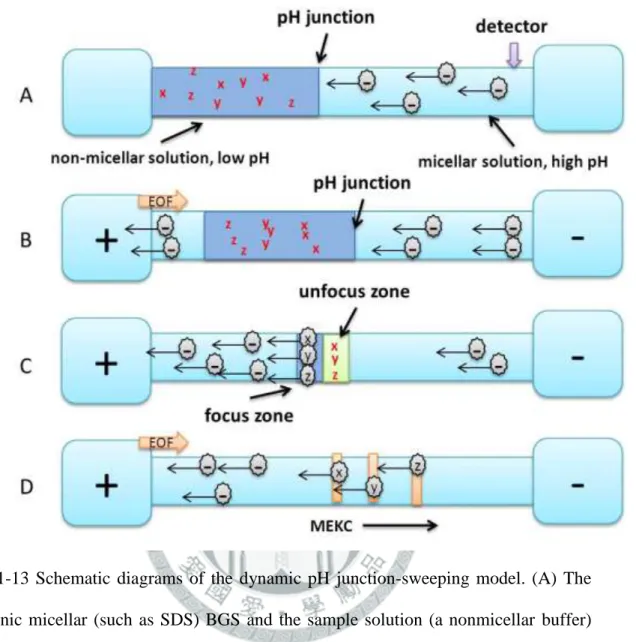
1-8-9 動力 pH 接合-掃集(dynamic pH junction-Sweeping) ... 21
1-9 毛細管電泳之未來展望 ... 22
第 2 章 文獻探討... 40
2-1 類黃酮類化合物 ... 40
2-1-1 基本介紹 ... 40
2-1-2 結構及分類 ... 40
2-1-3 我們選用之分析物 ... 42
2-2 類黃酮化合物之毛細管電泳相關文獻 ... 43
第 3 章 實驗設備與方法... 51
3-1 實驗藥品 ... 51
3-1-1 分析物 ... 51
3-1-2 緩衝溶液 ... 51
3-1-3 緩衝溶液添加劑 ... 52
3-1-4 其他試劑與藥品 ... 52
3-2 實驗設備及耗材 ... 52
viii
3-2-1 毛細管電泳儀 ... 52
3-2-2 毛細管柱 ... 53
3-2-3 實驗室型酸鹼度/氧化還原電位計(Laboratory pH Meter) ... 54
3-2-4 實驗室型導電度計(Laboratory conductivity meter) ... 54
3-3 實驗方法 ... 55
3-3-1 分析物之配製 ... 55
3-3-2 緩衝溶液之配置 ... 55
3-3-3 紅酒樣品之萃取 ... 56
3-3-4 蜂蜜樣品之萃取 ... 56
3-3-5 毛細管的處理 ... 56
3-3-6 實驗操作 ... 57
3-3-7 毛細管電泳之相關計算 ... 57
第 4 章 結果與討論... 61
4-1 線上濃縮模式之挑選 ... 61
4-2 一般模式-毛細管區帶電泳法(CZE) ... 62
4-3 大體積樣品堆積(LVSS) ... 62
4-4 正向大體積堆積掃集 (LVSS-Sweeping) ... 63
4-5 正向掃集模式 (sweeping with normal polarity) ... 64
4-5-1 pH 值 ... 64
4-5-2 注入時間 ... 64
4-6 逆向掃集模式 (sweeping with reverse polarity) ... 64
4-6-1 不同秒數的樣品注入變化 ... 64
4-6-2 120 秒樣品注入 ... 65
4-6-3 480 秒注入 ... 67
4-7 各方法比較 ... 68
ix
4-8 結論 ... 69 參考文獻... 111
圖目錄
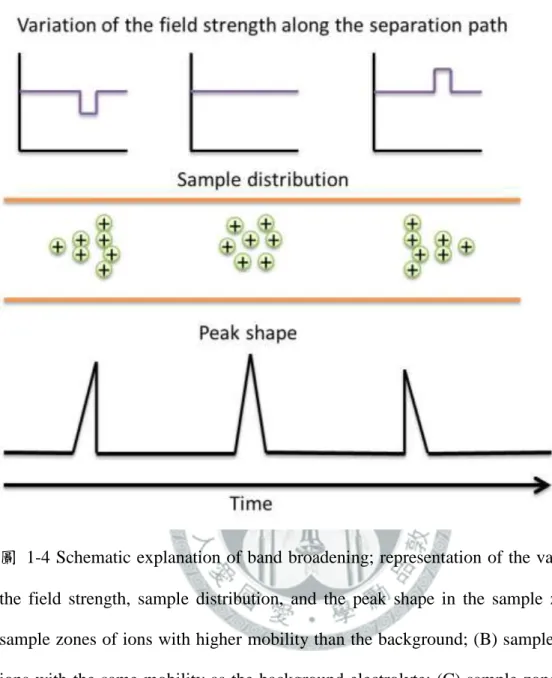
圖 1-1 Stern model of the electric double layer is occurring at the interface between an electrolyte solution and the surrounding surface. ... 24 圖 1-2 Flow profiles of HPLC (A) and CE (B), respectively. ... 25 圖 1-3 Instrumental set-up of a capillary electrophoresis system ... 26 圖 1-4 Schematic explanation of band broadening; representation of the variation of the field strength, sample distribution, and the peak shape in the sample zone: (A) sample zones of ions with higher mobility than the background;
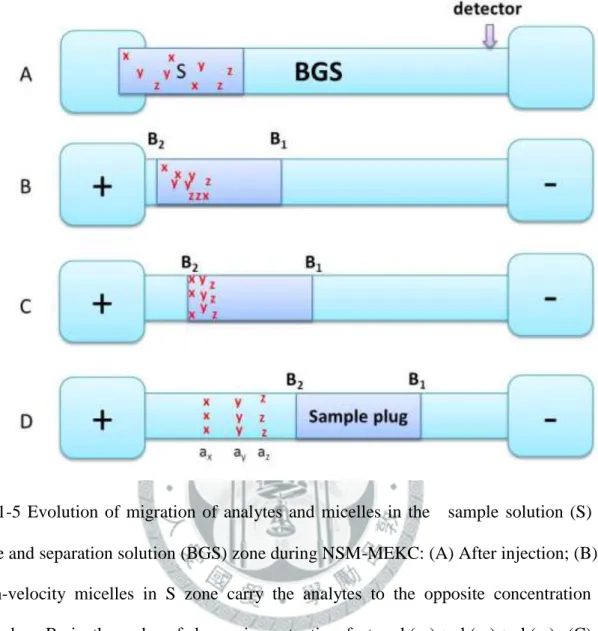
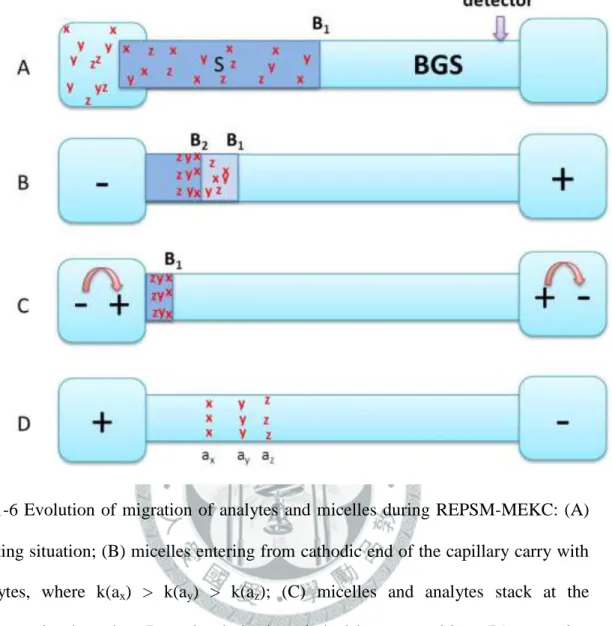
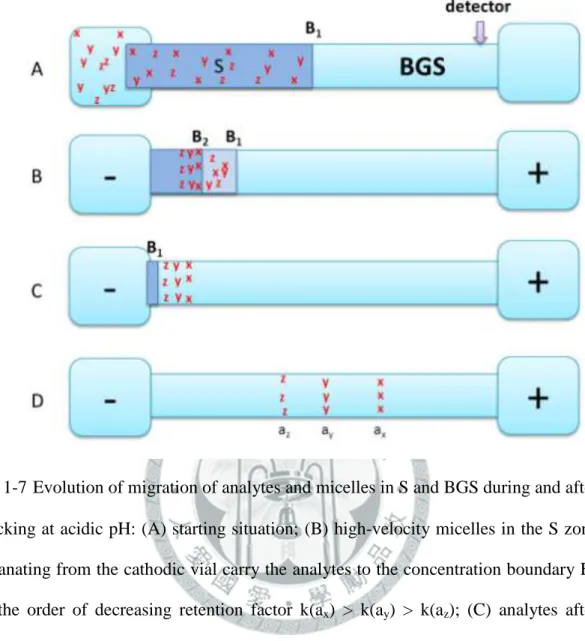
(B) sample zones of ions with the same mobility as the background electrolyte; (C) sample zones of ions with lower mobility than the background electrolyte. ... 27 圖 1-5 Evolution of migration of analytes and micelles in the sample solution (S) zone and separation solution (BGS) zone during NSM-MEKC: (A) After injection; (B) high-velocity micelles in S zone carry the analytes to the opposite concentration boundary B2 in the order of decreasing retention factor: k(ax) > k(ay) > k(az); (C) analytes stack in B2 into thin concentrated zones; (D) analytes zones separate by virtue of MEKC. ... 28 圖 1-6 Evolution of migration of analytes and micelles during REPSM-MEKC: (A) Starting situation; (B) micelles entering from cathodic end of the capillary carry with analytes, where k(ax) > k(ay) > k(az); (C) micelles and analytes stack at the concentration boundary B1 and polarity is switched later to positive; (D) separation and later detection of zones. ... 29 圖 1-7 Evolution of migration of analytes and micelles in S and BGS during and after stacking at acidic pH: (A) starting situation; (B) high-velocity micelles in the S zone emanating from the cathodic vial carry the analytes to the concentration boundary B1 in the order of decreasing retention factor k(ax) >
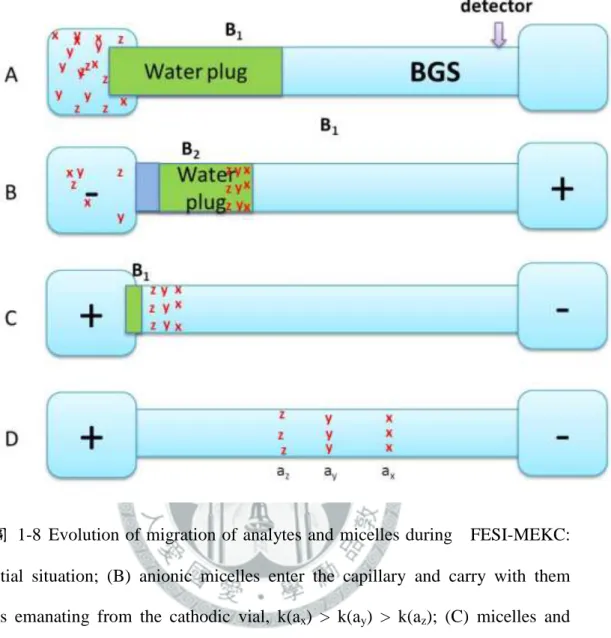
k(ay) > k(az); (C) analytes after stacking leave B1 prior to the removal of the sample matrix due to electrophoresis in the BGS zone; (D) analyte zones migrate toward the detector and continue to separate. ... 30 圖 1-8 Evolution of migration of analytes and micelles during FESI-MEKC: (A)
x
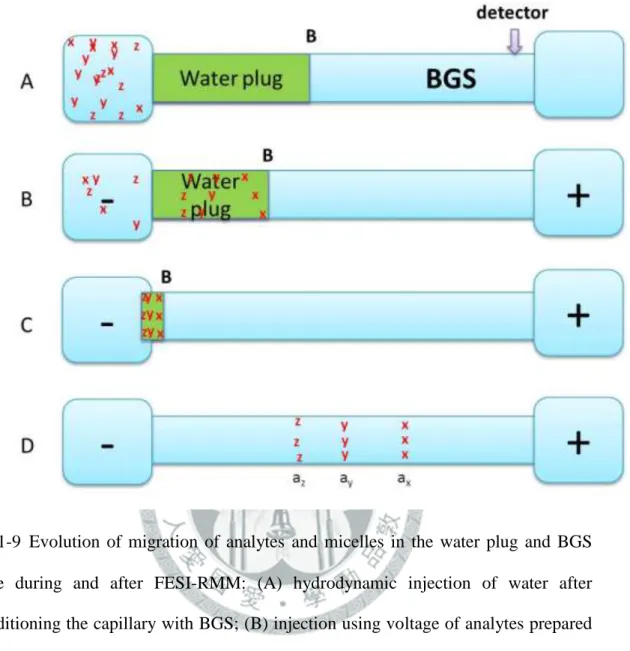
initial situation; (B) anionic micelles enter the capillary and carry with them analytes emanating from the cathodic vial, k(ax) > k(ay) > k(az); (C) micelles and analytes stacked at the concentration boundary B2, voltage is cut and the sample vial is replaced by another BGS vial when the measured current is approximately 97-99 % of the predetermined current, voltage is then applied at positive polarity; (D) separation of zones occurs. ... 31 圖 1-9 Evolution of migration of analytes and micelles in the water plug and BGS zone during and after FESI-RMM: (A) hydrodynamic injection of water after conditioning the capillary with BGS; (B) injection using voltage of analytes prepared in a micellar matrix found in the cathodic vial (analytes solubilized in these micelles enter the water plug in the order of decreasing retention factor k(ax) > k(ay) > k(az)); (C) voltage is shut, the sample vial is replaced by another BGS vial and voltage is applied at negative polarity again, and analytes stack at the concentration boundary B; (D) focused bands separate by virtue of MEKC. ... 32 圖 1-10 Evolution of migration of analytes and micelles in the water zone, S and BGS zones during SRW-MEKC: (A) Hydrodynamic injection of water followed by S after conditioning the capillary with BGS; (B) application of voltage at negative polarity with the BGS in the inlet and outlet vials (analytes solubilized in the micelles enter the water zone and stack in the concentration boundary in the order of decreasing retention factor k(ax) >
