國立交通大學
應用化學研究所
碩士論文
Joined Use of Oxazolidinone and Desymmetic Amino Protection in
Application of Oligosaccharide Synthesis
環胺基酸酯結合非對稱胺基保護在寡醣合成上的應用
學生:林士哲 (Shih-Che, Lin)
指導教授:蒙國光 博士 (Dr. Kwok-Kong, Tony, Mong)
Abstract
Joined use of N-benzyl oxazolidinone and N-benzyl-N-benzyloxycarbonyl (N-BnCbz) desymmetric amino-protecting function is reported. The new synthetic approach enables the facile preparation of type 1 and type 2 LacNAc disaccharide in satisfactory yield. One-pot deprotection of N-BnCbz and O-benzyl ether is achieved by hydrogenolysis under mild conditions. Further application of this protection strategy realizes the synthesis of trisaccharide H blood group substrate.
O OBn RO RO N(Cbz)Bn STol N-Bn, N-Cbz desymmetric amino protection O BnO OBn BnO O O O OBn HO N(Cbz)Bn O(CH2)6Cl O BnOOBn OBn one-pot global deprotection acetylation O AcO OAc AcO O O O OAc AcO NHAc O(CH2)6 Cl O AcOOAc OAc
摘要
本篇論文要報導芐基(benzyl group)取代的 oxazolidinone 和非對稱胺基保護 (desymmetric amino protection)的應用性。我們應用此合成策略製備了一型和二 型的乳糖胺,此方法不但容易操作而且有著極佳的產率。無論是在氮原子上面的 芐氧羰基(benzyloxycarbonyl group)和芐基,或是在氧原子上的芐基,都可以在 溫和條件下的氫解反應(hydrogenolysis)中一併去除。接著我們合成出 H 型血基 質(H blood group substrate)中的部分三醣結構,再度證明此一策略的應用性。
O OBn RO RO N(Cbz)Bn STol O BnO OBn BnO O O O OBn HO N(Cbz)Bn O(CH2)6Cl O BnOOBn OBn O AcO OAc AcO O O O OAc AcO NHAc O(CH2)6Cl O AcOOAc OAc N-Bn, N-Cbz 非對稱胺基保護 一鍋化 全去保護 乙醯基化
誌謝
從專題生就進入蒙老師實驗室,到現在碩班畢業,待了整整有三年之久,從 當時懞懂無知的大學生轉變為漸漸懂得實驗背後原理的碩班學生了。這段為期三 年的化學之旅,有歡笑也有淚水,在我的腦海裡留下了無法抹滅的回憶。 首先我衷心地感謝蒙國光老師,雖然老師對學生很嚴格,但他還是會很有耐 心地和我討論實驗上遇到的未知問題,並且一起尋找解決的方法。老師最常跟我 說的話就是:「遇到挫折不要太早放棄,要去面對甚至想辦法解決它。」儘管在 實驗上遇到許多挫折,但在老師的督導和鼓勵之下,慢慢地解決了一些問題,也 無形之中增強了自己的自信和堅毅,讓我從原本做事瞻前顧後的猶豫性格轉變為 計畫好就勇往直前的做事態度。即便將來遇到更大的挫折甚至是挑戰時,我也能 夠處之泰然,無所畏懼地去克服它。 其次感謝口試委員洪上程老師、林俊成老師、陳焜銘老師、孫仲銘老師撥空 前來參加我的口試,無論是對研究計畫或是論文的內容,都給我很多寶貴的意 見,從中學到很多東西,也了解到自己還有很多地方需要加強與努力。還有張秋 景小姐,不但協助我做變溫 NMR 實驗,平常有圖譜上的問題,也都會很熱心地幫 我解決。 再來就是感謝相處時間最久的實驗室夥伴了。首先是晉陞壆長,從我是專題 生時就是他帶我做實驗,感謝他的提攜與幫助,無論是實驗或生活上都是對我照 顧有加。再來是 diwi 學長和世聖學長,總是會不厭其煩地教導我做實驗的技巧 和相關軟體的操作,藉由跟他們相處的過程中,我慢慢地體會到追求知識的熱忱與渴望。振瑋和崑章是陪我一起打棒球的好伙伴,也是實驗室互相打氣的好戰 友。後來加入的璟妤、鈺芳、彥勳、哲豪、劭儒、桔程、郁惠、育賢和俊翰學弟 妹,也為實驗室添加了不少活力和笑聲。還有曾經在實驗室待過的專題生們,也 和我有愉快的互動。 另外感謝大學時代 418.5 的夥伴們,在實驗苦悶的時光裡,有可以傾訴的對 象,也藉著出去遊玩的時間,調適一下緊繃的心情。同樣也在求學生涯的博班學 長徐昀,除了給我許多實驗上寶貴的經驗之外,也不吝分享他個人實驗心得,幫 助我解決了不少實驗上碰到的難題,在此也祝福學長能順利取得博士學位喔! 最後,我要感謝我的父母,能夠體諒我不常回家,也因為有他們在背後的支 持和無怨無悔的付出,我才能無後顧之憂地勇往直前完成我的碩士學業。謹以此 論文獻給你們,表達我對你們的感謝。
List of Abbreviations
AgOTf silver triflate
BF3·OEt2 trifluoride etherate
BSM benzenesulfinylmorpholine
BSP benzenesulfinylpiperidine
Bu4NBr tetrabutyl ammonium bromide
t-BuOK potassium tert-butoxide
Cbz benzyloxycarbonyl
CH2Cl2 dichloromethane
CH3CN acetonitrile
d doublet
DMSO dimethyl sulfoxide
DMTST dimethyl(thiomethyl) sulfonium trifluoromethane sulfonate DTBMP 2,6-di-tert-butyl-4-methyl pyridine Et3SiH triethylsilane Hz hertz m multiplet NaBH3CN cyanoborohydride NIS N-iodosuccinimide Na2S2O3 sodium thiosulphate NEt3 triethylamine t triplet
PhSOTf (PST) phenylsulfenyltriflate
PTSA (TsOH) p-toluenesulfonic acid
s singlet
TFA trifluoroacetic acid
Tf2O triflic anhydride
THF tetrahydrofuran
TMSOTf trifluoromethanesulfonate
Table of Contents
Abstract...i
摘要...ii
誌謝... iii
List of Abbreviations...v
Table of Contents ...vii
