Poly(ether imide)s with Terminal Amino Groups
YAO-TE CHANG,1 CHING-FONG SHU,1 CHYI-MING LEU,2 KUNG-HWA WEI2 1Department of Applied Chemistry, National Chiao Tung University, Hsin-Chu, Taiwan 30035, Republic of China
2
Department of Materials Science and Engineering, National Chiao Tung University, Hsin-Chu, Taiwan 30035, Republic of China
Received 29 April 2003; accepted 26 July 2003
ABSTRACT: We synthesized an AB2-type monomer,
4-{4-[di(4-aminophenyl)methyl]-phenoxy}phthalic acid, which contained one phthalic acid group and two aminophenyl functionalities. The direct self-polycondensation of the AB2-type monomer in the
pres-ence of triphenylphosphite as an activator afforded a hyperbranched poly(ether imide) with a large number of terminal amino groups. This polymer was characterized with1H
NMR and IR spectroscopy. The degree of branching of the hyperbranched poly(ether imide) was approximately 56%, as determined by a combination of model compound studies and an analysis of1H NMR spectroscopy integration data. The terminal amino
groups underwent functionalization readily. The solubility and thermal properties of the resulting polymers depended on the nature of the chain end groups. In addition, the hyperbranched poly(ether imide) was grafted with polyhedral oligomeric silsesquioxane (POSS). Transmission electron microscopy analysis revealed that the grafted POSS molecules aggregated to form a nanocomposite material.© 2003 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem 41: 3726 –3735, 2003
Keywords: AB2 monomer; degree of branching; hyperbranched; nanocomposites;
poly(ether imide); TEM
INTRODUCTION
Hyperbranched polymers have been the subject of considerable interest because their unique highly branched structures and surface functionalities confer upon them some unusual properties.1– 4As
predicted theoretically by Flory,5these polymers
can be conveniently prepared in a single step by the one-pot polymerization of ABn-type monomers
to produce highly branched, irregular structures that contain a large number of terminal func-tional groups. Nevertheless, these polymers still maintain many of the architectural features and
properties found in their well-defined dendrimer counterparts,6 –10which are built up through syn-thetic sequences with isolation and purification at each step.11–14 The one-step synthesis is
attrac-tive because it allows hyperbranched polymers to be more readily prepared on a large scale for any potential applications.
The excellent thermal, mechanical, and electri-cal properties of aromatic poly(ether imide)s make them suitable for use as high-performance polymer materials.15,16The preparation of tradi-tional linear poly(ether imide)s has been carried out typically by two methods, the first of which is a synthesis involving the condensation of dianhy-dride and diamine monomers, thus generating poly(amic acid), followed by cyclodehydration to form the imide ring.17 The second method uses
Correspondence to: C.-F. Shu (E-mail: [email protected])
Journal of Polymer Science: Part A: Polymer Chemistry, Vol. 41, 3726 –3735 (2003) © 2003 Wiley Periodicals, Inc.
aromatic nucleophilic substitution reactions of phenoxide nucleophiles with nitro- or fluoro-sub-stituted phthalimides to form the ether linkage.18 Generally, hyperbranched polymers are prepared by the self-condensation of AB2-type monomers.
Because of the high reactivity of amine and an-hydride functionalities, there is no stable AB2
-type monomer containing one amino group and two anhydride groups (or one anhydride group and two amino groups) available for the prepara-tion of hyperbranched poly(ether imide)s. To over-come this problem, Moore and coworkers19 –21 reported a modification of the nucleophilic dis-placement method for the preparation of hyper-branched poly(ether imide)s that used a Krickel-dorf-type reaction involving tert-butyldimethylsi-lyl-protected benzenediol groups and an activated fluoride in the presence of a catalytic amount of CsF. Kakimoto et al.22–24 prepared hyper-branched poly(ether imide)s from a monomer con-sisting of an amino group and a phthalic acid monomethyl ester. In a previous study, we re-ported that hyperbranched poly(ether imide)s could be prepared by the aromatic nucleophilic substitution of an AB2 monomer containing two
phenol groups and a fluoro-substituted phthalim-ide unit.25More recently, the syntheses of hyper-branched aromatic polyimides with an A2⫹ B3(or
BB⬘2) approach have also been reported. 26 –29
This report describes an alternative synthetic approach for the preparation of hyperbranched poly(ether imide)s. In this procedure, the imide rings are formed by the direct self-polycondensa-tion of a substituted phthalic acid, which contains two aminophenyl groups in the same molecule, with triphenylphosphite (TPP) as an activator.30 Thus, an AB2-type monomer,
4-{4-[di(4-amino-phenyl)methyl]phenoxy}phthalic acid (3), was synthesized, and its subsequent self-polymeriza-tion led to the formaself-polymeriza-tion of a hyperbranched poly-(ether imide) possessing a large number of termi-nal amino groups. It is noteworthy that the NMR spectroscopy signals of the methine protons of the hyperbranched polymer obtained in this way can indicate the degree of branching (DB) of the poly-mer (as discussed later). Through the chemical modification of the terminal amino groups, a va-riety of different functional groups were intro-duced into the hyperbranched poly(ether imide). We investigated the effects that particular termi-nal functiotermi-nal groups had on the physical proper-ties of these hyperbranched poly(ether imide)s. Furthermore, a hyperbranched poly(ether imide) was grafted with polyhedral oligomeric
silsesqui-oxane/benzyl chloride (POSS–BzCl) through the terminal amino groups to form an organic–inor-ganic hybrid material.31The morphology of this hybrid material was examined by means of trans-mission electron microscopy (TEM), which indi-cated that the grafted polyhedral oligomeric sils-esquioxane (POSS) molecules probably aggre-gated to form a nanoscale composite material with a defined architecture.
