Autophagy 6:5, 1-12; July 1, 2010; © 2010 Landes Bioscience
BASIC ReSeARCh pApeR BASIC ReSeARCh pApeR
*Correspondence to: Wan-Wan Lin; Email: [email protected] Submitted: 11/29/09; Revised: 03/22/10; Accepted: 04/01/10
Previously published online: www.landesbioscience.com/journals/autophagy/article/11945 Introduction
Cell death can be largely divided into two classes, programmed cell death and necrosis. Programmed cell death can be further divided into several categories including apoptosis and autophagic death. Apoptosis is a cell-intrinsic mechanism for suicide regulated by cellular signaling pathways, morphologically characterized by caspase activation, cell shrinking, nuclear and cytoplasmic con-densation, DNA fragmentation and formation of apoptosomes.1
The morphological hallmark of autophagy is a double-membrane
zVAD-induced autophagic cell death
requires c-Src-dependent ERK and JNK activation
and reactive oxygen species generation
Szu-ying Chen,1 Ming-Chei Maa,2 Ling-Ya Chiu,1 Jang-Shiun Wang,1 Chung-Liang Chien3 and Wan-Wan Lin1,*
1Department of pharmacology; College of Medicine; National Taiwan University; 2Graduate Institute of Molecular Systems Biomedicine; China Medical University; 3Department of Anatomy and Cell Biology; College of Medicine; National Taiwan University; Taiwan
Key words: caspase 8, autophagy, zVAD, c-Src, PARP, ROS
Abbreviations: ASK1, apoptosis signal-regulating kinase; Atg, autophagy-related; BAPTA-AM, 1,2-bis(2-aminophenoxy)ethane-N,N,N’,N’-tetraacetic acid; BHA, butylated hydroxyanisol; CICD, caspase-independent cell death; DCFH2DA, 6-carboxy-2',7'-dichlorofluorescein diacetate; DED, death effector domains; DMSO, dimethyl sulfoxide; DR, death receptor; ECL, enhanced chemiluminescence; ERK, extracellular signal-regulated kinase; FACS, fluorescence-activated cell sorting; FBS, fetal bovine serum;
FCCP, carbonyl cyanide p-(trifluoromethoxy) phenylhydrazone; JNK, c-Jun-N-terminal kinase; LPS, lipopolysaccharide; 3-MA, 3-methyladenine; MAPK, mitogen-activated protein kinase; MEF, mouse embryonic fibroblasts; MNNG,
N-methyl-N’-nitro-N-nitrosoguanidine; MTT, 3-(4,5-dimethylthiazol-2-yl)2,5-diphenyltetrazolium; NFκB, nuclear factor-kappaB; NOX, NADPH oxidase; PARP, poly(ADP-ribose) polymerase; PARG, poly(ADP-ribose) glycohydrolase; PE,
phosphatidylethanolamine; PI3K, phosphatidylinositol 3-kinase; RIP, receptor interacting protein; ROS, reactive oxygen species; RT-PCR, reverse transcription-polymerase chain reaction; siRNA, small interfering RNA; TNF, tumor necrosis factor;
TNFR1, tumor necrosis factor receptor type 1; TRAF2, TNF receptor-associated factor 2; WM, wortmannin; zVAD, benzyloxycarbonyl-Val-Ala-Asp
This manuscr
ipt has been published online, pr
ior to pr
inting. O
nce the issue is complete and page numbers hav
e been assigned, the citation will change accor
dingly
.
vesicle called an autophagosome, which is formed in the cytosol, and encapsulates long-lived proteins, organelles and bulk cyto-plasm. Through fusing with the lysosome, the contents in the autophagosome are degraded, and thus autophagy is an impor-tant mechanism to maintain homeostasis and to sustain cell viability under starvation conditions.2 However, recent studies
have provided evidence that autophagy may lead to cell death.3
In contrast to apoptosis, necrosis has been traditionally thought to be a passive and accidental form of cell death, which is char-acterized by cytoplasmic swelling, irreversible plasma membrane
A previous report showed that the pan-caspase inhibitor zVAD can induce necrosis accompanied by autophagosome formation in L929 fibrosarcoma cells. Such autophagic cell death relies on caspase 8 inhibition and ROS accumulation. Since the connection of these molecules is still poorly understood, we explored the underlying signaling cascades in this event. First, we confirmed zVAD can stimulate LC3 cleavage, beclin 1 gene expression, autophagosome formation, and ROS accumulation in L929 cells. Antioxidants, Beclin 1 or Atg5 silencing, and class III ptdIns3K inhibitors all effectively blocked ROS production and cell death, suggesting ROS accumulation downstream of autophagy contributes to cell necrosis. zVAD also stimulated pARp activation, and the pARp inhibitor DpQ can reduce zVAD-induced cell death, but not affect ROS production, suggesting the increased ROS leads to pARp activation and cell death. Notably, our data also indicated the involvement of Src-dependent JNK and eRK in zVAD-induced autophagic cell death. We found caspase 8 is associated with c-Src at resting state, and upon zVAD treatment this association is decreased and accompanied by c-Src activation. These results provide new insight into the nonenzymatic function of caspase 8. In addition to initiating proteolytic activity for cell apoptosis, inactivated caspase 8 also functions as a signaling molecule for autophagic cell death.
cells,11 we cannot exclude other sources for ROS elevation. Most
importantly, how caspase inhibition links to autophagy forma-tion has not been fully investigated. PARP1 activaforma-tion is another issue not solved regarding its contribution in the autophagic cell death caused by zVAD in L929 fibrosarcoma. Therefore, in this study, taking L929 fibrosarcoma as a cell model, we elucidated the signaling pathways triggered by caspase inhibition and con-tributing to autophagosome formation.
Results
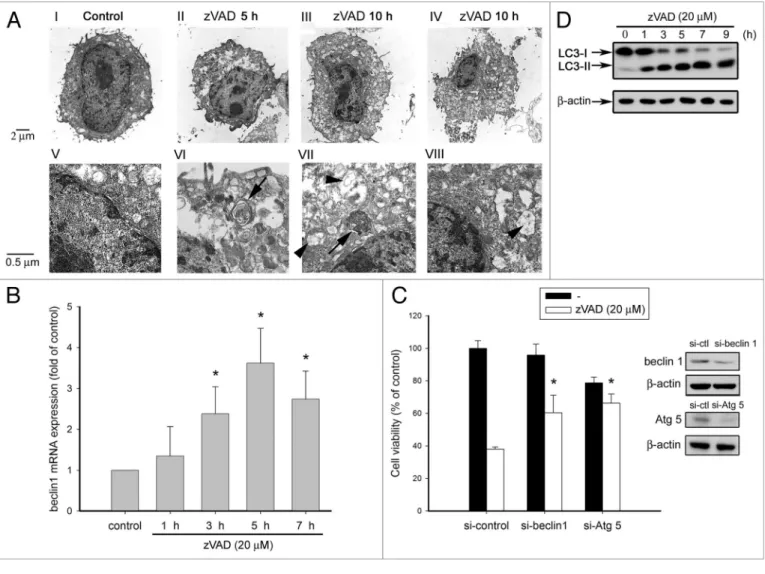
zVAD induces autophagic cell death in L929 fibrosarcoma. It has been reported that zVAD can induce autophagic cell death in L929 cells.10,11 Consistent with previous reports, we found that
zVAD can induce autophagic features under electron micros-copy observation. Compared with the control, zVAD induced autophagosome formation at 5 h and 10 h (Fig. 1A, parts VI, VII). Furthermore, some cells under zVAD treatment exhibited necrosis-like features, which were characterized by mitochondrial swelling (Fig. 1A, parts VII, VIII) and disruption of plasma membrane (Fig. 1A, parts II–IV). Before assessing the cell-death mechanism induced by zVAD in L929 cells, we first used bio-chemical and genetic approaches to confirm the death features of autophagy. Beclin 1 gene transcription, an initial step required for autophagosome formation, was determined by QPCR. We found that zVAD was able to induce a time-dependent increase of beclin 1 mRNA level, which was significantly increased at 3 h and peaked at 5 h (Fig. 1B). When Beclin 1 and Atg5 protein expres-sion were suppressed using an siRNA approach, zVAD-induced cell death was attenuated as compared to the group of cells receiv-ing control siRNA (Fig. 1C). Moreover, since LC3 conversion is a prerequisite for phagophore expansion and autophagosome formation, we measured the effect of zVAD on LC3-II expres-sion. Figure 1D showed a marked appearance of LC3-II after 1 h incubation of zVAD. These results together demonstrate that autophagy is a mediator for zVAD-induced cell death in L929 fibrosarcoma.