Index of Schemes...ix
Index of Tables...xi
Index of Figures ...xii
1 Introduction...1
1.1 Lewis blood group antigens...1
1.2 General mechanism for the 1,2-trans-glycosylation of 2-amino sugars...3
1.3 Amino protecting groups for 1,2-trans--glycosylation of 2-amino sugars ....5
1.3.1 Monosubstituted aminoprotecting groups...6
1.3.2 Disubstituted aminoprotecting groups...8
1.3.2.1 Symmetric disubstituted aminoprotecting group...9
1.3.2.2 Desymmetric disubstituted aminoprotecting group...9
1.3.3 Oxazolidinone aminoprotecting group...11
1.3.3.1 N-Unsubstituted oxazolidinone...11
1.3.3.2 N-Acetyl oxazolidinone ...15
1.3.3.3 N-Benzyl oxazolidinone...26
1.4 Motivation...30
2.1 Anomerization phenomenon of oxazolidinone protected thioglycoside 4...31
2.2 Glycosylation studies of the thioglycoside donor 5...34
2.3 Reactivity based chemoselective glycosylations of thioglycoside 4 ...36
2.4 Retrosynthetic analysis of tetrasaccharide Lewis Y ...38
2.5 Removal of oxazolidinone ring in monosaccharide...39
2.6 Synthesis and characterization of desymmetric aminoprotecting groups....40
2.7 Glycosylation studies of the glucosamine acceptor 25 ...44
2.8 Hydrogenolysis of oxazolidinone protected disaccharides...46
2.9 Synthesis of disaccharides using desymmetric aminoprotecting strategy...49
2.10 Synthesis of trisaccharide 41 via one-pot glycosylation and deprotection ..50
3. Conclusion ...52
4. Experimental...53
4.1 General procedures ...53
4.2 General procedure for glycosylations ...54
4.3 Procedures and experimental data ...55
5. References...78
Index of Schemes
Scheme 1. General mechanism for the glycosylation of amino sugars ...3 Scmeme 2. Glycosylation of 2-acetamido-2-deoxy glycosyl donors ...5 Scmeme 3. Intermediates in Lewis acid-activated glycosylation with D-glucosamine
derivatives ...8 Scheme 4. Oxazolidinone donors as versatile intermediates...12 Scheme 5. Orthogonal glycosylation reactions of N-unsubstituted thioglycosides...14 Scmeme 6. Glycosylation of N-acetyl oxazolidinone acceptors with thioglycosides .15 Scmeme 7. Preparation of the -configured methyl oxazolidinone acceptor...17 Scmeme 8. Preparation of the -configured methyl oxazolidinone acceptor...17 Scmeme 9. Removal of the oxazolidinone in - and -methyl oxazolidinone series .18 Scmeme 10. Stereoselectivity-controllable glycosylation of the N-acetyl oxazolidino-
ne donor ...22 Scmeme 11. Proposed mechanism for the anomerization via endocyclic cleavage ....23 Scmeme 12. Stereoselectivity-controllable glycosylation of the N-acetyl oxazolidino-
ne donor ...24 Scmeme 13. Synthesis of N-benzyl oxazolidinone protected donors and acceptors ...27 Scmeme 14. One-pot synthesis of trisaccharides via oxazolidinone glycosides ...28 Scmeme 15. The deprotection sequence of an anti-Helicobacter pyroli oligosaccharid- es ...29 Scmeme 16. Synthesis of N-benzyl oxazolidinone protected glucosamine donor 5 ...31 Scmeme 17. Proposed mechanism for the anomerization of cyclic carbonate or –carb-
Scmeme 19. Investigations of desymmetric glucosamine acceptors...40
Scmeme 20. Plausible mechanism for the formation of compound 23 ...41
Scmeme 21. Selective deprotection studies of the Cbz protecting group...45