EXPERIMENTAL
General Directions
Unless otherwise stated, all the reagents and sol-vents were used as received from commercial sources. N-Methyl-2-pyrrolidinone (NMP) was distilled over CaH2under reduced pressure.
Pyri-dine was dried by distillation after being heated under reflux in the presence of KOH. TPP was purified by distillation under reduced pressure. POSS–BzCl was purchased from Hybrid Plastics.
1
H and13C NMR spectra were recorded on a Var-ian Unity 300-MHz spectrometer or a Bruker-DRX 300-MHz spectrometer. IR spectra were ob-tained on a Nicolet 360 Fourier transform infra-red (FTIR) spectrometer. Mass spectra were recorded on a JEOL JMS-SX 102A mass spec-trometer. Gel permeation chromatography (GPC) was carried out on a Waters chromatography unit interfaced with a Waters 410 differential refrac-tometer. We used three 5-m Waters Styragel columns (300⫻ 7.8 mm), connected in series in decreasing order of pore size (105, 104, and 103Å), with dimethylformamide (DMF) as the eluent; standard samples of poly(methyl methacrylate) were used for calibration. Differential scanning calorimetry (DSC) was performed with a DuPont TA 2000 instrument, with a heating/cooling rate of 20 °C min⫺1. Samples were scanned from 30 to 300 °C, cooled to 30 °C, and then scanned a second time from 30 to 300 °C. The glass-transition tem-perature (Tg) was determined from the second
heating scan. Thermogravimetric analysis (TGA) was performed on a DuPont TGA 2950 instru-ment. The thermal stability of the samples was determined under a nitrogen atmosphere by the measurement of the weight loss during heating at a rate of 20 °C min⫺1. The TEM measurements were conducted on a JEOL-2000FX instrument with an acceleration voltage of 200 kV.
4-[Di(4-aminophenyl)methyl]phenol (1)
A mixture of 4-hydroxybenzaldehyde (5.00 g, 41.0 mmol), aniline (13.20 g, 141.9 mmol), and aniline
hydrochloride (0.31 g, 2.42 mmol) was heated at 150 °C under a nitrogen atmosphere for 1.5 h. The excess aniline was then evaporated under re-duced pressure. The residue was added to water (60 mL) and extracted with ethyl acetate (3⫻ 100 mL). The combined extracts were dried over MgSO4, and the solvent was evaporated. The
product was purified by recrystallization from ethyl acetate and then recrystallization from eth-anol to afford 1 (7.00 g, 58.9%).
1
H NMR [deuterated dimethyl sulfoxide (DMSO-d6),␦]: 4.80 (br, 4H), 5.06 (s, 1H), 6.48 (d, J⫽ 8.1 Hz, 4H), 6.65 (d, J ⫽ 8.4 Hz, 2H), 6.72 (d, J⫽ 8.1 Hz, 4H), 6.85 (d, J ⫽ 8.4 Hz, 2H), 9.28 (s,
1H).13C NMR (DMSO-d6, ␦): 54.1, 114.1, 115.0,
129.6, 130.0, 133.0, 136.3, 146.5, 155.3. High-res-olution mass spectrometry (HRMS): [M⫹] calcd. for C19H18N2O, 290.1419; found, 290.1423.
4-{4-[Di(4-aminophenyl)-methyl]phenoxy}phthalonitrile (2)
Potassium carbonate (5.00 g, 36.2 mmol) was added to a solution of 1 (5.00 g, 17.2 mmol) and 4-nitrophthalonitrile (2.99 g, 17.3 mmol) in DMF (20 mL). The mixture was stirred at 130 °C for 12 h under nitrogen. After cooling, the solution was poured slowly into water (120 mL). The pre-cipitated product was collected by filtration, dried
in vacuo, and purified by column chromatography
(1:1 hexane/ethyl acetate) to afford 2 (5.04 g, 70.4%). 1 H NMR (DMSO-d6, ␦): 4.91 (s, 4H), 5.22 (s, 1H), 6.49 (d, J⫽ 8.4 Hz, 4H), 6.76 (d, J ⫽ 8.4 Hz, 4H), 7.06 (d, J⫽ 8.7 Hz, 2H), 7.16 (d, J ⫽ 8.7 Hz, 2H), 7.32 (dd, J⫽ 8.7, 2.6 Hz, 1H), 7.76 (d, J ⫽ 2.6 Hz, 1H), 8.04 (d, J ⫽ 8.7 Hz, 1H). 13C NMR (DMSO-d6, ␦): 54.1, 108.0, 114.0, 115.5, 116.1, 116.7, 120.0, 121.8, 122.6, 129.5, 130.9, 131.8, 136.4, 143.5, 146.7, 151.6, 161.3. HRMS: [M⫹] calcd. for C27H20N4O, 416.1637; found, 416.1631.
4-{4-[Di(4-aminophenyl)methyl]phenoxy}phthalic Acid (3)
A solution of potassium hydroxide (9.0 g) in a mixture of water (60 mL) and ethanol (90 mL) was added to compound 2 (3.00 g, 7.21 mmol). The mixture was then heated under reflux for 12 h. The solution was diluted with water (200 mL) and acidified with 1 N HCl (aqueous) to pH⬃ 4. The precipitated product was filtered, washed thor-oughly with water, and dried to give 3 (3.09 g, 94.3%). 1 H NMR (DMSO-d6,␦): 5.22 (s, 1H), 6.51 (d, J ⫽ 8.4 Hz, 4H), 6.76 (d, J ⫽ 8.4 Hz, 4H), 7.00 (d, J ⫽ 8.7 Hz, 2H), 7.07 (dd, J ⫽ 8.7, 2.4 Hz, 1H), 7.10 (d, J⫽ 8.7 Hz, 2H), 7.33 (d, J ⫽ 2.4 Hz, 1H), 7.93 (d, J⫽ 8.7 Hz, 1H).13C NMR (DMSO-d6,␦): 54.0, 115.1, 117.3, 118.7, 119.6, 126.3, 129.5, 130.7, 132.3, 133.4, 136.8, 141.9, 144.5, 152.9, 159.4, 167.5, 168.4. HRMS: [M⫹ ⫹ H] calcd. for C27H23N2O5, 455.1607; found, 455.1599.
Synthesis of Model Compound 4
TPP (0.12 mL, 0.45 mmol) was added to a solution of 3 (200 mg, 440mol) and 4-n-butylaniline (660 mg, 4.43 mmol) in NMP/pyridine (2.5 mL, 4:1 v/v), and the mixture was then heated at 150 °C for 12 h. The mixture was poured into water (20 mL) and extracted with ethyl acetate (3⫻ 20 mL). The combined extracts were dried over MgSO4,
evap-orated to dryness, and purified by column chro-matography (1:1 hexane/ethyl acetate) to afford 4 (125 mg, 50.1%). 1 H NMR (DMSO-d6,␦): 0.90 (t, J ⫽ 7.5 Hz, 3H), 1.26 –1.37 (m, 2H), 1.53–1.63 (m, 2H), 2.62 (t, J ⫽ 7.8 Hz, 2H), 4.94 (s, 4H), 5.23 (s, 1H), 6.49 (d, J ⫽ 8.4 Hz, 4H), 6.76 (d, J ⫽ 8.4 Hz, 4H), 7.08 (d, J ⫽ 8.4 Hz, 2H), 7.16 (d, J ⫽ 8.4 Hz, 2H), 7.27–7.33 (m, 5H), 7.37 (dd, J⫽ 8.1, 1.8 Hz, 1H), 7.93 (d, J ⫽ 8.4 Hz, 1H).13 C NMR (DMSO-d6,␦): 13.8, 21.8, 33.1, 34.5, 53.9, 111.4, 113.8, 119.8, 122.6, 125.2, 125.8, 127.1, 128.7, 129.4, 129.4, 130.8, 131.7, 134.2, 142.4, 143.0, 146.7, 152.5, 163.0, 166.4, 166.5. HRMS: [M⫹] calcd. for C37H33N3O3, 567.2522; found, 567.2517.
Synthesis of Model Compounds 5 and 6
A solution of compound 4 (60 mg, 0.11 mmol) and phthalic anhydride (20 mg, 0.14 mmol) in dim-ethylacetamide (DMAc; 1.5 mL) was heated at 150 °C under nitrogen for 6 h. The resulting mix-ture was added to water (20 mL) and extracted with ethyl acetate (3 ⫻ 20 mL). The combined extracts were dried over MgSO4, and the solvent
was evaporated in vacuo. The products were pu-rified by column chromatography (1:1 hexane/ ethyl acetate). Compound 5 1 H NMR (DMSO-d6, ␦): 0.90 (t, J ⫽ 7.5 Hz, 3H), 1.27–1.36 (m, 2H), 1.53–1.61 (m, 2H) 2.62 (t, J ⫽ 7.5 Hz, 2H), 5.00 (s, 2H), 5.54 (s, 1H), 6.53 (d, J ⫽ 8.1 Hz, 2H), 6.84 (d, J ⫽ 8.1 Hz, 2H), 7.14 (d, J
⫽ 8.7 Hz, 2H), 7.20–7.42 (m, 12H), 7.84–8.00 (m, 5H). 13C NMR (DMSO-d6, ␦): 13.8, 21.8, 33.1, 34.5, 54.2, 111.6, 113.9, 120.0, 122.7, 123.4, 125.3, 125.8, 127.1, 128.7, 129.3, 129.5, 129.8, 130.2, 130.9, 131.5, 134.3, 134.7, 141.6, 142.4, 144.6, 147.1, 152.9, 162.8, 166.4, 166.5, 167.1. HRMS: [M⫹] calcd. for C45H35N3O5, 697.2577; found,
697.2603. Compound 6 1 H NMR (DMSO-d6, ␦): 0.90 (t, J ⫽ 7.2 Hz, 3H), 1.27–1.36 (m, 2H), 1.53–1.61 (m, 2H), 2.62 (t, J ⫽ 7.2 Hz, 2H), 5.87 (s, 1H), 7.20 (d, J ⫽ 7.8 Hz, 2H), 7.24 –7.50 (m, 16H), 7.80 – 8.0 (m, 5H). 13C NMR (DMSO-d6, ␦): 13.8, 21.8, 33.1, 34.5, 54.4, 111.9, 120.3, 122.8, 123.5, 125.4, 125.9, 127.1, 127.4, 128.7, 129.5, 130.2, 131.1, 131.5, 134.3, 134.7, 140.2, 142.4, 143.3, 153.3, 162.7, 166.3, 166.5, 167.0. HRMS: [M⫹] calcd. for C53H37N3O7, 827.2631; found, 827.2623.
Preparation of Polymer PEI-1
A solution of 3 (700 mg, 1.54 mmol) and TPP (400 L, 1.54 mmol) in NMP/pyridine (35 mL, 4:1 v/v) was heated at 150 °C under nitrogen for 12 h. After cooling, the resulting polymer was precipi-tated into methanol. The polymer was collected, purified twice by reprecipitation from DMF into methanol, and dried in vacuo to give PEI-1 (630 mg, 97.6%).
1
H NMR (DMSO-d6,␦): 4.98 (br, 2H), 5.19–5.86
(m, 1H), 6.42– 6.58 (m, 2H), 6.70 – 6.88 (m, 2H), 7.00 –7.40 (m, 10H), 7.92 (br, 1H). IR (KBr): 3457, 3365 (NOH), 1778, 1721 (CAO, imide ring), 1373 cm⫺1(CON, imide).
Preparation of Polymer PEI-2
Acetyl chloride (1.0 mL) was added dropwise un-der nitrogen to a solution of PEI-1 (100 mg, 240 mol) in DMAc (2.0 mL) at 0 °C. After the addi-tion was complete, the mixture was stirred at 0 °C for 2 h and then at 25 °C for 6 h. The resulting solution was added dropwise to methanol (50 mL) with agitation. The polymer was collected by fil-tration, purified twice by reprecipitation from DMF into methanol, and dried in vacuo to give PEI-2 (95 mg, 90%).
1
H NMR (DMSO-d6,␦): 2.00 (s, 3H), 5.50–5.85
(m, 1H), 6.98 –7.68 (m, 14H), 7.91 (br, 1H), 9.91 (br, 1H). IR (KBr): 3380 (NOH), 1778, 1721
(CAO, imide ring), 1670 (CAO, amide), 1373 cm⫺1(CON, imide).
Preparation of Polymer PEI-3
A solution of PEI-1 (100 mg, 240mol), benzoic acid (120 mg, 980mol), LiCl (20 mg), and TPP (60L, 240 mol) in NMP/pyridine (2.5 mL, 4:1 v/v) was heated at 110 °C under nitrogen for 12 h. The resulting solution was added dropwise to methanol (50 mL) with agitation. The polymer was collected by filtration, purified twice by repre-cipitation from DMF into methanol, and dried in
vacuo to give PEI-3 (120 mg, 92%). 1
H NMR (DMSO-d6, ␦): 5.59–5.84 (m, 1H),
6.95– 8.10 (m, 20H), 10.26 (br, 1H). IR (KBr): 3380 (NOH), 1778, 1721 (CAO, imide ring), 1665 (CAO, amide), 1373 cm⫺1(CON, imide).
Preparation of Polymer PEI-4
PEI-4 was prepared from PEI-1 (100 mg, 240 mol) and 4-n-octylbenzoic acid (170 mg, 726 mol) with the same procedure used for the prep-aration of PEI-3.
Yield: 144 mg, 95%. 1H NMR (DMSO-d6, ␦):
0.55–1.63 (m, 15H), 2.61 (br, 2H), 5.58 –5.84 (m, 1H), 7.02–7.42 (m, 12H), 7.63– 8.00 (m, 7H), 10.14 (br, 1H). IR (KBr): 3375 (NOH), 1778, 1721 (CAO, imide ring), 1665 (CAO, amide), 1373 cm⫺1(CON, imide).
Preparation of Polymer PEI-5
A solution of PEI-1 (100 mg, 240 mol) and phthalic anhydride (150 mg, 1.01 mmol) in DMAc (2.0 mL) was heated at 110 °C under nitrogen for 12 h. The resulting solution was added dropwise to a methanol solution (50 mL) with agitation. The polymer was collected by filtration, purified twice by reprecipitation from DMF into methanol, and dried in vacuo to give PEI-5 (117 mg, 89%).
1
H NMR (DMSO-d6,␦): 5.84 (br, 1H), 7.02–7.45
(m, 14H), 7.78 – 8.00 (m, 5H). IR (KBr): 1778, 1721 (CAO, imide ring), 1373 cm⫺1(CON, imide).
Preparation of Poly(ether imide)/Polyhedral Oligomeric Silsesquioxane (PEI–POSS)
A solution of POSS–BzCl (160 mg, 154mol) in tetrahydrofuran (THF; 2.0 mL) was added to a mixture of PEI-1 (60 mg, 0.14 mmol), K2CO3(133
mg, 0.96 mmol), and NaI (40 mg, 0.27 mmol) in DMF (2.0 mL). The reaction mixture was heated
at 70 °C for 12 h. The resulting solution was added dropwise to n-hexane (50 mL) with agita-tion to remove the ungrafted POSS–BzCl. The precipitate was collected by filtration, washed with n-hexane, purified twice by reprecipitation from THF into methanol, and dried in vacuo to give PEI–POSS (120 mg, 56%).
1
H NMR (THF-d8,␦): 0.70–2.15 (m, 77H), 4.33
(br, 2H), 5.20 –5.60 (m, 1H), 6.45– 8.00 (m, 19H). IR (KBr): 3390 (NOH), 1778, 1716 (CAO, imide ring), 1373 cm⫺1(CON, imide).
RESULTS AND DISCUSSION
Monomer Synthesis
The synthesis of the AB2-type monomer 3 is
out-lined in Scheme 1. The acid-catalyzed condensa-tion of 4-hydroxybenzaldehyde with an excess of aniline gave 1.32The nucleophilic substitution of the nitro function of 4-nitrophthalonitrile by the phenoxide group generated from 1 in the presence of K2CO3 in DMF furnished the phthalonitrile
compound 2, which subsequently, on hydrolysis with aqueous KOH in ethanol, afforded the de-sired 3. The AB2-type monomer contained one
phthalic acid group and two aminophenyl func-tionalities. The structures of the synthesized com-pounds were verified with1H and13C NMR spec-troscopy and mass specspec-troscopy.
Polymer Synthesis
The one-step polymerization of monomer 3 was performed (Scheme 2) in NMP/pyridine at 150 °C for 12 h, in the presence of equimolar amounts of TPP as an activator, to obtain quantitatively the
hyperbranched poly(ether imide) PEI-1 contain-ing terminal aminophenyl groups. The molecular weight of PEI-1 was determined by GPC with DMF/LiBr (0.05 M) as the eluent, calibrated against poly(methyl methacrylate) standards. GPC analysis shows that the weight-average mo-lecular weight (Mw) and polydispersity
[weight-average molecular weight/number-[weight-average mo-lecular weight (Mw/Mn)] of the polymer were
ap-proximately 7.5⫻ 104g/mol and 4.6, respectively. These values, however, were only indicative of the polymer size and weight distribution because the highly branched nature of PEI-1 may have caused it to deviate strongly when measured against lin-ear, coil-like poly(methyl methacrylate) stan-dards. The degree of polymerization was depen-dent on the reaction time. The wide molecular weight distribution of the hyperbranched poly-(ether imide) broadened with increasing conver-sion. For example, as the reaction time proceeded from 3 to 9 h, both Mwand Mw/Mnof the obtained
polymers increased from 1.8 to 3.8 ⫻ 104 g/mol and from 2.6 to 3.9, respectively. The broadening of the molecular weight distribution resembled that of the previous reports of other hyper-branched polymers and agreed with Flory’s pre-dictions concerning molecular weight distribution behavior for highly branched systems.5,33,34
The structure of PEI-1 was characterized by FTIR spectroscopy. The absence of the CAO vi-brational band at 1670 cm⫺1 of poly(amic acid) indicated that imide formation was complete. The polymer exhibited characteristic carbonyl absorp-tions corresponding to an imide ring at 1778 (CAO asymmetric stretching) and 1721 cm⫺1 (CAO symmetric stretching), as well as the typi-cal aromatic–imide CON stretching mode at 1373 cm⫺1. Furthermore, the peaks at 3457 and 3365 cm⫺1 were attributed to the terminal amino groups. The thermal properties of the hyper-branched poly(ether imide) were investigated by DSC and thermogravimetry measurements. From the DSC measurement, Tg of PEI-1 was
detected at 280 °C. TGA revealed that PEI-1 had high thermal stability, with a 5 wt % loss
ob-Scheme 1
served at 487 °C followed by a 10 wt % loss at 544 °C.
DB
Hyperbranched polymer PEI-1 was formed by a sequential condensation of the AB2monomer and
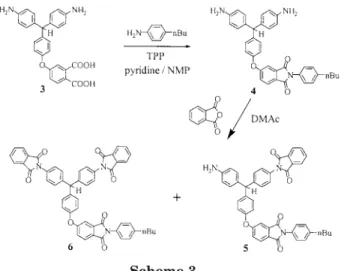
resulted in an irregular dendritic structure in which three different types of subunits (i.e., ter-minal, linear, and dendritic) could be present. DB, a typical characteristic often used to evaluate the structural irregularity of hyperbranched poly-mers, is defined as the sum of dendritic and ter-minal units relative to the total number of units.35,36A combination of studies of model com-pounds and NMR spectroscopy was used to quan-tify the different subunits present in the hyper-branched polymer and, subsequently, to deter-mine its DB.35 The preparation of model compounds used for NMR spectroscopy character-ization is illustrated in Scheme 3. Model com-pounds 4, 5, and 6 resemble the terminal unit, the linear unit, and the dendritic unit, respectively, and their1H NMR spectra are shown in Figure 1. The chemical shift of the methine proton is sensitive to the number of aminophenyl groups attached to the methine carbon atom. A distinct resonance of the methine proton for the terminal model compound 4 appears at 5.23 ppm (Ht),
whereas resonances of the corresponding protons of the linear (5) and dendritic (6) model com-pounds can be observed at 5.54 (Hl) and 5.87 (Hd)
ppm, respectively. In addition, two pairs of AB quartets, observed at ␦ ⫽ 6.49/6.76 ppm and ␦ ⫽ 6.53/6.84 ppm, can be attributed to the protons of the aminophenylene rings (Hb and Hc) for
model compounds 4 and 5, respectively. Figure 1 also shows the1H NMR spectrum of the hyper-branched poly(ether imide). A good correlation is found between the 1H NMR spectrum of PEI-1 and those of the model compounds. The reso-nances at 5.22, 5.52, and 5.82 ppm can be attrib-uted to the methine protons of the terminal, lin-ear, and dendritic subunits, respectively, in the hyperbranched polymer. The resonances at 6.42– 6.58 and 6.70 – 6.88 ppm are due to Hb and Hcof
the terminal and linear subunits. The integration of these well-resolved resonances allowed the rel-ative percentage of each subunit in PEI-1 to be determined. DB of the hyperbranched poly(ether imide) PEI-1 was estimated to be 56% on the basis of the relative integrations of these sig-nals.35 It should be noted that the percentage estimated for the terminal subunit was approxi-mately equal to that for the dendritic subunit. This result is consistent with the theoretical pre-diction that the number of dendritic units is equal to the number of terminal units for an AB2-type
hyperbranched polymer possessing a high molec-ular weight.5
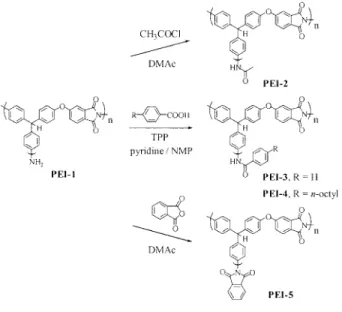
Chemical Modification
Hyperbranched polymers are characterized by a large number of chain-terminating groups. The
Scheme 3
Figure 1. 1H NMR spectra in DMSO-d
6of (a) PEI-1
terminal amino groups in PEI-1 were easily func-tionalized to yield hyperbranched polymers with a variety of functional end groups. As shown in Scheme 4, the amino end groups of PEI-1 were reacted with either an acid chloride or an acid in the presence of TPP as an activator to give the corresponding amide/imide derivatives PEI-2 to PEI-4. Upon reaction with phthalic anhydride, the amino groups were converted into phthalim-ide units to yield the imphthalim-ide derivative PEI-5. The successful modification of the end groups of PEI-1 was confirmed by the 1H NMR spectra of the derivatives. Resonances associated with the pro-tons of the aminophenyl groups disappeared en-tirely after end capping. The resonances of the protons of the amide groups of PEI-2 to PEI-4 appear in the region of 9.9 –10.2 ppm. We note that the signals corresponding to the methine protons of the terminal, linear, and dendritic sub-units of PEI-5 appear together as a broad peak at
5.84 ppm. We attribute this feature to the similar chemical environments experienced by the three different subunits, which resemble the dendritic subunit, after the modification reaction is com-plete. The analysis of the integrations of the 1H
NMR spectra of the derivatives indicates that the modification reactions proceeded almost quanti-tatively (⬎95%).
The nature of the end groups influences the physical and chemical properties of hyper-branched polymers.37 Table 1 summarizes the
solubility and thermal properties of the hyper-branched poly(ether imide)s PEI-1 to PEI-5. Be-cause of their highly branched structures, these poly(ether imide)s had enhanced solubility in or-ganic solvents and were highly soluble in polar solvents such as DMAc, DMF, NMP, and pyri-dine. The incorporation of different chain ends, however, led to differences in solubility in very polar and relatively nonpolar solvents. PEI-1 to PEI-3 and PEI-5 were soluble in DMSO, whereas PEI-4, which possessed flexible n-octyl chain-end-ing groups, was soluble in DMSO only upon heat-ing. Conversely, in solvents such as chloroform and dichloromethane, polymers PEI-1 to PEI-3 were completely insoluble, PEI-4 was soluble in chloroform and soluble in dichloromethane upon heating, and PEI-5 was soluble in both solvents. In THF, only PEI-4 was soluble. It is known that the transition from polar end groups to nonpolar end groups results in a decrease in the value of Tg
for hyperbranched polymers because of a reduc-tion in the extent of intermolecular interacreduc-tions between polymer molecules.38 As revealed by DSC measurements, Tgof PEI-1, which had polar
amino terminal groups, was 280 °C, whereas the
Tg values of PEI-3 and PEI-5, which had less polar terminal groups (amide and imide groups), were 240 and 267 °C, respectively. The lowering of Tg was caused by a decrease in the extent of Scheme 4
Table 1. Thermal and Solubility Properties of Hyperbranched Poly(ether imide)s
Polymer Tg(°C) Solubilitya CHCl3 CH2Cl2 THF DMF DMSO PEI-1 280 ⫺ ⫺ ⫺ ⫹ ⫹ PEI-2 214 ⫺ ⫺ ⫺ ⫹ ⫹ PEI-3 240 ⫺ ⫺ ⫺ ⫹ ⫹ PEI-4 192 ⫹ ⫾ ⫹ ⫹ ⫾ PEI-5 267 ⫹ ⫹ ⫺ ⫹ ⫹
hydrogen bonding. Further decreases in the val-ues of Tg, to 214 and 192 °C, were observed for
PEI-2 and PEI-4, respectively, brought about by the replacement of the phenyl group with a methyl group and the introduction of flexible n-octyl chain termini. Polymers PEI-1 to PEI-5 pos-sessed good thermal stability, as evidenced by TGA. The 10% weight loss occurred in the range of 486 –547 °C under a nitrogen atmosphere. PEI-4, which had long alkyl chain end groups, exhibited the lowest decomposition temperature.
PEI–POSS
Organic–inorganic nanocomposite materials have been the subject of considerable attention because of their potential to bridge the gap between or-ganic polymers and inoror-ganic ceramics.31,39,40 One class of inorganic materials (POSS) has a nanometer-sized structure with a high surface area and controlled porosity and is suitable for designing hybrid materials with various func-tions. POSS consists of a rigid, cubic silica core (Si8O12) surrounded by eight organic groups, of
which seven are inert and one is reactive.41 Through this one reactive site, POSS molecules have been successfully incorporated as nanosize inorganic pendants into linear organic polymers, such as poly(methyl methacrylate),42 poly(4-methylstyrene),43 polynorbornene,44,45 and poly-urethane.46
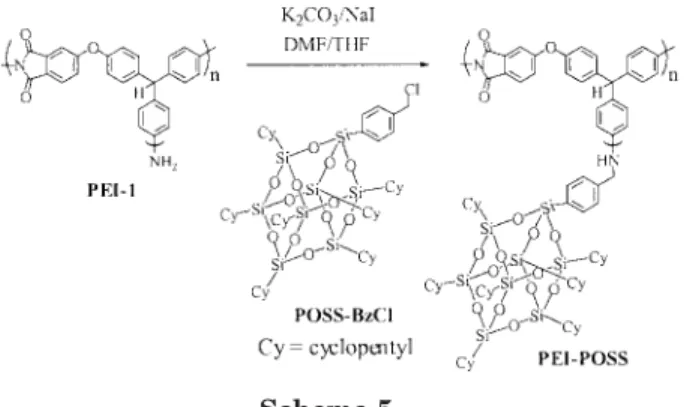
We introduced silsesquioxane cubes into the hyperbranched poly(ether imide) by the reaction of the amino end groups with POSS–BzCl to give an organic–inorganic hybrid material (PEI– POSS; Scheme 5). We used 1.1 equiv of the POSS reagent for the grafting reaction. When the reac-tion was complete, the resulting polymer solureac-tion was precipitated into n-hexane to remove the un-reacted POSS–BzCl. The degree of POSS
incorpo-ration in the hybrid material was estimated to be about 80 mol % by1H NMR spectroscopy, based on the ratio between the integrals of the cyclopen-tyl signals of the POSS unit and the sum of the aromatic signals. The morphology of the hybrid material PEI–POSS was examined by means of TEM. The high density of the Si atom of POSS allowed direct imaging without the need for con-ventional staining. Figure 2 shows the TEM mi-crograph of a sample prepared by the dripping of the polymer solution (5 wt % in THF) onto a 200-mesh copper net coated with a carbon film and drying in air. The hybrid material self-assem-bled to form defined core–shell-structured spheres with diameters ranging from 80 to 120 nm. The inner core was about 60 –100 nm wide, consisting of the hyperbranched poly(ether imide) and POSS molecules attached to the interior branches of poly(ether imide). The thickness of the outer shell of these spheres was about 10 –15 nm, which suggested that the aggregated POSS molecules were positioned at the peripheries of the exterior. Because the dipole– dipole interac-tions between imide segments were quite differ-ent from the interactions between POSS mole-cules, the grafting of POSS molecules to the ter-mini of the hyperbranched poly(ether imide) chains led to a phase-separated system. Addition-ally, the interchain and/or intrachain POSS– POSS interactions resulted in variations in the
Scheme 5
aggregate solubility. For example, PEI–POSS was completely soluble only in a mixture of THF and DMF and was partially soluble in THF. These results suggest that the grafted POSS molecules probably aggregated to form a nanoscale compos-ite material; this supports a method for preparing materials with well-defined architectures.
CONCLUSIONS
The direct self-polycondensation of a new AB2
-type monomer (3), which contained one phthalic acid group and two aminophenyl functional-ities, in the presence of TPP as an activator gave a hyperbranched poly(ether imide) with chain-ending amino groups. We estimated DB to be 56% on the basis of 1H NMR spectral analysis. The poly(ether imide) exhibited good solubility in common organic solvents, such as NMP, DMF, pyridine, and DMSO, which we attributed to its highly branched structure. The amino groups at the ends of the chains were readily accessible to reagents in solution and were converted into a variety of functional groups. The nature of the end groups signifi-cantly affected the solubility and thermal prop-erties of the hyperbranched poly(ether imide)s. Additionally, the chain-ending amino groups of the hyperbranched poly(ether imide) were grafted with POSS molecules. TEM analysis re-vealed that the resulting hybrid material self-assembled to form core–shell structures with total diameters ranging from 80 to 120 nm and shell thicknesses of 10 –15 nm. This result indi-cated that the grafted POSS molecules aggre-gated to form a nanocomposite material with a well-defined architecture.
The authors thank the National Science Council of the Republic of China for its financial support.
REFERENCES AND NOTES
1. Malmstro¨m, E.; Hult, A. J Macromol Sci Rev Mac-romol Chem Phys 1997, 37, 555.
2. Kim, Y. H. J Polym Sci Part A: Polym Chem 1998, 36, 1685.
3. Voit, B. J Polym Sci Part A: Polym Chem 2000, 38, 2505.
4. Jikei, M.; Kakimoto, M. Prog Polym Sci 2001, 26, 1233 and references therein.
5. Flory, P. J. J Am Chem Soc 1952, 74, 2718.
6. Tomalia, D. A.; Naylor, A. M.; Goddard, W. A., III. Angew Chem Int Ed Engl 1990, 29, 138.
7. Fre´chet, J. M. J. Science 1994, 263, 1710.
8. Newkome, G. R.; Moorefield, C. N.; Vo¨gtle, F. Den-dritic Molecules: Concepts, Syntheses, Perspec-tives; VCH: Weinheim, 1996.
9. Hecht, S.; Fre´chet, J. M. J. Angew Chem Int Ed Engl 2001, 40, 74.
10. Tomalia, D. A.; Fre´chet, J. M. J. J Polym Sci Part A: Polym Chem 2002, 40, 2719.
11. Tomalia, D. A.; Baker, H.; Dewald, J.; Hall, M.; Kallos, G.; Martin, S.; Roeck, J.; Ryder, J.; Smith, P. Polym J 1985, 17, 117.
12. Newkome, G. R.; Yao, Z.-Q.; Baker, G. R.; Gupta, V. K. J Org Chem 1985, 50, 2003.
13. Hawker, C. J.; Fre´chet, J. M. J. J Am Chem Soc 1990, 112, 7638.
14. Xu, Z. F.; Moore, J. S. Angew Chem Int Ed Engl 1993, 32, 1354.
15. Polyimides; Wilson, D.; Stenzenberzer, H. D.; Hergenrother, P. M., Eds.; Blackie: New York, 1990.
16. Polyimides: Fundamentals and Applications; Ghosh, M. K.; Mittal, K. L., Eds.; Marcel Dekker: New York, 1996.
17. Harris, F. W. In Polyimides; Wilson, D.; Stenzen-berger, H. D.; Hergenrother, P. M., Eds.; Blackie: New York, 1990; Chapter 1.
18. White, D. M.; Takekoshi, T.; Williams, F. J.; Relles, H. M.; Donahue, P. E.; Klopfer, H. J.; Louks, G. R.; Manello, J. S.; Matthews, R. O.; Schluenz, R. W. J Polym Sci Polym Chem Ed 1981, 19, 1635.
19. Thompson, D. S.; Markoski, L. J.; Moore, J. S. Macromolecules 1999, 32, 4764.
20. Markoski, L. J.; Thompson, J. L.; Moore, J. S. Mac-romolecules 2000, 33, 5315.
21. Orlicki, J. A.; Thompson, J. L.; Markoski, L. J.; Sill, K. N.; Moore, J. S. J Polym Sci Part A: Polym Chem 2002, 40, 936.
22. Yamanaka, K.; Jikei, M.; Kakimoto, M. Macromol-ecules 2000, 33, 1111.
23. Yamanaka, K.; Jikei, M.; Kakimoto, M. Macromol-ecules 2000, 33, 6937.
24. Yamanaka, K.; Jikei, M.; Kakimoto, M. Macromol-ecules 2001, 34, 3910.
25. Wu, F.-I.; Shu, C.-F. J Polym Sci Part A: Polym Chem 2001, 39, 2536.
26. Fang, J.; Kita, H.; Okamoto, K. Macromolecules 2000, 33, 4639.
27. Hao, J.; Jikei, M.; Kakimoto, M. Macromolecules 2002, 35, 5372.
28. Chen, H.; Yin, J. J Polym Sci Part A: Polym Chem 2002, 40, 3804.
29. Liu, Y.; Chung, T.-S. J Polym Sci Part A: Polym Chem 2002, 40, 4563.
30. Im, J. K.; Jung, J. C. J Polym Sci Part A: Polym Chem 1999, 37, 3530.
31. Sanchez, C.; Soler-Illia, G. J. de A. A.; Ribot, F.; Lalot, T.; Mayer, C. R.; Cabuil, V. Chem Mater 2001, 13, 3061.
32. Ueda, M.; Nakayama, T. Macromolecules 1996, 29, 6427.
33. Gong, Z.-H.; Leu, C.-M.; Wu, F.-I.; Shu, C.-F. Mac-romolecules 2000, 33, 8527.
34. Wu, F.-I.; Shu, C.-F. J Polym Sci Part A: Polym Chem 2001, 39, 3851.
35. Hawker, C. J.; Lee, R.; Fre´chet, J. M. J. J Am Chem Soc 1991, 113, 4583.
36. Ho¨lter, D.; Burgath, A.; Frey, H. Acta Polym 1997, 48, 30.
37. Hawker, C. J.; Chu, F. Macromolecules 1996, 29, 4370.
38. Schmaljohann, D.; Ha¨uler, L.; Po¨tschke, P.; Voit, B. I.; Loontjens, T. J. A. Macromol Chem Phys 2000, 201, 49.
39. Tsukruk, V. V. Prog Polym Sci 1997, 22, 247. 40. Klok, H. A.; Lecommandoux, S. Adv Mater 2001,
13, 1217.
41. Feher, F. J.; Weller, K. J.; Schwab, J. J. Organo-metallics 1995, 14, 2009.
42. Lichtenhan, J. D.; Otonari, Y. A.; Carr, M. J. Mac-romolecules 1995, 28, 8435.
43. Haddad, T. S.; Lichtenhan, J. D. Macromolecules 1996, 29, 7302.
44. Mather, P. T.; Jeon, H. G.; Romo-Uribe, A.; Haddad, T. S.; Lichtenhan, J. D. Macromolecules 1999, 32, 1194.
45. Zheng, L.; Waddon, A. J.; Farris, R. J.; Coughlin, E. B. Macromolecules 2002, 35, 2375.
46. Fu, B. X.; Hsiao, B. S.; Pagola, S.; Stephens, P.; White, H.; Rafailovich, M.; Sokolov, J.; Mather, P. T.; Jeon, H. G.; Phillips, S.; Lichtenhan, J.; Schwab, J. Polymer 2001, 42, 599.