國 立 交 通 大 學
生 物 科 技 學 院
生物科技研究所
碩 士 論 文
開發對原發性肝癌具有專一性之抗體及其應用
Preparation of Heptocellular Carcinoma Specific
Antibodies and Their Application
研 究 生:王富生
指導教授:吳東昆 博士
開發對原發性肝癌具有專一性之抗體及其應用
Preparation of Heptocellular Carcinoma Specific Antibodies and Their
Application
研 究 生:王富生 Student:Fu-Sheng Wang 指導教授:吳東昆 Advisor:Tung-Kung Wu Ph.D. 國 立 交 通 大 學 生 物 科 技 學 院 生物科技研究所 碩 士 論 文 A ThesisSubmitted to Institute of Biological Science and Technology College of Biological Science and Technology
National Chiao Tung University In partial Fulfillment of the Requirements
For the Degree of M.S. In
Biological Science and Technology Aug. 2012
Hsinchu, Taiwan
開發對原發性肝癌具有專一性之抗體及其應用
研究生:王富生 指導教授:吳東昆 博士 國 立 交 通 大 學 生 物 科 技 學 院 生物科技研究所 碩 士 論 文中文摘要
肝癌是全球十大常見癌症中的第四位,每年約有 662000 人死於肝癌。尤其在台灣 因為 B 型肝炎的流行,肝癌是十大常見癌症中的第一位。由於現行的肝癌檢測方式價格 昂貴且無法有效檢測出初期的腫瘤,因此我們希望能開發出一個能檢測肝癌的系統。 近年來有研究指出在肝癌病患的血漿中有些生物標計分子會不正常的增加。在 2005 年的文獻中指出,將其中三種生物標計分子組合起來應用於肝癌檢測時,不論肝癌 發生至哪一階段都能百分之百的檢測出肝癌,它們分別是胎兒球蛋白(Alpha-fetoprotein, AFP) 、 岩 藻 醣 苷 水 解 酶 (Alpha-fucosidase, AFU) 以 及 血 管 內 皮 增 生 因 子 (Vascular endothelial growth factor, VEGF)。因此,我們希望能發展生物辨識分子能專一的辨認這 些生物標計分子。我們選定抗體來做為生物辨識分子,因為抗體具有高度的靈敏度以及 辨識能力。在本研究中我們利用分子生物學的技術將生物辨識分子利用大腸桿菌大量表達並 利用管住層析純化,這些生物辨識分子被用做老鼠的免疫並且發展出多株對於這些生物
辨識分子具專一性的單株抗體。
為了能將這些抗體應用於肝癌檢測,我們將鹼性磷酸水解酶結合至抗體上,並應用 三明治免疫法發展檢測平台。而發展出的平台其偵測範圍在 10 μg/ml 到 1 mg/ml 間。
Preparation of Heptocellular Carcinoma Specific Antibodies and Their
Application
Graduate student: Fu-Sheng Wang Advisor: Tung-Kung Wu Ph. D. Institute of Biological Science and Technology
College of Biological Science and Technology National Chiao Tung University
Abstract
Heptocellular carcinoma (HCC) is the primary cancer of liver. It causes about 66,200 deaths per year worldwide and ranks the forth of the most common cancer. HCC is hard to be detected in its early stage by present diagnostic techniques. Recent researches indicate that the concentration of some biomolecules is abnormal in serum of HCC patients. In this study, we aim to develop biorecognition molecules specific to these target biomarkers. We chose antibody as our biorecognition molecules, based on the high sensitivity and specificity of the antibody. In this study, three proteins, alpha-fetoprotein (AFP), alpha-fucosidase (AFU), and vascular endothelial growth factor (VEGF) were chosen as target biomolecules to develop monoclonal antibodies. The cDNA of AFU and VEGF were expressed directly as fusion proteins in vector pET22a and pET28a, respectively. For AFP, two truncated regions were overexpressed. The recombinant biomarkers were used to generate monoclonal antibodies. The monoclonal antibodies, which are specific to AFU and VEGF and AFP, respectively, have been developed.
To develop HCC detection system the monoclonal antibodies were conjugated to alkaline
conjugated antibodies was from 10 μg/ml to 1 mg/ml.
Keywords: Hepatocelluar carcinoma, alpha-fetoprotein, alpha-fucosidase, vascular
謝 誌
首先誠摯感謝指導教授吳東昆 老師在這兩年的教導,讓我不論在學問上,處事上 以及生活態度上都更加成熟,同時也感謝老師給予我很多磨練的機會,讓我在我的碩士 生涯中有所成長。此外也非常感謝口試委員兼召集人清華大學張大慈 教授,以及本校 張家靖 教授百忙中能撥冗前來擔任口試委員並給予學生論文審閱,使本論文能更加的 完整。 首先最要感謝文鴻學長這兩年在研究與生活上的指導與協助。謝謝學長能耐心且細 心的帶領生物知識匱乏又實驗技巧拙劣的我,如果沒學長在實驗上巨細靡遺的教導我想 我不會有機會能完成這份論文。再來感謝實驗室的大哥程翔學長,學長豐富的知識不論 在研究上,生活上以及論文上都給予我很大的幫忙。感謝裕國學長淵博的學問以及犀利 的眼光,常常能在我實驗瓶頸上一針見血的給我意見或是血淋淋的指出我的錯誤。感謝 電腦通的晉豪學長,在電腦以及軟體上學長給予了許多幫助。感謝晉源學長在做 construction 時候的鼓勵,真的是只要做成功過一次以後就很順手。再來感謝實驗室的大 姐們媛婷學姊和聖慈學姊在生活上給予的鼓勵以及幫忙。感謝 mili 學姊常為實驗室帶來 歡笑。感謝世穎學長,欣怡學姊,欣芳學姊,以及怡臻學姊在我們碩一時起給予的幫助。 感謝陪我一起奮鬥兩年的同學孟兒,欣樺以及婉婷在我的碩士生活中不斷的加油打氣並 分享許多快樂。感謝書磊,文茜,奕汝,家豪以及宛珊在重要時刻給予的幫忙。感楔子 傑與玫華為實驗室帶來新的活力。 最後,由衷感謝我的家人與唯婷,與我分享喜樂與憂愁,包容我的缺點、並在我失 意的時候給予鼓勵與支持,謝謝你們對我的付出,在此對你們獻上無限的謝意。 王富生 謹誌 國立交通大學生物科技研究所 中華民國一百零一年八月List of Contents
中文摘要 ... i
Abstract …………..………iii
謝誌
………..……….v
List of Figures ... ix
Literature Review ... 1
1-1. Hepatocellular Carcinoma and its diagnosis ... 1
1-2. Alpha Fetoprotein ... 3
1-3. Vascular Endothelial Growth Factor ... 8
1-4. Alpha-L- Fucosidase ... 15
1-5. Alkaline Phosphatase ... 20
Specific Aim... 25
Materials and Methods ... 29
3-1. Apparatus ... 29
3-2. Materials ... 29
3-3. Vectors ... 30
3-4. Bacteria Strain, Gene, and Recombinant DNA methods... 32
3-5. Preparation of competent cells ... 34
3-6 Transforming of bacteria ... 34
3-7. Expression of Recombinant AFP and small fragments of AFP by E. coli ... 35
3-8. Expression of Recombinant VEGFA by E.coli ... 35
3-9. Expression of Recombinant AFU by E.coli ... 36
3-10. Resolublization and Refolding of the Fragments of AFP ... 36
3-11. Purification of the Fragments of AFP ... 37
3-12. Resolublization and Refolding of the Fragments of AFU ... 38
3-13. Purification of AFU ... 38
3-14. Resolublization and Refolding of VEGFA ... 39
3-16. Gel Electrophoresis ... 40
3-17. Enzyme Linked Immunosorbent Assay ... 41
3-18. Animal Care and Use ... 42
3-19. Immunization of Mice ... 42
3-20. Production of Monoclonal Antibody ... 42
3-21. Purification of monoclonal antibody ... 44
3-22. Measurement of Antibody titer ... 45
3-23. Western Blot Analysis ... 45
3-24. Conjugation of AP to antibodies ... 46
3-25. Sandwich ELISA Immunoassay ... 46
Results and Discussion ... 47
4-1. Construction of AFP and VEGF expression vectors ... 47
4-3. Expression and purification of the partial sequence of AFP ... 51
4-4. Expression and purification of recombinant VEGFA ... 57
4-5. Expression of recombinant AFU ... 61
4-6. Generation of AFP-specific monoclonal antibodies ... 63
4-7. Generation of VEGFA-specific monoclonal antibodies ... 68
4-8. AFU-specific monoclonal antibodies ... 72
4-9. Purification of monoclonal antibodies ... 74
4-10. Immunoassay of AP-conjugated antibodies. ... 75
Conclusion ... 79
Future Perspective ... 80
References ... 81
Appendix ... 90
8-1. Gene sequence of alpha fetoprotein ... 90
8-2. Gene sequence of alpha fetoprotein (fragment head) ... 91
8-3. Gene sequence of alpha fetoprotein (fragment mid) ... 91
8-4. Gene sequence of alpha fetoprotein (fragment tail) ... 91
List of Tables
Table 1-1. Common biomarkers of HCC……… 2 Table 1-2. Accuracy of HCC diagnosis depends on AFP, AFU, and VEGF and their combinations in different stages ………...3
List of Figures
Figure 1-1. Primary, secondary, and tertiary structure of alpha-fetoprotein……….5
Figure 1-2. Location of ligands binding residues in AFP………..6
Figure 1-3. Selective internal iliac angiography of control rabbit………... 9
Figure 1-4. Molecular interactions with vascular endothelial growth factor (VEGF)…..11
Figure 1-5. Structure of VEGF. ……… 12