Scmeme 22. Hydrogenolysis reactions of disaccharides 30 and 33 ...48
Scmeme 23. Removal studies of N-benzyl oxazolidinone ring in disaccharide 16 ...49
Scmeme 24. Synthesis of trisaccharide 41 via one-pot glycosylation and deprotection strategy...51
Index of Tables
Table 1. Stereoselective formation of -linked glycosides in high yields...12
Table 2. Alternative coupling methods for the formation of disaccharide...16
Table 3. Stereoselective coupling reactions between N-acetyl oxazolidinone donors and acceptors...19
Table 4. Glycosylation of oxazolidinone thioglycoside donor under BSP/Tf O activation conditions 2 ...21
Table 5. Couplings of various oxazolidinone protected donors...25
Table 6. The coupling of donor B and acceptor G ( both in Table 5) under different conditions...26
Table 7. Optimizations of the reductive benzylidene ring opening of thioglycoside 333 Table 8. Glycosylation studies of N-benzyl oxazolidinone protected donor 5 and acceptor 7...35
Table 9. Glycosylation studies of N-benzyl oxazolidinone acceptor 4...37
Table 10. Removal studies of the N-benzyl oxazolidinone ring in monosaccharide 19 ...39
Table 11. Glycosylation studies of the desymmetric glucosamine acceptor 26...44
Table 12. Deprotection of desymmetric glucosamine acceptor 26 ...46
Table 13. Glycosylation studies of donor 38 and acceptor 7 ...50
Table 14. The amounts of glycosyl donor, glycosyl acceptor, NIS, and TMSOTf used in glycosylation...54
Index of Figures
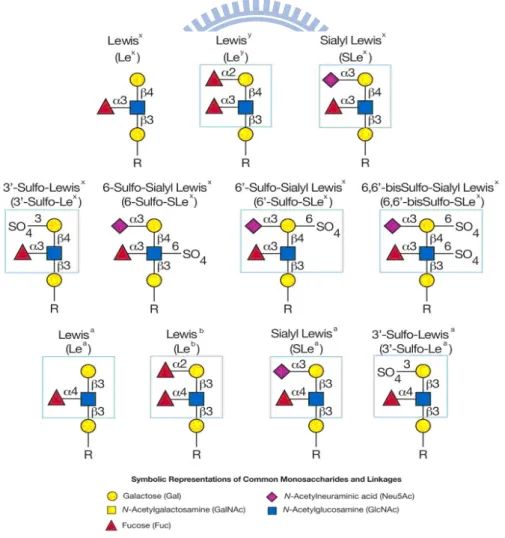
Figure 1. Type-1 and type-2 Lewis determinants ...1
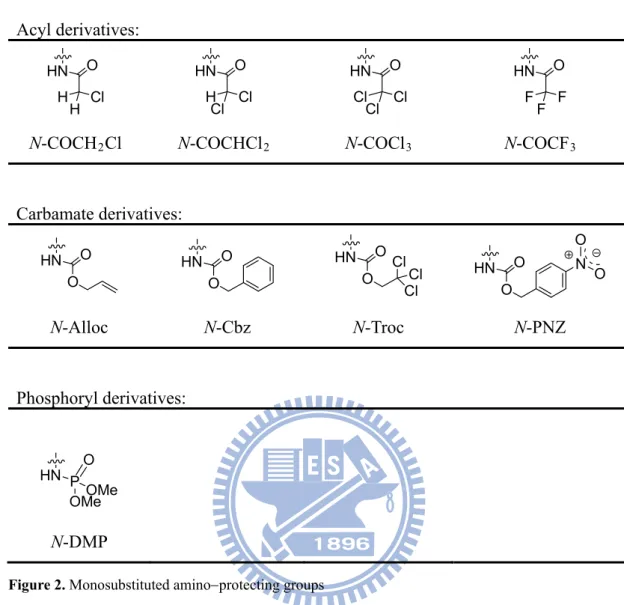
Figure 2. Monosubstituted aminoprotecting groups...7
Figure 3. Disubstituted aminoprotecting groups...10
Figure 4. N-Unsubstituted and N-acetyl oxazolidinone protected thioglycoside donors ...20
Figure 5. Chemoselective manipulation of N-acetyl protected oxazolidinone ...22
Figure 6. H NMR spectra of compound A at different temperatures1 ...43
1 Introduction
1.1 Lewis blood group antigens
The involvement of carbohydrates and their conjugates in numerous biological processes are recognized.1-4 An important calss is the series of Lewis blood group antigens which contain either Gal-(1→3)-GlcNAc (type 1 LacNAc) or Gal- (1→4)-GlcNAc (type 2 LacNAc) backbone.5 The nomenclature of these Lewis blood group antigens is dependent on the linkages of fucose (Fuc), N-acetyl-neura- minic acid (Neu5Ac) or sulfate to the backbones (Figure 1).6
Some members of the Lewis blood group antigen family have been proven to be specific tumor makers for cancer diseases; thus, they are attractive targets for various biomedical investigations. For example, Lewis Y antigens have some relations with apoptosis in gastric and colorectal carcinomas.7 Synthetic studies not only provide the evidence for chemical structures of the oligosaccharides but also supply sufficient amount of desired molecules or glycoconjugates for various study. Since nearly all Lewis blood group antigens contain the N-acetyl glucosamine which is connected to galactose in 1,2-trans--glycosidic linkage. To develop a facile method for the construction of this glycosidic linkage is necessary and desirable.