JNK- and ERK-regulated ROS production from mitochon-dria contributes to zVAD-induced cell death. Previously, it was shown that zVAD-induced autophagic cell death is JNK- and ROS-dependent.10,11 Thus, we were interested in understanding
the causal relationship of both death mediators, as well as the involvement or not of ERK and p38. Using antioxidant (trolox, which is a water soluble vitamin E analogue), ROS scavenger (BHA), mitochondrial respiratory chain inhibitors (rotenone, FCCP), and MAPK inhibitors (U0126, SP60015, SB203580), we found that zVAD-induced cell death can be attenuated by antiox-idants, mitochondrial respiratory chain inhibitors, JNK inhibi-tor and MEK/ERK inhibiinhibi-tor, whereas SB203580 (p38 inhibiinhibi-tor) had no effect (Fig. 2A). We also detected the intracellular ROS level with the fluorescent dye DCFH2. Figure 2B showed that zVAD can induce intracellular ROS production in a time-depen-dent manner, and this effect was attenuated by the ROS scav-enger BHA, and the JNK inhibitor SP600125. Likewise, trolox, U0126, but not SB203580, dramatically reduced the ROS pro-duction induced by zVAD (Fig. 2C). These results suggest that damage, and organelle breakdown. The release of intracellular
content after plasma membrane rupture is the cause of inflam-mation upon cell necrosis.
The activation of autophagy involves a series of steps: induc-tion, expansion, compleinduc-tion, fusion, and degradation.2 The
induction of autophagy requires Beclin 1 and its interact-ing partner, class III PtdIns3K, resultinteract-ing in the generation of PtdIns(3,4,5)P3 on the endomembrane. In the induction stage, conversion of LC3 from LC3-I to LC3-II and then the recruit-ment of lipidated LC3-II to the phagophore are prerequisite steps for phagophore expansion.4 Once the autophagosome is
com-pleted, it then fuses with the lysosome and exhibits the punctate structure of the autolysosome. The sequestered cargo together with the LC3-II trapped in the lumen of the autophagosome is degraded within the autolysosome. Autophagy contributes to the elimination of misfolded proteins, and abnormal autophagy may lead to pathogenesis of some neurodegenerative disorder such as Alzheimer, Huntington and Parkinson diseases.3,5 The regulation
of autophagy is also important during the progression of cancer. Autophagy may protect tumor cells from apoptosis in response to treatment with anticancer agents, but may be a mechanism for cell death, especially in tumor cells with defective apoptotic machinery.6,7
Because caspase activation is necessary for apoptosis induc-tion, benzyloxycarbonyl-Val-Ala-Asp (zVAD), a cell-permeable pan-caspase peptide inhibitor that irreversibly binds to the cata-lytic site of caspase proteases and inhibits caspase-mediated apop-tosis, is a widely used inhibitor in characterizing apoptotic cell death. However, recent studies have revealed a greater complex-ity of findings with zVAD, and suggest that caspases might be involved in other types of cell death. In the presence of zVAD to block apoptotic machinery, many death insults lead to necrotic cell death, which was termed caspase-independent cell death (CICD).8 No matter whether autophagy is involved or not,
reac-tive oxygen species (ROS) were demonstrated to contribute to CICD.8,9
In murine L929 fibrosarcoma cells, Lenardo’s group found that zVAD alone could lead to cell necrosis involving autophagosome formation and ROS production. Both actions of zVAD depend on caspase 8 inhibition.10,11 Moreover, they found the vacuolated
cell formation is blocked by c-Jun siRNA, indicating that c-Jun N-terminal kinase (JNK) is involved in autophagic cell death induced by zVAD.10 Besides L929 cells, such autophagic CICD
was also shown in lipopolysaccharide and zVAD co-treated mac-rophages.12 In that study, they found that such autophagy requires
PARP1 activation, and involves a ROS-PARP1-autophagy pathway. Caspase-independent autophagy is not only detected in zVAD-treated cells where an apoptotic pathway cannot be achieved, but also in staurosporine, etoposide or photodamage-treated mouse embryonic fibroblasts (MEF) from Bax/Bak dou-ble knockout mice, which are resistant to apoptosis.13,14
Even though autophagic cell death following caspase inhi-bition by zVAD has been identified in L929 fibrosarcoma, the molecular mechanism for ROS induction and JNK activation upon zVAD treatment is ambiguous. Although catalase degra-dation contributes to ROS accumulation in zVAD-treated L929
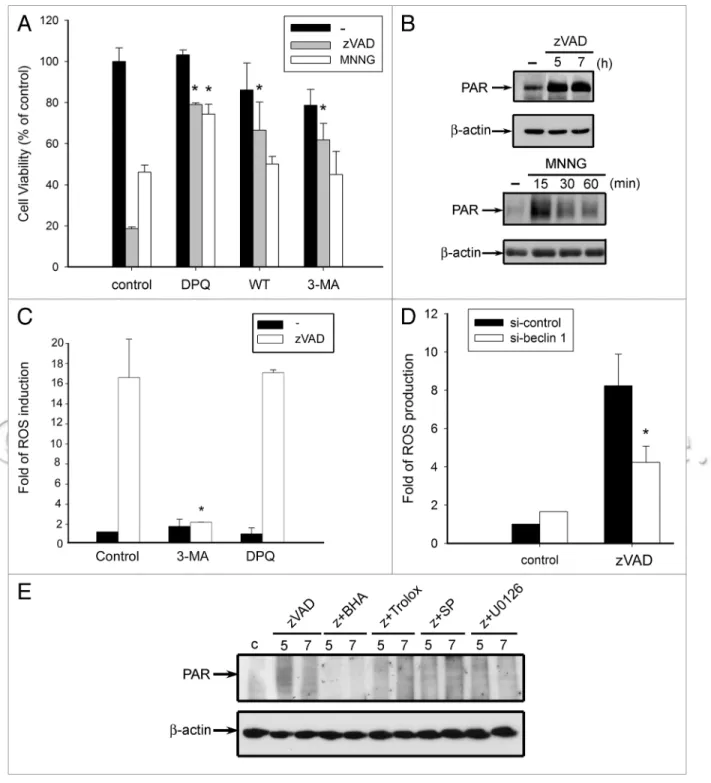
to find out if autophagy is upstream or downstream of ROS pro-duction under zVAD treatment, and if PARP1 is also involved in this event. To this end, we compared the death features of N-methyl-N’-nitro-N’-nitrosoguanidine (MNNG), a PARP1 activator,16,17 with zVAD. In L929 cells, MNNG (250 µM)
treat-ment for 12 h could induce cell death. Pretreating cells with the PARP inhibitor DPQ (10 µM) could effectively diminish cell death in response to zVAD and MNNG (Fig. 3A), suggesting the involvement of PARP1 activation in death signals of both agents. Confirming this point, we found zVAD and MNNG could stimulate PARP1 activation as assessed by increasing PAR accumulation (Fig. 3B). In contrast to the rapid increase of PAR JNK and ERK signaling pathways are upstream of ROS increase.