Figure 1-6. Stereo representation of the receptor-binding face of VEGF………….…… 13
Figure 1-7.Sequence alignment of VEGF, PLGF, VEGF-B, VEGF-C, PDGF-A and PDGF-B. ………..………14
Figure 1-8.Stability and activity of AFU in different temperature and pH…...…………16
Figure 1-9. Overall view of α-fucosidase. ……….…………17
Figure 1-10.Structural comparison with family GH27………17
Figure 1-11. The catalytic pocket AFU….……….………18
Figure 1-12. Fucisylation of AFP in HCC patient. ………..……19
Figure 1-13. Overall structure of alkaline phosphatase. ………21
Figure 1-14. Active site region of AP, including bound phosphate, magnesium ion, and two zinc ions. ………..………22
Figure 1-15. Proposed mechanism of two-metal ion catalysis in the hydrolysis of phosphate monoesters by AP. ………23
Figure 3-1. Interaction between nickel ion and poly-histidine tag……….…………37
Figure 4-1. Agarose gel analysis of restriction enzyme mapping pET28a- AFP plasmid……..………..48
Figure 4-2. Agarose gel analysis of restriction enzyme ma pping pET28a-VEGF……. 48 Figure 4-3. Agarose gel analysis of restriction enzyme mapping pET28a- AFP-head… 49
Figure 4-4. Agarose gel analysis of restriction enzyme mappi ng pET28a- AFP-mid… 49 Figure 4-5. Agarose gel analysis of restriction enzyme mapping pET 28a- AFP-Tail… 50 Figure 4-6. 12.5% SDS-PAGE analysis of protein purification from E. coli cells containing AFP-mid ……….53 Figure 4-7. Protein ID confirmation by MALDI-TOF for the upper position band in purified AFP-mid fragment……….…… 54 Figure 4-8. Protein ID confirmation by MALDI-TOF for the lower position band in purified AFP-mid fragment……….……… 55 Figure 4-9. 12.5% SDS-PAGE analysis of protein purification from E. coli cells containing AFP-tail ………56 Figure 4-10. Protein ID confirmation by MALDI-TOF for the purified AFP-tail fragment ……….…………57 Figure 4-11. 12.5% SDS-PAGE analysis of protein purification from E. coli cells containing VEGF ……….…………59 Figure 4-12. Protein ID confirmation by MALDI-TOF for the purified VEGF……… 60 Figure 4-13. Protein ID confirmation by MALDI-TOF for the purified VEGF ………60 Figure 4-14. 12.5% SDS-PAGE analysis of protein purification from E. coli cells containing AFU………..………61 Figure 4-15. Protein ID confirmation by MALDI-TOF for the purified AFU …………62 Figure 4-16. Protein ID confirmation by MALDI-TOF for the purified AFU………… 63 Figure 4-17.Titer of anti-AFP antibody………64 Figure 4-18. Identification of specificity of monoclonal antibodies 3C2F4 against serum sample spiked with AFP-mid proteins by Western blotting (right) and corresponding SDS PAGE (left). ………65
Figure 4-19. Identification of specificity of monoclonal antibodies 2C3B1 against serum sample spiked with AFP-tail proteins by Western blotting (right) and corresponding SDS PAGE (left). ………66 Figure 4-20. Identification of specificity of monoclonal antibodies 3D8D6 against serum sample spiked with AFP-mid proteins by Western blotting (right) and corresponding SDS PAGE (left) ……….………67 Figure 4-21. Identification of specificity of monoclonal antibodies 1D6E4 against serum sample spiked with AFP-mid proteins by Western blotting (right) and corresponding SDS PAGE (left) ……….………68 F i g u r e 4 - 2 2 . T i t e r o f V E G F s p e c i f i c a n t i b o d i e s 3 H 6 F 8 a n d 1E2B3. ……….………69 Figure 4-23. The Identification of specificity of monoclonal antibodies 3H6F8 against serum sample spiked with VEGF proteins by Western blotting (right) and corresponding SDS PAGE (left) ……….……70 Figure 4-24. The Identification of specificity of monoclonal antibodies 1E2B3 against serum sample spiked with VEGF proteins by Western blotting (right) and corresponding SDS PAGE (left) ……….………71 Figure 4-25.Titer of AFU specific antibodies 3-65-3 and 1-52. The titer of 1-52 was 1:5000, 3-65-3 was 1:2000………...72 Figure 4-26.The Identification of specificity of monoclonal antibodies 1-52 against serum sample spiked with AFU proteins by Western blotting (right) and corresponding SDS PAGE (left) ………..73 Figure 4-27. The Identification of specificity of monoclonal antibodies 3-65-3 against serum sample spiked with AFU proteins by Western blotting (right) and corresponding SDS PAGE (left)………..………74
Literature Review
1-1. Hepatocellular Carcinoma and its diagnosis
Hepatocellular carcinoma (HCC) is the primary cancer of liver. It is the most common type of liver cancer. . It causes about 66,200 deaths per year worldwide and ranks the third of the most common cancer. The risk factors of HCC vary from country to country. In country where hepatitis B widely spread such as Taiwan, hepatitis B is the major cause of HCC. Other risk factors such as chronic cirrhosis, non-alcoholic fatty liver disease, and alcohol-induced liver disease also play important roles (Pang, Joh et al. 2008). Treatment of HCC and prognosis are dependent on many factors especially on tumor size and staging. Tumor grade is also important. High-grade tumors have a poor prognosis. The clinical manifestations of HCC include abdominal pain, hepatomegaly, and weight loss. The diagnosis of HCC is usually based on the laboratory screening including index of hepatic damage, the index of cholestasis, the tumor markers, and instrumental tests including hepatic ultrasonography, computed tomography (CT), and nuclear magnetic resonance (NMR) (El-Serag, Marrero et al. 2008).
CT scans use contrast agents, which are typically composed of iodine or barium. Some patients are allergic to these contrast agents. Usually the allergic reaction is manageable. An alternative to CT imaging study is MRI, but machines of MRI's are more expensive and not as available. More importantly MRI is just beginning to be applied in tumor detection and fewer facilities are skilled at MRI studies when it is used as a screening device. MRI is mostly used to do a secondary study to look at an area where a tumor has already been detected. Besides, MRI's also use contrast agent which means it can also face allergic problem as CT.
A more general method to screen HCC is using ultrasound. However many patients with either large HCC (>5 cm) or multifocal HCC (more than three lesions) may not be screened out. Besides, the limitations of ultrasound include its operator dependence and its poor ability to differentiate malignant from benign nodules in the small cirrhotic liver. Although with some improvement of CT and MRI the diagnostic accuracy can be raised, these techniques are too expensive for widespread screening (Zinkin, Grall et al. 2008).