1.2 General mechanism for the 1,2-trans-glycosylation of 2-amino sugars
The so-called glycosylation involves the formation of a carbon-oxygen bond via a nucleophilic substitution reaction. This reaction involves the anomeric position (C-1) of a glycoside carrying a leaving group (X), namely the glycosyl donor A (Scheme 1). This glycosyl donor would react with the hydroxyl group of an alcohol (R1OH, glycone or aglycone). The glycone acceptor can be a mono- or oligosaccharide (Scheme 1).8 The glycosylation is promoted by an electrophilic activator, namely the promoter. The amount of promoter used can be varied from catalytic to stoichiometric
O R1R2NX shielding of the -face ex: Ag2O O R1R2N OR' O R1R2N X ROH slow Insoluble Promoter Partly Soluble Promoter O R1R2N Y +Y-, -X+ +X-, -Y+ O R1R2N + R'OH R'OH fast O R1R2N OR' b b a if R1 is a participating group O N R2 R1 + R'OH 1, 2-trans opening a O R1R2N OR' E F A B C G D F Ag R'OH
dependent on the types of leaving groups. The function of the promoter is to facilitate the departure of the anomeric leaving group (X), and then usually a mixtures of and -glycosides (path a and b respectively) are produced due to a lack of stereoselectivity.
In order to obtain 1,2-trans--glycosidic bond, two approaches are generally employed. The most widely used method is to protect the amino function with a neighboring group which results in a cyclic intermediate (D) shielding the “-face” of the donor by participating effect.9 In this way, the hydroxyl group of the acceptor can only access the “-face” and afford the 1,2-trans-glycoside (F). Another method for the synthesis of 1,2-trans--glycoside (F) includes the use of 1,2-cis-2-azido-2- deoxy--D-glycopyranosyl halides (A) (having a nonparticipating aminoprotecting
group) and an insoluble promoter such that the “-face” of the donor is shielded from reaction during the substitution with the acceptor alcohol (E). Similar to this concept is the use of 2-azido-2-deoxy--D-glycopyranosyl diphenyl phosphates or imidates in
nitrile solvent, which promotes the formation of -nitrilium intermediate with the oxocarbenium ion and shields the “-face” of the donor.10 The above non-participating methods used mainly with 2-azido-2-deoxy glycosyl donors, and the 1,2-trans--stereoselectivities obtained are often lower than those given by the use of donors containing the participating groups. As a result, to mask the amino function with a participating protecting group apparently is a better choice for the construction of the 1,2-trans--glycosidic linkage of 2-amino sugars.
1.3 Amino protecting groups for 1,2-trans--glycosylation of 2-amino sugars
2-Acetamido-2-deoxy-glucopyranose is an important constituent in all Lewis blood group antigens, and is present in 1,2-trans-glycosidic linkage. Apparently, a glycosylation strategy by using GlcNAc donors should give the simplest synthetic route in oligosaccharide synthesis. However, the reactions of these donors often suffer from the formation of relatively stable oxazoline intermediates (D) due to neighboring group participation. As a consequence, the glycosylation yield is moderate to the best.8,11,12 Although the GlcNAc oxazoline derivatives can react with acceptor alcohols to afford 2-acetamido-2-deoxy--glycosides (C) via oxazolinium ion intermediates (B or E) in the presence of Brnsted or Lewis acids. The harsh reaction conditions required precluded their wide application (Scheme 2).13-15 Therefore, a wide variety of amino-protecting groups for amino sugars have been developed. To be a suitable amino-protecting group, it should be stable to a wide range of reaction
O AcHN LG ROH promoters O HN O HOR O N O O N O HOR LA O AcHN OR ROH Lewis acid A B D E LA = Lewis Acid LG = leaving group C
conditions. In addition, this amino protection provides desirable stereoselectivity and acceptable yield in glycosylations. Moreover, it should be removed under mild conditions after glycosylations. Based on literature search, the aminoprotecting groups for 2-amino-2-deoxy sugars are briefly classified into three types according to the substituent patterns including (1) monosubstituted, (2) disubstituted, and (3) oxazolidinone amino-protecting group. The above three categories are described hereafter. This thesis mainly focuses on the discussion of oxazolidinone protecting group and its derived desymmetric protecting function.