k(ay) > k(az)); (C) separation of stacked zones prior to the total removal of the low conductivity zones; (D) focused bands separate by virtue of MEKC.
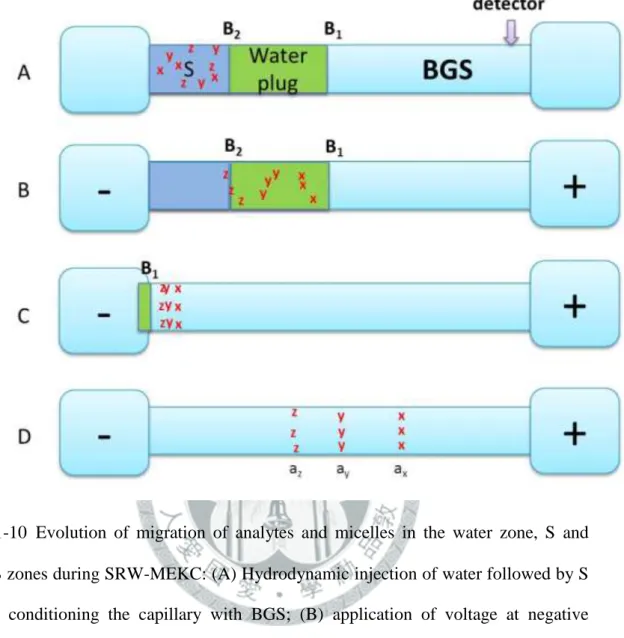
... 33 圖 1-11 Evolution of migration of analyte zones in Sweeping-MEKC: (A) Starting situation, injection of sample solution S with length l inj after conditioning the capillary with BGS (found at both electrode vials); (B) micelles from the cathodic vial enter the S zone or capillary (area enclosed by blue lines) and sweep the analytes into narrower bands depending on the retention factor (ka1 , ka2 , depicted by red and green lines, respectively); (C) First batch of micelles that entered the S zone reaches the interface I between S and BGS zones; (D) Separation of zones based on MEKC. ... 34 圖 1-12 Evolution of migration of analyte zones in CSEI-Sweeping-MEKC: (A)
starting situation; (B) electrokinetic injection at positive polarity of cationic analytes prepared in a low-conductivity matrix or water, nonmicellar background buffer found in the outlet end, cationic analytes focus or stack at the interface between the water zone and high-conductivity buffer
xi
void of organic solvent zone; (C) application of voltage at negative polarity, micelles entering from the cathodic vial into the capillary and sweep the
analytes to narrower bands; (D) separation of zones based on MEKC. ... 35
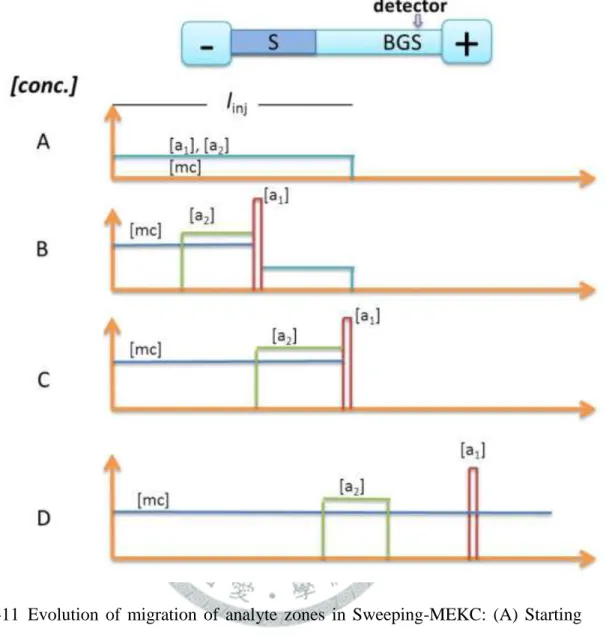
圖 1-13 Schematic diagrams of the dynamic pH junction-sweeping model. (A) The anionic micellar (such as SDS) BGS and the sample solution (a nonmicellar buffer) are injected into the capillary, respectively; (B) when the injection is complete, a positive polarity is applied (if a negatively charged SDS surfactant is used) to power the CE separation; (C) the neutral analytes are converted to anions and are swept by the SDS micelles; (D) separation occurs by the MEKC mode. ... 36
圖 2-1 類黃酮化合物基本架構 ... 41
圖 2-2 類黃酮類化合物之結構及分類 ... 46
圖 3-1 左一為智利紅酒梅若紅,左二為台灣紅麴紅酒,左三為美國加州紅酒, 左四為西班牙克里斯提娜紅酒... 51
圖 3-2 Beckman P/ACE MDQ 毛細管電泳儀 ... 53
圖 3-3 毛細管 ... 53
圖 3-4 酸鹼度計 ... 54
圖 3-5 導電度計 ... 55
圖 3-6 四個分析物之結構及 pKa 值... 58
圖 3-7 Beckmann 軟體 120 sec 樣品注入的計算 ... 59
圖 3-8 Beckmann 軟體 480 sec 樣品注入的計算. ... 59
圖 4-1 Effect of pH of running buffer: (A) pH 9.0; (B) pH 9.5; (C) pH 10.0. Separation buffer: 50 mM borate buffer. Sample: 10 μg/mL flavonoids. Injection pressure: 1 psi, applied voltage: +25 kv. Injection time: 10 sec. .. 70
圖 4-2 Effect of injection time of sample on the stacking efficiency: 1 μg/mL mix flavonoids dissolved in water, separation buffer, 50 mM borate buffer at pH 9.0; Injection pressure, 1 psi; applied voltage, +25 kv. Injection time: 5-60 sec. ... 71
圖 4-3 (A) LVSS with 480 sec analyte injection, (B) LVSS with 120 sec analyte injection, (C) CZE with 5 sec sample injection, analyte concentration : 1 μg/mL for (A) and (B), 10 μg/mL for (C). Peak identification, 1 = hesperetin, 2 = naringenin, 3 = quercetin, 4 = kaempferol. ... 72
圖 4-4 The calibration curves of hesperetin and naringenin, LVSS with polarity switch at 0.4 min. ... 73
圖 4-5 The calibration curves of quercetin and kaempferol, LVSS with polarity switch at 0.4 min. ... 74 圖 4-6 Effect of glycol and SDS in the running buffer: (A) 10 % glycol and 20 mM
xii
SDS, (B) 10 % glycol and 50 mM SDS, (C) 20 % glycol and 20 mM SDS and (D) 20 % glycol and 50 mM SDS, Separation buffer: 50 mM borate buffer at pH 9.0. Sample: 1 μg/mL flavonoids. Injection pressure: 1 psi, applied voltage: +25 kv. Injection time: 120 sec. ... 75 圖 4-7 Effect of pH of running buffer electrolyte: sample solution, flavonoids (10 μg/mL) dissolved in 20 mM phosphate buffer, separation buffer, 20 mM phosphate buffer containing 25 mM SDS at different pH: (A) pH 6.5 (B) pH 7.0 (C) pH 7.5. Injection time:120 sec. Peak identification, 1 = hesperetin, 2 = naringenin, 3 = quercetin, 4 = kaempferol... 76 圖 4-8 The variation of peak height of analytes as a function of pH in the range 6.5~7.5 in a phosphate (20 mM) buffer. Injection time:120 sec. Analyte identification, 1 = hesperetin, 2 = naringenin, 3 = quercetin, 4 = kaempferol.