Next, we suspect that ROS production is derived from mito-chondria, because BHA, a ROS scavenger specifically targeting mitochondria,15 is effective in blocking zVAD-induced ROS
pro-duction. To verify this point, we used a mitochondria-specific dye MitoSox for ROS measurement. Results showed that zVAD still can induce mitochondrial ROS production in a time-dependent manner (Fig. 2D), indicating mitochondria is the major source for ROS production.
ROS production mediates PARP1 activation and autophagic cell death induced by zVAD. After observing that ROS and autophagy are involved in zVAD-induced necrosis, we attempted
Figure 1. zVAD induces autophagic cell death in L929 fibrosarcoma. (A) L929 cells were treated with zVAD (20 µM) for 5 or 10 h as indicated, and cells
were collected and then prepared for electron microscopy (TeM) analysis as described in Materials and Methods. The photos shown in (V–VIII) are the amplification of (I–IV), respectively. The arrows indicate the appearance of autophagosomes, and arrowheads indicate the swelling mitochon-dria, which contain the double-membrane structures. (B) zVAD was added for the time periods as indicated. Total RNA was transcribed to cDNA and subjected to real-time pCR by using a specific primer for beclin 1. Values were normalized to β-actin gene expression, and expressed as fold of control. Data are mean ± S.e.M. from three independent experiments. *p < 0.05, indicating significant increase of beclin 1 mRNA by zVAD. (C) L929 cells were transfected with specific beclin 1 and Atg5 siRNA to knock down endogenous expression of beclin 1 and Atg5 or non-targeting siRNA as a control. After 48 h of transfection, cells were treated with zVAD (20 µM) for 12 h, and then cell viability was measured by MTT assay. When stimulating with indicated agents, we also collected cell lysates at the same time to determine the silencing efficiency by immunoblotting with anti-Beclin 1 and Atg5 antibod-ies. Data are mean ± S.e.M. from three independent experiments. *p < 0.05, indicating significant attenuation of zVAD-induced cell death by beclin 1 siRNA. (D) zVAD was added for the time periods as indicated, and total cell lysates were prepared and immunoblotted with LC3 and β-actin antibodies. Traces are representative from two independent experiments.
activation is not sufficient to induce cell autophagy, and vice versa it might be a downstream event of autophagy.
Furthermore, in order to understand the roles of autophagy and PARP1 in zVAD-induced ROS production, PARP1 and autophagic inhibitors were tested in the ROS productive response of zVAD. As shown in Figure 3C, L929 cells pretreated with an autophagy inhibitor (3-MA) could inhibit zVAD-induced intra-cellular ROS production, while a PARP1 inhibitor (DPQ) could not. Similar inhibition by 3-MA was observed in zVAD action on mitochondrial ROS production (data not shown). Likewise, formation by MNNG, which is a PARP-1 activator, the onset of
the zVAD response is slower. All these results suggest that PARP1 activation is involved in zVAD-induced autophagic cell death. To clarify the participation of autophagy in MNNG-induced cell death, we treated cells with two autophagic inhibitors which can achieve this action via inhibition of PtdIns3K.18 As expected,
wortmannin (WT, 1 µM) and 3-methyladenine (3-MA, 10 mM) can effectively protect cells against zVAD-induced cell death. In contrast, both autophagic inhibitors have no effects on MNNG-induced cell death (Fig. 3A). These results suggest that PARP1
Figure 2. ROS production from mitochondria mediates zVAD-induced cell death. (A) L929 cells were pretreated with trolox (1 mM), BhA (100 µM),
rotenone (Rot, 3 µM), FCCp (10 µM), U0126 (10 µM), Sp600125 (10 µM) or SB203580 (3 µM) for 30 min, followed by zVAD (20 µM) incubation for 12 h. Cell viability was measured by MTT assay and expressed as percentages of control. Data are mean ± S.e.M. from three independent experiments. *p < 0.05, indicating significant attenuation of zVAD-induced cytotoxicity by trolox, BhA, rotenone, FCCp, U0126 and Sp600125. (B) L929 cells were pre-treated with or without BhA or Sp600125 for 30 min, and then stimulated with zVAD (20 µM) for the indicated time periods. After treatment, cells were harvested and followed by intracellular ROS measurement. (C) L929 cells were pretreated with trolox (1 mM), BhA (100 µM), U0126 (10 µM), Sp600125 (10 µM) or SB203580 (3 µM) for 30 min, and then stimulated with zVAD (20 µM). After 10 h incubation, cells were harvested, followed by measurement of intracellular ROS. Data are mean ± S.e.M. from three independent experiments. *p < 0.05, indicating significant attenuation of zVAD-induced ROS production by trolox, BhA, U0126 and Sp600125. (D) zVAD (20 µM) was added for the time periods as indicated. After treatment, cells were harvested followed by measurement of mitochondrial ROS.
of autophagy, but upstream of PARP1 activation. To further support previous suggestions, we analyzed the effects of antioxi-dants on zVAD-induced PAR formation. As shown in Figure 3E, silencing beclin 1 also achieved the inhibitory effect on
zVAD-induced ROS production (Fig. 3D). These results together sug-gest that zVAD-induced ROS production occurs downstream
Figure 3. zVAD-mediated ROS production occurs downstream of autophagy, but upstream of pARp activation. (A) L929 cell were pretreated with
DpQ (10 µM), 3-MA (10 mM) or wortmannin (WT, 1 µM) for 30 min, followed by zVAD (20 µM) or MNNG (250 µM) incubation for 12 h. Cell viability was measured by MTT assay and expressed as percentages of control. (B) zVAD (20 µM) and MNNG (250 µM) were added for the time as indicated, and total cell lysates were prepared and immunoblotted with pAR and β-actin antibodies. (C) L929 cells were pretreated with 3-MA or DpQ for 30 min, and then stimulated with zVAD (20 µM) for 10 h. After treatment, cells were harvested followed by measurement of intracellular ROS. (D) L929 cells were transfected with specific beclin 1 siRNA. After 48 h transfection, cells were treated with zVAD (20 µM) for 10 h, then intracellular ROS was measured. Data represent the mean ± SeM of three independent experiments. *p < 0.05, indicating significant attenuation of zVAD-induced cytotoxicity (A) and ROS production (C and D). (e) Cells were treated with BhA (100 µM), trolox (1 mM), Sp600125 (10 µM) or U0126 (10 µM) for 30 min, followed by zVAD (20 µM) for the time as indicated, and total cell lysates were prepared and immunoblotted with pAR and β-actin antibodies.
inhibit caspase 8 activation.23,24 Moreover, novel enzymatic
inde-pendent actions of caspase 8 in adhesion and epidermal growth factor-induced activation of the ERK pathway were reported.25,26
In order to link caspase 8 inhibition and c-Src activation upon zVAD treatment, we determined the binding condition of both proteins. Results of co-immunoprecipitation using c-Src anti-body revealed that caspase 8 can bind to c-Src at resting state. Such constitutive binding was reduced after zVAD incubation for 10 and 30 min (Fig. 6A). zVAD treatment within 4 h does not change the expression levels of caspase 8 or c-Src (Fig. 6B). In addition, cleaved active caspase 8 was not detected following zVAD treatment. Intriguingly, zVAD is able to induce c-Src phos-phorylation at Tyr 418, indicating the activation of this kinase (Fig. 6C).