1.3.1 Monosubstituted aminoprotecting groups
Monosubstituted protection is referred to those aminoprotecting groups in GlcNAc that contains one acyl/alkoxycarbonyl function (Figure 2). Acyl derivatives include N-monochloroacetyl (N-COCH2Cl),16 N-dichloroacrtyl (N-COCHCl2),17 N- trichloroacetyl (N-COCl3),18 and N-trifluoroacetyl (N-COCF3).19 Carbamate derivatives include N-allyloxycarbonyl (N-Alloc),20,21 N-benzyloxycarbonyl (N- Cbz),22 N-2,2,2-trichloroethoxycarbonyl (N-Troc),23,24 and N-p-nitrobenzyloxycarbon- yl (N-PNZ).25 Recently, phosphoryl derivatives such as N-dimethylphosphoryl (N-DMP) and others were also repated.26,27 Among of the monosubstituted aminoprotecting groups mentioned above, the N-Troc group has wide application in oligosaccharide synthesis, and it is fairly stable to glycosylation conditions. However, the carbamate proton can be abstracted under standard basic conditions that leads to the formation of byproducts.
Acyl derivatives: HN O Cl H H HN O Cl H Cl HN O Cl Cl Cl HN O F F F
N-COCH2Cl N-COCHCl2 N-COCl3 N-COCF3
Carbamate derivatives: HN O O HN O O HN O O ClCl Cl HN O O N O O N-Alloc N-Cbz N-Troc N-PNZ Phosphoryl derivatives: HN PO OMeOMe N-DMP
1.3.2 Disubstituted aminoprotecting groups
Disubstituted amino protection is referred to those aminoprotecting groups of GlcNAc that contains two acyl substitutions which are usually presented as cyclic or acyclic structure. In order to avoid the stable oxazoline intermediate D (Scheme 3), the abstractable amide proton may be substituted by a functional group that survives the glycosylation conditions.28 This concept is manifested by installation of the bivalent group, or by blocking the amino group with two monovalent protecting groups. Electrophilic activation of the glycosyl donor E results an oxocarbenium ion
F which can further form an oxazolinium ion G. The reactive intermediate G can only
be attacked from the -face by a nucleophile without forming a stable oxazoline.
O AcO OAc AcO N L H O R Lewis acid O AcO OAc AcO N H O R +C- AcO O OAc AcO N O RCH3 --H+ H O AcO OAc AcO N O R O AcO OAc AcO N L X X O O Lewis acid O AcO OAc AcO + C -N X X O O O AcO OAc AcO N X X O OC
-R = Me (Ac) L = leaving group tBuO (Boc) C = counter ion AlO (Aloc) etc. X X = (Phth) S S (Dts) A B C E F G D
(i) Monosubstituted amino protection:
(ii) Disubstituted amino protection:
1.3.2.1 Symmetric disubstituted aminoprotecting group
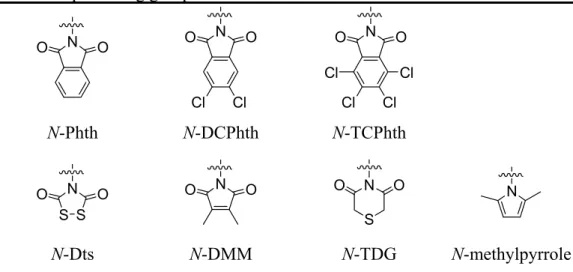
Most of the disubstituted symmetric aminoprotecting groups are either by installation of the bivalent group or by protection of two symmetric monovalent protecting groups (Figure 3). Bivalent protecting groups include N-phthaloyl (N-Phth),29,30N-4,5-dichlorophthaloyl (N-DCPhth),31 N-tetrachlorophthaloyl (N- TCPhth),32 N-dithiasuccinoyl (N-Dts),28,33,34 N-dimethylmaleoyl (N-DMM),35 N- thiodiglycoloyl (N-TDG),36 and N-methylpyrrole37. Protection of two symmetric monovalent protecting groups include N,N-diacetyl (N-Ac2)38 and N-dibenzyl (N-Bn2).39 Among of the disubstituted amino-protecting groups mentioned above, N-phthaloyl (N-Phth) thioglycosides are widely employed in oligosaccharide synthesis; however, the cleavage of N-phthaloyl moiety requires strongly basic conditions and high temperature that precludes its application to molecules with base-labile functions such as glycopeptides and glycolipids.40
1.3.2.2 Desymmetric disubstituted aminoprotecting group
Disubstituted desymmetric aminoprotecting groups are rarely used in the glycosylations of oligosaccharide synthesis. Only N-acetyl-N-2,2,2-trichloroethoxy- carbonyl (N-AcTroc) thioglycoside has been exploited as a donor for oligosaccharide synthesis involving 1,2-trans--glycosylations (Figure 3).41
Bivalent protecting groups: N O O O N O Cl Cl N O O Cl Cl Cl Cl N-Phth N-DCPhth N-TCPhth S S N O O O N O N S O O N N-Dts N-DMM N-TDG N-methylpyrrole
Symmetric monovalent protecting groups:
N
O O N
N-Ac2 N-Bn2