... 77 圖 4-9 Effect of injection time of sample on the stacking efficiency: mix flavonoids 10 μg/mL for (A), 1 μg/mL for (B)~(E) dissolved in 20 mM phosphate buffer at pH 6.5, separation buffer, 20 mM phosphate buffer containing 25 mM SDS at pH 6.5; Injection pressure, 1 psi; applied voltage, +20 kV.
Injection time: (A) 5 sec, (B) 60 sec, (C) 120 sec, (D) 180 sec, (E) 240 sec.
... 78 圖 4-10 Plots of peak height versus injection time with a mixture of four flavonoids at a sample concentration of 1 μg/mL dissolved in 20 mM phosphate buffer containing methanol (1 %, v/v) at pH 2.0; separation buffer, 20 mM phosphate containing 20 mM SDS at pH 2.0; applied potential, - 15 kV.... 79 圖 4-11 Plots of peak area versus injection time with a mixture of four flavonoids at a sample concentration of 1 μg/mL dissolved in 20 mM phosphate buffer containing methanol (1 %, v/v) at pH 2.0; separation buffer, 20 mM phosphate containing 20 mM SDS at pH 2.0; applied potential, - 15 kV.... 80 圖 4-12 Effect of applied voltage on the stacking efficiency and separation of the mix flavonoids: Sample solution, mix naringenin, hesperetin, quercetin and kaempferol (1 μg/mL for each) dissolved in 20 mM phosphate buffer containing methanol (1 %, v/v) at pH 2.0; Injection pressure, 1 psi; injection time, 120 sec. Applied voltage: (A) -25 kV, (B) -22 kV, (C) -20 kV, (D) -18 kV, (E) -15 kV ... 81 圖 4-13 The variation of peak area of analytes as a function of applied voltage (-15 kV, -18 kV, -20 kV, -22 kV, -25 kV) in a phosphate (20 mM) buffer at pH 2.0. Injection time : 120 sec. Analyte symbol, 1 = hesperetin, 2 = naringenin, 3 = quercetin, 4 = kaempferol. ... 82
xiii
圖 4-14 The variation of peak height of analytes as a function of applied voltage(-15 kV, -18 kV, -20 kV, -22 kV, -25 kV) in a phosphate (20 mM) buffer at pH 2.0. Injection time: 120 sec. Analyte symbol, 1 = hesperetin, 2 = naringenin, 3 = quercetin, 4 = kaempferol. ... 83 圖 4-15 Effect of acetonitrile concentration in the buffer on the separation efficiency and separation of the mix flavonoids: mix naringenin, hesperetin, quercetin and kaempferol (1 μg/mL for each) dissolved in 20 mM phosphate buffer containing methanol (1 %, v/v) at pH 2.0; Injection pressure, 1 psi; injection time, 120 sec; applied voltage, -15 kv. Acetonitrile concentration: (A) 0 %, (B) 4 %, (C) 6 %, (D) 10 % ... 84 圖 4-16 Effect of methanol concentration in the buffer on the separation efficiency and separation of the mix flavonoids: mix naringenin, hesperetin, quercetin and kaempferol (1 μg/mL for each) dissolved in 20 mM phosphate buffer containing methanol (1 %, v/v) at pH 2.0; Injection pressure, 1 psi; injection time, 120 sec; applied voltage, -15 kv. Methanol concentration: (A) 0 %, (B) 4 %, (C) 8 %, (D) 10 %, ... 85 圖 4-17 Effect of SDS concentration in the buffer on the stacking efficiency and separation of the mix flavonoids: mix naringenin, hesperetin, quercetin and kaempferol (1 μg/mL for each) dissolved in 20 mM phosphate buffer containing methanol (1 %, v/v) at pH 2.0; Injection pressure, 1 psi; injection time, 120 sec; applied voltage, -15 kv. SDS concentration: (A) 25 mM, (B) 50 mM. ... 86 圖 4-18 The variation of peak area of analytes as a function of SDS concentration in the range 25~100 mM in a phosphate (20 mM) buffer at pH 2.0. Injection time:120 sec. Peak identification, 1 = hesperetin, 2 = naringenin, 3 = quercetin, 4 = kaempferol. ... 87 圖 4-19 The variation of peak height of analytes as a function of SDS concentration in the range 25~100 mM in a phosphate (20 mM) buffer at pH 2.0. Injection time : 120 sec. Peak identification, 1 = hesperetin, 2 = naringenin, 3 = quercetin, 4 = kaempferol. ... 88 圖 4-20 Effect of sample matrix on separation and peak height of flavonoids: Sample solution, flavonoids (1 μg/mL) dissolved in (A) 10 mM, (B) 20 mM, (C) 50 mM, (D) 75 mM, (E) 100 mM phosphate buffer containing methanol (1 %, v/v) at pH 2.0; separation buffer, 20 mM phosphage containing 50 mM SDS at pH 2.0; applied potential, -15 kV. Peak identification, 1 = hesperetin, 2 = naringenin, 3 = quercetin, 4 = kaempferol. ... 89 圖 4-21 The variation of peak height of analytes as a function of phosphate
xiv
concentration in the range 10~100 mM in the sample matrix at pH 2.0.