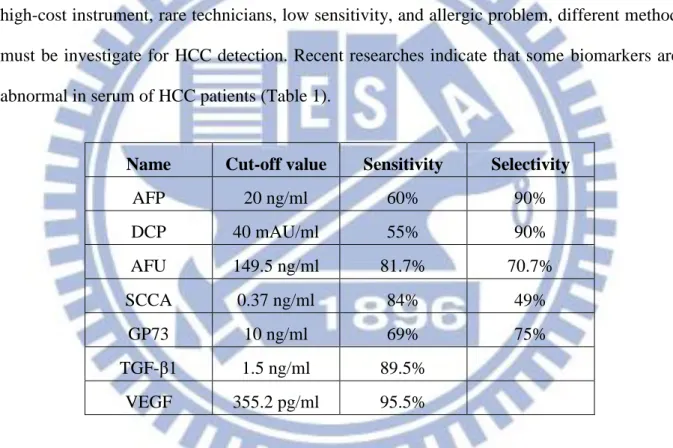
Since that the application of imaging on HCC diagnosis has some restrictions such as high-cost instrument, rare technicians, low sensitivity, and allergic problem, different method must be investigate for HCC detection. Recent researches indicate that some biomarkers are abnormal in serum of HCC patients (Table 1).
Name Cut-off value Sensitivity Selectivity
AFP 20 ng/ml 60% 90% DCP 40 mAU/ml 55% 90% AFU 149.5 ng/ml 81.7% 70.7% SCCA 0.37 ng/ml 84% 49% GP73 10 ng/ml 69% 75% TGF-β1 1.5 ng/ml 89.5% VEGF 355.2 pg/ml 95.5%
Table 1-1. Common biomarkers of HCC. These molecules have been suggested to be
involved in HCC in recent researches (Zhou, Liu et al. 2006; Gomaa, Khan et al. 2009; Malaguarnera, Giordano et al. 2010).
Some of these biomarkers are related to hepatic disease (such as AFP, AFU), and some of them are related to tumorigenesis (such as TGFβ, VEGF). The most commonly used serum marker of HCC is AFP. It has a reported sensitivity of 39% to 65% and selectivity of
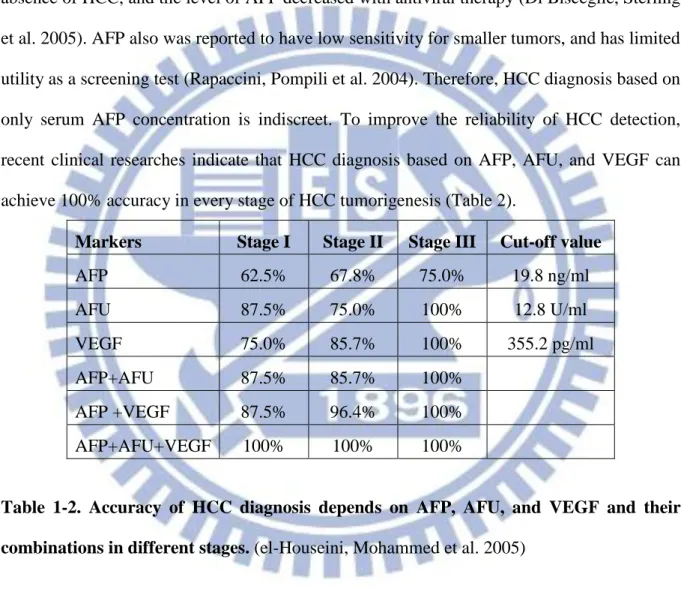
65% to 94% (Daniele, Bencivenga et al. 2004). However, not only HCC but other hepatic diseases also induce the increase of AFP serum level (Arrieta, Cacho et al. 2007). Recently, high serum levels of AFP have been reported in HCV patients. (Mousa, Gad et al. 2012). Patients from the Hepatitis C Antiviral Long-term Treatment against Cirrhosis study, there were 27% of patients with HCV and cirrhosis had an high AFP serum level (20 ng/ml) in the absence of HCC, and the level of AFP decreased with antiviral therapy (Di Bisceglie, Sterling et al. 2005). AFP also was reported to have low sensitivity for smaller tumors, and has limited utility as a screening test (Rapaccini, Pompili et al. 2004). Therefore, HCC diagnosis based on only serum AFP concentration is indiscreet. To improve the reliability of HCC detection, recent clinical researches indicate that HCC diagnosis based on AFP, AFU, and VEGF can achieve 100% accuracy in every stage of HCC tumorigenesis (Table 2).
Markers Stage I Stage II Stage III Cut-off value
AFP 62.5% 67.8% 75.0% 19.8 ng/ml AFU 87.5% 75.0% 100% 12.8 U/ml VEGF 75.0% 85.7% 100% 355.2 pg/ml AFP+AFU 87.5% 85.7% 100% AFP +VEGF 87.5% 96.4% 100% AFP+AFU+VEGF 100% 100% 100%
Table 1-2. Accuracy of HCC diagnosis depends on AFP, AFU, and VEGF and their combinations in different stages. (el-Houseini, Mohammed et al. 2005)
1-2. Alpha Fetoprotein
Alpha fetoprotein (AFP) is a glycoprotein containing up to 35% carbohydrates with molecular mass varied from 68 to 73 kD depending on carbohydrate content and biological species. AFP is synthesized in yolk sac during early fetal life and later on in fetal liver. After birth, its role is replaced by serum albumin and its serum level drops from 3 mg/ml to 10
ng/ml.
AFP is a member of serum albumin family. It shows high identity in structure, physical property, and chemical property between AFP and albumin. Besides, the primary structure of AFP is conserved in human, rat, and mouse. For example, primary structures of human and mouse AFP share 66% identity (Morinaga, Sakai et al. 1983). Interestingly, some domains of AFP and albumin from the same animal species exhibit lower similarity than corresponding domains of different animal species (Gorin, Cooper et al. 1981). Analysis of the primary structure of AFP and albumin from different animal species indicate that identity degree between these two proteins reduced from man to dog, horse, mouse, and rat. For human AFP and albumin the number of identical amino acid residues is 236 (38.8%), for dog AFP and albumin this value is a bit lower, 228 (37.5%), and for other animals: horse (205, 33.7%), mouse (203, 33.5%), and rat (193, 31.7%). Consequently, the divergence of AFP and albumin from a common ancestor increases. This demonstrates the highest conservation for the AFP and albumin in human. AFP isolated from various tissues of one animal species (both embryonic and tumor) is characterized. They have the same amino acid sequence and are also immunologically identical (Terentiev and Moldogazieva 2006).
AFP has an albumin-like structure. Its primary structure shares 39% identity with albumin. The amino acid sequence of human AFP was encoded by nucleotide sequence of its mRNA, and the translation product of this mRNA contained 609 residues (Morinaga, Sakai et al. 1983). It was demonstrated that AFP becoming mature form after the first 18 residue being cleaved therefore the mature AFP molecule contains 591 residues. AFP contains approximately 4% carbohydrates (Peters, Nishi et al. 1979). Proteins of serum albumin family also have similar α-helical secondary structure but lack β-structure. The proteins have similar spatial organization. They were consisted of three homologous domains. The domain consists
of two globular subdomains and linked with disulfide bonds (Tomasi 1977). Figure 1-1 shows the structure of AFP.
Figure 1-1. Primary, secondary, and tertiary structure of alpha-fetoprotein. (a)AFP is
separated into three domains. Domain I and domain III are connected by domain II. (b) AFP are composed mainly by α-helix. UNSW Embryology.
The structure of AFP is separated into three domains by the aid of electron microscopy. It revealed a U shaped structure. The secondary structures of these domains are very similar, but they differ by parameters of tertiary structure. Domains I and III have rigid tightly packed tertiary structure, and joined together by flexible domain II. The C-terminal part of domain II could be considered as a hinge region, responsible for conformation flexibility of all domains, and therefore it promotes the interaction of AFP with ligands and other proteins. This site is characterized by lack of disulfide bonds and has been found only in the AFP molecule but not in other albumin-like proteins (Mizejewski 2001).
The function of AFP is still not well investigated, but scientists believe that it plays an important role in binding and transporting a multitude of ligands such as flavonoids, bilirubin, fatty acids, etc (Deutsch 1991). Figure 1-2 shows some AFP residues that interact with ligands.
Figure 1-2. Location of ligands binding residues in AFP. AFP interacts with a variety of
ligands. Reds stand for fatty acid binding site. Purples stand for estrogen binding residues. Blues stand for insulin-like segment. Oranges stand for cell adhesion motifs. An introduction
to alpha fetoprotein and the growth of inhibitory peptide. Akul Y. Mehta. PharmaXChange.info
. The serum level of AFP was first correlated with HCC in 1963 by Tatarinov et al. They used immunoprecipitate to identify the existence of AFP in the liver tissue and serum of HCC patients (Abelev 1971). Scientists then try to figure out the role of AFP in HCC tumorgenesis. But until today there is still no obvious correlation between the serum AFP level in human primary hepatocellular cancer and any of the clinical or biochemical parameters of the disease, the size and stage of the tumor, or survival time after diagnosis. The relation between the degree of differentiation of the tumor to the presence of the globulin was conflict (Kew 1974). The functional role of AFP in HCC is still unclear. But according to clinical researches, 70% HCC patients have high concentration of AFP in serum. This makes
AFP still being a useful biomarker for HCC screening.
