Cite this:
Mol. BioSyst
., 2011, 7, 3366–3374
High performance screening, structural and molecular dynamics analysis
to identify H1 inhibitors from TCM Database@Taiwanw
Su-Sen Chang,
aHung-Jin Huang
band Calvin Yu-Chian Chen*
acdefReceived 2nd August 2011, Accepted 29th September 2011 DOI: 10.1039/c1mb05320e
New-type oseltamivir-resistant H1N1 influenza viruses have been a major threat to human health since the 2009 flu pandemic. To resolve the drug resistance issue, we aimed to identify a new type of inhibitors against H1 from traditional Chinese medicine (TCM) by employing the world’s largest TCM database (www.tcm.cmu.edu.tw) for virtual screening and molecular dynamics (MD). From the virtual screening results, sodium (+)-isolaricireinol-2 alpha-sulfate, sodium 3,4-dihydroxy-5-methoxybenzoic acid methyl ester-4-sulfate, sodium
(E)-7-hydroxy-1,7-bis(4-hydroxyphenyl)hept-5-ene-3S-sulfonate, and 3-methoxytyramine-betaxanthin were identified as potential drug-like compounds. MD simulation of the binding poses with the key residues Asp103 and Glu83, as well as other binding site residues, identified higher numbers of hydrogen bonds than N-Acetyl-D-Glucosamine (NAG), the natural ligand of the esterase domain in H1. Ionic bonds, salt bridges, and electrostatic energy also contribute to binding stability. Key binding residues include Lys71, Glu83, Asp103, and Arg238. Structural moieties promoting H-bond or salt bridge formations at these locations greatly contribute to a stable ligand–protein complex. An available sodium atom for ionic interactions with Asp103 can further stabilize the ligands. Based on virtual screening, MD simulation, and interaction energy evaluation, TCM candidates demonstrate good potential as novel H1 inhibitors. In addition, the identified stabilizing features can provide insights for designing highly stable H1 inhibitors.
Introduction
Influenza A virus is one of the most infectious flu viruses that targets human and livestock alike, and has caused large scale pandemics in 1918, 1957, and 1968. The most recent influenza strain H1N1/09 has caused nearly 20 000 deaths during the 2009 pandemic period1 and at least 52 deaths in 2011.2–7
Despite cessation of the 2009 world pandemic, H1N1 viruses remain active worldwide and virus activity monitored through FluNet totals 53 739 in 2010 and 78 916 cases in 2011 as of Sep. 2011.8Glycoproteins on influenza viral coating include hemagglutinin (HA; H1–H16) and neuraminidase (NA; N1–N9).
The most infectious subtypes are H1, H2, H3, N1, and N2. The notorious H1N1 strain is comprised of H1 and N1 glycoproteins. HA is critical for viruses to attach to host cell membranes for infection.9 The infection process is initiated when the HA esterase cleaves through the protective host cell mucus layer, allowing virus attachment to host membrane sialic acid through the HA1 domain and incorporation into host cells via HA2-mediated membrane-fusion activity.10 Influenza viral strains share common infection routes and have conserved active domains. Nevertheless, evolution studies on pandemic strains revealed sequence and structural mutations in HA or NA.11–14Mutations in N1 were considered to be the
cause of drug resistance observed in the H1N1/09 pandemic.15 Viral invasion can be prevented by inhibiting the esterase activity required for host cell entrance. We aimed for an in silico approach for identifying novel H1 inhibitors from traditional Chinese medicine (TCM). TCM has been a popular medical practice in East Asia for thousands of years. This class of medicine has been introduced as alternative medicine complementary to western medicine. TCM has high therapeutic values such as anti-tumor growth,16–19anti-diabetes,20–23and anti-metastasis functions.24,25 In addition, high throughput
in silico pharmacology has been employed for TCM drug discovery against chronic disorders such as insomnia, allergy, stroke, cancer, and many more.26–34This approach has been
a
Laboratory of Computational and Systems Biology, School of Chinese Medicine, China Medical University, Taichung, 40402, Taiwan
bSciences and Chinese Medicine Resources, China Medical University,
Taichung, 40402, Taiwan
cChina Medical University Beigang Hospital, Yunlin, 65152, Taiwan d
Department of Bioinformatics, Asia University, Taichung, 41354, Taiwan
eDepartment of Systems Biology, Harvard Medical School, Boston,
MA 02115, USA
fComputational and Systems Biology, Massachusetts Institute of
Technology, Cambridge, MA 02139, USA. E-mail: [email protected]; Tel: +1-617-353-7123
w Electronic supplementary information (ESI) available. See DOI: 10.1039/c1mb05320e
Molecular
BioSystems
Dynamic Article Links
well adapted in our laboratory and employed in H1 related research.24,35,36Recent studies have reported diverse sequences
in the H1 esterase domain in H1N1 strains.37,38Our investiga-tion focuses on the latest H1 sequence from influenza virus strain A/Jiangsu/1/2009 (GenBank: ADK25944.1).