Injection time : 120 sec. ... 90 圖 4-22 The calibration curves of hesperetin and naringenin in the sweeping mode with 120 sec sample injection. ... 91 圖 4-23 The calibration curves of quercetin and kaempferol in the sweeping mode with 120 sec sample injection. ... 92 圖 4-24 Electropherogram of honey obtained with addition of SDS at 50 mM in 20 mM phosphate buffer at pH 2.0. Injection pressure, 1 psi; injection time, 120 sec; applied voltage, -15 kV... 93 圖 4-25 Electropherogram of red wine 1 obtained with addition of SDS at 50 mM in 20 mM phosphate buffer at pH 2.0. Injection pressure, 1 psi; injection time, 120 sec; applied voltage, -15 kV... 94 圖 4-26 Electropherogram of red wine 5 obtained with addition of SDS at 50 mM in 20 mM phosphate buffer at pH 2.0. Injection pressure, 1 psi; injection time, 120 sec; applied voltage, -15 kV... 95 圖 4-27 Effect of acetonitrile concentration in the buffer on the stacking efficiency and separation of the mix flavonoids: mix naringenin, hesperetin, quercetin and kaempferol (1 μg/mL for each) dissolved in 20 mM phosphate buffer containing methanol (1 %, v/v) at pH 2.0; Injection pressure, 1 psi; injection time, 480 sec; applied voltage, -15 kv. Acetonitrile concentration: (A) 10 %, (B) 15 %, (C) 20 %, ... 96 圖 4-28 Effect of SDS concentration in the buffer on the stacking efficiency and separation of the mix flavonoids: naringenin, hesperetin, quercetin and kaempferol (1 μg/mL for each) dissolved in 20 mM phosphate buffer containing methanol (1 %, v/v) at pH 2.0; Injection pressure, 1 psi; injection time, 480 sec; applied voltage, -15 kv. SDS concentration: (A) 25 mM, (B) 50 mM, (C) 75 mM, (D) 100 mM. ... 97 圖 4-29 The variation of peak area of analytes as a function of SDS concentration in the range 25~100 mM in a phosphate (20 mM) buffer at pH 2.0. Injection time : 480 sec. Peak identification, 1 = hesperetin, 2 = naringenin, 3 = quercetin, 4 = kaempferol. ... 98 圖 4-30 The variation of peak height of analytes as a function of SDS concentration in the range 25~100 mM in a phosphate (20 mM) buffer at pH 2.0. Injection time: 480 sec. ... 99 圖 4-31 The calibration curves of hesperetin and naringenin at the sweeping mode with reverse polarity. Sample injection time: 480 sec. ... 100 圖 4-32 The calibration curves of quercetin and kaempferol at the sweeping mode
xv
with reverse polarity. Sample injection time: 480 sec. ... 101
圖 4-33 Electropherogram of different stacking mode (A) in normal mode (B) LVSS (C) Sweeping (D)Sweeping. Sample concentration: (A) 10 μg/mL, (B)~(D) 1 μg/mL. Injection: (A) 1 psi;5 sec; (B)and (C)1 psi; 120 sec; (D) 1 psi, 480 sec. ... 102
表目錄 表 1-1 Injection techniques in capillary electrophoresis ... 37
表 1-2 Comparisons of CE and HPLC ... 38
表 1-3 Conditions of on-line preconcentration techniques in MEKC using SDS micelles ... 39
表 2-1 The references about MEKC (part I). ... 47
表 2-2 The references about MEKC (part II). ... 48
表 2-3 The references about MEKC (part III) ... 49
表 2-4 The references about MEKC (part IV). ... 50
表 3-1 MEKC 常用界面活性劑之臨界微胞濃度值(CMC)及單體聚集數(N)[137]. ... 60
表 4-1 The conductivity of the phosphate buffer matrix ... 103
表 4-2 The conductivity of running buffer ... 104
表 4-3 LOD and reproducibility of flavonoids for 120 sec injection in the LVSS mode(n=3) ... 105
表 4-4 LOD and reproducibility of flavonoids for 120 sec injection in the sweeping mode (n=6) ... 106
表 4-5 Recovery of two kinds of wine ... 107
表 4-6 The flavonoid contents for different species of wines ... 108
表 4-7 LOD and reproducibility of flavonoids for 480 sec injection in the sweeping mode(n = 3) ... 109
表 4-8 Comparison of the enhancement effect of different modes ... 110
1
第1章 序論
1-1 引言
近年來,毛細管電泳為一已成熟之分離技術,其利用加電壓於毛細管兩端,
使得帶電荷之分析物擁有不同的電泳速度而分離,相較於傳統的 HPLC 分離技術,
其特色為分析樣品量少,時間短,廣泛運用於分離各種藥物或環境汙染物之鑑定 上。但因毛細管管徑狹小,使得其靈敏度不佳,更使得偵測極限較高,故各式各 樣的離線濃縮方法及線上濃縮方式陸續地被開發出來。
許多研究指出類黃酮化合物具有抗氧化性、抗心血管疾病、以及抗癌的功效,