The likely site of production of AFP in HCC is the tumor itself. Research indicated that malignant hepatocytes synthesize AFP. Two possible mechanisms have been considered. First, it has been suggested that with neoplastic transformation the hepatocytes dedifferentiate to a stage of development at which the AFP expression is not repressed, and production of the protein is resumed. The second possibility is that the tumor arises from those cells which have the AFP gene a non-repressed form. With either view, the production of AFP reflects that hepatic cells are less mature than hepatocytes. The reason that some tumors secrete AFP while others do not hasn’t been established (Kew 1974).
In HCC diagnosis, the concentration 20 ng/ml of AFP serum level is the most commonly used cut-off value to differentiate HCC patients from healthy adults in clinical researches. However, the proper cut-off value is different depending in ethnic (Gomaa, Khan et al. 2009).
Since AFP has been the most important biomarker of HCC, how to gain pure AFP becomes an important issue. Because of the similarity between AFP and serum albumin it is hard to isolate AFP. At the end of 20 century scientist used immunoaffinity procedure on achieving complete purification of AFP from cell culture or human tissue. The first recombinant AFP expressed by Escherichia coli was reported in 1997. They generated complete human AFP cDNA from a fetal liver cDNA library by PCR then inserted the AFP gene fragment into fusion vector pTrp. The recombinant AFP was expressed by Escherichia
coli strain BL21. The overexpressed protein would aggregate and form inclusion body. With
refolding process and column chromatography pure AFP could be gained (Boismenu, Semeniuk et al. 1997). Lately on 2007 AFP was purified form serum free HepG2 cell culture (Carlini, Ferranti et al. 2007).
In previous researches, AFP would form polymer and lost its function in vitro easily (Wu and Waterhouse 1982).Therefore, the storage condition of AFP is critical. James T. Wu et al in 1985 tried to find out the optimal AFP storage condition. In the research they concluded the effects of AFP stability including concentration of AFP, storage temperature, addition of serum proteins, and time. It shows that while under frozen condition AFP form polymer easily, and with the addition of serum protein AFP turn to be unstable. The best storage way of AFP is at 4 degree Celsius (Wu and Knight 1985).
1-3. Vascular Endothelial Growth Factor
Vascular endothelial growth factor (VEGF) is a mitogen produced by vascular endothelial cells. These cells may be derived from arteries, veins, and lymphatics. It is a homodimeric glycoprotein of 45 kDa. In 1983 Senger et al noted that VEGF induce the vascular leakage. Later on it was found that VEGF is a survival factor for cultured endothelial cells and immature retinal vessels (Alon, Hemo et al. 1995). Moreover, VEGF induces the expression of serine protease in human endothelial cells (Unemori, Ferrara et al. 1992). These evidences indicate that VEGF may participate in angiogenesis.
In 1994, Takeshita et al verified the function of VEGF in angiogenesis by intraarterial injectionVEGF administration. The density of capillary is significantly high compared with control (Figure 1-3), which directly demonstrated that VEGF can induce the growth of blood vessel (Takeshita, Zheng et al. 1994).
Figure 1-3. Selective internal iliac angiography of control rabbit. Direct and linear
extension of internal iliac artery can be barely observered in control group (a, b, c) but in VEGF-treat group (d, e, f). (J Clin Invest, 1994, 93(2):662-670).
There are several other members in VEGF gene family, including placenta growth factor, VEGF-B, VEGF-C, and VEGF-D. The most-characterized one in this family is VEGF (also refers to VEGFA), and its function is discussed in previous section. VEGF-B plays a less role in angiogenesis. Its main function is to maintain newly formed blood vessels (Zhang, Tang et al. 2009). Besides, VEGF-B plays roles in protection of neurons in the retina (VEGF-B inhibits apoptosis via VEGFR-1–mediated suppression of the expression of BH3-only protein genes in mice and rats) and the cerebral cortex during stroke (Sun, Jin et al. 2004). The function of VEGF-C and VEGF-D is mediating lymphangiogenesis (Orpana and Salven 2002). The overexpress of VEGF-C can cause lymphedema. Placental growth factor (PGF) is a key molecule in angiogenesis and vasculogenesis, in particular during embryogenesis. Placental growth factor-expression within human atherosclerotic lesions is associated with plaque inflammation and neovascular growth (Hauser and Weich 1993).
VEGF reveal its function in angiogenesis via several different receptors which can be classified into two types: kinase domain receptor (KDR) and Fms-like tyrosine kinase 1 (Flt-1) Two tyrosine kinase receptors, VEGFR I (KDR) and VEGFR II (Flt-1), were found on endothelial cells. The members of VEGF family have different affinity toward each receptor. The main function of VEGFR1 is in developmental angiogenesis, including recruiting of endothelial cell progenitors, monocyte migration, increasing the adhesive properties of natural killer cells, and inducing growth factors from liver sinusoidal endothelial cells (Lamszus, Ulbricht et al. 2003). VEGFR2 mediates the downstream effects of VEGF in angiogenesis, including microvascular permeability enhancement, endothelial cell proliferation, and tumor cell migration. Neuropilin-1 and Neuropilin-2 are transmembrane glycoproteins that interact with several members of the VEGF family Researches indicated that Neuropilin serve as coreceptors for VEGF. They enhance the binding affinity of VEGF family VEGF receptors and affect subsequent intracellular signaling. VEGF binding to Neuropilin-1 and Neuropilin -2 leads to the increase of endothelial mitogenesis and chemotaxis (Wang, Zeng et al. 2003; Sulpice, Plouet et al. 2008). The interaction of VEGF with their receptors and the signal pathway is shown in figure 1-4.
Figure 1-4. Molecular interactions with vascular endothelial growth factor (VEGF) are shown. VEGF family triggers various signal pathways, which lead to tumor angiogenesis,
Figure 1-5. Structure of VEGF. Monomer of VEGF is composed of 2 α-helix and 7
beta-sheets. One of the monomer is colored in blue and the other in red. The structure was folded by the aid of disulfide bonds (shown in white). The sulfur atoms are shown in yellow. (Structure, 1997, 5:1325-1338)
The crystal structure of VEGF was published in 1997, which is shown in figure 1-5. As early mentioned, native VEGF reveals as a homodimer. The monomers are joined together by disulfide bond. According to mutagenesis, the residues important for KDR binding map are located on the same face of the molecule, spanning across the interface of dimer (shown in figure1-6). The main feature in structure of the receptor-binding face is a short three-stranded, antiparallel β-sheet in one subunit, which packs against the N-terminal α-helix from the other subunit. This interaction is important for the stability of the receptor-binding face. It accounts for 65% of the total surface buried within the dimer. The loop connecting strand β5 to β6 is important for variability of VEGF. This loop plays role in binding to both KDR and Flt-1,
therefore, the flexibility of the loop might be functionally important in accessing different conformations required for binding to these distinct receptors.
Figure 1-6. Stereo representation of the receptor-binding face of VEGF. The
receptor-binding face of VEGF crosses the dimer. By mutagenesis, there are several residues playing important roles in the receptor-binding face: Phe17 and Gln79; and Ile46, Glu64 and Ile83 (shown in yellow). Other residues (shown in gray) are responsible for receptor-binding specificity among VEGF and its homologs. (Structure, 1997, 5:1325-1338)
The function of VEGF homologs, including PGF, VEGF-B, VEGF-C, in regulation of angiogenesis is still not clearly known. Sequence alignment has been used to help to explain the differences in specificity of the VEGF homologs towards KDR and Flt-1. It shows that in VEGF family, the residues on the receptor binding residues are highly conserved. This may suggest that VEGF binds in a similar manner to both receptors. This phenomenon is also found in the binding of human growth hormone to both the growth hormone receptor and the prolactin receptor. From the crystal structure of these proteins although there are significant
differences in primary structure, the overall arrangement shows high identity.