A structure-based approach to identify and evaluate all possible protein–ligand binding poses was utilized. High throughput virtual screening was performed on the world’s largest TCM database39 to maximize the identification of potential H1 inhibitors. Molecular dynamics (MD) simulation was used to investigate protein–ligand binding stabilities. The protein–ligand binding features can hence be characterized from the simulation analysis.
Materials and methods
TCM database
The world’s largest traditional Chinese medicine database TCM Database@Taiwan (http://tcm.cmu.edu.tw/)39was used to screen for potential candidates from 38 877 TCM com-pounds. Energy minimization and proper protonation at physiological pH was applied to prepare compounds prior to screening.
Homology modeling and validation
The H1 structure from influenza virus strain A/Darwin/2001/ 2009 (PDB: 3M6S)40was selected as the template for the new H1 protein sequence of influenza virus strain A/Jiangsu/1/ 2009 (GenBank: ADK25944.1). Sequence alignment was per-formed by the ClustalW method using progressive pairwise alignment algorithm. The homology model of H1 was built by MODELLER in Discovery Studio 2.5 (DS 2.5). Five distinct loops were generated for each of the 20 resulting models. The structures of H1 model were evaluated by Ramachandran plot and Profile-3D to confirm structure folding.
Docking
Potential candidates were screened from 38 877 compounds in TCM Database@Taiwan39 using the LigandFit41 algorithm and CHARMm force field.42 The binding site was obtained from the natural ligand N-Acetyl-D-Glucosamine (NAG), and
identified during H1 homology modeling. Monte Carlo trials were performed with the fixed protein. The ligands were initially flexible and then fixed for energy minimization once docked. A maximum of five docking poses were generated. Default setting for LigandFit was used for the other para-meters. Scoring functions in DS 2.5 were used to evaluate each docking result. Top ranking compounds were visually inspected for binding poses, binding location and interacting residues. Candidates having similar binding characteristics and locations with the control NAG were selected for further investigation. Molecular dynamics
The molecular dynamics (MD) simulation was performed with the DS 2.5 Molecular Dynamics package Standard Dynamics Cascade Module and Dynamics (Production) Module under CHARMm force field.42 Relevant parameters were set as follows: [minimization] 500 cycles each of Steepest Descent43
and Conjugate Gradient;44[target temperature] 310 K within
50 ps; [equilibration] 200 ps; [production] 20 ns with NVT canonical ensemble and trajectory frames saved every 20 ps. SHAKE algorithm was applied to immobilize all bonds with
Table 1 Virtual screening results of H1 candidates
Name Dock score
Sodium (+)-isolaricireinol-2 alpha-sulfate 215.847 Sodium 3,4-dihydroxy-5-methoxybenzoic acid
methyl ester-4-sulfate 211.120 Sodium (E)-7-hydroxy-1,7-bis (4-hydroxyphenyl)hept-5-ene-3S-sulfonate 210.630 3-Methoxytyramine-Betaxanthin 137.174 Radicamine B 127.690 Noradrenaline 127.258 Canavanine 125.611 Flavaspidic acid Pb 125.151 Noradrenaline 125.024 Dopamine 123.716 NAGa 49.688 aNAG:
N-acetyl-D-glucosamine; denotes control compound.
Fig. 1 The docking poses of (a) Top 1: sodium (+)-isolaricireinol-2 alpha-sulfate, (b) Top 2: sodium 3,4-dihydroxy-5-methoxybenzoic acid methyl ester-4-sulfate, (c) Top 3: sodium (E)-7-hydroxy-1,7-bis(4-hydroxyphenyl) hept-5-ene-3S-sulfonate, (d) Top 4: 3-methoxy-tyramine-Betaxanthin, (e) NAG in the H1 esterase binding site. The residues involved in polar and van der Waals interactions are repre-sented by magenta and green circles, respectively. Hydrogen bonds (blue dash), pi–cation interactions (brown), and polar interactions (magenta) are also shown.
hydrogen atoms. The time step was set to 2 fs for all MD stages. The Berendsen thermal coupling method was used and the temperature coupling decay time set at 0.4 ps. Post processing of the trajectories was conducted with Analyze Trajectory.
Results and discussion
Homology modeling and docking
Sequence alignment between the new H1 sequence (ADK25944) and the template H1 (3M6S) indicated high sequence identity and similarities of 96.8% and 97.4%, respectively (Fig. S1, ESIw). Ramachandran plot identified 94.2% amino acid c and j angles of the new H1 sequence resided within the favored region (Fig. S2, ESIw). Profile 3D further indicated positive scores for binding site residues Lys71, Asp103, Asn104 and Arg238 (Fig. S3, ESIw). The two validation methods suggest a reliable H1 homology model.