Discussion
zVAD-induced autophagic necrosis in L929 fibrosarcoma. zVAD, a pan-caspase inhibitor, is widely used to determine whether cell death occurs by apoptosis. Nevertheless, to maintain cellu-lar viability under proapoptotic conditions, recent studies have increased the complexity of findings with zVAD. In this context, zVAD alone or in the presence of extrinsic death receptor ligands, including TNFα, FasL, TRAIL and LIGHT, can drive necrotic cell death.8,9,27-29 Among these cases that were collectively defined
as CICD, it is intriguing to note that zVAD alone is sufficient to induce L929 cell death, and in particular, unlike other necrosis induced by death receptor activation, it involves autophagosome formation.10-12 This unusual and cell-specific effect of zVAD in
L929 cells becomes a valuable model to elucidate the link between caspase inhibition and autophagy formation.
In this study, in agreement with previous observations,10 our
results showed that zVAD can exhibit autophagic features in L929 cells, such as autophagosome formation, LC3 cleavage and beclin 1 gene expression (Fig. 1A, B and D). All these features were markedly induced within 3–7 h, earlier than the necrotic cell death, which occurred at 10 h and was characterized mor-phologically by organelle swelling and disintegration, nuclear degradation and breakdown of the plasma membrane (Fig. 1A). Consistent with the previous notion that autophagy is involved in zVAD-induced cell necrosis, we confirmed this cell death was blocked by autophagy inhibitors (the class III PtdIns3K inhibi-tor 3-MA, and the classical PtdIns3K inhibiinhibi-tor wortmannin) as well as by knocking down beclin 1 and Atg5 (Figs. 1C and 3A). Nevertheless, Wu’s work in L929 cells shows that zVAD (10 µM) may block autophagy through the inhibition of cathepsin B activity, and autophagy may play a protective role in zVAD-induced necrotic cell death.30 In that work, 10 µM zVAD was
used and it induced a rather delayed cell death as compared to our findings using 20 µM zVAD. We currently have no answer for this discrepancy, but we speculate that the switched role of autophagy in the control of cell survival and death might depend on the strength of the stimulus.
Mitochondrial ROS production is a downstream signal of autophagy. Several lines of evidence shown in this study sup-port the essential role of mitochondria-derived ROS production both trolox and BHA treatment abolished PAR induction caused
by zVAD. Since ERK and JNK were shown to regulate zVAD-induced ROS production (Fig. 2C), we tested their roles in this aspect. Consistent with our scenario, U0126 and SP600125 reduced zVAD-induced PAR expression (Fig. 3E).
c-Src is involved in zVAD-induced autophagic cell death. The relationship between c-Src and autophagy is still unclear. Previously it has been shown that insulin-induced cell swelling is sensed by integrins and thus transduces a signal for p38 activation via c-Src. This effect leads to the inhibition of autophagic prote-olysis in rat liver cells.19 To understand if c-Src plays a crucial role
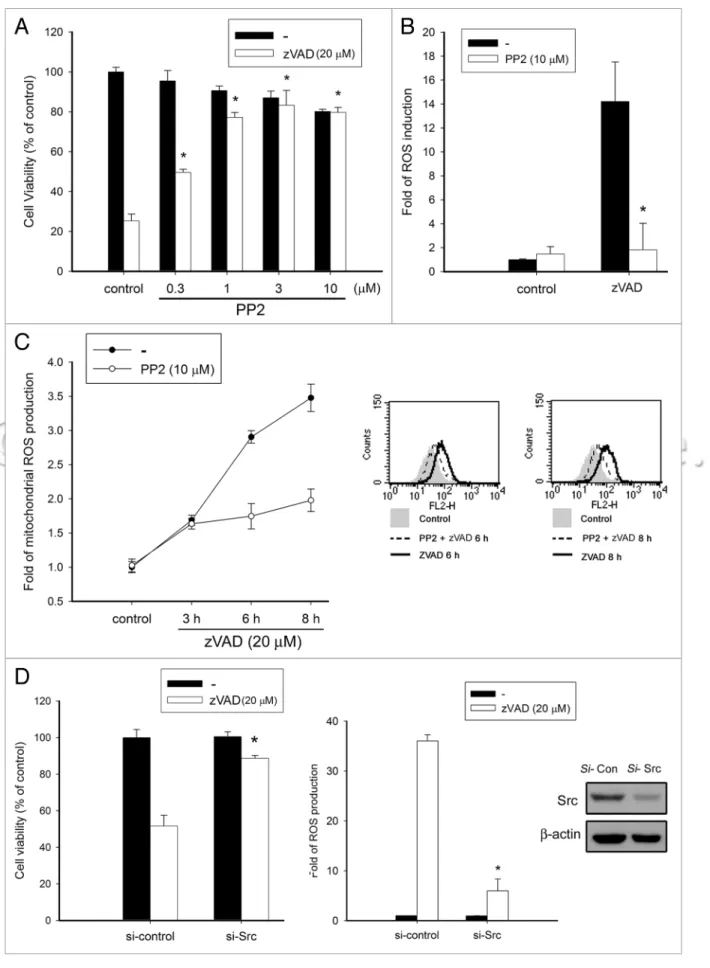
in zVAD-induced autophagic cell death in L929 fibrosarcoma, we examined the effects of the specific c-Src inhibitor PP2. In Figure 4A, we found that PP2 treatment in a concentration-dependent manner confers cell protection against zVAD-induced cytotoxic-ity. Concomitantly, PP2 markedly reduced zVAD-induced ROS production in cytosol (Fig. 4B) and in mitochondria (Fig. 4C), suggesting that c-Src activity might mediate ROS-dependent autophagic cell death induced by zVAD. To further elucidate this event, we knocked down c-Src expression using an siRNA tech-nique. Under efficient silencing of c-Src, we found that zVAD-induced cell death and ROS production were attenuated (Fig. 4D). These results highlight a new role played by c-Src in an autophagic cell death model of zVAD.