Figure 1-7.Sequence alignment of VEGF, PLGF, VEGF-B, VEGF-C, PDGF-A and PDGF-B. Binding determinants of VEGF for the KDR receptor are shown in black. Residues
accessible on the same receptor-binding face as the KDR-binding determinants in VEGF are shown in gray. Mutations in these residues result in differences in affinity and selectivity within the VEGF group. (Structure, 1997, 5:1325-1338)
Tumerigenesis always couple with angiogenesis. Researches indicated that VEGF is frequently express in HCC patient. In a quantitative study VEGF expression demonstrating by immunohistochemistry was observed in 63.9% of encapsulated and 78.3% of nonencapsulated HCCs, and 90.9% of HCCs with extrahepatic metastasis (Yao, Wu et al. 2005). Besides definite correlation between tumor VEGF expression and microvessel density, which is a marker of tumor angiogenesis, has been demonstrated (Wada, Nagano et al. 2006; Tseng, Tai et al. 2008). Some researchers suggested that tissue VEGF expression increased according to the stepwise development of HCC (Park, Kim et al. 2000) because of that VEGF levels were noted to progressively increase through the tumorigenesis in early HCC. These clinical
researches suggested that VEGF is a possible biomarker for HCC.
The serum level of VEGF in HCC diagnosis has been investigated. In a study of 63 patients, patients with HCC had significantly higher serum VEGF levels (median, 245 pg/mL) compared with healthy volunteers (median, 180 pg/mL) (Seo, Park et al. 2010). The most widely used cut-off value of VEGF serum level in HCC diagnosis is 355 pg/ml, which provide HCC screening sensitivity to 95.5% (el-Houseini, Mohammed et al. 2005).
1-4. Alpha-
L- Fucosidase
Alpha fucosidase (AFU) is a 156 kDa lysosomal enzyme found in all mammalian cells.. Its main function is the degradation of a variety of L-fucose containing glycoconjugates.
L-fucose is a monosaccharide that is a common component of many N- and O-linked glycans and glycolipids produced by mammalian cells. L-fucosylated glycanconjugates have various functions in mammalians. L-fucose plays an important in immune respond: it involves in the composition of ABO blood group antigens (Lowe 1993) and it contributes to selectin-dependent leukocyte adhesion (Kansas 1996; VESTWEBER and BLANKS 1999).
L-fucose also helps the interaction between host and micro: the gastric pathogen Helicobacter
pylori is capable of attachment to the gastric epithelium via host expression of the Lewis
antigen, a structure containing fucose (Hooper and Gordon 2001). Besides, L-fucose is involved in ontogenic events: the stage-specific embryonic antigen-1, which is a fucosylated glycan, is expressed during early embryogenesis (Solter and Knowles 1978).
The degradation of L-fucose containing glycanconjugates are catalyzed by AFU. AFU was found because of fucosidosis. Fucosidosis was first described in 1966. This disease
causes progressive mental retardation and neurologic deterioration. Later on in 1969 Loeb et al showed that these patients lack the activity of lysosomal alpha-L-fucosidase, which led the accumulation of undegraded polyglycans in neuron cell.
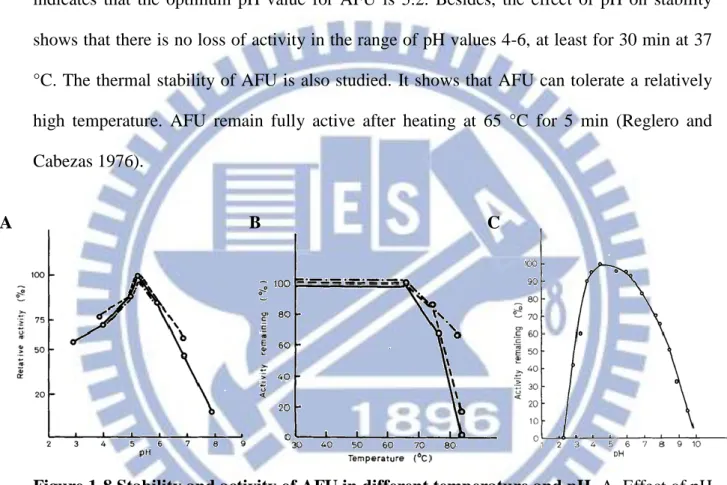
Fig 1-8 shows some basic biochemical property of AFU. The pH-activity profile indicates that the optimum pH value for AFU is 5.2. Besides, the effect of pH on stability shows that there is no loss of activity in the range of pH values 4-6, at least for 30 min at 37 °C. The thermal stability of AFU is also studied. It shows that AFU can tolerate a relatively high temperature. AFU remain fully active after heating at 65 °C for 5 min (Reglero and Cabezas 1976).
Figure 1-8.Stability and activity of AFU in different temperature and pH. A. Effect of pH
on the purified AFU in different pH. B. Thermostability of AFU after 5 min at temperatures ranged between 25 °C and 83 °C. C. Stability of AFU as a function of pH in 0.1 M Trisodium citrate buffe at 37 °C for 30 min. (Eur. J. Biochem. 1976, 66: 379- 387)
The structure of AFU is composed of subunits of molecular weight 50 kDa and the native enzyme is a homotrimer of molecular weight 156 kDa (Shown in figure 1-9).
Figure 1-9. Overall view of α-fucosidase. A. Stereo ribbon diagram colored of α-fucosidase.
It shows the secondary and tertiary structure if AFU. B. Figure shows the overall structure of AFU and bound fucose. The trimers are colored magenta and white. Bound fucose molecules are shown in green. (The Journal of Biological Chemistry, 2004, 279:13119-13128)
Compare the structure of AFU with glucoside hydrolase 27 revealing high identity. Some residues of the active sites are conserved, and the location of bounded ligand is overlap. This may suggest that AFU and GH27 have same revolution ancestor.
Figure 1-10.Structural comparison with family GH27. Comparison of structure between
AFU (left) and N-acetylgalactosaminidase (NAGA, right). The catalytic domains are shown in yellow and blue, the C-terminal domains in red and magenta, and the secondary structure elements carrying the catalytic residues and bound ligands in green and gray for AFU and NAGA, respectively. (The Journal of Biological Chemistry, 2004, 279:13119-13128)
The catalytic pocket of AFU is shown in figure 1-11. It can be observed that the pocket is composed of aromatic and acidic amino acid. The aromatic residues can stabilize L-fucose by π-π interaction, whether the acidic residues can interact with L-fucose by hydrogen bonds.
Figure 1-11. The catalytic pocket AFU. Multiple interactions are involved in the
stabilization of fucose (shown in boldface). Hydrogen bonds are shown as dashed lines, and van der Waals contacts are shown as pink dotted lines. (The Journal of Biological Chemistry, 2004, 279:13119-13128)
The amount and activity of AFU was first correlated with HCC in 1984 (Deugnier, David et al. 1984). In the research they studied in sera from 30 controls, 32 patients with primary hepatic carcinomas, 24 patients with secondary metastatic liver carcinomas and 36 patients with cirrhosis. They found out that with the cut-off value 110 nkat/L, the diagnosis of HCC depending on AFU can achieve 75% sensitivity and 90% selectivity (Giardina, Matarazzo et al. 1998). From further researches, not only activity but also the mount of AFU becomes higher in HCC patients than in healthy individuals and in chronic hepatic disease patients. Today, the most common cut-off value of AFU in HCC diagnosis is set to 870nmole/L/hr. With the cut-off value AFU shows a sensitivity of 81.7% and a selectivity of
70.7% (Malaguarnera, Giordano et al. 2010). For serum level of AFU, laboratory tests showed cut-off value for plasma alpha-l-fucosidase level of 145.9 ng/ml (Ai-Ping, Yue et al. 2007)
The role of AFU in HCC tumorigenesis is still unclear. However in some HCC patients, serum AFP is fucosylated whether it is not in liver cirrhosis patients. Therefore these fucosylated AFP may induce the raise of the amount and activity of AFU, although there is no correlation between AFU serum concentration and AFP levels activity (Cheng, Chang et al. 2007).
Figure 1-12. Fucisylation of AFP in HCC patient. Western blotting was used to identify the
form of AFP in liver tissue of HCC patients and liver cirrhosis patients. Fucosylated AFP was stained by anti-AFP antibody. In HCC patient AFP-L3 was observed. (Journal of the Chinese Medical Association, 2007, 70:310-317)
In clinic, AFU measurement is useful in association with AFP in the early diagnosis of HCC. Moreover, there is a positive correlation between AFU levels and tumor size in HCC patients (Takahashi, Saibara et al. 1994). The AFU increase has been observed in non-cancerous extrahepatic disease such as diabetes, pancreatitis, and hypothyroidism.