Considering high variability in HA’s sialic binding site,45–47 we took an alternative approach by targeting the more con-served esterase activity site. Docking results were ranked based on the Dock Scores (Table 1). All ligands with higher Dock Scores than NAG are listed. The top four candidates, sodium (+)-isolaricireinol-2 alpha-sulfate (Top 1), sodium 3,4-dihydroxy-5-methoxybenzoic acid methyl ester-4-sulfate (Top 2), sodium (E)-7-hydroxy-1,7-bis(4-hydroxyphenyl)-hept-5-ene-3S-sulfonate (Top 3), and 3-methoxytyramine-Betaxanthin (Top 4), were selected for further analysis.
The Top 1 and Top 2 candidates are found in Polygonum cuspidatum.48TCM classified this herb as a dampness-resolving
medicinal, which remove excessive phlegm from flu symptom. Top 3 originated from ginger (Zingiber officinale),49a common wind-cold-dispersing medicine used for relieving flu symptoms. Top 4 is a constituent of Celosia argentea,50which has been used for heat clearing in TCM. Though literature describing the anti-influenza effect of the candidates per se is lacking, anti-anti-influenza effects of their respective TCM sources have been reported.51–54
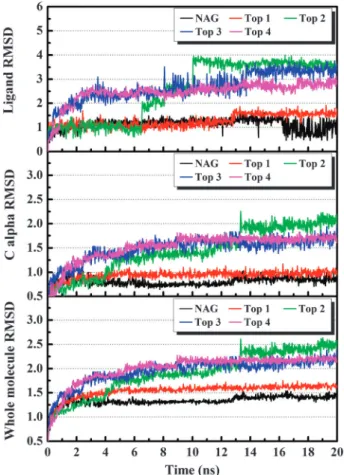
Fig. 2 The RMSD (A˚) trajectories of top four candidates and NAG in H1 complexes during 20 ns MD simulation.
Table 2 H-bond occupancies for top four candidates and NAG (control) during 20 ns molecular dynamics (MD) simulation
Name Ligand atom Amino acid Occupancy
Top 1 O21 Arg238 : HH11 7.20%
O22 Arg238 : HH12 7.00% O13 Arg238 : HH12 0.20% O13 Arg238 : HH22 50.80% O14 Lys71 : HZ1 37.50% O14 Lys71 : HZ2 57.30% O14 Lys71 : HZ3 16.70% H53 Asp103 : OD1 34.40% H53 Asp103 : OD2 10.10% Top 2 O9 Arg238 : HH22 0.30% O14 Lys71 : HZ1 0.10% O18 Asn104 : HD22 0.10% O12 Lys71 : HZ1 7.50% O16 Lys71 : HZ1 0.20% O7 Lys71 : HZ1 41.90% O12 Lys71 : HZ2 10.40% O16 Lys71 : HZ2 0.20% O7 Lys71 : HZ2 40.80% O12 Lys71 : HZ3 13.40% O16 Lys71 : HZ3 0.20% O7 Lys71 : HZ3 37.60% H28 Glu83 : OE1 67.00% H28 Glu83 : OE2 0.70% Top 3 O7 Arg238 : HH12 34.40% O7 Arg238 : HH22 97.90% O16 Lys71 : HZ1 39.60% O25 Lys71 : HZ1 7.80% O16 Lys71 : HZ2 52.20% O25 Lys71 : HZ2 1.00% O16 Lys71 : HZ3 46.40% O25 Lys71 : HZ3 0.60% H46 Ala152 : O 0.30% H47 Glu83 : OE1 4.20%
Top 4 H43 Glu83 : OE1 93.00%
H43 Glu83 : OE2 94.10%
O19 Arg238 : HH12 98.60% O19 Arg238 : HH22 99.80%
O26 His155 : HE2 81.60%
O22 Lys71 : HZ1 45.30% O23 Lys71 : HZ1 29.60% O22 Lys71 : HZ2 65.80% O23 Lys71 : HZ2 39.30% O22 Lys71 : HZ3 53.30% O23 Lys71 : HZ3 20.70% NAG O7 Arg238 : HH12 99.68% O3 Arg238 : HH22 95.40% O7 Arg238 : HH22 7.53% H12 Asn104 : OD1 16.13% H15 Asp103 : OD1 28.60% H15 Asp103 : OD2 71.03%
Top 1: sodium (+)-isolaricireinol-2 alpha-sulfate; Top 2: sodium 3,4-dihydroxy-5-methoxybenzoic acid methyl ester-4-sulfate; Top 3: sodium (E)-7-hydroxy-1,7-bis(4-hydroxyphenyl) hept-5-ene-3S-sulfonate; Top 4: 3-methoxytyramine-Betaxanthin; NAG: N-acetyl-D-glucosamine. H-bond cutoff: 2.5 A˚.