且廣泛地存在於各種水果、植物及食物中,例如蜂蜜及紅酒或一些天然藥材都含 有類黃酮化合物,本篇論文研究類黃酮化合物在不同條件下之線上濃縮效果,並 試著應用到真實樣品(蜂蜜或紅酒)之檢測。
1-2 毛細管電泳發展史
電泳(Electrophoresis)定義為溶液中之帶電荷分子,在電場作用下受到吸引力 或排斥力,而朝向相反電荷之兩極方向遷移的現象。利用帶電荷粒子的帶電性、
帶電量及粒子的大小差異形成不同的遷移速度而達到分離的效果,這種分離技術 即稱為電泳分離法。早先電泳實驗大多在親水性凝膠或其他支撐介質上進行,通 常用以分離較大的分子物質如蛋白質或其他生化物質,但是凝膠的製備不僅費時 費力,實驗結果的再現性亦不佳。然而,電泳分離法在化學或生化的領域中被視 為是一種非常重要的分析技術。
1897 年,Kohlraush[1]首先提出帶電荷粒子在電解質中的遷移方程式,利用 物理定律來解釋離子在電解質中的遷移行為。直到 1937 年,瑞典科學家 Tiselius[2]
以”移動邊界(moving boundary)”方法將人體血清中的 α、β、γ 球蛋白以及胺基酸 分離出來,由於他在電泳技術發展與應用有卓越貢獻,使他在 1948 年榮獲諾貝
2
爾化學獎,從此電泳分離技術逐漸受到大家重視。
理論上電泳的分離效率與外加電場的大小成正比,但施加高電壓時會產生焦 耳熱(Joule heating)而導致熱擴散與對流的現象,此熱效應會使介質產生黏度梯度 及速度梯度而導致樣品區帶變寬,使得分離的效率受到熱擴散和對流的限制[3]。
此種焦耳熱效應會隨著電場的強度增加而增加,早期為了克服這項限制,使用許 多介質如濾紙、聚丙烯醯胺(polyacrylamide)、澱粉(strach)或瓊脂(agarose)凝膠來 穩定電解質的擾動。另外以較小內徑的管柱來乘載電解質,也可以減少中心到兩 側或管柱內徑向的黏度差異與速度差異。
1967 年 Hjerten[4]使用內徑僅 1~3 mm 的石英管以減少熱效應,且以甲基纖 維素(methylcellulose)修飾毛細管管壁,然後將毛細管繞著管柱中心軸緩慢的旋轉 以防止電滲流的產生,成功地分離無機離子、核酸的水解物、蛋白質、核酸與濾 過性病毒等。1974 年 Virtanen 和 Mikkers[5, 6]使用 200~500 μm 內徑的玻璃毛細 管和聚四氟乙烯管進行電泳分離的實驗,可有效地控制焦耳熱產生的對流問題。
目前大家所論及的毛細管電泳技術,又稱為高效能毛細管電泳法(High performance capillary electrophoresis, 簡稱 HPCE),其為在內徑約 25~75 μm 的毛 細管中進行電泳分離實驗,因口徑小的毛細管內壁表面積大,散熱率好,使得理 論板數大為提高。然而毛細管與傳統電泳的主要差別為前者是在毛細管中進行的,
後者為在平板上進行,毛細管電泳是由 Jorgenson 和 Lukacs[7-9]在 1981 年所提 出的,他們以內徑 75 μm 的毛細管柱來分離衍生化的胺基酸,在充滿緩衝溶液的 毛細管兩端施加 30 kV 的高電壓進行分離實驗,並且用螢光偵測器做線上偵測,
成功地分離胺基酸的衍生物。他們詳細探討各種影響分離效率的因素,並且建
立毛細管電泳的理論和實驗基礎。
毛細管區帶電泳法(Capillary zone electrophoresis)於一般情況下只能分離帶 電荷離子,而無法應用於中性分析物,中性分析物通常隨電滲流一起遷移。但是
3
Terabe 在 1984 年 [10] 提 出 微 胞 電 動 力 毛 細 管 層 析 法 (Micellar electrokinetic capillary chromatography),使得毛細管電泳法能夠同時分離帶電荷粒子與中性分 析物。此技術為在緩衝溶液中添加濃度大於臨界微胞濃度值(CMC)的界面活性劑 而形成微胞,利用分析物在微胞相與水相有不同的分配係數來進行分離。1985 年 Hjerten[11]將傳統的等電聚焦技術應用到毛細管電泳中,進而提出毛細管等電 聚焦法(Capillary isoelectric focusing)。1987 年 Cohen 和 Karger[12]亦將傳統的凝 膠電泳法應用到毛細管中以分離胜肽與蛋白質,因而提出毛細管凝膠電泳 (Capillary gel electrophoresis)的技術。同年,Tsuda[13]結合高效能液相層析管柱 與電泳技術提出毛細管電層析法(CEC)。此外,還有其他的毛細管電泳技術被發 展出來,如毛細管等速電泳法(Capillary isotachophoresis)。1988 年 Rose 和 Jorgenson[14]首先提出毛細管電泳作微量分析的可行性,且商業化的毛細管電泳 儀亦於此年上市,加速毛細管電泳的應用與發展。
1-3毛細管電泳的分離原理
1-3-1電泳遷移率(Electrophoretic mobility)
電泳是溶液中的帶電荷粒子在電場的作用下,以不同的速度朝兩極移動的現 象,其電泳速率υep可表示如下:
υep = μepE (1-1)
其中μep為電泳遷移率,E 為外加電場。
一個帶 zi電荷的粒子,在固定電場 E 作用下所受到的靜電力為:
Fi = zieE (1-2)
其中 e 為一個電子的電量。
這個力量會使此粒子進行加速運動直到與反向摩擦力 Fd達成平衡:
Fi = Fd (1-3)
此摩擦力與粒子的運動速度υi及介質黏滯係數成正比[15]。假設粒子為球型離子,
則摩擦力可表示為:
4
Fi = 6πηriυi (1-4)
其中 ri為球型分子的離子半徑。
因此在電泳過程中,當靜電力與摩擦力達到穩態平衡時,兩者大小相等而方向相 反,可知帶電粒子 i 在電場 E 作用下的移動速率為 υi :
(1-5)
這種帶電粒子的遷移行為即為電泳,我們定義電泳遷移率為單位電場下的電泳速 率為μi或μep:
(1-6)
1.2.2 電雙層(Electric double layer)與 Zeta 電位
在毛細管電泳中,不論是帶電荷粒子或毛細管電泳的內壁均會有電雙層的形 成,電雙層是浸於溶液中的物質表面都會發生的一種情形。一般毛細管的材料常 採用熔融矽膠(fused silica),在 pH > 3 的情況下,毛細管表面受到鹼液活化後,
毛細管表面的矽醇基(Si-OH)解離而形成帶負電荷的 Si-O-,使溶液中存在過多的 正電荷;有些被不可逆地吸附在毛細管的內壁,稱為固定層(Stern layer);還有一 些分佈在距離毛細管較遠的區域,其電荷密度隨著距離管壁愈遠而成指數趨勢急 劇下降,此層稱為擴散層(Diffusion layer),此兩部分電荷所組成的離子層稱為電 雙層(Electric double layer),如圖 1-1[16]所示。由於毛細管溶液中電荷分佈不均 勻,使溶液與管壁間有電位差的存在,若將溶液的中央部分(Bulk solution)的電位 定為ψ1,管壁的電位定為ψ2,此時固定層的電位下降φ,而在擴散層中電位由 ψ2
降至ψ1,毛細管內溶液各處與(ψ2 - φ)的差距稱為 Zeta 電位(ζ potential),其方程 式可表示如下:
(1-7)
式中δ 為電雙層的厚度,e 為每單位表面積的電荷,ε 為溶液的介電常數。
5
1-3-2 電滲流(Electroosmotic flow, EOF)與流型(Flow profile)
廣義地說,電滲流是緩衝液相對於毛細管內壁表面移動的電泳現象,在電場 的作用之下,毛細管內壁形成電雙層,其中水合陽離子集體向負極移動所引起的 電滲流與帶電粒子在溶液中的電泳遷移行為類似,可用下列方程式表示之:
υeo = μeoE (1-8)
(1-9)
其中υeo為電滲流速率,μeo為電滲流遷移率,η 為緩衝液的黏度,ε 為介電常數,
ζ 為電滲流的 Zeta 電位。
由此方程式可以看出電滲流與 Zeta 電位及介電常數成正比,與黏度成反比。
一般而言,電滲流強度受 Zeta 電位、電雙層的厚度和緩衝液黏度的影響。Zeta 電位愈大、電雙層愈厚、黏度愈小,將使得電滲流值愈大。由於 Zeta 電位在擴 散層中急劇下降,其寬度僅數十微米,故毛細管中接近內管壁的大部分溶液,其 Zeta 電位均為定值,因此毛細管在電場作用下,除了靠近毛細管管壁微米區域之
外,其他區域皆以同一個速度前進,此時的電泳速率 υeo即造成穩定的電滲流。
影響電滲流的大小與方向除了緩衝液組成與濃度、pH 值、有機添加劑、毛細管 管壁修飾與否之外,毛細管樣品區帶的組成亦是影響的主要因素。
毛細管電泳中的電滲流是電解質相對於毛細管管壁移動的現象,此由距離管 壁表面甚近的正電荷離子所引起的,當施加電場後,這些正電荷粒子因電場作用 之下而集體向負極移動,同時也帶動附近水分子一起移動,當此種電滲流與電解 質黏滯力達到平衡時,整個流體會像一個扁平板塞狀而向負極移動,與一般 HPLC 的拋物線流型是不相同的,如圖 1-2[17]所示。因為 HPLC 的驅動力是靠 壓力系統,受黏滯力的影響,中央處的速度大約是平均速度的 2 倍。換言之,即
6
使樣品在管柱中不滯留,也會因為本身流型的影響造成區帶變寬的現象,然而毛 細管電泳的流型是扁平型,因此樣品不會有區帶變寬的現象發生。