Numerous mammalian tissues, for example liver (Opheim and Touster 1977), semen (Khunsook, Alhadeff et al. 2002), and serum, can extract AFU. AFU can be also purified from some microorganism: Thermotoga maritime (Sulzenbacher, Bignon et al. 2004), and
Streptomyces Species (Reglero and Cabezas 1976). The most efficient material to extract AFU
is human semen. AFU has been reported that plays an important role in sperm-egg interaction. This may cause by the fucosylation participating in the modification of surface of sperm cell, interaction of sperm with female tissue, and sperm-oocyte interaction. But the actual function of AFU in seminal plasma is still unclear.
Despite of extracting native AFU from organism, recombinant protein is also applied in AFU researches. In 1984, Hisao et al combine the anti sera of AFU and the technique of phage display to sequence the cDNA of AFU. The recombinant AFU expressed by eukaryotic and prokaryotic cells is achieves by Fukushima et al and Alejandro et al (Fukushima, Nishimoto et al. 1990; de Carlos, Montenegro et al. 2003).
1-5. Alkaline Phosphatase
Alkaline phosphatase (AP) is one of the most frequently referenced enzymes and has been widely investigated. This is because of its enzymatic activity was a signal for a variety of disease states particularly the liver and bone. AP was first defined in 1912. It shows that AP present in variety of tissue, especially in intestinal mucosa and can hydrolyze glycerophosphate and fructose 1-6 diphosphate. In 1960s AP was found out that not only presenting in mammalian tissue but also microorganism. Investigators discovered that E. coli possessed AP that was derepressible by phosphate starvation. AP is a 160 kDa dimeric glycoprotein. Its main function is the hydrolysis of phosphmonoester. In humans, AP can be found in all tissues throughout the entire body and present in several isoforms. It has been
also reported that AP is a metalloenzyme. A native AP contains four zinc ions and two magnesium ions.
The structure of AP is shown in figure 1-13. Each monomer has an N-terminal α-helix, which forms an arm that embraces the other monomer. Besides, a “crown domain” is formed on the interface by the insertion of a 60-residue segment from each monomer. This domain consists of two small interacting β-sheets, each composed of three parallel strands and surrounded by six large and flexible loops containing a short α-helix and is the location of active site. There is also a “metal binding domain” which comprised of 76 residues and folded into two β-strands flanked by two α-helices. It has been verified that this domain coordinate with calcium, but the function is still unknown (Le Du, Stigbrand et al. 2001).
Figure 1-13. Overall structure of alkaline phosphatase. (The Journal of Biological
Chemistry, 2001, 276:9158-9165). The overall structure of AP is shown in ribbon. Side chain of the three extra domains were represented in ball and stick.
The function of AP is similar in mammalian cell or bacteria (Schwartz 1963). Although the identity of primary structure of AP between these two species is low, which is about 30%, the residues of active site are highly conserved (Kam, Clauser et al. 1985). The residues of the active site of AP were shown in figure 1-14.
Figure 1-14. Active site region of AP, including bound phosphate, magnesium ion, and two zinc ions. Water molecules are represented in an encircled letter w. Hydrogen bonds are
shown as broken lines. (FEBS Letters, 1999, 462:7-11)
From the structure of active site, Arg-166 plays an important role in to stabilizing the transition state and to directly interact with the substrates or products of the reaction because of its guanidinium group. This hypothesis is confirmed by mutagenesis. Mutations of Arg-166 (R166Q, R166S, and R166A) lower the substrate binding affinity of AP over 50-fold (Butler-Ransohoff, Kendall et al. 1988; Chaidaroglou, Brezinski et al. 1988). In spite of Arg-166, Asp-101, Asp-153, and Lys-328, form secondary interactions to phosphate through either a water molecule or Arg-166. Site-specific mutagenesis reveals that disruption results in enzymes with reduced phosphate affinity (Mandecki, Shallcross et al. 1991; Xu and Kantrowitz 1991; Chen, Neidhart et al. 1992; Janeway, Xu et al. 1993; Murphy, Xu et al.
To understand the mechanism of hydrolysis of phosphatemonoester by AP, the crystal structure of native E. coli AP complexed with inorganic phosphate (Pi), which is a strong competitive inhibitor for AP has been solved (Kim and Wyckoff 1991) and the possible mechanism has been proposed.
Figure 1-15. Proposed mechanism of two-metal ion catalysis in the hydrolysis of phosphate monoesters by AP. (FEBS Letters, 1999, 462:7-11)
In the free enzyme (noted as E), three water molecules fill the active site by the aid of zinc and Arg-166. Meanwhile, the hydroxyl group of Ser102 forms a hydrogen bond with the Mg-coordinated hydroxide ion. Ser102 is then deprotonated for nucleophilic attack with the
concomitant transfer of the proton to the Mg-coordinated hydroxide group. This activated Ser102 is stabilized by Zn and can attacks the phosphorus center of phosphomonoester (ROP) making AP binding with substrate to form a covalent serine-phosphate intermediate. Zn then coordinates the bridging oxygen atom of the substrate and facilitating the departure of the alcohol group (RO), leaving a nucleophilic hydroxide ion on Zn. This nucleophilic hydroxide ion attacks the phosphorus atom, result in the hydrolysis of the covalent serine-phosphate intermediate to form the non-covalent enzyme-phosphate product complex and regenerate the nucleophilic Ser102, which then attack the coordinated water on Mg and release phosphate (Holtz and Kantrowitz 1999).
AP has a variety application. In molecular biology, AP was used to prevent self ligation. DNA normally has phosphate groups on the 5' end. The removal of these phosphates prevents the DNA from ligating, keeping DNA molecules linear until the next step of the process (Maxam and Gilbert 1980). AP also can be used as a label of enzyme in immunoassay. Besides AP can be a marker of pasteurisation in cows' milk. This is because if the denature of AP by elevated temperatures found during pasteurisation (Aschaffenburg and Mullen 1949).
The most wildly used substrate in immunoassay applying AP was p-Nitrophenyl Phosphate (pNPP). It was hydrolyzed to p-Nitrophenyl by the aid of AP and turned its color from yellow to dark purple. Besides, AP catalyzed a verity of substrate which contained phosphate, including 4-methylumbelliferyl phosphate, O-methyl-O-(N-butylfluorescein), cAMP, cGMP, d-luciferin 6’-O-phosphate etc, which became chemiluminescent or fluorescent.
Specific Aim
Our aim is to develop a low-cost HCC diagnosis system. HCC diagnosis based on a combination test of three biomarkers can achieve 100% accuracy. Therefore, we want to generate antibodies specific for AFP, VEGF, and AFU. Antibodies have high specificity and selectivity toward antigens. These antibodies will act as biorecognition molecules. To quantify the amount of biomarkers, AP will be conjugated to antibodies as a reporter. AP catalyzes a variety of substrate. One of these substrates is 4-methylumbelliferyl phosphate (4-MUP). AP catalyzes the removal of phosphate from 4-MUP to make 4-methylumbelliferone. 4-methylumbelliferone is a photophore. It is excited at 360 nm and has emitted at 460. We believe this platform can achieve our goal.
To generate the biomarker specific antibodies, pure biomarkers are needed. As previously described, we attempted to obtain proteins from mammalian tissue, cultured cells, and bacteria. We choose bacteria as our protein source because of its convenience. Therefore, our first goal is to generate bacteria that express the target proteins.
To obtain the HCC biomarkers we clone the genes into an expression vector. These vectors are used to transform the bacteria and express the proteins. With IPTG induction, recombinant biomarkers are overexpressed in the form of an inclusion body. These insoluble
proteins can be recovered by refolding. The proteins are purified by column chromatography (Scheme 1). The process of generating antibodies has been well established. Purified proteins are used to immune mice. These biomarkers act as antigens that triggering the immune system of mice, making their spleen generate B cells. These B cells can secret antibodies that are against biomarkers. Because a B cell is well differentiated, its life is limited. To preserve the B cell, we fuse the cell with a cancer cell (Scheme 2).
Scheme 2-1. Recombinant HCC biomolecules were expressed by Escherichia coli. The
DNA sequence of the antigens were cloned into expression vector pET28a and transformed into E. coli BL21 and BL21(C43). Proteins were then expressed by aid of isopropyl β-D-1-thiogalactopyranoside. Proteins were further purified by column chromatography.
Scheme 2-2. Procedure for producing HCC-specific monoclonal antibodies: purified
proteins were injected into mice. After injecting three times, the anti-sera of mouse was tested for titer. The spleen of the mice would be separated out if the titer was higher than 5000. Further, B cells would be extracted and fused with FO cell, which makes the B cell undead. Then the cell that can secrete the antigen-specific antibody would be screened out.