The ability of the candidates to dock into the H1 esterase binding site and inhibit initiation of viral invasion could be one of the underlying mechanisms for the observed anti-influenza effects reported for these TCM.
Based on the docking poses, all four candidates formed polar interactions with Lys71 and Glu83 at the esterase active site. The top three candidates also formed hydrogen bonds (H-bonds) with Arg238. In addition, the sodium ion from each of the top three compounds formed an ionic bond with the
carboxyl group on Asp103 (Fig. 1(a)–(c)). Notably, Top 1 also formed two pi–cation interactions that further contribute to binding affinity (Fig. 1(a)). By comparison, only H-bonds and
Fig. 3 Time dependent H-bond distances (A˚) for (a) sodium (+)-iso-laricireinol-2 alpha-sulfate, (b) sodium 3,4-dihydroxy-5-methoxy-benzoic acid methyl ester-4-sulfate, (c) sodium (E)-7-hydroxy-1,7-bis(4-hydroxyphenyl) hept-5-ene-3S-sulfonate, (d) 3-methoxytyramine-betaxanthin, (e) NAG in the H1 binding site during 20 ns MD simulation.
Fig. 4 Snapshots of sodium (+)-isolaricireinol-2 alpha-sulfate (a, b), sodium 3,4-dihydroxy-5-methoxybenzoic acid methyl ester-4-sulfate (c, d), sodium (E)-7-hydroxy-1,7-bis(4-hydroxyphenyl) hept-5-ene-3S-sulfonate (e, f), 3-methoxytyramine-betaxanthin (g, h) and NAG (i, j) during 20 ns MD simulation. H-bonds are represented by green dash lines.
van der Waals (vdW) interaction were observed in H1-NAG, suggesting less binding affinity than the candidates.
Top 4 has a distinctive intermolecular binding pattern where three stabilizing salt bridges were observed between the polar NH+ on the ring structure and Lys71 and Glu83, as well as between the lone oxygen atom on the phenyl group and Arg238 (Fig. 1(d)). These interactions indicate stronger protein–ligand binding affinities than the NAG (Fig. 1(e)). All candidate com-pounds have higher binding affinities than the control compound based on docking poses. Amino acids Lys71, Glu83, Asp103, and Arg238 are key interaction residues.
Molecular dynamics simulation
Root mean square deviations (RMSDs) were assessed for each complex, each ligand, and each respective C-alpha protein structures of all candidates during the 20 ns simulation (Fig. 2). All candidates and the control RMSDs stabilized at 13 ns during MD, suggesting that the minimum energy states were reached. Most ligands were stable except for Top 2 according to ligand RMSD trajectories. Significant changes in RMSDs during 6–10 ns were observed in Top 2. These were the result of the stabilizing process which may lead to changes in the protein–ligand interactions.
Based on H-bond analysis (Table 2), Top 1 had significantly high H-bond occupancies with Lys71 (50.80%) and Arg238 (57.30%). Furthermore, the H-bond with Asp103 reached equilibrium below 3 A˚ after 13 ns, suggesting the formation of a stable H-bond (Fig. 3(a)). The corresponding H-bond occupancy of 34.40% matched the observation. Top 2 formed H-bonds with Glu83 and Lys71. The H-bond with Glu83 appeared after 6 ns and remained at 2–3 A˚ throughout the simulation period (Fig. 3(b)). On the other hand, the H-bonds with Lys71 fluctuated between 2–4 A˚. Nevertheless, the distance between oxygen O7 on Top 2 and Lys71 remained relatively constant. In contrast, the H-bond between Top 2 and Arg238 was only present for a short period and is speculated to be an intermediate interaction leading to a lower complex energy state. For Top 3, three distinct H-bonds with Arg238 were presented in the complex. The H-bond associated with the oxygen O22 dissipated almost immediately at the beginning of the MD simulation. Comparatively, the H-bonds between Arg238 and oxygen O7 were extremely stable with occupancies as high as 97.90% and distances of approximately 2 A˚ (Fig. 3(c)). The three H-bonds between Top 3 and Lys71 fluctuated throughout the simulation. However, at least one H-bond with Lys71 was present within any given time point. This implied a structurally stable pose between Top 3 and Lys71. In addition, a longer H-bond present between Top 3 and Glu83 at approximately 3 A˚ further suggested the binding stability between H1 and Top 3 (Fig. 3(c)). Similar to the other TCM candidates, Top 4 formed H-bonds with Lys71. The 2–3 A˚ bond distance between Top 4 and Lys71 suggests a tighter binding affinity. Other H-bonds with Top 4 at Arg238, Glu83, and His155 were also stable within the H-bond forming range of 2–3 A˚ (Fig. 3(d)).