c-Src mediates zVAD-induced JNK, ERK activation and autophagy. After observing the inhibitory effects of PP2 on ROS production and cell death, we were interested to under-stand the role of c-Src in zVAD-mediated upstream signaling cascades. Despite some studies that have demonstrated the roles of JNK and ERK in autophagy formation,20-22 and c-Src in the
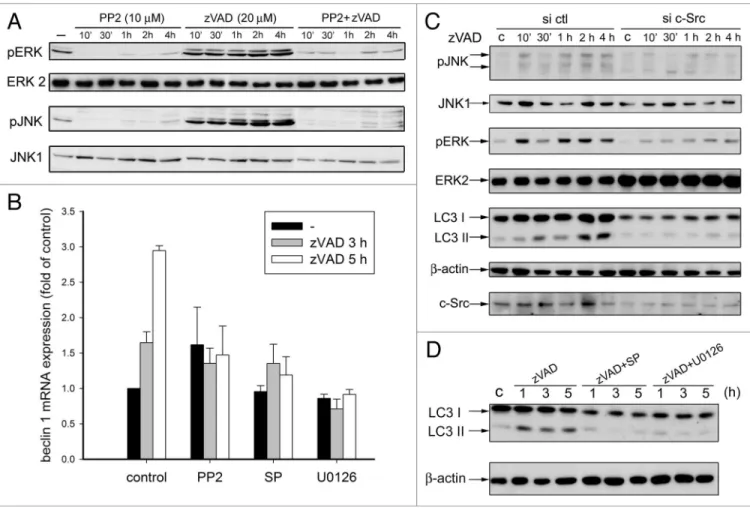
activation of both kinases, the function of c-Src in autophagy remains unknown. To clarify how c-Src crosstalked with ERK and JNK, we determined the effects of PP2 on zVAD-elicited ERK and JNK. Figure 5A showed that zVAD can induce a rapid and sustained activation of JNK and ERK within 4 h incubation. Moreover, both effects of zVAD were abolished by PP2, indicat-ing c-Src is functionindicat-ing upstream to JNK and ERK signalindicat-ing. Next, to verify how c-Src, ERK and JNK activation contributes to autophagy, we determined the effects of kinase inhibitors on zVAD-induced beclin 1 gene expression. As shown in Figure 5B, zVAD-elicited beclin 1 gene transcription at 3 and 5 h was inhib-ited by PP2, U0126 and SP600125. These results suggest that c-Src activation by zVAD leads to ERK and JNK signal path-ways and that the activation of both kinases in turn contributes to autophagy. We also used siRNA to knock down c-Src expres-sion for further validation of its role in zVAD-induced autophagy, JNK and ERK signaling. Figure 5C showed that zVAD-induced LC3-II conversion, and JNK and ERK activation were inhibited after c-Src silencing. JNK and ERK inhibition after SP600125 and U0126 pretreatment, respectively, also blocked zVAD-induced LC3-II conversion (Fig. 5D). These results support the idea that c-Src mediates zVAD-induced JNK and ERK activation and autophagy.
Caspase 8 inhibition leads to c-Src activation. Recent stud-ies identified caspase 8 as a c-Src substrate, and demonstrated that such tyrosine phosphorylation provides a new mechanism to
and found it could be inhibited by the autophagy inhibitor 3-MA (data now shown). Fifth, NADPH oxidase, which is responsible for TNFα-induced generation of O2- in L929 fibrosarcoma,31 did
not participate in the action of zVAD. Our results showed that the NADPH oxidase inhibitor DPI (1 µM) is not able to inhibit zVAD-elicited ROS production (data not shown). Therefore, we conclude that under zVAD treatment ROS production in L929 cells from mitochondria is downstream of autophagy, and medi-ates cell necrosis. In this aspect, an amplification loop between autophagy formation and ROS production might exist. It has downstream of autophagy in zVAD-induced cell necrosis. First,
there is a correlated attenuation of zVAD-induced cytotoxicity (Fig. 2A) and ROS production (Fig. 2B and C) by ROS scav-engers (trolox, BHA). Second, the autophagic inhibitor (3-MA) (Fig. 3C) and siRNA to beclin 1 (Fig. 3D) can effectively reduce ROS induction. Third, inhibitors of the mitochondrial respira-tory chain (i.e., rotenone and FCCP) remarkably inhibit ROS production and cell death, suggesting mitochondria are the major source for the zVAD-elicited ROS increase. Fourth, we detected a mitochondrial ROS increase under zVAD stimulation (Fig. 2D)
Figure 4 (See previous page). c-Src is involved in zVAD-induced autophagic cell death. (A) L929 cells were pretreated with pp2 at concentrations
indi-cated for 30 min, followed by zVAD (20 µM) stimulation. After 12 h incubation, cell viability was measured by MTT assay. (B) L929 cells were pretreated with pp2 (10 µM) for 30 min, followed by zVAD (20 µM) stimulation for 10 h, and then cells were harvested for intracellular ROS measurement. (C) L929 cells were pretreated with pp2 (10 µM) for 30 min, followed by zVAD stimulation at the indicated time periods, and then cells were harvested for mitochondrial ROS measurement. The panel at the right hand shows the raw data. (D) L929 cells were transfected with Src-targeted siRNA, followed by stimulation with zVAD (20 µM). Cell viability (left) and intracellular ROS (right) were determined. Upon stimulating with the indicated agents, cell lysates were collected to determine the silencing efficiency by immunoblotting with anti-Src antibody. Data represent the mean ± SeM of three inde-pendent experiments. *p < 0.05, indicating significant attenuation of zVAD-induced cytotoxicity and ROS production.
Figure 5. c-Src mediates JNK and eRK activation caused by zVAD. (A) L929 cells were treated with pp2 and zVAD for the indicated time periods. Cell
lysates were harvested for immunobloting of JNK-p, JNK1, eRK-p and eRK2. (B) Cells were treated with pp2 (10 µM), U0126 (10 µM) or Sp600125 (10 µM) for 30 min, and then stimulated with zVAD (20 µM). After 3 or 5 h incubation, cells were harvested, followed by measurement of beclin 1 mRNA. (C) L929 cells were transfected with specific c-Src siRNA to knock down endogenous expression of c-Src or non-targeting siRNA as a control. After 48 h of transfection, cells were treated with zVAD (20 µM) for the indicated time. Cell lysates were harvested for immunobloting of JNK-p, JNK, eRK-p, eRK, LC3, c-Src and β-actin. (D) Cells were treated with Sp600125 (10 µM) or U0126 (10 µM) for 30 min, and then stimulated with zVAD (20 µM) for the indicated time. Cell lysates were harvested for immunobloting of LC3 and β-actin.
against zVAD-induced cytotoxicity (Fig. 4A) and intracellular ROS production (Fig. 4B and C), but it has no effects on the similar responses caused by TNFα or MNNG in L929 cells (data not shown). Consistently, when silencing c-Src with an siRNA approach, both zVAD-induced cell death and ROS production are attenuated, while it still has no effect on TNFα-induced ROS production (data not shown). Such an insult-specific difference might be due to the distinct cell death pathways stimulated by these inducers. TNFα and MNNG, as we mentioned above, induce necrosis without containing autophagy in L929 cells. Therefore, it is suggested that c-Src may mediate signaling cas-cades responsible for autophagosome formation.
A previous study has shown the involvement of caspase 8 inhi-bition in zVAD-induced autophagic cell death in L929 cells.10
One mechanism possibly contributing to this event is due to the accumulation of Beclin 1, a novel caspase substrate, lead-ing to an increase in autophagy.40 Despite this, Finlay and Vuori
(2007) strikingly demonstrated a novel enzymatic-independent action of procaspase 8. They showed that in neuroblastoma cells upon EGF stimulation, procaspase 8 forms a complex with c-Src via its death effector domain.41 Subsequently c-Src-mediated
phosphorylation of caspase 8 at Tyr380 leads to caspase inhibi-tion,23,24 and then converts caspase 8 from a pro-apoptotic factor
to a novel signaling molecule that can regulate cell adhesion and motility.25,26 Extending the above findings showing that c-Src
been demonstrated that starvation induces complex formation between class III PtdIns3K and Beclin 1, which in turn, together with other signals, leads to a local rise in H2O2 in the vicinity of mitochondria. This oxidative signal inactivates Atg4, thereby promoting lipidation of Atg8, an essential step in the process of autophagy.32
However, unlike the case in zVAD-treated L929 cells, TNFα, which also induces ROS-dependent necrosis in L929 cells,33 does
not trigger autophagy formation based on morphological and biochemical analyses (data not shown). MNNG action is another case that can induce ROS production, but not autophagy forma-tion (data not shown). Moreover, 3-MA and wortmannin have no effects on MNNG- (Fig. 3A), TNFα- and H2O2-induced cyto-toxicity (data not shown). Therefore, even though autophagy is induced by ROS in various pathophysiological conditions,12,32,34
such as during nutrient starvation, mitochondrial toxins, isch-emia and reperfusion, it is not a general phenomenon. At least in L929 fibrosarcoma, it does not happen in the treatment with MNNG, TNFα and H2O2. Alternatively the autophagy induc-tion by zVAD might rely on an addiinduc-tional specific upstream regu-lating signal.