由於帶電荷粒子在毛細管內的緩衝溶液中有自己的電泳遷移率(μep);而分離
緩衝溶液本身亦有電滲流遷移率(μeo)的存在,因此帶電荷粒子在沒有其它因素影
響下的視遷移率(μap)為這兩者向量的加成:
μap = μep + μeo (1-10)
在毛細管電泳實驗中,可由中性物質(如氰甲烷)的遷移時間 t0,以及帶電荷分析
物的遷移時間 tm,來求得分析物的電泳遷移率(μep):
(1-11)
其中 Ld為毛細管進樣端到偵測器的有效長度,Lt為毛細管的總長度,V 為施加
的總電壓。
在毛細管電泳分離的過程中,分離的機制就是藉著各種樣品分析物在緩衝溶 液中之電泳遷移率差異性來達成分離。因此藉由改變緩衝條件如成分、濃度、pH 值、添加有機修飾劑等來達成分離的目的。
1-3-3分離效率(Separation efficiency)
毛細管電泳(CE)與高效能液相層析(HPLC)同為液相分離技術,因此 CE 在功 能和結果上與 HPLC 十分相似,所以毛細管電泳的分離效率可以用理論板數(N) 及解析度(RS)當作指標:
(1-10)
√ √ (1-11)
其中 H 為理論板高,σ2為分析物的變異係數,D 為分析物在介質中的擴散係數,
7
Δt 為二分析物的遷移時間差,Wi為分析物 i 的吸收峰半高寬,Δμ 為二分析物的 視電泳遷移率差。
由於毛細管電泳是藉著電泳的現象流動,其流動層面為扁平板面,而不像高 效能液相層析是藉由壓力系統來達成,其流動層面為拋物面,所以在毛細管電泳 中,理論板數常常可以高達每米數十萬以上,甚至數百萬之上,其分離效率相當 可觀。
此外,在毛細管電泳中造成吸收峰波形變寬而影響分離效率的因素主要有下 列幾種:
(1) 焦耳熱造成管內溫度分佈不均,形成介質各處的黏度不一樣;
(2) 溶質吸附於管壁而造成吸收峰拖尾;
(3) 溶質與緩衝溶液的導電度相差太大而造成峰行畸變;
(4) 溶質在介質中的擴散現象;
(5) 樣品進樣太多。
因此在毛細管電泳分離過程中,必須特別注意以上這些現象,才能適當地克 服這些缺點,進而增加分離效率。
8
1-4毛細管電泳的分離模式
毛細管電泳技術發展至今,已有數種不同分離模式,不同分離模式的分離機 制各不相同。目前已發展出來的方法是依照分離原理來命名的:
毛細管區帶電泳法
(Capillary zone electrophoresis, CZE)
微胞電動力層析法
(Micellar electrokinetic chromatography, MEKC)
毛細管凝膠電泳法
(Capillary gel electrophoresis, CGE)
毛細管等電聚焦電泳法
(Capillary isoelectric focusing, CIEF)
毛細管等速電泳法
(Capillary isotachophoresis, CITP)
毛細管電層析法
(Capillary electrochromatography, CEC) 1-4-1 毛細管區帶電泳法(CZE)
毛細管區帶電泳法是電泳技術中最簡單的一種,其分離機制是藉由施加電壓 的條件下,毛細管緩衝溶液中的各種樣品分析物因質量與有效電荷量的不同,使 得電泳遷移率有差異性,而達到分離的效果,因此又可以稱為自由區帶電泳(free zone electrophoresis)。由於有電滲流的存在,所以可用來分離陰、陽離子,而中 性溶質本身在電場中不移動,故隨電滲流一起流出毛細管。 此方法可藉由改變 緩衝液的成份、濃度、pH 值或添加有機修飾劑等方法來改善分離效率。因為這 個方法操作簡單和多樣化,所以毛細管區帶電泳法的應用範圍很廣,包括胺基酸 的分析、多肽分析、蛋白質的純度分析、陰陽離子分析等等[18-21]。
9
1-4-2 微胞電動力層析法(MEKC)
微胞電動力層析法是由 Terabe 等人[10]於 1984 年所提出的一種創新的毛 細管電泳分析法,毛細管區帶電泳分離技術受限於化合物必須會解離成離子形式,
然而微胞電動力層析法的特色就是能同時對中性物質與帶電物種進行分離。此方 法為添加界面活性劑在緩衝溶液中,藉由分析物在水相和微胞相中有不同的分配 作用,進而將樣品分析物分離的一種方法。添加大於臨界微胞濃度(Critical Micellar Concentration, CMC)的界面活性劑於緩衝溶液中,界面活性劑單體分 子會聚集在一起而形成微胞(micelle)。微胞在水溶液中的結構為界面活性劑單體 的疏水性端向內聚集形成微胞中心,親水性端朝外分佈於緩衝溶液中。微胞電動 力層析法有兩相,一為緩衝液所組成的水相,另一為微胞所組成的微胞相,此兩 相同時存在。微胞電動力層析法分離的原理即是利用樣品分析物在微胞相與水相 之間的分配係數不同而達成分離。 在微胞動力層析法中,分析物分離的機制不 僅是光靠其本身所帶的電荷數與電性的差別,並且利用分析物疏水性及親水性的 大小差異而達到分離,所以不帶電荷的分析物也可以在這個方法中被分離。因此 微胞動力毛細管層析法的引入,對於毛細管電泳法提供了一種很有價值的分離技 術,也使得應用的領域更加寬廣[10, 22-24]。
1-4-3 毛細管凝膠電泳法(CGE)
毛細管凝膠電泳法是目前所有的分析技術中,具有最高分離效率的一種,其 理論板數可達每米數百萬的範圍。因為膠體本身具有分子篩的功能,可以用來當 作電泳分離的介質,而且膠體亦為非傳導性的介質,它能降低溶質擴散所造成的 區帶變寬現象,及減少溶質吸附在毛細管管壁上,通常將管壁覆蓋水溶性聚合物,
以消除電滲流。其主要的分離機制為帶電的樣品分析物在一種網狀構造中,藉由 施加一外電場作用,使樣品從毛細管的進樣端向偵測器端移動,由於樣品分析物 的分子大小或構形不同,因而受到不同程度的網狀阻力,較大分子所受的阻力愈 大,其遷移時間愈長,進而達到分離的效果[25-28]。
10
1-4-4 毛細管等電聚焦電泳法(CIEF)
毛細管等電聚焦法是於 1985 年由 Hjerten 和 Zhu[11]首先提出,是一種依樣 品的等電點(pI)不同而達到分離。在這個分析技術中,將酸性溶液放置於毛細管 的正極端,而於負極端放置鹼性溶液,當分析物與兩性介質溶液導入毛細管中時,
會建立一個 pH 梯度。在外加電場的作用下,兩性的分析物在介質中會移動到 其等電點的位置而停止,此時分析物的淨電荷為零。各分析物被聚焦於很窄的區 帶裡,再藉由其他方式將分析物推出毛細管偵測分析[29-31],進行 CIEF 實驗 時必須設法抑制或消除電滲流,以免聚焦步驟結果之前,兩性電解質即被沖出毛 細管。可將毛細管壁覆蓋或共價鍵合或加入高黏度的水溶性聚合物(如甲基纖維 素)以提高緩衝液的黏度,均可達到抑制或完全消除電滲流的效果。
1-4-5 毛細管等速電泳法(CITP)
Isotachophoresis(ITP)是一種等速移動界面的電泳技術,iso-tacho 是等速的意 思。等速電泳是在各領域使用相當廣泛的一種分析技術。它的主要特色是使用不 連續的緩衝溶液系統,樣品成份聚集在前導電解質(leading electrolyte)和尾端電解 質(terminating electrolyte)組成的緩衝溶液中以相等的速度移動,各個成份依據電 泳速率的不同而形成連續的樣品區帶,所選用的緩衝溶液,其前導電解質的陰離 子遷移率,必須大於樣品的遷移率,而尾端電解質的陰離子遷移率,則必須小於 樣品的遷移率,所以毛細管等速電泳法的分離機制即是利用分析物電泳速度的不 同來分離。而它的最大的優點在於分析物的濃縮效應,並且提供毛細管電泳法線 上濃縮技術的發展,改善了傳統毛細管電泳法偵測靈敏度不佳的缺點[32-34]。
1-4-6 毛細管電層析法(CEC)
毛細管電層析法是使用類似液相層析法中的填充物於毛細管中[35-37]。將平 均 2 μm ~ 10 μm 大小的特殊塗佈或鍵結化學官能基的顆粒填充到毛細管中,利 用分析物在這種特殊填充物與溶質間的分佈平衡,而產生分離的一種機制。在外 加電場的作用下,電滲流是由填充粒子的表面所產生的,但是因為受到填充粒子
11
的影響,流動的方向較為曲折,但是和壓力驅動的系統比較,實際上它的流動性 仍較為一致,很接近平板式的流動圖型。因此使用相同的填充管柱,在毛細管電 層析法中比液相層析法能夠得到較高的分離效率。
1-5 毛細管電泳的注入方式
樣品注入的再現性是毛細管電泳法的一個主要困難的問題,樣品區帶愈小愈 好而不致於變成為區帶變寬的主要來源。為達到此目的,非常小的樣品體積(2~20 nL)須具有再現性的注射,過量的樣品體積導致峰形迅速扭曲及損失解析度。為 達如此高的要求和小體積較好處理,樣品的自動化和微小化是必需的。大部份商 業自動化儀器的進樣型態列於表 1-1。
由於毛細管電泳使用的毛細管內徑小於 100 μm,一般 HPLC 的注射針器十 分困難使用,從進樣角度而言,一般電泳技術的進樣量必須小於 100 nL,否則 會造成過載(overloading)。毛細管電泳的進樣方法主要分為水動力注入法、水靜 力注入法及電動力注入法等三種,以下就毛細管電泳技術的進樣方法加以說明:
1-5-1 水動力注入法(Hydrodynamic injection)