To develop a HCC diagnosis based on an antibody, a pair of antibodies binding with different epitopes is needed. One of them is conjugated with AP, the other one is used to recognize the biomarker in serum (Scheme 3).
AP is conjugated to an antibody through the aid of glutaraldehyde. Glutaraldehyde is an amine-reactive homobifunctional crosslinker that catalyzes the formation of covalent bond between AP and antibody.
Scheme 2-3. HCC diagnosis system applying sandwich immunoassay and alkaline phosphatase. The capture antibody was coated on the plate. It recognizes and captures the
biomarkers. The recognition antibody conjugated with AP was then added to bind with the biomarker. AP hydrolyzes 4-MUP to methylumbellifer. The signal was tested by exitation 360 nm and emittion at 460 nm.
Materials and Methods
3-1. Apparatus
UVS400, Thermo Savant SPD SpeedVac, BECKMAN COULTER Allegra 21R Centrifuge, ORBITOR SHAKER, Centrifuge 5415R, eppendorf, Orbital shaking incubator Model S300R, FIRSTEK SCIENTIFIC, 550 sonic dismembrator, FIRSTEK SCIENTIFIC, Mini Trans-Blot Cell, Bio-Red Fusion Universal Microplate Analyzer, Packard
3-2. Materials
All chemicals were of analytical grade and were used without further purification.
Restriction enzymes and T4 ligase were purchased from NEB. Agrose gel, sodium chloride were purchased from USB. Gel Band Purification Kit, DEAE resin was from Amershame. IPTG, HY medium, HAT supplement, PEG/DMSO, sodium carbonate, kanamysin, imidazole, alkaline phosphatase, glutaraldehyde were purchased sigma. Nickel resin was purchased from Novagen. Dulbecco’s modified Eagle Medium (DMEM), penicillin/streptomycin (P/S) was purchased from Gibco. Fetal bovine serum (FBS), PVDF membrane was purchased from Hyclone. Trypton, methanol, acetic acid, yeast extract, coomassie brilliant blue, β-mercaptoethanol, EDTA, glycerol were purchased from Merck.
3-3. Vectors
3-3-1. pET28a (Novagen)
3-4. Bacteria Strain, Gene, and Recombinant DNA methods
E. coli strains XL1-Blue was from our bacteria stock. E.coli strain BL21(C43) was kindly provide by. Expression vector pET28a was from our plasmid stock. All genes were gained from Mammalian Gene Collection gene bank of NCTU. Mammalian Gene Collection (MGC) is a project that provides researchers with unrestricted access to sequence-validated full-length protein-coding cDNA clones for human, mouse, and rat genes. The cDNA clones of MGC are obtained by screening of cDNA libraries, transcript-specific RT-PCR cloning, and DNA synthesis of cDNA inserts. The library number of AFP gene we used is NIH_MGC_122. The RNA source of NIH_MGC_122 is from anonymous pool of 24 week female lung, 16 week female spleen, and 20-22 week male spleens. Library is constructed by primer of oligo-dT nd directionally cloned. The average insert size 1.4 kb, insert size range 1-3 kb. Library is normalized and enriched for full-length clones and was constructed in pCMV-SPORT6 by C. Gruber. The library number of VEGF gene we used is NIH_MGC_428. This gene is constructed by DNA synthesis. The accession number is BC172307. All the information of these two genes is from MGC web site (http://mgc.nci.nih.gov/). The gene of AFU constructed into expression vector of pET28a is kindly provided by Y.K. Lee’s lab, Department of Applied Chemistry, NCTU, Hsinchu, Taiwan.
The genes from MGC were cloned into the vector pCMV.SPORT6. To extract these genes and construct AFP, VEGF expression vector, we designed 1 pair and 4 pairs of primers for the constructions of VEGF and AFP respectively. The primers of VEGF are . The forward one contains the first nucleotides of VEGF gene and an EcoRI restriction site, where the reverse one contains the last nucleotides and a NotI restriction site. The primers for full length of AFP gene are 5’–gAATTC ATgAAgTgggTggAATCA-3’ and 5’– CTCgAg AACTC CCAAAgCAgCAC -3’. The forward one contains the first 18 nucleotides of AFP gene and an
restriction site. The primes for first 600 nucleotides, named as AFP-head, are 5’–gAATTC ATgAAgTgggTggAATCA-3’and 5’– CTCgAg TTCAACTgCATTTTCAg -3’. The forward one contains the first 18 nucleotides of AFP gene and an EcoRI restriction site, where the reverse one contains the nucleotides 583-600 and a XhoI restriction site. The primes for nucleotides 601-1243, named as AFP-mid, are 5’–gAATTC TgCTTCCAAACAAAggCA -3’and 5’– CTCgAg TCgCTTTgCCAATgCTT -3’. The forward one contains the nucleotides 612-618 of AFP gene and an EcoRI restriction site, where the reverse one contains the nucleotides 1225-1243 and a XhoI restriction site. The primes for nucleotides 1244-1859, named as AFP-tail, are 5’–gAATTC TTggCAAAgCgAAgCTgC -3’and 5’– CTCgAg AACTCCCAAAgCAgCAC -3’. The forward one contains the nucleotides 1245-1262 of AFP gene and an EcoRI restriction site, where the reverse one contains the last 18 nucleotides and a XhoI restriction site.
PCR was used to amplify the gene fragments. Considering the difference of annealing temperature for different primer, we use different condition to amplify the genes. The amplified fragments are ligated into pCR2.1-TOPO by the aid of TOPO TA cloning kit for convenience of stocking.
To construct the AFP-head, AFP-mid, AFP-tail and VEGF expression vector we first digest the fragments from the TOPO vectors. The procedure is described below:
1 mg/ml TOPO vector containing desire gene
5 μl EcoRI buffer 2 μl EcoRI 0.5 μl NotI/XhoI 0.5 μl ddH2O 10 μl BSA solution 2μl
Mix the reagents and incubate at 37 °C for overnight. The digested fragment can be purified with gel filtration by 1.5% agarose gel and PCR product cleaning kit. The expression vector pET28a is treated with the same digestion process. Purified fragments are ligated into expression with procedure described below:
Purified fragments 7.5 μl Purified pET28a 0.5μl T4 lgase 0.5μl T4 ligas buffer 1μl
ddH2O 1μl
The mixed solution is incubated at 37 °C for 3 hrs. All of the constructed expression vectors are confirmed by sequencing and stock at -20 °C.
3-5. Preparation of competent cells
All the solutions were prepared by distill water and autoclaved. Three milliliter of overnight cultured medium was 1:100 diluted into 300 milliliter LB and incubated at 37°C until OD600=0.3~0.5. Cells were harvested by centrifuge at 3000 g for 15 min and resuspended by 60 ml 100mM magnesium chloride. Following centrifuge at 3000 g for 10 mins, cells were resuspended by 150 ml 100 mM calcium chloride. Cells were harvested by centrifuge at 3000 g for 15 min and resuspended by 5 ml 85 mM calcium chloride 25% glycerol. Stock the cells at -80 °C.
3-6 Transforming of bacteria
The frozen competent cell is first thawing on ice. 1 μl of 1 mg/ml plasmid solution is added in to cell solution and incubate on ice for 30 mins, which makes plasmid attaching on the cell surface. The solution is then incubated at 42 °C for 45 secs. This heat-shock process
lowers the fluidity of cell membrane and speed up the exchange between intracellular and extracellular materials. After heat-shock, the solution is incubated on ice for another 5 mins to recover the cell and then plate the cells. The plate is incubated at 37 °C for 12~16 hrs and stock at 4 °C.
3-7. Expression of Recombinant AFP and small fragments of AFP by E. coli
Bacteria cells transformed with AFP, pET28a-AFP-head, pET28a-AFP-mid, pET28a-AFP-tail were grown at 37 °C for 16hr in 3 ml LB medium containing 25 μg/ml kanamycin. The culture was diluted 100 fold in 500 ml kanamycin supplement LB medium. Induction was performed in LB medium prepared as follows: 10 g trypton, 5 g yeast extract, and 10 g sodium chloride in per liter Milli-Q water are adjusted to pH=7.4. And the solution is autoclaved. The cells are then incubated at 37 °C to an OD 600 =0.5, which usually takes two
and half hours. To induce the expression of the proteins, IPTG was added to final concentration of 0.5 mM, and the bacteria solution was incubated an additional 4 hrs. The cells were then harvest by centrifugation at 3500 g. The pellet was stored at -20 °C. AFP expression was evaluated in E. coli BL21, as well as in protease-deficient strains.