c-Src-dependent ERK and JNK are required for autophagy formation. Intriguingly, our study also showed a novel enzymatic activity-independent action of caspase 8 in Src, JNK and ERK activation in L929 cells. It has been reported that MAPK signal-ing pathways are able to modulate autophagy in different man-ners. ERK1/2 could phosphorylate G interacting protein (GAIP) and stimulate autophagy.20 Conversely, amino acids stimulate the
sustained phosphorylation of GAIP at Ser259, which is involved in the negative regulation of Raf-1, and thus inhibits Ras/Raf-1/ ERK1/2-mediated autophagy.35 In contrast, it was shown that
p38 might limit the constitutive autophagy activity by imped-ing the fusion of autophagosomes and lysosomes. This blockade, however, could be relieved by transiently activated ERK induced by autophagic stimuli such as starvation.36 The relationship
between JNK and autophagy has also been reported recently. In this context, ER stress induced IRE1-JNK activation was demonstrated to be required for autophagosome formation.21
Subsequent studies further identified the molecular links between JNK and autophagy. Phosphorylation of Bcl-2, which disrupts the Bcl-2-Beclin 1 complex in turn leading to autophagy, was demonstrated.37,38 Except Bcl-2 phosphorylation, JNK-mediated
beclin 1 expression and p53 phosphorylation were demonstrated to contribute to autophagic cell death in cancer cells.39 Moreover,
since Src has been reported in the upstream signaling of MAPKs, including ERK and JNK, our results suggest the necessity of ERK and JNK for zVAD-induced autophagy, and provide evi-dence that Src is a key player for the activation of both kinases.
Caspase 8 inhibition releases c-Src activation under zVAD treatment. Currently, the relationship between Src and autophagy is still unclear. Previously, Schliess et al. showed that insulin-induced cell swelling is sensed by integrins, which trans-duce signaling via Src into p38 activation, and lead to inhibi-tion of autophagic proteolysis in rat liver cells.19 Interestingly,
here we found Src is involved in zVAD-induced autophagic cell death. Src inhibitor (PP2) pretreatment can protect L929 cells
Figure 6. zVAD induced c-Src dissociation from caspase 8 and
activa-tion. (A) After treating with zVAD (20 µM) for different periods, cell lysates were subjected to immunoprecipitation with c-Src antibody, fol-lowed by immunobloting with caspase 8 and c-Src antibodies. In (B and C), cell lysates were subjected to immunobloting with caspase 8, Thy418 phosphorylated Src, Src and β-actin antibodies.
caspase 8 can dissociate and release c-Src for activation, which then transduces signals to ERK and JNK. The activation of both MAPKs in turn sequentially triggers autophagy, ROS accumula-tion, PARP1 activation and necrotic cell death.
Materials and Methods
Cell culture. Murine L929 fibrosarcoma cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) (v/v), 100 U/ml penicillin, 100 µg/ml streptomycin, and incubated at 37°C in a humidified atmosphere of 5% CO2 in air.
Reagent. DMEM (12100-046), FBS (04001-1A), penicillin and streptomycin (03031-1B) were purchased from Gibco BRL. Polyclonal antibodies specific for ERK2 154) and JNK1 (sc-474) horseradish peroxidase-conjugated anti-mouse (sc-2969) and anti-rabbit (sc-2004) antibodies were purchased from Santa Cruz Biotechnology. Polyclonal antibody specific for β-actin (MAB1501) was obtained from Upstate. PP2 (529573), U0126 (662005), SB203580 (559389), trolox (6-hydroxy-2,5,7,8-tetram-ethylchroman-2-carboxylicacid) (648471), SP600125 (420119), and wortmannin (681675) were obtained from Calbiochem. Phosphorylated antibodies of ERK (Thr202/Tyr204, 9101) and JNK (Thr183/Tyr185, 9251) were purchased from Cell Signaling Technology. Beclin 1 (612112) and PAR (51811) antibodies were obtained from BD Pharmingen. 3-Methyladenine (3-MA, M9281), carbonyl cyanide p-(trifluoromethoxy) phenylhydrazone (FCCP, C2920), rotenone (R8875), zVAD-fluoromethylketone (zVAD-FMK, H8787), butylated-hydroxyanisol (BHA, B6655), 3-(4,5-dimethylthiazol-2-yl)2,5-dephenyltetrazoliumbromide (MTT, M2128), and 2'7'-dichloro-dihyrofluorescein diacetate (DCFH2-DA, D6883) were from Sigma-Aldrich. N-methyl-N’-nitro-N-nitrosoguanidine (MNNG, O2392) was purchased from Chem Service. LC3 (PM036) antibody was purchased from MBL. MitoSOXTM (M36008) was purchased from
Invitrogen-Molecular Probes. Atg5 antibody (GTX102361) was purchased from Gentex.
Measurement of cell viability by MTT assay. Cells (104/
ml) grown in 96-well plates were incubated with the indicated drugs. For MTT assay, MTT (5 mg/ml) was added for 1 h, then the culture medium was removed; the formazan granules gener-ated by live cells were dissolved in 100% DMSO and shaken for 10 min. The OD at 550 nm and 630 nm was measured using a microplate reader. The net absorbance (OD550 minus OD630) indicates the enzymatic activity of mitochondria and implicates cell viability.
RNA interference. The siRNA duplexes specific for mouse c-Src (LU-040877-00-0002), beclin 1 (L-055895-00-00005) and Atg5 (L-064838-00-0005) were obtained from Dharmacon RNA Technologies. The siRNA contained four RNA sequences in a SmartPool selected from the NCBI RefSeq Database by a proprietary algorithm. The control nontargeting pool contains four functional nontargeting siRNAs with guanine and cyto-sine content comparable to that of the functional siRNA but lacking specificity for known gene targets. The 100 nM RNAi was transfected by means of MicroPorator (Promega), and the activation provides a new role for caspase 8 in signal transduction,
we surprisingly observed the binding of c-Src and caspase 8 under resting conditions in L929 fibrosarcoma (Fig. 6A). Moreover, it is interesting to note that such an interaction is reduced when caspase 8 is in an inhibitor-bound inactive state, leading to the dissociation and activation of c-Src (Fig. 6A and C). Therefore, our study again strengthens the crucial crossregulation between caspase 8 and c-Src, and provides an additional mechanism to limit c-Src activation at the basal state. According to previous findings as well as ours, caspase 8 is a new signaling adaptor for c-Src under resting conditions, and it can initiate catalytic activity-independent signaling pathways. In L929 fibrosarcoma, upon caspase inhibitor binding to caspase 8, recruited c-Src can be released for activation and then initiation of autophagic cell death. Nevertheless in some EGF-stimulated cancer cells, c-Src activation leads to phosphorylation of the recruited caspase-8, preventing its autocatalysis and switching its function as an adap-tor for cell migration and adhesion. In L929 cells, we suggest the binding between c-Src and caspase 8 can be modulated by the caspase 8 conformation; possibly the binding of zVAD to the cat-alytic groove of procaspase 8 results in conformational changes and hinders the accessibility of c-Src to the death effector domain of procaspase 8.