水動力注入法可細分成壓力差(pressure)和抽真空(vacuum)兩種。採用水動力 注入法所注入的樣品量與樣品本身的電泳遷移率無關,比電動力注入法較不受樣 品基質的影響,較適合作定量分析但其精確度可能較電動力注入法差。本論文所 使用的水動力注入法為在毛細管入口端(inlet)施加一壓力差,以此壓力差將入口 端的樣品推入毛細管中。進樣量可經由 Hagen-Poiseuille 方程式求出:
(1-12)
其中ΔP 為施加在毛細管兩端的壓力差(mbar),d 為毛細管內徑(μm),C 為分析 物的濃度,t 為分析物的進樣時間,η 為緩衝液的黏度以及 L 為毛細管的總長。
1-5-2 水靜力注入法(Hydrostatic injection) 若是以虹吸進樣,方程式中的壓力差可以表示為:
12
(1-13)
其中ρ 為緩衝溶液的密度,g 為重力常數,Δh 為毛細管兩端的高度差。由上述
的公式可得知採用水動力注入法及水靜力注入法的進樣量不受樣品基質的影響,
也與樣品本身的電泳遷移率無關,因此較適合作定量分析。
1-5-3 電動力注入法(Electrokinetic injection)
電動力(或電遷移)注入法是施加一電壓將分析物自樣品溶液瓶中注入毛細 管中,電動力注入法所注入分析物的量與分析物的遷移率有關,遷移率愈小則進 樣量就愈少,故不適合定量,且其再現性亦較差,因而在應用上受到某些限制。
但是當毛細管中有黏性介質或凝膠時,無法採用水動力注入,則需使用電動力注 入法。
進樣量 Q (g 或 moles)由 Jorgenson 提出,可用下式計算之:
(1-14)
其中μep為分析物的有效電泳遷移率,μeo為電滲流的電泳遷移率,V 為電壓,
r 為毛細管內徑,C 為分析物的濃度,t 為進樣時間及 L 為毛細管的總長。
由上式可知分析物的進樣量與本身有效電泳遷移率及電滲流有關,視遷移率 (μep + μo)快的分析物進樣量會比遷移率慢的分析物進樣量多。但是使用電場放大 法對微量樣品作分析時,電動力注入法則有其優勢所在[15, 21, 22]。
13
1-6毛細管電泳的儀器裝置
一般毛細管電泳儀的儀器組件包括一根毛細管柱(含窗口)、兩個緩衝溶液瓶、
穩定高壓電源、一組光源及偵測器(如 UV 或 PAD)和資料處理器(如積分器),如 圖 1-3 所示[16]。
1-7 線上樣品濃縮的原理與相關計算
毛細管區帶電泳法只能對離子型分析物進行分離,因此 CZE 模式的樣品堆 積技術只能對離子物種進行濃縮;然而適當的選擇微胞相與控制相關的分析參數,
使得微胞電動力毛細管層析法的樣品濃縮技術可應用於中性分析物,這是因為在 分離與濃縮的過程中,分析物溶入微胞的程度不同,使得分析物的有效遷移率不 同,進而達到濃縮與分離的效果。
在 MEKC 模式中,已知常用的四種線上樣品濃縮技術分別為樣品堆積 (sample stacking)[38-45] 、 掃 集 (sweeping)[46-50] 、 瞬 間 等 速 電 泳 (transient isotachophoresis, t-ITP)[51-55] 與 動 力 pH 接 合 - 掃 集 (dynamic pH junction-sweeping)[56-62]。樣品堆積發生在毛細管內高電場區帶(如低導電度之樣 品溶液(S 區帶)或水區帶(water))與低電場區帶(如高導電度之分離緩衝液(BGS 區 帶))的濃度邊界,分析物在此兩區帶的有效遷移率(或速度)相差甚大,利用分析 物的有效遷移率驟降而濃縮在兩種溶液間的濃度邊界。然而掃集是樣品基質與分 離緩衝溶液的導電度是維持相等的,因此毛細管內 S 區帶與 BGS 區帶的電場強 度大約是相等,利用假靜相(如微胞)挑選(picking)與聚積(accumulating)分析物,
且 S 區帶為不含微胞的區帶。瞬間的等速電泳是緩衝溶液內使用兩種具不同遷 移率的電解質如前導與末端電解質,以聚焦低濃度且具中度遷移率的離子型分析 物。一般而言,毛細管的線上濃縮技術應用與分析物的性質與樣品基質有關。動 力 pH 接合是另一種基於分析物在多區段緩衝液的離子化程度(遷移率)不同的線 上聚焦方法。動力 pH 接合初始用以描述弱酸性分析物聚焦在樣品區帶與緩衝 液區帶接合處形成一段移動的 pH 邊界(moving pH boundary)。當使用 UV 偵側器
14
時,已有數篇文獻證實弱酸性與兩性分析物的動力 pH 接合的濃度靈敏度可增強 毫微莫耳級數[56],然而,動力 pH 接合無法應用在強酸性與中性分析物,因分 析物的遷移率與緩衝液 pH 無關。
在以流體動力注入的樣品濃縮技術中,先將整支毛細管充滿一段含高導電度 的 BGS 區帶,再進樣一段長時間且低導電度的 S 區帶。當在毛細管兩端施加 一高電壓後,在 S 區帶與 BGS 區帶的濃度邊界,微胞速度降低以致濃縮成一 段非常窄的區帶,此時的微胞速率υ (mc) 可表示如下[63]:
υ(mc) = υeo + υep(mc) (1-1)
其中υeo與υep(mc)分別為電滲流速度與微胞速度。
在一個連續性的均相系統中,微胞速度υmc可表示如下:
υ(mc) = [μeo + μep(mc)] · E (1-2)
其中μeo與μep(mc)分別為電滲流遷移率與微胞遷移率。若 ES與 EBGS分別表示在 S
區帶與 BGS 區帶的電場強度,κs與κb分別表示在 S 區帶與 BGS 區帶的導電度,
並且其電阻分別為 RS與 RBGS,分析物的增強因子(enhancement factor, γ)可依下 式表示之[95]:
(1-3)
假設 S 區帶與 BGS 區帶的電場強度在電泳分離實驗過程中皆保持固定[96],
Chien 和 Burgi [69]提出毛細管的平均電滲流速率 υeo(ave)可表示如下:
(1-4)
其中υeof (S)為毛細管只填充 S 的電滲流速率;υeof (BGS)為毛細管只填充 BGS 的電
滲流速率;x 為樣品長度佔毛細管總長的分率,其值介於 0 與 1 之間,所以可將 方程式(2-1)重新表示為:
υ(mc) = υeo(ave) + υep(mc) (1-5)
因為 S 區帶與 BGS 區帶的電場強度不同,所以微胞在此兩區的遷移速度亦不同,
15
微胞在 S 區帶與 BGS 區帶的遷移速度分別可表示為 υ(mc)(S)與υ(mc)(BGE),若假設微 胞在此兩段的有效遷移速率μep(mc)相同,υ(mc)(S)與υ(mc)(BGE)可表示如下:
υ(mc)(S) = υeo(ave) + υep(mc,S) (1-6a)
υ(mc)(BGS) = υeo(ave) + υep(mc,BGS) (1-6b)
υep(mc,S) = μep(mc) ES (1-7a)
υep(mc,BGS) = μep(mc) EBGS (1-7b)
其中υep(mc,S)為微胞在 S 區帶的遷移速度,υep(mc,BGS)為微胞在 BGS 區帶的遷移速
度。
(1-8a)
(1-8b)
Eo(=V/L)為施加在毛細管兩端的總平均電場強度, V 為施加在毛細管兩端的總 電壓,L 為毛細管的總長。依方程式(1-8a)和(1-8b)可知當樣品區帶長度 x 減小 時,ES、EBGS增加,ES為 EBGS的γ 倍,亦使 υ(mc)(BGS)與υep(mc,BGS)增加。實際上,
υep (mc,S)、υep (mc,BGS)與υeof隨 x 改變而改變,但 υeo 與 υep (mc,BGS)的變化沒有υep (mc,S)
變化大。 堆積效率(Stacking efficiency, SE)為表示分析物的樣品濃縮程度,
10~1000 倍的濃縮效率相當於增加分析物 10~1000 倍的偵測靈敏度(detection sensitivity),或是相當於降低一~三個級數的濃度偵測極限(如濃度偵測極限從 10-8 M 降低至 10-9~10-11 M)。常用 SE height 與 SE area 來表示吸收峰高度與吸收峰面 積的堆積效率,分別表示如下:
(1-9)
其中 Hstack為長時間進樣堆積後的吸收峰高,Hstandard為短時間進樣所得的吸收峰
高,Cstandard為短時間進樣的濃度,Cdiluted為稀釋後的濃度。
(1-10)
16
其中 Acorr,stack 為長時間進樣後堆積的相對面積,Acorr,standard為短時間進樣的相對
面積(1 或 2 sec)。
(1-11)
其中 A 為樣品的面積,tr為樣品的遷移時間。
一般主要是以 SEheight來表示分析物的堆積程度,是因為電滲流的再現性為 影響分析物吸收峰面積的因素,而利用遷移時間修正分析物的吸收峰面積,是為 了消除在 MEKC 模式的電滲流之再現性問題。
1-8 毛細管電泳之線上樣品濃縮技術
已有數十篇的文獻探討樣品線上濃縮的機制[24, 46, 64-70],為了有一套完整 性的說法,本論文以陰離子界面活性劑 SDS 為例子,將分析物在 MEKC 模式的 線上樣品濃縮分成數種濃縮技術,其命名原則乃根據實驗中變異的因素來命名:
正向堆積模式 (Normal stacking mode, NSM)
電極極性反向堆積模式 (Reversed electrode polarity stacking mode, REPSM)
反向遷移微胞堆積 (Stacking with reverse migration micelles, SRMM)