Desymmetric monovalent protecting groups:
N
O O
O ClCl Cl
N-AcTroc
1.3.3 Oxazolidinone aminoprotecting group
In 1969, Gross et al. first applied the cyclic carbamate (oxazolidinone) to the synthesis of the trans-2,3-N,O-carbamate protected glucosamine monosaccharide.42 It was not noticed until Kerns et al. reported on the use of oxazolidinone protected glucosamine as donors in 2001.43 This draws the attention of other chemists to exploit the use of the oxazolidinone protected glycosides in carbohydrate chemistry. The N-unsubstituted oxazolidinone protected glycosyl donors for 1,2-cis--glycosylations were further elaborated to N-acetyl and N-benzyl oxazolidinone derivatives. Both of them were applied to the oligosaccharide synthesis for either 1,2-cis--glycosylations or 1,2-trans--glycosylations.
1.3.3.1 N-Unsubstituted oxazolidinone
In 2001, Kerns et al. first applied the use of 2,3-N,O-carbamate protected glucosamine donors in glycosylations.43 In their studies, using phenylsulfenyltriflate (PhSOTf) as the activating agent in dichloromethane at 78 oC, the N-unsubstituted oxazolidinone GlcNAc donor was condensed with an acceptor to afford -linked glycosides in excellent yields (Table 1).44 Another advantage of using N-unsubstituted oxazolidinone is that the mild condition for ring-opening deprotection of disaccharide
AF (Table 1) did not promote -elimination of the 4-linked uronic acid, a significant limitation of many base-labile protecting groups in glycosaminoglycan (GAG) synthesis. In addition to the stereoselective formation of -linked glycosides, the ring-fused oxazolidinone provided a versatile protecting group for 2-amino sugar synthesis. Further elaborations of these oxazolidinone protected glycosyl donors to
Table 1. Stereoselective formation of -linked glycosides in high yields O SPh AcOO NH O OAc O O O HO O O O OCH3 HO BnO N OBn O O O BnO BnO HO OCH3 O O O HO OCH3 O O Ph O OBn HO CO2CH3 BzO OBz A B C D E F
Entry Glycosyl donor Glycosyl acceptor Disaccharides Isolated yield (%)
1 A B AB 97 2 A C AC 75 3 A D AD 90 4 A E AE 95 5 A F AF 75 O SPh R2O R1O O NH O O SPh HO BnO O NH O O SPh O NH O OBn AcO O SPh R2O R1O HO NH2 O SPh R2O R1O HO NHCO2R3 A: R1 = R2 = Ac B: R1 = R2 = PhCH C (from A): R1 = R2 = H D (from B): R1 = R2 = PhCH (a) (b)
E (from A): R1 = R2 = H, R3 = Methyl
F (from B): R1 = R2 = PhCH, R3 = Trichloroethyl (c) (d) G from B H
Scheme 4. Oxazolidinone donors as versatile intermediates
Reagents and conditions: (a) NaOH, H2O/THF, 7580%. (b) Cs2CO3, R-OH, 7580%. (c) NaCNBH3,
Further investigations of the same research focused on the utility of oxazolidinone protected thioglycosides as glycosyl acceptors. The accepter compatibility was demonstrated in different orthogonal glycosylation strategies (Scheme 5).45 The yield was satisfactory; however, the following activation of the resulting disaccharides H-J using PST as previous procedure was not efficient at all. The activation was also unsuccessful at low temperatures despite the use of other promoter systems. Incompleteness of the reactions or loss of stereocontrol were observed at higher reaction temperatures (20 oC to room temperature). This phenomenon is similar to that reported by Boons, trans-2,3-cyclic carbonate protected glycosyl donors have significantly lower anomeric reactivities than fully acylated and N-acyl protected donors.46 When the phenyl thioglycoside was changed into p-methylphenyl thioglycoside which was anticipated more readily activated (Scheme 5), the yield was still moderate. What’s even worse is that the oxazolidnone nitrogen was more readily glycosylated with the donor than an amide nitrogen, and this may complicate the iterative linking of sugar units. One way to avoid this off-target glycosylation was to protect the oxazolidinone nitrogen. Hence, to eliminate N-glycosylation and aim at improving the -stereoselectivity, two types of N-protected oxazolidinone protected glycosides had been developed.