PARP1 mediates ROS-dependent necrosis. PARP1 hyper-activation-induced necrosis has been implicated in several pathophysiological conditions. Overactivation of PARP1 results in unregulated PAR synthesis and widespread cell death. Previous studies, in most cases using MNNG as a potent PARP1 activa-tor, have revealed that the generation of PAR can trigger intra-cellular ATP depletion, mitochondrial dysfunction, AIF release, calpain activation and eventually caspase-independent necrotic cell death.17 Even though it has been demonstrated that PARP
activation could modulate autophagic cell death,12 the signaling
pathway between PARP and autophagy is unclear.
Currently bidirectional interactions between ROS production and PARP1 activation have been documented. In this aspect, some studies showed that ROS production can trigger PARP1 activation followed by autophagic cell death.12 Vice versa, PARP1
activation also was reported to induce ROS generation from mitochondria.17 In this study, we found that the PARP1 inhibitor
(DPQ) can attenuate cell death induced by zVAD and MNNG (Fig. 3A), but not by TNFα (data not shown), suggesting PARP1 activation is also involved in zVAD-induced autophagic cell death in L929 cells. Conversely, DPQ has no effect on zVAD-induced ROS production (Fig. 3C), while it can block that induced by MNNG (data not shown). These findings suggest that upon zVAD stimulation of L929 cells, PARP1 activation is a down-stream event of ROS production. Recent studies also have impli-cated the close relationship between ER stress and autophagy. ER stress-induced IRE1-JNK activation is required for autophago-some formation.21 Based on this, we examined whether an ER
stress response occurs in zVAD-treated cells. We did not observe any features of ER stress, for example GRP78 and CHOP induc-tion, in zVAD-treated L929 cells (data not shown).
In summary, we propose signaling pathways for zVAD-induced cell death in L929 cells. zVAD-zVAD-induced inhibition of
Quantitative real-time PCR analysis. Total RNA was extracted by acid guanidinium thiocyanate-phenol-chloroform extraction. RT was performed using a Promega kit, and 5 µg of total RNA was reverse transcribed to cDNA. Real-time PCR was performed in 96-well plates and each 35 µl PCR reaction contained cDNA, platinum SYBR green QPCR superMix-UDG (Invitrogen), primers (mouse beclin forward primer 5'-AAA CTG GAC ACG AGC TTC AAG AT-3'; mouse beclin reverse primer 5'-TCG CCT GGG CTG TGG TA-3'; β-actin forward primer 5'-CGG GGA CCT GAC TGA CTA CC-3'; β-actin reverse primer 5'-AGG AAG GCT GGA AGA GTG C-3'), and the pas-sive reference dye to normalize the SYBR green/double stranded DNA complex signal during analysis to correct for well-to-well variations. A no RT control RNA sample was used with each real-time PCR experiment containing β-actin primers to verify no genomic DNA contamination. Amplification products were detected by an ABI Prism 7900 (Applied Biosystems), checked using dissociation curve software (Perkin-Elmer Life Sciences) and by gel electrophoresis to confirm the size of the DNA frag-ment and that only one product was formed.
Electron microscopy detection of autophagosomes. Cells were collected and fixed with 4% paraformaldehyde in 0.1 M cacodylate buffer at 4°C overnight. Cells were then rinsed with cacodylate buffer and post-fixed with 1% osmium tetroxide in 0.1 M cacodylate buffer and subsequently dehydrated in a graded series of ethanol (75%, 85%, 95% and 100%), and embedded in an Epon-araldite mixture. After the resin had solidified, the plastic culture dish was broken up to release the resin containing the embedded cells. Specimens were sectioned with a Diatome diamond knife on a Reichert Ultracut E ultramicrotome (Leica). Ultrathin sections were stained with uranyl acetate and lead cit-rate and viewed with an H-7100 electron microscope (Hitachi). All images were taken by an ORCA-ER CCD digital camera sys-tem (Advanced Microscopy Techniques).
Statistical evaluation. Values are expressed as the mean ± S.E.M. of at least three experiments. Analysis of variance was used to assess the statistical significance of the differences, with a p value of <0.05 being considered statistically significant.
Acknowledgements
This work was supported by the Frontier and Innovative Research of National Taiwan University (98R0335) and the cooperative research program of NTUCM and CMUCM (97F008-103). condition was PulseVoltage (V):1600; Pulse Width (ms):10;
PulseNumber: 4. To achieve gene silencing, we transfected the cells with the siRNA for 48 h and followed by the drug treat-ment; then the gene silencing effects were evaluated by western blot analysis.
Immunoblot analysis and immunoprecipitation. After stimulation, cells were lysed in 1% Triton X-100 lysis buffer (20 mM Tris-HCl, pH 7.5, 125 mM NaCl, 1% Triton X-100, 1 mM MgCl2, 25 mM β-glycerophosphate, 50 mM NaF, 100 µM Na3VO4, 1 mM PMSF, 10 µg/ml leupeptin, and 10 µg/ml apro-tinin). The cell lysates were resolved by SDS-PAGE and analyzed by western blotting with specific antibodies. To determine the binding between caspase 8 and c-Src, cells treated with zVAD were washed twice with PBS, lysed in 500 µl RIPA lysis buffer (containing 150 mM NaCl) and centrifuged at 14,000 rpm, at 4°C for 30 min. The supernatant fraction was collected and pre-cleaned by normal IgG and 10 µl protein A/G-agarose beads for 1 h at 4°C. After centrifugation, supernatant was incubated with c-Src antibody at 4°C with rocking overnight and then 10 µl pro-tein A/G-agarose beads were added and the sample was rotated at 4°C for another 30 min. The immunocomplexes were washed three times with cold lysis buffer (containing 150 mM NaCl) and twice with 300 mM NaCl-containing lysis buffer. The precipi-tated complexes were added to 50 µl 2X Laemmli sample buffer and the samples were heated to 95°C for 5 min. After centrifuga-tion, the sample supernatants were resolved by 10% SDS-PAGE, and then the proteins detected by immunoblotting with specific primary antibody.
Cytosolic and mitochondrial ROS detection. For measur-ing cytosolic and mitochondrial ROS, we used DCFH2DA and MitoSOXTM, respectively. DCFH
2DA can readily enter cells and
be cleaved by esterase to yield a polar, nonfluorescent product, DCFH. ROS in the cells promotes the oxidation of DCFH to yield the fluorescent product DCF. MitoSOXTM is a live-cell
per-meant and is rapidly and selectively targeted to the mitochondria. Once in the mitochondria, MitoSOXTM Red reagent is oxidized
by superoxide and exhibits red fluorescence (Ex/Em: 510/580). After treating for the indicated time periods, cells were collected, then incubated in PBS containing fluorescent reagent (5 µM) for 30 min at 37°C. After incubation, cells were washed with PBS twice, trypsinized, resuspended in 0.5 ml PBS, and immedi-ately submitted to flow analysis using a FACScan flow cytometer (Becton Dickinson).
References
1. Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell 2004; 116:205-19.
2. Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol 2007; 8:741-52. 3. Levine B, Kroemer G. Autophagy in the pathogenesis
of disease. Cell 2008; 132:27-42.
4. Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell 2004; 6:463-77.