增強電場樣品進樣 (Field enhanced sample injection, FESI)
以反向遷移微胞增強電場樣品進樣 (Field enhanced sample injection with reverse migrating micelles, FESI- RMM)
利用反向遷移微胞與水之堆積 (Stacking using reverse migrating micelles and a water, SRW)
掃集 (Sweeping)
陽離子選擇性注射法與掃集之結合模式 (Cation-Selective Exhaustive Injection and Sweeping, CSEI-Sweeping)
動力 pH 接合-掃集 (dynamic pH junction-Sweeping)
17
1-8-1 正向堆積模式(NSM)
本章所論及的線上樣品濃縮模式皆以分離緩衝液中添加陰離子界面活性劑 (如 SDS)為例子,而添加陽離子界面活性劑於分離緩衝液的線上樣品濃縮模式為 類似的情況,一般只需將施加高電壓時的電極極性方向反轉。Quirino 和 Terabe[64]
詳述微胞與分析物在 NSM 的堆積過程,其遷移行為與堆積如圖 1-5 所示。NSM 為最簡單的樣品堆積技術,衍生自 MEKC 的一般技術。首先,利用水動力注入 方式將整隻毛細管填充高導電度且含微胞之 BGS 區帶,再長時間進樣低導電度 且含分析物的 S 區帶(圖 1-5(A)),另外分析物 ax, ay, az 的滯留因子(retention factor)大小為 k(ax) > k(ay) > k(az);最後施加一正向高電壓進行線上濃縮與分離(圖 1-5 (B) ~ (D))。以下所討論的數種濃縮模式,若無特別標示分析物的種類,則分 析物可適用於中性分子或帶正電荷的離子。
將樣品溶液的導電度製備成比緩衝液之導電度更低,導致帶電荷物種(如微 胞)在 S 區帶的電場強度較 BGS 帶區的電場強度增加,使得帶電荷物種在 S 區帶 有相對較高的遷移速率。因此微胞從 BGS 區帶經濃度邊界 B1進入 S 區帶時,微 胞的遷移率增加;微胞從 S 帶區經濃度邊界 B2進入另一段 BGS 區帶時,微胞的 遷移率減少。同理可知分析物從濃度邊界 B1到濃度邊 B2的有效速度 υep(S)比在 BGS 帶區的有效速度 υep(BGS)更大。當分析物進入相對電場強度較低的 BGS 區 帶之後,由於電場強度變小,使得遷移速率變小,因此樣品分析物在濃度邊界
B2不斷地受到擠壓而達到濃縮的效果。
18
1-8-2 電極極性反向堆積模式(REPSM)
微胞與分析物在 REPSM 過程的遷移行為如圖 1-6 所示[65],製備一低導電 度且不含微胞的樣品溶液,如此會使得微胞進入 S 區帶的電泳速率比進入 BGS 區帶來得快。首先將毛細管充滿含微胞的緩衝液,然後注入比 NSM 更長的樣品 長度(圖 1-6 (A)),S 區帶為含分析物(ax , ay , az)的深色區帶,BGS 區帶是淺色區 帶,並於毛細管兩端施加一所需的負電壓(圖 1-6 (B)),以使電動力注入的微胞將 中性分析物從負極帶到偵測端的正極。高滯留因子的化合物比低滯留因子的化合 物移動快,緩衝液中的陰離子亦進入 S 區帶。同時毛細管中的樣品基質(如水)被 電滲流趕至負極瓶中,並由正極瓶中含微胞的緩衝液遞補。移除樣品基質可減少 高低導電區帶間的電滲流的不匹配,使層流(laminar flow)的分散效應減小。當大
部份的中性分析物已濃縮聚集在濃度邊界 B1時,此時電流為施加負電壓之電流
的 97 ~ 99 %,再將電極的極性改變成正電壓,直到完成整個分離過程(圖 1-6 (C), (D))。若注入的樣品體積大於一般操作模式 10 倍以上的時候又稱為大體積樣品 堆積模式(large volume sample stacking, LVSS)。
1-8-3 反向遷移微胞濃縮(SRMM)
SRMM 是在毛細管充滿含微胞的緩衝液後,從負極之進樣端進樣一定長度 的樣品溶液,如圖 1-7 (A)所示[66],施加一負電壓以使微胞遷移至偵測端的正 極,此時微胞的電泳速率大於電滲流速率[71],此乃因毛細管內壁表面的矽醇基 質子化而降低了電滲流。深灰色區帶為含中性分析物的 S 區帶,淺灰色區帶為含 微胞的 BGS 區帶。經過一段時間之後,如圖 1-7 (B)所示,負極瓶中的微胞進入 毛細管中且帶動樣品區帶中的中性分析物至偵測端的正極。在中性分析物濃縮至
濃度邊界 B1後,分析物聚集區帶從 B1開始分離,且樣品基質不斷地被電滲流推
出負極瓶中,而且由正極瓶中的緩衝液所遞補。在樣品基質完全移除之前,中性 分析物已分離且進入含微胞的緩衝液區帶(圖 1-7 (C)),之後樣品基質從管柱中完 全移除,然後繼續分離與被偵測器所偵測之,此模式不需改變電極極性(圖
19
1-7(D))。
1-8-4 增強電場樣品進樣(FESI)
FESI 的機制如圖 1-8 所示[67],首先將毛細管充滿含微胞的緩衝液,然後進 樣一定長度的水區帶,再將負極瓶子換成含有微胞的樣品溶液之瓶子(圖 1-8 (A))。於毛細管兩端施加一負電壓(圖 1-8 (B)),因為有水區帶的增強電場,使得 以電動力注入的樣品之電泳速率比電滲流、微胞及在毛細管中的中性分析物還來
得快,中性分析物移動至濃度邊界 B2,B2為水區帶中含微胞與不含微胞的邊界。
當中性分析物被帶至濃度邊界 B1,水區帶開始被電滲流推出毛細管外。在電流
為施加負電壓之總電流的 97 ~ 99 %時,再將進樣端瓶子換成裝含有微胞的緩衝 液之瓶子,並施加一正電壓(圖 1-8 (C)),然後繼續分離與被偵測器所偵測之(圖 1-8 (D))。若以電動力注入,樣品進樣量會受到其本身之解離度的影響。中性分 析物的遷移順序與滯留因子有關,可知遷移時間隨滯留因子增大而增長。若滯留 因子大小為 k(ax) > k(ay) > k(az),則其遷移時間為 t(az) > t(ay) > t(ax)。
1-8-5 以反向遷移微胞增強電場樣品進樣(FESI-RMM)
圖 1-9 為描述 FESI-RMM 的過程[68],首先將毛細管充滿含微胞的緩衝液,
然後進樣一定長度的水區帶(圖 1-9 (A))。再將負極瓶子換成含有微胞的樣品溶 液之瓶子,於毛細管兩端施加一負電壓(圖 1-9 (B)),因為有水區帶的增強電場,
使得以電動力注入的樣品之電泳速率比電滲流、微胞及在毛細管中的中性分析物 還來得快,中性分析物堆積至濃度邊界 B, B 為水區帶中含微胞與緩衝液的邊 界,水區帶開始被電滲流推出毛細管外。在電流為施加負電壓之總電流的 70 ~ 90
%時,再將進樣端瓶子換成含有微胞緩衝液之瓶子,再施加一負電壓(圖 1-9 (C)),
然後繼續分離與被偵測器所偵測之(圖 1-9 (D))。
1-8-6 利用反向遷移微胞濃縮與水之堆積(SRW)
圖 1-10 為 SRW 模式之堆積過程[69],為了提供一比水更高的電場,將一定 量的水比樣品優先進樣於毛細管中,如此可消除由樣品區帶中的微胞所引起的擴
20
散效應[65](圖 1-10 (A))。其樣品置於一低導電度的微胞基質中,此使得在樣品 區帶的分析物可以較快的速率進入水區帶。採用低 pH 值的樣品溶液是為了降低 電滲流與減少樣品吸附於毛細管的表面,低的電滲流可防止樣品掉入進樣端瓶中 而減少損失。若分析物在樣品區帶與水區帶的有效電泳速率比電滲流快,分析物 終會到達偵測器的正極,故施加一負電壓以進行樣品濃縮與移除樣品基質與水區 帶(圖 1-10 (B), (C)),樣品區帶與水區帶被電滲流推至入口端瓶中,然後由出口 端瓶中含微胞的緩衝液所遞補。在樣品移除的過程中,電滲流一直在遞減,造成 微胞遷移速率的方向改變,即低導電度的樣品區帶與水區帶的長度減小而增加緩
衝液的電場。 水區帶與緩衝液區帶之間的濃度邊界 B1的遷移速率等於電滲流的
速率且向負極移動[72](圖 1-10 (B)),樣品區帶與水區帶之間的濃度邊界 B2的遷
移速率假設等於電滲流的速率且向負極移動。分析物濃縮在濃度邊界 B2沒有比
濃縮在濃度邊界 B1上來得重要,大部份的正極含微胞的緩衝液中的陽離子會濃
縮在濃度邊界 B2,且對全部過程沒有太大的影響。最後微胞繼續帶動中性分析
物區帶遷移至偵測器與分離(圖 1-10 (D))。
1-8-7 掃集(Sweeping)
掃集為另一種不同於樣品堆積的線上濃縮技術,此技術為 Terabe 及其學生 Quirino 於 1998 年所發表[46],掃集與樣品堆積的最大不同在於樣品基質與緩衝 溶液的導電度是維持相等的,因此毛細管內的電場強度是一定的。其機制為先在 毛細管內填充低 pH 值酸性緩衝溶液,可抑制電滲流且其值趨近於零。再利用水 動力注射法注入一段和緩衝液導電度相近的樣品溶液,最後將管柱兩端置於含 SDS 微胞的酸性緩衝溶液中,並施加一負電壓(圖 1-11(A))。管柱進樣端的陰離 子微胞在電場的作用下進入管柱中並朝偵測器方向移動,在微胞進入樣品區帶內 後,SDS 微胞會慢慢地採集和積聚不同分佈係數的中性分析物。與微胞作用力大 的中性分析物堆積而成的濃縮區帶會比與微胞作用小的分析物濃縮區帶較為狹