In comparison to NAG in which the H-bonds with Arg238 and Asp103 were weak (Fig. 3(e)), the TCM candidates showed stronger binding affinities and higher stability during MD analysis. All candidates formed stable H-bonds with Lys71 and additional H bonds with either Asp103 or Glu83, which further increased protein–ligand binding stability.
Table 3 Interaction energy of TCM candidates and NAG after 20 ns simulation
van der Waals Electrostatic Interaction energy
Top 1 9.478 160.395 169.873
Top 2 4.727 129.670 124.943
Top 3 2.697 164.825 167.522
Top 4 12.461 326.846 339.307
NAG 12.754 52.733 65.487
Unit of energy: kcal mol 1; interaction energy = van der Waals + electrostatic; Top 1: sodium (+)-isolaricireinol-2 alpha-sulfate; Top 2: sodium 3,4-dihydroxy-5-methoxybenzoic acid methyl ester-4-sulfate; Top 3: sodium (E)-7-hydroxy-1,7-bis(4-hydroxyphenyl) hept-5-ene-3S-sulfonate; Top 4: 3-methoxytyramine-Betaxanthin; NAG:
N-acetyl-D-glucosamine.
These observations could provide insights to designing stron-ger inhibitors against the H1 esterase site.
Binding analysis during MD simulation
Snapshots during MD were taken to identify significant struc-tural changes contributing to stability. A significant rotation on
an ethyl (–CH2OH) functional group in Top 1 enabled the
formation of an additional stabilizing H-bond with Asp103 (Fig. 4(a) and (b)). Top 2 did not interact with Arg238 through-out most of the simulation. However, multiple H-bonds were present with Lys71 and Glu83 during MD (Fig. 4(c) and (d)). Intriguingly, the three H-bonds present on the 8.02 ns snap-shot were formed with the same oxygen atom on the ligand,
Fig. 6 Residue positions relative to (a) sodium (+)-isolaricireinol-2 alpha-sulfate, (b) sodium 3,4-dihydroxy-5-methoxybenzoic acid methyl ester-4-sulfate, (c) sodium (E)-7-hydroxy-1,7-bis(4-hydroxyphenyl) hept-5-ene-3S-sulfonate, and (d) 3-methoxytyramine-betaxanthin, and (e) NAG based on the center of mass.
suggesting a critical moiety for binding stability. In Top 3, Glu83 maintained stable distance with the ligand hydroxyl group despite the inability of DS 2.5 to show an H-bond due to the 2.5 A˚ cutoff. The same hydroxyl group also formed fluctuating H-bonds with Lys71 (Fig. 4(e) and (f)). In addition, the sodium atoms had strong ionic interactions with the negatively charged Asp103 and contributed to bind-ing stabilities. Snapshots indicated that Top 4 remained relatively stable with the formed H-bonds during the 20 ns MD simulation (Fig. 4(g) and (h)). Salt bridges were obser-ved between Top 4 and Lys71, Glu83, and His155. Compara-tively, NAG had stable binding with Arg238, Asp103, and Asn104 (Fig. 4(i) and (j)) despite fluctuations in its ligand RMSD (Fig. 2). While the snapshots suggest stability of NAG within the H1-ligand binding site, the TCM candidates demonstrate higher stability when bound within the H1 esterase binding site.
Interaction energy
Protein–ligand interaction energy was used to quantify the binding stabilities of TCM candidates after MD simulation (Table 3). Interaction energy collectively evaluates the vdW interaction and electrostatic interactions. Re-ranking of the TCM candidates based on their interaction energies after MD is as follows: Top 4 ( 339.307 kcal mol 1), Top 1 ( 169.873 kcal mol 1), Top 3 ( 167.522 kcal mol 1), and Top 2 ( 124.943 kcal mol 1). On the other hand, NAG only obtained an interaction energy of 65.487 kcal mol 1, indicating lesser stability than the TCM candidates.