5. Winslow AR, Rubinsztein DC. Autophagy in neuro-degeneration and development. Biochim Biophys Acta 2008; 1782:723-9.
6. Hippert MM, O’Toole PS, Thorburn A. Autophagy in cancer: good, bad or both? Cancer Res 2006; 66:9349-51.
7. White E, DiPaola RS. The double-edged sword of autophagy modulation in cancer. Clin Cancer Res 2009; 15:5308-16.
8. Tait SW, Green DR. Caspase-independent cell death: leaving the set without the final cut. Oncogene 2008; 27:6452-61.
9. Chen TY, Chi KH, Wang JS, Chien CL, Lin WW. Reactive oxygen species are involved in FasL-induced caspase-independent cell death and inflammatory responses. Free Radic Biol Med 2009; 46:643-55. 10. Yu L, Alva A, Su H, Dutt P, Freundt E, Welsh S, et al.
Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science 2004; 304:1500-2. 11. Yu L, Wan F, Dutta S, Welsh S, Liu Z, Freundt E, et al.
Autophagic programmed cell death by selective catalase degradation. Proc Natl Acad Sci USA 2006; 103:4952-7.
12. Xu Y, Kim SO, Li Y, Han J. Autophagy contributes to caspase-independent macrophage cell death. J Biol Chem 2006; 281:19179-87.
13. Shimizu S, Kanaseki T, Mizushima N, Mizuta T, Arakawa-Kobayashi S, Thompson CB, Tsujimoto Y. Role of Bcl-2 family proteins in a non-apoptotic pro-grammed cell death dependent on autophagy genes. Nat Cell Biol 2004; 6:1221-8.
14. Buytaert E, Callewaert G, Vandenheede JR, Agostinis P. Deficiency in apoptotic effectors Bax and Bak reveals an autophagic cell death pathway initiated by pho-todamage to the endoplasmic reticulum. Autophagy 2006; 2:238-40.
15. Festjens N, Kalai M, Smet J, Meeus A, Van Coster R, Saelens X, Vandenabeele P. Butylated hydroxyanisole is more than a reactive oxygen species scavenger. Cell Death Differ 2006; 13:166-9.
34. Scherz-Shouval R, Elazar Z. ROS, mitochondria and the regulation of autophagy. Trends Cell Biol 2007; 17:422-7.
35. Pattingre S, Bauvy C, Codogno P. Amino acids inter-fere with the ERK1/2-dependent control of mac-roautophagy by controlling the activation of Raf-1 in human colon cancer HT-29 cells. J Biol Chem 2003; 278:16667-74.
36. Corcelle E, Djerbi N, Mari M, Nebout M, Fiorini C, Fenichel P, et al. Control of the autophagy maturation step by the MAPK ERK and p38: lessons from environ-mental carcinogens. Autophagy 2007; 3:57-9. 37. Wei Y, Pattingre S, Sinha S, Bassik M, Levine B.
JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell 2008; 30:678-88.
38. Wei Y, Sinha S, Levine B. Dual role of JNK1-mediated phosphorylation of Bcl-2 in autophagy and apoptosis regulation. Autophagy 2008; 4:949-51.
39. Park KJ, Lee SH, Lee CH, Jang JY, Chung J, Kwon MH, Kim YS. Upregulation of Beclin-1 expression and phosphorylation of Bcl-2 and p53 are involved in the JNK-mediated autophagic cell death. Biochem Biophys Res Commun 2009; 382:726-9.
40. Cho DH, Jo YK, Hwang JJ, Lee YM, Roh SA, Kim JC. Caspase-mediated cleavage of ATG6/Beclin-1 links apoptosis to autophagy in HeLa cells. Cancer Lett 2009; 274:95-100.
41. Finlay D, Vuori K. Novel noncatalytic role for cas-pase-8 in promoting SRC-mediated adhesion and Erk signaling in neuroblastoma cells. Cancer Res 2007; 67:11704-11.
26. Finlay D, Howes A, Vuori K. Critical role for caspase-8 in epidermal growth factor signaling. Cancer Res 2009; 69:5023-9.
27. Vercammen D, Beyaert R, Denecker G, Goossens V, Van Loo G, Declercq W, et al. Inhibition of cas-pases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J Exp Med 1998; 187:1477-85.
28. Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, et al. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol 2000; 1:489-95. 29. Wilson CA, Browning JL. Death of HT29
adenocar-cinoma cells induced by TNF family receptor activa-tion is caspase-independent and displays features of both apoptosis and necrosis. Cell Death Differ 2002; 9:1321-33.
30. Wu YT, Tan HL, Huang Q, Kim YS, Pan N, Ong WY, et al. Autophagy plays a protective role during zVAD-induced necrotic cell death. Autophagy 2008; 4:457-66.
31. Kim YS, Morgan MJ, Choksi S, Liu ZG. TNF-induced activation of the Nox1 NADPH oxidase and its role in the induction of necrotic cell death. Mol Cell 2007; 26:675-87.
32. Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J 2007; 26:1749-60.
33. Schulze-Osthoff K, Bakker AC, Vanhaesebroeck B, Beyaert R, Jacob WA, Fiers W. Cytotoxic activity of tumor necrosis factor is mediated by early damage of mitochondrial functions. Evidence for the involvement of mitochondrial radical generation. J Biol Chem 1992; 267:5317-23.
16. Haince JF, Rouleau M, Hendzel MJ, Masson JY, Poirier GG. Targeting poly(ADP-ribosyl)ation: a promising approach in cancer therapy. Trends Mol Med 2005; 11:456-63.
17. Xu Y, Huang S, Liu ZG, Han J. Poly(ADP-ribose) polymerase-1 signaling to mitochondria in necrotic cell death requires RIP1/TRAF2-mediated JNK1 activa-tion. J Biol Chem 2006; 281:8788-95.
18. Rubinsztein DC, Gestwicki JE, Murphy LO, Klionsky DJ. Potential therapeutic applications of autophagy. Nat Rev Drug Discov 2007; 6:304-12.
19. Schliess F, Reissmann R, Reinehr R, vom Dahl S, Haussinger D. Involvement of integrins and Src in insulin signaling toward autophagic proteolysis in rat liver. J Biol Chem 2004; 279:21294-301.
20. Ogier-Denis E, Pattingre S, El Benna J, Codogno P. Erk1/2-dependent phosphorylation of Gα-interacting protein stimulates its GTPase accelerating activity and autophagy in human colon cancer cells. J Biol Chem 2000; 275:39090-5.
21. Ogata M, Hino S, Saito A, Morikawa K, Kondo S, Kanemoto S, et al. Autophagy is activated for cell sur-vival after endoplasmic reticulum stress. Mol Cell Biol 2006; 26:9220-31.
22. Shinojima N, Yokoyama T, Kondo Y, Kondo S. Roles of the Akt/mTOR/p70S6K and ERK1/2 signaling pathways in curcumin-induced autophagy. Autophagy 2007; 3:635-7.
23. Cursi S, Rufini A, Stagni V, Condo I, Matafora V, Bachi A, et al. Src kinase phosphorylates Caspase-8 on Tyr380: a novel mechanism of apoptosis suppression. EMBO J 2006; 25:1895-905.
24. De Toni EN, Kuntzen C, Gerbes AL, Thasler WE, Sonuc N, Mucha SR, et al. P60-c-src suppresses apoptosis through inhibition of caspase 8 activation in hepatoma cells, but not in primary hepatocytes. J Hepatol 2007; 46:682-91.
25. Barbero S, Barila D, Mielgo A, Stagni V, Clair K, Stupack D. Identification of a critical tyrosine residue in caspase 8 that promotes cell migration. J Biol Chem 2008; 283:13031-4.