3-8. Expression of Recombinant VEGFA by E.coli
Bacteria cells transformed with pET28a-VEGF were grown at 37 °C for 16 hrs in 3 ml LB medium containing 25μg/ml kanamycin. The culture was diluted 100 fold in 500 ml kanamycin supplement LB medium. Induction was performed in LB medium prepared as follows: 10 g trypton, 5 g yeast extract, and 10 g sodium chloride in per liter Milli-Q water are adjusted to pH=7.4. And the solution is autoclaved. The cells are then incubated at 37 °C to an OD 600 = 0.4, which usually takes two and half hours. To induce the expression of the proteins,
IPTG was added to final concentration of 0.5 mM, and the bacteria solution was incubated an additional 4 hrs. The cells were then harvest by centrifugation at 3500 g. The pellet was stored
at -20 °C. VEGF expression was evaluated in E. coli BL21(C43), as well as in protease-deficient strains.
3-9. Expression of Recombinant AFU by E.coli
Bacteria cells transformed with pET22a-AFU were grown at 37 °C for 16 hr in 3ml LB medium containing 25 μg/ml ampicillin. The culture was diluted 100 fold in 500 ml ampicillin supplement LB medium. Induction was performed in LB medium prepared as follows: 10 g trypton, 5 g yeast extract, and 10 g sodium chloride in per liter Milli-Q water are adjusted to pH=7.4. And the solution is autoclaved. The cells are then incubated at 37 °C to an OD 600 = 0.4, which usually takes two and half hours. To induce the expression of the proteins,
IPTG was added to final concentration of 0.5 mM, and the bacteria solution was incubated an additional 4 hours, harvested by centrifugation at 3500 g. The pellet was stored at -20 °C. AFU expression was evaluated in E. coli BL21, as well as in protease-deficient strains.
3-10. Resolublization and Refolding of the Fragments of AFP
All procedures were carried out at 4℃. Each frozen cell solution was thawed and resuspend by TBS buffer (20 mM Tris-Hcl pH=8, 0.5 M sodium chloride, 20 mM imidazole) the cell was lysed by sonication with amplitude=4, duty cycle =50%. The cell lysate was centrifuged at 13000 g for 30 mins to harvest inclusion body. The supernatant was discard and the pellet was resuspended by 10ml denature buffer (20 mM Tris-HCl, 0.5 M NaCl, 8 M urea, pH = 8.0) and stir for further 2hrs. After suspension, the solution is centrifuged at 16500 g for 30 mins to remove dissoluble impurities.
To refold the denatured proteins, the solution was added to TGE buffer (20 mM Tris-HCl, 25% glycerol, 0.5 mM EDTA, pH=8.0) with gentle stirred. After the denatured protein is
totally added, the solution is stirred for overnight. The insoluble impurity is eliminated by centrifuge at 16500 g for 30 mins.
3-11. Purification of the Fragments of AFP
Nickel sepharose were used to purify AFP. There are six histidines on the C terminal of recombinant protein. The imidazole ring of histidine can coordinate with nickel ion, resulting in the binding of the protein and nickel-containing resin and separate target protein from others.
We first recharge nickel containing resin. To remove remaining nickel ions in the resin 0.1 M EDTA is used to chelate nickel ions. After washing with ten column volumes EDTA solution we add another ten column volumes water to remove EDTA. To recharge the water-washed column, we load 0.5 column volumes of 0.1 M nickel chloride in distilled water. Wash with another ten column volumes water to remove unbound nickel ions.
Figure 3-1. Interaction between nickel ion and poly-histidine tag. Nickel ions bind to
Since that the TGE buffer contains EDTA, calcium chloride is needed to neutralize EDTA. The solution was then mix with 3 ml recharged nickel sepharose and incubate for 1 hr in room temperature. The mixture is poured into column and flowthrough is discarded. To remove non-specific binding protein 5 column volumes TBS containing 0.1M imidazol is added. After washing another 5 column volumes TBS containing 0.5 M imidazol is added to elute target protein. The purity of the protein is verified by gel electrophoresis. Protein solution is stock at 4 °C.
3-12. Resolublization and Refolding of the Fragments of AFU
All procedures were carried out at 4 °C. Each frozen cell solution was thawed and resuspend by TBS buffer (20 mM Tris-Hcl pH=8, 0.5 M sodium chloride, 20 mM imidazole) the cell was lysed by sonication with amplitude=4, duty cycle =50%. The cell lysate was centrifuged at 13000 g for 30 mins to harvest inclusion body. The supernatant was discard and the pellet was resuspended by 10 ml denature buffer (20 mM Tris-HCl, 0.5 M NaCl, 8 Murea, pH = 8.0) and stir for further 2 hrs. After suspension, the solution is centrifuged at 16500 g for 30 mins to remove dissoluble impurities.
To refold the denatured proteins, the solution was dialyze against TBS buffer containing 6 M, 4 M, 2 M, 1 M, and 0 M urea. Each step takes 6 hrs. The insoluble impurity is eliminated by centrifuge at 16500 g for 30 mins.
3-13. Purification of AFU
DEAE and Nickel sepharose were used to purify AFU. First 1 ml DEAE spharose was balanced with PB buffer (20 mM phosphate pH =6.0). Refolded Protein was poured into the column. PBS containing different concentration of sodium chloride (0 M, 0.1 M, 0.2 M, 0.3
M, 0.4 M, and 0.5 M) is used to separate out target protein from others. AFU was collected in phosaphate buffer containing 0.1 M sodium chloride.
For further purification nickel sepharose is used. We first recharge nickel containing resin. To remove remaining nickel ions in the resin 0.1 M EDTA is used to chelate nickel ions. After washing with ten column volumes EDTA solution we add another ten column volumes water to remove EDTA. To recharge the water-washed column, we load 0.5 column volumes of 0.1 M nickel chloride in distilled water. Wash with another ten column volumes water to remove unbound nickel ions.
The solution was then mix with 3ml recharged nickel sepharose and incubate for 1 hour in room temperature. The mixture is poured into column and flowthrough is discarded. To remove non-specific binding protein 5 column volumes TBS containing 0.1 M imidazol is added. After washing another 5 column volumes TBS containing 0.5 M imidazol is added to elute target protein. The purity of the protein is verified by gel electrophoresis. The protein is stocked at -20 °C.
3-14. Resolublization and Refolding of VEGFA
All procedures were carried out at 4 °C. Each frozen cell solution was thawed and resuspend by TBS buffer (20 mM Tris-Hcl pH=8, 0.5 M sodium chloride, 20 mM imidazole) the cell was lysed by sonication with amplitude=4, duty cycle =50%. The cell lysate was centrifuged at 13000 g for 30 mins to harvest inclusion body. The supernatant was discard and the pellet was resuspended by 10 ml denature buffer (20 mM Tris-HCl, 0.5 M NaCl, 8 M urea, pH = 8.0) and stir for further 2hrs. After suspension, the solution is centrifuged at 16500 g for 30 mins to remove dissoluble impurities.
To refold the denatured proteins, the solution was dialyze against TBS buffer containing 6 M, 4 M, 2 M, 1 M, and 0 M urea. Each step takes 6 hours. The insoluble impurity is eliminated by centrifuge at 16500 g for 30 mins.
3-15. Purification of VEGFA
DEAE and Nickel sepharose were used to purify VEGFA. First 5 ml DEAE spharose was balanced with Tris buffer (20 mM Tris-HCl pH =8.0). Refolded proteins were poured into the column. Protein was collected in flowthrough. For further purification nickel sepharose is used. We first recharge nickel containing resin. To remove remaining nickel ions in the resin 0.1 M EDTA is used to chelate nickel ions. After washing with ten column volumes EDTA solution we add another ten column volumes water to remove EDTA. To recharge the water-washed column, we load 0.5 column volumes of 0.1 M nickel chloride in distilled water. Wash with another ten column volumes water to remove unbound nickel ions.
The solution was then mix with 3ml recharged nickel sepharose and incubate for 1 hr in room temperature. The mixture is poured into column and flowthrough is discarded. To remove non-specific binding protein 5 column volumes TBS containing 0.1 M imidazol is added. After washing another 5 column volumes TBS containing 0.5 M imidazol is added to elute target protein. The purity of the protein is verified by gel electrophoresis. The protein is stocked at -20 °C.
3-16. Gel Electrophoresis
Sodium dodecyl sulfate-PAGE (SDS-PAGE) containing 13.5% (wt/vol) polyacrylamide was used for the characterization of the recombinant proteins. The gel is prepared as follow: