行政院國家科學委員會專題研究計畫 成果報告
(總計畫與子計畫一)建立評估抗 SARS 病毒中草藥療效的
技術平台
計畫類別: 整合型計畫 計畫編號: NSC92-2751-B-039-002-Y 執行期間: 92 年 07 月 01 日至 93 年 06 月 30 日 執行單位: 中國醫藥大學醫學研究所 計畫主持人: 張建國 報告類型: 完整報告 處理方式: 本計畫可公開查詢中 華 民 國 93 年 8 月 19 日
行政院國家科學委員會補助專題研究計畫
⌧ 成 果 報 告
□期中進度報告
治療嚴重型急性呼吸道症候群中草藥之開發-(總計畫與子計
畫一)建立評估抗 SARS 病毒中草孳療效的技術平台
計畫類別:□ 個別型計畫
⌧ 整合型計畫
計畫編號:NSC92-2751-B-039-002-Y
執行期間:92 年 7 月 1 日至 93 年 6 月 31 日
計畫主持人:張建國
共同主持人:
計畫參與人員:
成果報告類型(依經費核定清單規定繳交):□精簡報告 ⌧完整報告
本成果報告包括以下應繳交之附件:
□赴國外出差或研習心得報告一份
□赴大陸地區出差或研習心得報告一份
□出席國際學術會議心得報告及發表之論文各一份
□國際合作研究計畫國外研究報告書一份
處理方式:除產學合作研究計畫、提升產業技術及人才培育研究計畫、列
管計畫及下列情形者外,得立即公開查詢
□涉及專利或其他智慧財產權,⌧一年□二年後可公開查詢
執行單位:中國醫藥大學醫學研究所
中 華 民 國 九十三 年 八 月 十 日
行政院家科學委員會專題研究計畫成果報告
計畫名稱:治療嚴重型急性呼吸道症候群中草藥之開發-(總計
畫與子計畫一)建立評估抗 SARS 病毒中草藥療效的技術平台
計畫編號:
NSC92-2751-B-039-002-Y
執行期限:
92 年 7 月 1 日至 93 年 6 月 31 日
主 持 人:張建國
執行機構及單位名稱:
中國醫藥大學 醫學研究所
聯絡方式:張建國
中國醫藥大學附設醫院 分子醫學科
台中市 404 育德路 2 號
Tel:04-22052121 ext.7075
Fax:04-22033295
E-mail:[email protected]
本研究已被國際期刊 Intervirology 接受, 印刷中。
中文摘要 嚴重急性呼吸道症候群 (SARS) 是由一種新的冠狀病毒引起。藉由分 析該病毒基因體的序列將可了解其流 行病學及建立疫苗。在本計畫中, 我 們建立了一種快速直接定序臨床檢體 體的方法。我們總共分析了三個檢體, 結果顯示有 17 種單一鹼基的變異及 兩種二個鹼基缺及一種一個鹼基缺失 的突變, 而言些變異並無改變所 coding 的胺基酸。在這 17 種變異中, 在 26203 及 27812 兩個鹼必定為 T:T 或 C:C 兩型。又經由基因演化樹分析 得知, SARS 在台地區的感染可分為兩 群。因此, 我們認為本地區的感染源 至少有二種。 關鍵詞:嚴重急性呼吸道症候群, 多 組即時聚合酶連鎖反應, 基因組分析, 基因演化樹 Abstract
Objective: Severe acute respiratory
syndrome (SARS) is caused by a new coronavirus. Genomic sequence analysis will provide the molecular epidemiology and help to develop vaccines. Methods: We developed a rapid method to amplify and sequence the whole SARS-CoV genome from clinical specimens. The technique employed one step multiplex RT-PCR to amplify the whole SARS-CoV genome, and then nested PCR was performed to amplify a 2 kb region separately. The PCR products were sequenced. Results: We sequenced the genomes of SARS-CoV from 3 clinical specimens obtained in Taiwan. The sequences were similar to those reported by other groups, except that 17 single nucleotide variations and two two-nucleotide deletions, and a one-nucleotide deletion were found. All the variations in the clinical specimens did not alter the amino acid sequence. Of these 17 sequenced variants, two loci (position 26203 and 27812) were segregated together as a specific genotype-T:T or
C:C. Phylogenetic analysis showed two major clusters of the SARS patients in Taiwan. Conclusions. We developed a very economical and rapid method to sequence the whole genome of SARS-CoV, which can avoid cultural influence. From our results, SARS patients in Taiwan may be infected from two different origins.
Key words: SARS-CoV, multiplex
RT-PCR, genome analysis, phylogenetic tree
Introduction
An atypical pneumonia with high contagiousness first appeared in the Guangdong province of People’s Republic of China on November, 2002, which infected 792 cases and caused 31 deaths. This disease spread to Hong Kong first, and caused an outbreak in many countries including Vietnam, Canada, Singapore, Taiwan and other countries. Nearly nine thousand individuals have been infected and over 900 have died from the disease since February, 2003 [1]. This new and deadly syndrome was firstly brought to the attention of the World Health Organization (WHO) by Dr. Carlo Urbani, and was named as severe acute respiratory syndrome (SARS) [2]. The pathogenesis of SARS is believed to be a novel coronavirus (SARS-CoV), which was first found by Peiris et al in Hong Kong [3] and was confirmed by other groups [4-8]. The analysis of genomic sequences showed that the virus is different from previously known coronavirus [9-11], and serological studies also confirmed that the virus has not been found in human [12].
Coronaviridae family is a diverse group of large, enveloped and positive-stranded RNA virus, and these viruses may cause respiratory and enteric infections in human and animals. The previously known human coronaviruses are frequent causes of the common cold.
Life-threatening pneumonia caused by coronaviurses are uncommon. Generally speaking, the genome of coronavirus is the largest (about 30 kb in length) found in any RNA viruses, which encodes 23 putative proteins, including 4 major structure proteins: nucleocapsid (N), spike (S), membrane (M) and envelope (E) proteins. The N, S and M mature proteins contribute to generating the host immune response [13-14]. Similarly, the genome of SARS-CoV is a 29729-nucleotide with polyadenylated RNA and about 41% GC content. The genomic organization of SARS-CoV is similar to that of a typical coronvirus, containing the characteristic gene order (5’-replicase, spike, envelope, membrane, and nucleocapsid-3’) and short untranslated regions at both termini [9-11].
The RNA viruses have high rates in genetic mutations, resulting in evolution of new viral strains and escaping from host defenses [15]. It is very important to understand the mutation rate of the SARS virus spreading through the population for the development of effective vaccines. Meanwhile, several studies have shown that adaptation of a virus to non-natural host cells induces genetic changes in viral genome, and these culture-mediated mutations may produce bias and affect evolutionary studies [16-20]. In order to avoid these causations, we sequenced the entire SARS-CoV directly using clinical specimens. Using these approaches, we were able to analyze the orgin of SARS-CoV infection more accurately.
Materials and Methods
Patients. We collected clinical
specimens including nasal and pharyngeal swabs, blood and stool from three SARS patients. All patients fitted the WHO definition for probable SARS case: fever of 38 ℃ or higher, respiratory symptoms, hypoxia, chest
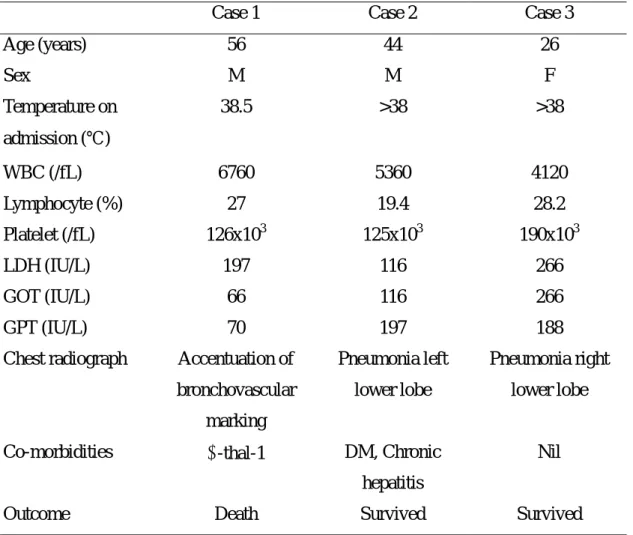
radiograph changes suggestive of pneumonia, and history of close contact with SARS patients. The clinical features of three cases were shown in the Table 1.
Methods. Total RNA was extracted
from the nasopharyngeal samples of the patients using a commercial kit (QIAamp Viral RNA Mini kits, QIAGEN Inc., CA, USA). For RT-PCR analysis of SARS-CoV, the upstream primer 5’-CTAACATGCTTAGGATAATGG-3’ and the downstream primer 5’-CAGGTAAGCGTAAAACTCATC-3’ were used to amplify part of polymerase gene, the methods used were identical to Ksiazek TG et al [7].
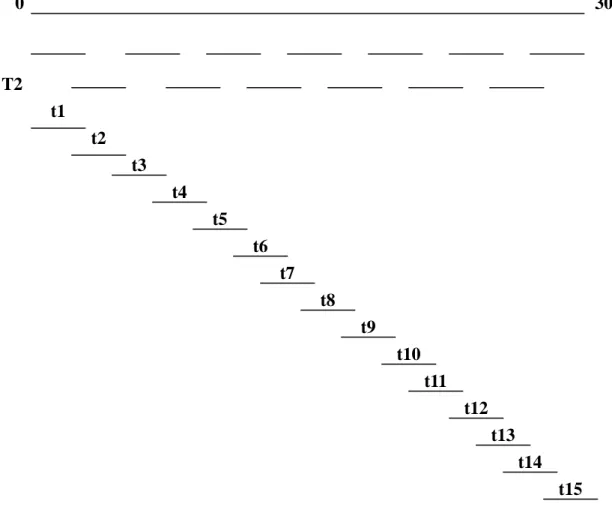
In order to have enough PCR products of SARS-CoV genome for sequencing, we utilized a multiplex one-step RT-PCR method to amplify the entire genome followed by nested PCR to separately amplify each region of the genome. The strategy was shown in Fig 1. The PCR products were then purified and sequenced. The whole SARS-CoV genome was amplified in 2 reactions (the primers used were shown in Table 2) using multiplex one-step RT-PCR method; the reaction T1 amplified 8 regions of the genome, which are nucleotide (nt) 29 to 2194, 3852 to 6070, 7841 to 10050, 11767 to 14164, 15948 to 18119, 19836 to 22177, 23771 to 25864, and 27551 to 29700, respectively; the reaction T2 amplified 7 regions including nt 1941 to 4058, 5908 to 8044, 9869 to 12006, 14021 to 16129, 17804 to 20040, 21956 to 23961, and 25647 to 27794, respectively. The PCR products of T1 were subjected to nested PCR amplification using primer pairs t1, t3, t5, t7, t9, t11, t13, and t15 separately, and T2 products were amplified in a different reaction using primer pair t2, t4, t6, t8, t10, t12 and t14, respectively. The primers of t1, t2, t3, t4……t15 were shown in Table 3.
was performed using a commercial kit and the procedures used was recommended by the manufacturer (QIAGEN One-step RT-PCR kit, QIAGEN Inc., CA, USA). Briefly, 50µl of reaction solution contains 1-2µl of RNA solution from the clinical extract, 0.6µM each of primers, 1X buffer, 0.6mM of each dNTP, 3µl enzyme mix, 6 units Rnase inhibitor. Sample mixtures were placed in a thermal cycler with temperature at 45℃ for 30 min, and then were incubated at 95℃ for 15 min before PCR amplification. The PCR amplification consisted of 3 steps including denaturation at 94℃ for 10 seconds, annealing at 50℃ or 54℃ for 1 min, and extension at 68℃ for 2.5 minutes. The 3 steps were repeated for 40 cycles. The nested PCR was performed as follows: 50µl of reaction solution contains 1µl of multiplex PCR product, 0.2µM of each nested PCR primer, 0.2mM of each dNTP, 1X Taq buffer, and 1 unit Taq polymerase. The PCR condition included denaturation at 94℃ for 1 minute, annealing at 56℃ for 1 min, and extension at 72℃ for 2 min, and these 3 steps were repeated for 35 cycles. The PCR products were resolved on agarose gels, and the proper PCR fragments were excised from gels and then purified using a commercial kit (Qiaex II gel extraction kit, QIAGEN Inc., CA, USA). DNA sequencing was performed using the di-deoxy chain termination method as described in Big DyeTM Terminator cycle sequencer and using an ABI 310 machine (Applied Biosystems Inc., CA, USA). The primers used for sequencing will be provided on request.
Phylogenetic analysis of the SARS-CoV genomes was carried out by ClustalW
(http://www.ebi.ac.uk/clustalw/index.ht ml).
Results
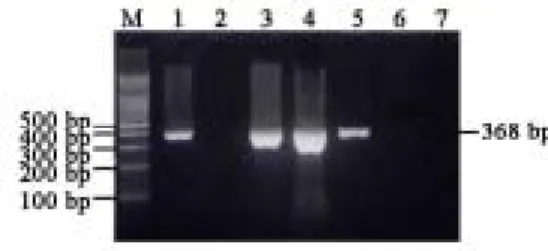
RT-PCR analytic strategy for SARS-CoV was shown in Fig. 1. The PCR product of SARS-CoV is a fragment of 368bp using specific primers (Fig. 2). Among 87 cases of SARS suspects examined, 3 were positive for the SARS-CoV. These three cases were further used for sequencing analysis of entire genomes.
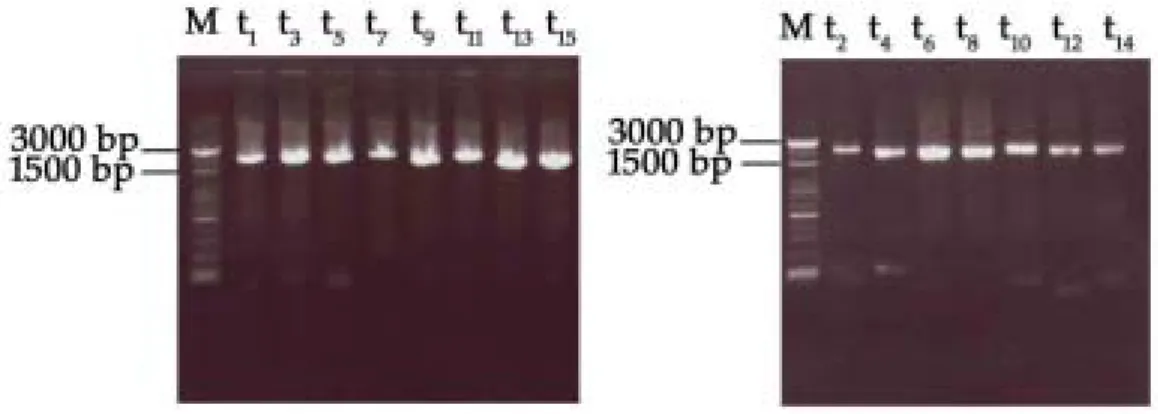
The whole SARS-CoV genome was amplified by multiplex RT-PCR in two separate reactions, followed by 15 different nested PCRs to amplify a 2-kb fragment in each reaction that cover the whole SARS-CoV genome (Fig. 3). These 15 nested PCR products were subjected for direct sequencing. The results were submitted to GenBank, and the accession numbers are AY348314 for TC3, AY338175 for TC2 and AY338174 for TC1.
The genomic sequences of these clinical specimens were similar to those reported from other groups. We compared those genomic and found there were 20 differences, which are 17 single nucleotide variations, two two-nucleotide deletions and a one-nucleotide deletion (Table 4). We further compared the complete genomic sequences of four clinical specimens with the complete genomic sequences of three cultural isolates in Taiwan. The differences were shown in Table 5. In total, there were 11 single nucleotide sequence variations, and one deletion of two nucleotides in the non- coding region. Of the 11 base substitutions, 9 did not alter its original amino acid coding, and the other 2 changed the amino acid sequence. The two missense mutations were all located at the M protein coding area, which were Cys 27 Phe for nt 26477 G→T, and Ala 68 Val for nt 26600 C→T, and were appeared only in the cultural isolates. In addition, the two-base deletion mutations were found in the cultural isolates. Of these 11 variants in these samples, two loci (position 26203 and 27812) were
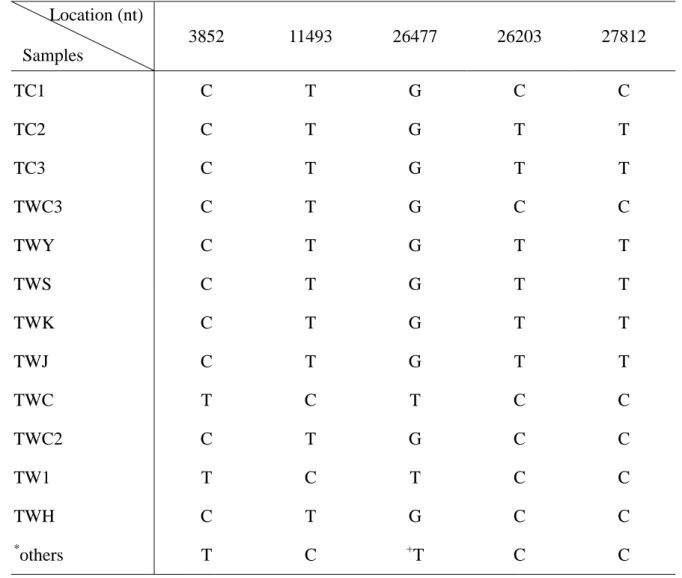
identified, and they were segregated together as a specific genotype, T:T or C:C (Table 6). The C:C genotype was linked to infection originated at the Amoy Garden in Hong Kong; the T:T genotype was linked to infection acquired from Hoping Hospital in Taipei, Taiwan.
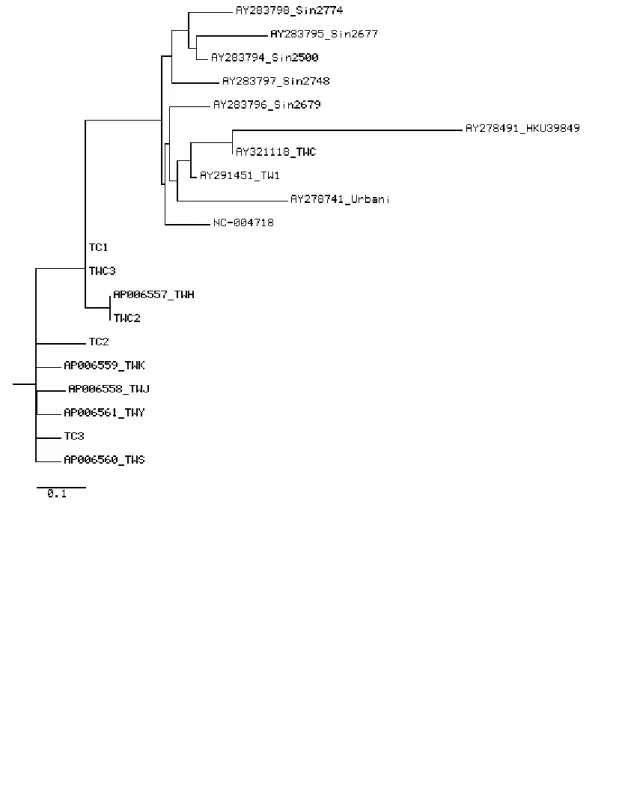
Phylogenetic analysis were shown in Figure 4. We analyzed the cases associated with the Hotel M of Hong Kong and the cases in Taiwan. Two major clusters were observed after the cases were analyzed.
Discussion
We developed a rapid method to sequence and analyze the whole genome of SARS-CoV from clinical specimens. It has been reported that some pathogenic microbes undergo adaptation in response to laboratory culturation. Host-mediated mutations in several viruses have been documented, such as HIV, Japanese encephalitis, hepatitis A, Sendai and influenza A viruses [17-20]. Molecular evolution studies using such sequences may be at risk of the data containing laboratory factors [16]; the analytic data that do not represent random samples of natural pathogen populations, or the sampling design is unknown. In this study, we can directly detect the whole genome of SARS-CoV from clinical samples, so that it will avoid the bias mentioned above. Although TC1 (a clinical specimen) and TWC (an isolate) were from the same patient, phylogenetic analysis revealed that they were located at different positions of the phylogenetic tree. Our results confirmed that the sequences of a cultural isolate may be different from these of a clinical specimen although they are derived from the same origin. We analyzed the sequences of the variant observed in cultural isolates and clinical specimens which were published in GenBank. Interestingly, we found that the nt 3852 C, 11493 T and
26477 G appeared in all the clinical specimens, but only in two of 23 cultural isolates. The nt 3852 T, 11493 C and 26477 T were found in all the cultural isolates except TWC2 and in none of clinical specimens (Table 5). From these discrepancies, these mutations may be most likely due to cultural adaptation, but we are unable to completely excluding the possibility of the shifting of two original different viruses during culturing process.
In clinical specimens or cultural isolates, two variants segregated tightly, nt 26203 C with 27812 C (C:C) and 26203 T with 27812 T (T:T). The C:C type appeared in TC1 and TWC3 and all the isolates in other infection area of the world. The T:T type appeared only in Taiwan area. From these results, the SARS infection in Taiwan could be from two origins. One is from either the Amoy Garden or Hotel M in Hong Kong, the other is yet to be identified.
There were 4 sequence variations (nt 8160,13098,26203,27812) in clinical specimens. All of the variations did not change the amino acid coding, indicating that the SARS-CoV maintains a stable structure, and it may favor the development of SARS-CoV vaccine. In the cultural isolates, we found 2 missense mutations that were all located at the M protein coding region. These mutations may be necessary for cultural adaptation. They may also appear in the clinical specimens, caused by other factors such as immunological adaptation. The M protein of SARS-CoV may play an important role in the immune response of infected patients.
Multiplex RT-PCR methods have been used to study the expression of more than ten genes simultaneously [21], to detect pathogens or subtypes [22-23], and to verify multiple chromosome translocations [24]. In order to increase the specificity, a nested PCR [22] or a probe [25], enzyme hybridization methods [26], or microarray [27] have
been used. In this study, we used multiplex RT-PCR to amplify the whole SARS genome in two separate reactions, and the PCR products were further amplified using 15 pairs of nested primers so that the products covered the whole genome in 15 separate tubes. The 15 PCR products were then sequenced. A Similar approach has been used to analyze the HN gene of human parainfluenza virus [28], however, only a small portion of human parainfluenza genome was analyzed. In contrast to this report, we analyze the whole genome of the biggest RNA virus. Although multiple RT-PCR to amplify the whole SARS genome can be used as a detection method, it is difficult to evaluate the quantitative change in the clinical specimens. So, we firstly amplified the Pol region of SARS virus to detect the virus and evaluate the quantitative changes of SARS virus, and then using multiplex RT-PCR and direct sequencing to analyze the whole genome.
Acknowledgments
We thank Dr. Shih-Feng Tsai for helpful discussion on the SARS-CoV sequences. This work was supported by a grant from China Medical University and by special SARS grant from the National Science Council, Taiwan (2751-B039-002-Y, NSC92-2751-B-039-007-Y).
References
1. WHO. Summary table of SARS cases by country, 1 November
2002-7 August 2003. http://www.who.int/csr/sars/country/
2003-07-11/en/
2. Reilley B,Van Herp M, Sermand D, Dentico N: SARS and Carlo Urbani. N Engl J Med 2003;348:1951-1952. 3. Peiris, J. S., S. T. Lai, L. L. Poon, Y.
Guan, L. Y. Yam, W. Lim, J. Nicholls, W. K. Yee, W. W. Yan, M. T. Cheung, V. C. Cheng, K. H. Chan,
D. N. Tsang, R. W. Yung, T. K. Ng, K. Y. Yuen, SARS study group: Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet 2003;361:1319-1325.
4. Lee N, Hui D, Wu A, Chan P, Cameron P, Joynt GM, Ahuja A, Yung MY, Leung CB, To KF, Lui SF, Szeto CC, Chung S, Sung JJ: A major outbreak of severe acute respiratory syndrome in Hong Kong. N Engl J Med 2003;348:1986-1994. 5. Tsang KW, Ho PL, Ooi GC, Yee
WK, Wang T, Chan-Yeung M, Lam WK, Seto WH, Yam LY, Cheung TM, Wong PC, Lam B, Ip MS, Chan J, Yuen KY, Lai KN: A cluster of cases of severe acute respiratory syndrome in Hong Kong. N Engl J Med 2003;348:1977-1985.
6. Poutanen SM, Low DE, Henry B, Finkelstein S, Rose D, Green K, Tellier R, Draker R, Adachi D, Ayers M, Chan AK, Skowronski DM, Salit I, Simor AE, Slutsky AS, Doyle PW, Krajden M, Petric M, Brunham RC, McGeer AJ; National Microbiology Laboratory, Canada; Canadian Severe Acute Respiratory Syndrome Study Team: Identification of severe acute respiratory syndrome in Canada. N Engl J Med 2003;348:1995-2005.
7. Ksiazek, TG, D. Erdman, C. S. Goldsmith, S. R. Zaki, T. Peret, S. Emery, S. Tong, C. Urbani, J. A. Comer, W. Lim, P. E. Rollin, S. F. Dowell, A. E. Ling, C. D. Humphrey, W. J. Shieh, J. Guarner, C. D. Paddock, P. Rota, B. Fields, J. DeRisi, J. Y. Yang, N. Cox, J. M. Hughes, J. W. LeDuc, W. J. Bellini, L. J. Anderson; SARS Working Group: A novel coronavirus associated with severe acute respiratory syndrome. N Engl J Med 2003;348:1953-1966.
8. Drosten, C., S. Gunther, W. Preiser, S. van der Werf, H. R. Brodt, S. Becker, H. Rabenau, M. Panning, L.
Kolesnikova, R. A. Fouchier, A. Berger, A. M. Burguiere, J. Cinatl, M. Eickmann, N. Escriou, K. Grywna, S. Kramme, J. C. Manuguerra, S. Muller, V. Rickerts, M. Sturmer, S. Vieth, H. D. Klenk, A. D. Osterhaus, H. Schmitz, H. W. Doerr: Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med 2003;348:1967-1976.
9. Rota, P. A., M. S. Oberste, S. S. Monroe, W. A. Nix, R. Campagnoli, J. P. Icenogle, S. Penaranda, B. Bankamp, K. Maher, M. H. Chen, S. Tong, A. Tamin, L. Lowe, M. Frace, J. L. DeRisi, Q. Chen, D. Wang, D. D. Erdman, T. C. Peret, C. Burns, T. G. Ksiazek, P. E. Rollin, A. Sanchez, S. Liffick, B. Holloway, J. Limor, K. McCaustland, M. Olsen-Rasmussen, R. Fouchier, S. Gunther, A. D. Osterhaus, C. Drosten, M. A. Pallansch, L. J. Anderson, W. J. Bellini: Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science 2003;300:1394-1399.
10. Marra, M. A., S. J. Jones, C. R. Astell, R. A. Holt, A. Brooks-Wilson, Y. S. Butterfield, J. Khattra, J. K. Asano, S. A. Barber, S. Y. Chan, A. Cloutier, S. M. Coughlin, D. Freeman, N. Girn, O. L. Griffith, S. R. Leach, M. Mayo, H. McDonald, S. B. Montgomery, P. K. Pandoh, A. S. Petrescu, A. G. Robertson, J. E. Schein, A. Siddiqui, D. E. Smailus, J. M. Stott, G. S. Yang, F. Plummer, A. Andonov, H. Artsob, N. Bastien, K. Bernard, T. F. Booth, D. Bowness, M. Czub, M. Drebot, L. Fernando, R. Flick, M. Garbutt, M. Gray, A. Grolla, S. Jones, H. Feldmann, A. Meyers, A. Kabani, Y. Li, S. Normand, U. Stroher, G. A. Tipples, S. Tyler, R. Vogrig, D. Ward, B. Watson, R.C. Brunham, M. Krajden, M. Petric, D. M. Skowronski, C. Upton, R. L. Roper: The genome
sequence of the SARS-associated
coronavirus. Science 2003;300:1399-1404.
11. Ruan, Y. J., C. L. Wei, A. L. Ee, V. B. Vega, H. Thoreau, S. T. Su, J. M. Chia, P. Ng, K. P. Chiu, L. Lim, T. Zhang, C. K. Peng, E. O. Lin, N. M. Lee, S. L. Yee, L. F. Ng, R. E. Chee, L. W. Stanton, P. M. Long, E. T. Liu: Comparative full-length genome sequence analysis of 14 SARS coronavirus isolates and common mutations associated with putative origins of infection. Lancet 2003;361:1779-1785.
12. World Health Organization Multicentre Collaborative Network for Severe Acute Respiratory Syndrome (SARS) Diagnosis. A multicentre Collaboration to investigate the cause of severe acute respiratory syndrome. Lancet 2003;361:1730-1733.
13. Horzinek MC: Molecular evolution of corona- and toroviruses. Adv Exp Med Biol 1999;473:61-72.
14. Lai MM, Cavanagh D: The molecular biology of coronaviruses. Adv Virus Res 1997;48:1-100. 15. Domingo E, Escarmis C, Sevilla N,
Moya A, Elena SF, Quer J, Novella IS, Holland JJ: Basic concepts in RNA virus evolution. FASEB J 1996;10:859-864.
16. Bush RM, Smith CB, Cox NJ, Fitch WM: Effects of passage history and sampling bias on phylogenetic reconstruction of human influenza A evolution. Proc Natl Acad Sci USA 2000;97:6974-6980.
17. Graff J, Normann A, Feinstone SM, Flehmig B: Nucleotide sequence of wild type hepatitis A virus GBM in comparison with two cell culture-adapted variants. J Virol 1994;68:548-554.
18. Itoh M, Isegawa Y, Hotta H, Homma M: Isolation of an avirulent mutant of Sendai virus with two amino acid mutation from a highly virulent field
strain through adaptation to LLC-MK2 cells. J Gen Virol 1997;78:3207-3215.
19. Sawyer LS, Wrin MT, Crawford-Miksza L, Potts B, Wu Y, Weber PA, Alfonso RD, Hanson CV: Neutralization sensitivity of human immunodeficiency virus type 1 is determined in part by the cell in which the virus is propagated. J Virol 1994;68:1342-1349.
20. Ni H, Chang GJ, Xie H, Trent DW, Barrett AD: Molecular basis of attenuation of neurovirulence of wild-type Japanese encephalitis virus strain SA14. J Gen Virol 1995;76:409-413.
21. Yamamoto M, Yamamoto F, Luong TT, Williams T, Kominato Y, Yamamoto F: Expression profiling of 68 glycosyltransferase genes in 27 different human tissues by the systematic multiplex reverse transcription-polymerase chain reaction method revealed clustering of sexually related tissues in hierarchical clustering algorithm
analysis. Electrophoresis 2003;24:2295-2307.
22. Coiras MT, Perez-Brena P, Garcia ML, Casas I: Simultaneous detection of influenza A, B, and C viruses, respiratory syncytial virus, and adenoviruses in clinical samples by multiplex reverse transcription nested-PCR assay. J Med Virol 2003;69:132-144.
23. Podder SK, Espina R, Schnurr DP: Evaluation of a single-step multiplex RT-PCR for influenza virus type and subtype detection in respiratory samples. J Clin Lab Anal 2003;16:163-166.
24. Shi RZ, Morrissey JM, Rowley JD: Screening and quantification of multiple chromosome translocations in human leukemia. Clin Chem. 2003;49:1066-1073.
25. Peter M, Gilbert E, Delattre O: A multiplex real-time PCR assay for
the detection of gene fusions observed in solid tumors. Lab Invest 2001;81:905-912.
26. Liolios L, Jenney A, Spelman D, Kotsimbos T, Catton M, Wesselingh S: Comparison of a multiplex reverse transcription-PCR-enzyme hybridization assay with conventional viral culture and immunofluorescence techniques for the detection of seven viral respiratory pathogens. J Clin Microbiol 2001;39:2779-2783.
27. Li J, Chen S, Evans DH: Typing and subtyping influenza virus using DNA microarrays and multiplex reverse transcriptase PCR. J Clin Microbiol 2001;39:696-704.
28. Echevarria JE, Erdman DD, Meissner HC, Anderson L: Rapid molecular epidemiologic studies of human parainfluenza viruses based on direct sequencing of amplified DNA form a multiplex RT-PCR assay. J Virol Methods 2000;88:105-109.
Figure legend
Fig. 1. The strategy of sequencing analysis of SARS-CoV from clinical specimens.
We used two separate multiplex RT-PCR [one contains 8 pairs of primers (T1), and the other contains 7 pairs of primers (T2)] to amplify the whole SARS-CoV genome. The PCR products were used as templates for 15 nested PCRs (t1, t2,t3….t15) to amplify a 2 kb region, respectively, to ensure the whole SARS-CoV was sequenced. The 15 nested PCR products were further sequenced and analyzed.
0 30000 bp T2 t1 t2 t3 t4 t5 t6 t7 t8 t9 t10 t11 t12 t13 t14 t15
Fig. 2. The results of RT-PCR analysis of SARS-CoV are shown. The positive case
had a PCR fragment of 368-bp. Lane 1 is a positive control; lane 2 is a negative control; lanes 3, 4 and 5 are the three positive cases collected in Taichung, Taiwan; lanes 6 and 7 are negative cases. M: 100 bp ladder markers.
Fig. 3. The results of nested PCR analysis of a 2-kb region in 15 different reactions,
which cover the whole SARS-CoV genome, are shown. The representative markers of t1,t2….t15 are similar to Fig. 1.
Fig. 4. Molecular relationships between 20 SARS-CoV genomes which linked to
infection acquired at the Hotel M in Hong Kong are shown. Phylogenetic trees were obtained by applying ClustalW to complete genome sequences of all 20 SARS-CoV published in GenBank.
Table 1. The clinical features of three SARS patients.
Case 1 Case 2 Case 3
Age (years) 56 44 26 Sex M M F Temperature on admission (℃) 38.5 >38 >38 WBC (/fL) 6760 5360 4120 Lymphocyte (%) 27 19.4 28.2 Platelet (/fL) 126x103 125x103 190x103 LDH (IU/L) 197 116 266 GOT (IU/L) 66 116 266 GPT (IU/L) 70 197 188
Chest radiograph Accentuation of bronchovascular marking Pneumonia left lower lobe Pneumonia right lower lobe
Co-morbidities α-thal-1 DM, Chronic hepatitis
Nil
Table 2. The primers used for multiplex RT-PCR.
Reaction Sequences Locations (nt)
Reaction T1 5’-GCCAACCAACCTCGATCTCTTG-3’ (F) 29 to 50 5’-CCTTGAGAAATTCAACTCCTGC-3’ (R) 2194 to 2173 5’-TGTCGTACAGAAGCCTGTCG-3’ (F) 3852 to 3871 5’-ATGTGACAGATAGCTCTCGT-3’ (R) 6070 to 6051 5’-GCGACGAGTCTGCTTCTAAG-3’ (F) 7841 to 7860 5’-ACAGGTTACTTGTACCATGC-3’ (R) 10050 to 10031 5’-GTACAGTCTAAAATGTCTGACG-3’ (F) 11767 to 11788 5’-AGTGGTTTTGCGAGATCAGC-3’ (R) 14164 to 14145 5’-GTGTCACTGGCTATTGATGC-3’ (F) 15948 to 15967 5’-GGTCATGTCCTTTGGTATGC-3’ (R) 18119 to 18100 5’-GTATCTACAATAGGTGTCTGC-3’ (F) 19836 to 19856 5’-GTAATGTTAATACCAAGAGGC-3’ (R) 22177 to 22157 5’-ACGTGAAGTGTTCGCTCAAG-3’ (F) 23771 to 23790 5’-GTCTTTAACACCTGAGTGCC-3’ (R) 25864 to 25845 5’-TCAACAAGAGCTCTACTCGC-3’ (F) 27551 to 27570 5’-CACATGGGGATAGCACTACT-3’ (R) 29700 to 29681
Table 2. The primers used for multiplex RT-PCR. (Continued)
Reaction Sequences Locations (nt)
Reaction T2 5’-TCCTGATTTGCAAAGAGCAGC-3’ (F) 1941 to 1962 5’-TCACCTACCATGTAAGGTGC-3’ (R) 4058 to 4039 5’-GCTTACTATACAGAGCAGCC-3’ (F) 5908 to 5927 5’-GAGCTGTAGCAACAAGTGCC-3’ (R) 8044 to 8025 5’-GCTATCGTGAAGCAGCTTGC-3’ (F) 9869 to 9888 5’-GTTATCGAGCATTTCCTCGC-3’ (R) 12006 to 11987 5’-CGATTTCGGTGATTTCGTAC-3’ (F) 14021 to 14040 5’-AACTCAGGTTCCCAGTACCG-3’ (R) 16129 to 16110 5’-CACACAAACTACTGAAACAGC-3’ (F) 17804 to 17824 5’-CATTGACGCTAGCTTGTGCT-3’ (R) 20040 to 20021 5’-TGCATTTAATTGCACTTTCGAG-3’ (F) 21956 to 21977 5’-CTAGGCATTCGCCATATTGC-3’ (R) 23961 to 23942 5’-GTTGGCTTTGTTGGAAGTGC-3’ (F) 25647 to 25666 5’-CAATGAGAAGTTTCATGTTCG-3’ (R) 27794 to 27774 nt: nucleotide; F: forward primer; R: reverse primer
Table 3. The SARS-CoV primers used for nested-PCR.
Name Sequences Location (nt)
t1 5’-GTAGATCTGTTCTCTAAACG-3’ (F) 5’-TCCATTCAAAGATAGGCCTG-3’ (R) 50 to 69 2155 to 2136 t2 5’-GTCATTACGTCTTGTCGACG-3’ (F) 5’-GTTCTGAGAATCATGGTAAACC-3’ (R) 1992 to 2001 3999 to 3978 t3 5’-TGAGGTTACCACAACACTGG-3’ (F) 5’-GAAGCTGGCTTTGTGAAGCC-3’ (R) 3903 to 3922 6050 to 6031 t4 5’-CTCAACCATTACCAAATGCG-3’ (F) 5’-AGTACTATCTCCAACGTCTG-3’ (R) 5945 to 5964 7947 to 7928 t5 5’-CTACAGTCAGCTGATGTGCC-3’ (F) 5’-ACCACTCTGCAGAACAGCAG-3’ (R) 7875 to 7894 9990 to 9971 t6 5’-GACTTTAGCAACTCAGGTGC-3’ (F) 5’-GAAACCATCTTCTCGAAAGC-3’ (R) 9913 to 9932 11933 to 11914 t7 5’-GTGCACATCTGTGGTACTGC-3’ (F) 5’-AAGTGAGGATGGGCATCAGC-3’ (R) 11793 to 11802 14109 to 14090 t8 5’-AGGCTGCGGAGTTCCTATTG-3’ (F) 5’-CAACATGTGGCCAGTAAGCT-3’ (R) 14051 to 14060 16070 to 16051 t9 5’-CATCCTAATCAGGAGTATGC-3’ (F) 5’-TGTAATGTAGCCACATTGCG-3’ (R) 15984 to 16003 17968 to 17949 t10 5’-GTCAACCGCTTCAATGTGGC-3’ (F) 5’-ACCTTTGACTGAACCTTCTG-3’ (R) 17838 to 17857 20000 to 19981 t11 5’-CAAGAAACCTACTGAGAGTGC-3’ (F) 5’-CCAGAAGGTAGATCACGAAC-3’ (R) 19874 to 19894 22126 to 22107 t12 5’-CATATCTGATGCCTTTTCGC-3’ (F) 5’-TCATGAAGCCAGCATCAGCG-3’ (R) 21980 to 21999 23940 to 23921 t13 5’-GGTCTTTTATTGAGGACTTGC-3’ (F) 5’-TGCCGTCACCTTCAGTAACG-3’ (R) 23881 to 23901 25790 to 25771 t14 5’-CCAACTACTTTGTTTGCTGGC-3’ (F) 5’-TCTAGATCCTGGATTTCGAG-3’ (R) 25695 to 25715 27750 to 27731 t15 5’-CTCATTGTTGCTGCTCTAGT-3’ (F) 5’-TAGGGCTCTTCCATATAGGC-3’ (R) 27579 to 27598 29662 to 29643 nt: nucleotide; F: forward primer; R: reverse primer
Table 4. The sequence variants of SARS-CoV in Taiwan area. 1782 3165 3852 8160 11493 13098 13347 16325 19361 20083 21581 25652 26203 26477 26600 26615 27167 27168 27808 27809 27812 28302 TWC2 T A C C T C C A T A C C C G C A A T T T C C TWC3 C A C C T C C A T A C C C G C A A T T T C C TWY C A C C T C T A T A C C T G C A A T T T T C TWS C A C C T C C A T A T C T G C A A T T T T C TWK C A C C T C C A T A C T T G C A A T T T T T TWJ C A C C T C C A C - C T T G C A - - T T T C TWH T A C C T C C A T A C C C G C A A T T T C C TC1 C A C C T C C A T A C C C G C A A T T T C C TC2 C A C T T T C A T A C C T G C A A T T T T C TC3 C A C C T C C A T A C C T G C C A T T T T C TWC C A T C C C C G T A C C C T T A A T - - C C TW1 C G T C C C C A T A C C C T C A A T T T C C
TC1 (clinical specimen) and TWC (cultural specimen) is the first case in Taiwan area; TC2 and TC3 from mid-Taiwan area; TW1, TWC2, TWC3, TWY, TWS, TWK, TWJ and TWH from Northern part of Taiwan.
Location (nt) Origin
Table 5. Comparison of the sequence variants between cultural isolates and clinical specimens in Taiwan area. 1782 3165 3852 8160 11493 13098 16325 26203 26477* 26600* 27812 Clinical specimen TC1 C A C C T C A C G C C TC2 C A C T T T A T G C T TC3 C A C C T C A T G C T TWC3 C A C C T C A C G C C Cultural isolates + TWC C A T C C C G C T T C TWC2 T A C C T C A C G C C TW1 C G T C C C A C T C C
*The base substitution causes amino acid change. nt 26477 G→T (Cys 27 Phe, M protein); nt 26600 C→T (Ala 681/Va1, M protein).
+
A two base-deletion (TT) at 27808-27809 was not shown. Location (nt)
Table 6. Comparison of sequence variants among the SARS-CoV genomes of SARS-CoV Taiwan area. 3852 11493 26477 26203 27812 TC1 C T G C C TC2 C T G T T TC3 C T G T T TWC3 C T G C C TWY C T G T T TWS C T G T T TWK C T G T T TWJ C T G T T TWC T C T C C TWC2 C T G C C TW1 T C T C C TWH C T G C C * others T C +T C C *
The SARS-CoV from other area. +
Except AY282752 (26477G) Location (nt)