O CO2CH3 AcO AcO OAc O NH CCl3 O CO2CH3 AcO AcO OAc O CO2CH3 AcO AcO OAc O AcO AcO OAc O NH CCl3 N N N N Bn O CO2CH3 LevO BzO OBz O NH CCl3 Br F A B C D E O CO2CH3 AcO AcO OAc O SPh HOO NH O OBn O SPh OO NH O OBn O AcO AcO OAc O SPh OO NH O OBn N N N N Bn O CO2CH3 LevO BzO OBz O SPh OO NH O OBn O CO2CH3 AcO AcO OAc O STol OO NH O OBn O CO2CH3 AcO AcO OAc O STol OO N O OBn O OAc OAc OAc H3CO2C + O STol HO O NH O OBn G F H I J K L (a) (b) (c) (d) (e) (f)
Scheme 5. Orthogonal glycosylation reactions of N-unsubstituted thioglycosides
Reagents and conditions: (a) F, BF3·OEt2, CH2Cl2, sieves, 0 oC to rt, 88%. (b) F, AgOTf, CH2Cl2,
030% for L. sieves, 0 oC to rt, 98%. (c) F, AgClO
4SnCl2, CH2Cl2, sieves, 0 oC to rt, 70%. (d) F, BF3·OEt2,
CH2Cl2, sieves, 0 oC to rt, 91%. (e) F, BF3·OEt2, CH2Cl2, sieves, 0 oC to rt, 81%. (f) A, BF3·OEt2,
1.3.3.2 N-Acetyl oxazolidinone
In 2003, Crich et al. first reported that oxazolidinone protected N-acetyl glucosamine 4-OH derivative was a highly reactive glycosyl acceptor.47 It is believed that the 4-hydroxyl group of N-acetyl glucosamine derivative is a poor glycosyl acceptor due to steric hindrance. This phenomenon is common to most pyranose 4-OH’s.48 In addition, the N-acetyl group is engaged in intermolecular hydrogen-bonded network that further decreases the nucleophilicity.49 To overcome these situations, a suitable N-acetyl oxazolidinone A (Scheme 6) was prepared and subjected to couplings with a range of thioglycosides.
O R'O SR (i) BSP, TTBP, Tf2O, CH2Cl2, -60 oC (ii) , -60 oC to rt O R'O O OBn O O N O AcOMe O OBn HOO N O AcOMe A
(i) Ba(OH)2, EtOH, △,
(ii) Ac2O O R'O O OBn O HO HN OMe Ac
Scmeme 6. Glycosylation of N-acetyl oxazolidinone acceptors with thioglycosides
The donors were exposed to the combination of 1-benzenesulfinylpiperidine (BSP),50 2,4,6-tri-tert-butylpyrimidine (TTBP),51 and triflic anhydride (Tf2O) in CH2Cl2 at 60 oC before addition of the acceptor and followed by warming the reaction mixture to room temperature. Meanwhile, they tested the acceptor’s generality by three other coupling methods such as Kahne’s sulfoxide method,52 Gin’s dehydrative coupling sequence,53 and Schmidt’s trichloroacetimidate protocol (Table
2).54 Even though the acceptor performed well in most cases, the selectivities and yields were varied with glycosyl donors and reaction conditions.
Table 2. Alternative coupling methods for the formation of disaccharide
O BnO OBn BnO BnO X + O OBn O O N O AcOMe O BnO OBn BnO BnO Promoters, CH2Cl2 O OBn HO O N O AcOMe B: X = -SPh A F C: X = -S(O)Ph D: X = , -OH E: X = -OC(=NH)CCl3
Donor Promoters Anomeric ratio (/) Yield (%)
B BSP; TTBP, Tf2O; 60 oC 6.2:1 78
C TTBP, Tf2O; 60 oC only 63
D Ph2SO, Tf2O, TTBP; 40 oC only 59
E TMSOTf, 30 oC 3.5:1 82
Since there are reports on dependence of nucleophilicity of glycosyl acceptors on their anomeric configurations, Crich et al. wanted to synthesize both - and -methyl glycosides to compare their nucleophilicities in 2005.55-57 At first, -methyl glycosides were successfully obtained (Scheme 7). However, their attempt to prepare these glycosides met with some issues.58 When the same reaction sequence was applied to -thioglycosides, -anomers were obtained instead of the expected -anomers. → anomerization occurred during reductive ring opening of benzylidene. Repeated attempts at overcoming this epimerization by controlling the acidity of the medium were unsuccessful and consequently they turned to an alternative reaction sequence (Scheme 8). Because the typical reductive cleavage of benzylidine acetals to benzyl ethers under conditions using cyanoboro-