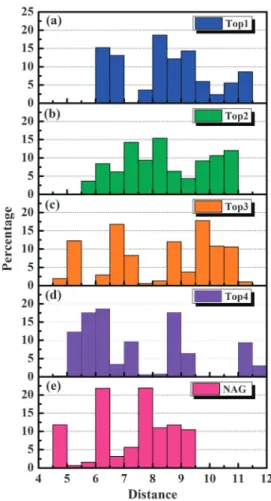
Ionic interactions and their corresponding bond lengths within each candidate are illustrated in Fig. 5. Top 1–3 each has a sodium ion that contributed to stable ionic interaction. Comparatively, Top 4 was surrounded by three ionic inter-actions in the form of salt bridges that had close binding length from 2.5 to 3.5 A˚. The presence of three stable ionic interactions could be the reason Top 4 has an ionic interaction strength three-folds higher than the other candidates. NAG did not have any ionic interaction with the H1 ligand binding site. To characterize binding positions throughout MD simula-tion, the distances between a ligand and its surrounding residues were calculated by the average distance between their centers of mass. As illustrated in Fig. 6, the residues within 3.5 A˚ for the top three candidates were the same. In addition, key residues Glu83, Lys71, and Arg238 were common surround-ing residues for all candidates. For Top 1, the average distances were near 6–7 A˚ and 8–9.5 A˚ (Fig. 7(a)). Top 2 had wider distance distributions with slightly higher occupancy between 7–8.5 A˚ (Fig. 7(b)). Top 3 has four distance distribution peaks at 5.25 A˚, 6.75 A˚, 8.75 A˚, and 9.75 A˚ (Fig. 7(c)). Compara-tively, Top 4 has shorter ligand–residue distances, primarily around 5–6.5 A˚ and 8.75 A˚ (Fig. 7(d)). NAG had a slightly different distance distribution pattern with three distance distribution peaks at 4.75 A˚, 6.25 A˚, and 7.75 A˚ (Fig. 7(e)). As summarized in Fig. 8, the distance distributions for all ligands were within 12.5 A˚, suggesting close distances which imply tight binding to H1. In particular, Top 4 was most closely bound to H1. The intermolecular distance analysis further supports interaction energy results.
In this study, the potential of TCM candidates as poten-tial H1 inhibitors was validated through different virtual computational methods. Dr Mark Fisher and Dr Jamie Lee at Massachusetts Institute of Technology shall be conducting
Fig. 7 Distribution of intermolecular residue distances (A˚) adjacent to (a) sodium (+)-isolaricireinol-2 alpha-sulfate, (b) sodium 3,4-dihydroxy-5-methoxybenzoic acid methyl ester-4-sulfate, (c) sodium (E)-7-hydroxy-1,7-bis(4-hydroxyphenyl) hept-5-ene-3S-sulfonate, (d) 3-methoxytyramine-betaxanthin and (e) NAG.
Fig. 8 Relative distance (A˚) distribution of surrounding amino acid residues to candidate ligands and NAG.
biological experiments to further assess and verify the potency of these TCM candidates in actual biological systems.
Conclusions
We identified four potential H1 inhibitors from the TCM database. The docking poses suggest higher binding affinity than NAG. The 20 ns MD simulation further identified stabilized docking conformations and stabilizing bonds. The binding features for each candidate as well as NAG are summarized in Fig. 9. Key binding residues include Lys71, Glu83, Asp103, and Arg238. Lys71 formed fluctuating H-bonds, but the distance remained constant. Glu83 formed stable H-bonds. While Arg238 formed stable H-bonds in general, some H-bonds dissipated during the MD simulation. Asp103 favored ionic interactions and had major contribution to the binding stability of each TCM candidate. This was demon-strated through interaction energy calculations. Intriguingly, Top 4 (3-methoxytyramine-betaxanthin) has significantly lower interaction energy and closer intermolecular distances than other candidates due to the formation of four stable salt bridges.
All four candidates had high interaction energy and low intermolecular distance within the binding site. In summary, virtual screening, MD simulation, interaction energy, and bind-ing distance analysis identified not only potential H1 inhibitors from TCM database, but also characterized the binding features targeting the residues Lys71, Glu83, Asp103, and Arg238. In addition to potential H1 inhibitor ligands and molecular features for drug design, the results of this study suggest that Polygonum cuspidatum, Zingiber officinale, and Celosia argentea may have potential as health foods with antiviral benefits.
Acknowledgements
The research was supported by grants from the National Science Council of Taiwan (NSC 99-2221-E-039-013), Committee on Chinese Medicine and Pharmacy (CCMP100-RD-030), China Medical University and Asia University (CMU98-TCM, TCM, S-02, ASIA-25, CMU99-ASIA-26 CMU99-ASIA-27 CMU99-ASIA-28). This study is also supported in part by Taiwan Department of Health Clinical Trial and Research Center of Excellence (DOH100-TD-B-111-004)
Fig. 9 The key binding features of (a) sodium (+)-isolaricireinol-2 alpha-sulfate, (b) sodium 3,4-dihydroxy-5-methoxybenzoic acid methyl ester-4-sulfate, (c) sodium (E)-7-hydroxy-1,7-bis(4-hydroxyphenyl) hept-5-ene-3S-sulfonate, (d) 3-methoxytyramine-betaxanthin, and (e) NAG. Interacting amino acids are represented in orange.
and Taiwan Department of Health Cancer Research Center of Excellence (DOH100-TD-C-111-005). We are grateful to the Asia University cloud-computing facilities.
References
1 World Health Organization, 2010.
2 XinHuaNet, http://news.xinhuanet.com/english2010/health/2011-01/ 28/c_13710292.htm, accessed on Sep. 17, 2011. 3 XinHuaNet, http://news.xinhuanet.com/english2010/health/2011-01/ 25/c_13706275.htm, accessed on Sep. 17, 2011. 4 XinHuaNet, http://news.xinhuanet.com/english2010/world/2011-01/ 24/c_13703835.htm, accessed on Sep. 17, 2011. 5 XinHuaNet, http://news.xinhuanet.com/english2010/health/2011-01/ 22/c_13703009.htm, accessed on Sep. 17, 2011. 6 XinHuaNet, http://news.xinhuanet.com/english2010/health/2011-01/ 19/c_13698298.htm, accessed on Sep. 17, 2011. 7 XinHuaNet, http://news.xinhuanet.com/english2010/china/2011-01/ 26/c_13708574.htm, accessed on Sep. 17, 2011.
8 World Health Organization. FluNet, http://gamapserver.who.int/ GlobalAtlas/DataQuery/browse.asp?catID=120000000000&lev=2, accessed on Sep. 17, 2011.
9 C. M. Carr, C. Chaudhry and P. S. Kim, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 14306–14313.
10 R. J. Russell, S. J. Gamblin, L. F. Haire, D. J. Stevens, B. Xiao, Y. Ha and J. J. Skehel, Virology, 2004, 325, 287–296.
11 Y. Bao, P. Bolotov, D. Dernovoy, B. Kiryutin, L. Zaslavsky, T. Tatusova, J. Ostell and D. Lipman, J. Virol., 2008, 82, 596–601. 12 A. Kilander, R. Rykkvin, S. G. Dudman and O. Hungnes, Euro.
Surveill., 2010, 15, 6–8.
13 World Health Organization, Wkly. Epidemiol. Rec., 2010, 85, 21–22.
14 S. Maurer-Stroh, R. T. Lee, F. Eisenhaber, L. Cui, S. P. Phuah and R. T. Lin, PLoS Curr., 2010, 2, RRN1162.
15 J. D. Bloom, L. I. Gong and D. Baltimore, Science, 2010, 328, 1272–1275.
16 K. C. Lai, S. C. Hsu, C. L. Kuo, S. W. Ip, J. S. Yang, Y. M. Hsu, H. Y. Huang, S. H. Wu and J. G. Chung, J. Agric. Food Chem., 2010, 58, 11148–11155.
17 J. Lin, L. Wei, W. Xu, Z. Hong, X. Liu and J. Peng, Mol. Med. Rep., 2011, 4, 1283–1288.
18 G. Tian, L. Guo and W. Gao, Curr. Drug Discovery Technol., 2010, 7, 32–36.
19 Y. Su, M. Loos, N. Giese, E. Metzen, M. W. Buchler, H. Friess, A. Kornberg and P. Buchler, Cancer, 2011, DOI: 10.1002/cncr.26344. 20 H. H. Chan, H. D. Sun, M. V. Reddy and T. S. Wu, Phytochemistry,
2010, 71, 1360–1364.
21 K. He, X. Li, X. Chen, X. Ye, J. Huang, Y. Jin, P. Li, Y. Deng, Q. Jin, Q. Shi and H. Shu, J. Ethnopharmacol., 2011, DOI: 10.1016/j.jep.2011.07.033.
22 W. Xie, Y. Zhao and Y. Zhang, Evidence-Based Complementary Altern. Med., 2011, 2011, 726723.
23 P. Lan, M. Q. Xie, Y. M. Yao, W. N. Chen and W. M. Chen, J. Comput. Aided Mol. Des., 2010, 24, 993–1008.
24 H. L. Yang, Y. H. Kuo, C. T. Tsai, Y. T. Huang, S. C. Chen, H. W. Chang, E. Lin, W. H. Lin and Y. C. Hseu, Food Chem. Toxicol., 2011, 49, 290–298.
25 F. Zhao and P. X. Liu, Zhongguo Zhongxiyi Jiehe Zazhi, 2007, 27, 178–181.
26 H. J. Huang, K. J. Lee, H. W. Yu, C. Y. Chen, C. H. Hsu, H. Y. Chen, F. J. Tsai and C. Y. C. Chen, J. Biomol. Struct. Dyn., 2010, 28, 23–37.
27 C. Y. Chen and C. Y. C. Chen, J. Mol. Graphics Modell., 2010, 29, 21–31.
28 C. Y. C. Chen, J. Mol. Graphics Modell., 2009, 28, 261–269. 29 C. Y. Chen, Y. H. Chang, D. T. Bau, H. J. Huang, F. J. Tsai,
C. H. Tsai and C. Y. C. Chen, J. Biomol. Struct. Dyn., 2009, 27, 171–178.
30 H. J. Huang, K. J. Lee, H. W. Yu, H. Y. Chen, F. J. Tsai and C. Y. C. Chen, J. Biomol. Struct. Dyn., 2010, 28, 187–200.
31 R. Chen, M. Chen, J. Xiong, F. Yi, Z. Chi and B. Zhang, Trials, 2010, 11, 121.
32 F. P. Chen, M. S. Jong, Y. C. Chen, Y. Y. Kung, T. J. Chen, F. J. Chen and S. J. Hwang, Evidence-Based Complementary Altern. Med., 2011, 2011, 236341.
33 S. Fu, J. Zhang, F. Menniti-Ippolito, X. Gao, F. Galeotti, M. Massari, L. Hu, B. Zhang, R. Ferrelli, A. Fauci, F. Firenzuoli, H. Shang, R. Guerra and R. Raschetti, PLoS One, 2011, 6, e19604.
34 A. S. Y. Lau, T. C. T. Or, C. L. H. Yang, A. H. Y. Law and J. C. B. Li, Neuropharmacology, 2011, 60, 823–831.
35 T. T. Chang, M. F. Sun, H. Y. Chen, F. J. Tsai, M. Fisher, J. G. Lin and C. Y. C. Chen, Mol. Simul., 2011, 37, 361–368. 36 T. T. Chang, M. F. Sun, H. Y. Chen, F. J. Tsai, M. Fisher,
J. G. Lin and C. Y. C. Chen, J. Biomol. Struct. Dyn., 2011, 28, 773–786.
37 J. Shen, J. Ma and Q. Wang, PLoS One, 2009, 4, e7789. 38 R. Xu, D. C. Ekiert, J. C. Krause, R. Hai, J. E. Crowe Jr. and
I. A. Wilson, Science, 2010, 328, 357–360. 39 C. Y.-C. Chen, PLoS One, 2011, 6, e15939.
40 H. Yang, P. Carney and J. Stevens, PLoS Curr., 2010, 2, RRN1152.
41 C. M. Venkatachalam, X. Jiang, T. Oldfield and M. Waldman, J. Mol. Graphics Modell., 2003, 21, 289–307.
42 B. R. Brooks, C. L. Brooks 3rd, A. D. Mackerell Jr., L. Nilsson, R. J. Petrella, B. Roux, Y. Won, G. Archontis, C. Bartels, S. Boresch, A. Caflisch, L. Caves, Q. Cui, A. R. Dinner, M. Feig, S. Fischer, J. Gao, M. Hodoscek, W. Im, K. Kuczera, T. Lazaridis, J. Ma, V. Ovchinnikov, E. Paci, R. W. Pastor, C. B. Post, J. Z. Pu, M. Schaefer, B. Tidor, R. M. Venable, H. L. Woodcock, X. Wu, W. Yang, D. M. York and M. Karplus, J. Comput. Chem., 2009, 30, 1545–1614.
43 R. Fletcher, Optimization, Academic Press, New York and London, 1969.
44 R. Fletcher and C. M. Reeves, Comput. J., 1964, 7, 149–154. 45 S. J. Gamblin, L. F. Haire, R. J. Russell, D. J. Stevens, B. Xiao,
Y. Ha, N. Vasisht, D. A. Steinhauer, R. S. Daniels, A. Elliot, D. C. Wiley and J. J. Skehel, Science, 2004, 303, 1838–1842. 46 S. J. Gamblin and J. J. Skehel, J. Biol. Chem., 2010, 285,
28403–28409.
47 T. Lin, G. Wang, A. Li, Q. Zhang, C. Wu, R. Zhang, Q. Cai, W. Song and K. Y. Yuen, Virology, 2009, 392, 73–81.
48 K. Xiao, L. Xuan, Y. Xu, D. Bai and D. Zhong, Chem. Pharm. Bull., 2002, 50, 605–608.
49 W. Sun and J. Sneng, Medicinal Science and Technology, Press of China, 1998.
50 Y. Cai, M. Sun, W. Schliemann and H. Corke, J. Agric. Food Chem., 2001, 49, 4429–4435.
51 H. Z. Zheng, Z. H. Dong and J. She, Beijing Yixueyuan Xuebao, 1998, 3, 2800–2820.
52 Y. Kim, S. Narayanan and K. O. Chang, Antiviral Res., 2010, 88, 227–235.
53 M. Mukhtar, M. Arshad, M. Ahmad, R. J. Pomerantz, B. Wigdahl and Z. Parveen, Virus Res., 2008, 131, 111–120.