hydridehydrogen chloride (NaBH3CNHCl) was suitable for numerous -glycosides, this anomerization was unexpected.59,60 They presumed that the strain imposed on the pyranose ring by the presence of the trans-fused oxazolidinone ring promoted the ring opening by endocyclic cleavage of the C1O5 bond (detailed discussion in result & discussion P.3233). O O O Ph HO OMe H2N
(a) 4-O2NC6H4OCOCl
(b) Amberlyst IR 120, 76% O O O Ph O OMe NH O AcCl EtN(i-Pr)2, 92% O O O Ph O OMe N O Ac NaCNBH3, HCl, Et2O, 80% O HO O OMe N O Ac OBn
Scmeme 7. Preparation of the -configured methyl oxazolidinone acceptor
O O O Ph HO H2N
(a) 4-O2NC6H4OCOCl
(b) Amberlyst IR 120, 81% O O O Ph O NH O AcCl EtN(i-Pr)2.89% O O O Ph O N O Ac Pd/C, H2 THF, 82% O HO O N O Ac OH OMe OMe OMe OMe NaCNBH3 HCl/Et2O 53% TDSCl, Imidazole DMF, 92% O HO O N O Ac OBn O HO O N O Ac OTDS OMe OMe TDS = thexyldimethylsilyl
With the - and -anomers in hand, a series of glycosylation couplings were examined with a variety of thioglycosides and they behaved analogously in glycosylation reactions. A major difference between these anomers was the deprotection of their resulting disaccharides (Scheme 9). The selective cleavage of oxazolidinone rings in the -anomer was possible and led to N-acetyl-glucosamine- based disaccharide. While for the -glycoside acceptor, complete removal of oxazolidinone and acetyl functions occurred, therefore reacetylation of amine was needed. O O O RO O N O AcOMe
OBn (i) Ba(OH)2, EtOH, Δ
(ii) Ac2O O O O RO HO AcHN OMe OBn O O O RO O N O Ac
OTDS Ba(OH)2, EtOH, Δ
O O O RO HO AcHN OTDS OMe OMe
Scmeme 9. Removal of the oxazolidinone in - and -methyl oxazolidinone series
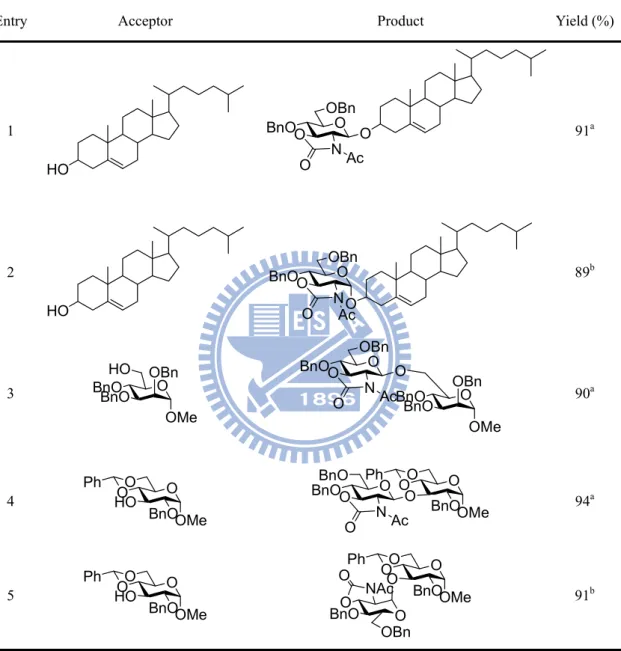
Shortly after Crich’s report, Oscarson et al. used N-acetyl oxazolidinone protected thioglycosides as glycosyl donors to glycosylate with a steroid and glycosyl acceptors using N-iodosuccinimide (NIS) and catalytic amount of silver triflate (AgOTf) as promoters, high yields of -linked glycosides were obtained. Surprisingly, when a larger quantity of AgOTf (0.4 equiv) was used in the coupling reactions, similar high yields of -linked glycosides were obtained (Table 3).61 This contrasting result may probably be related to in situ anomerization which was similar to Crich’s
observation for reductive opening of the benzylidene ring in oxazolidinone protected N-acetyl glucopyranosides.
Table 3. Stereoselective coupling reactions between N-acetyl oxazolidinone donors and acceptors
Entry Acceptor Product Yield (%)
1 HO O BnO OBn O N O Ac O 91a HO O BnO OBn O N O AcO 2 89b 3 O OBn BnO BnO O BnO OBn O N O Ac O O OBn BnO BnO OMe OMe HO 90a 4 O O O Ph BnOOMe HO O O O Ph BnOOMe O BnOO N O Ac O BnO 94a 5 O O O Ph O O NAc O OBn O O O Ph BnOOMe O BnO 91b BnOOMe HO
Afterwards, also at the same year, Kerns et al. realized their hypothesis proposed in 2003 that substitution of the oxazolidinone nitrogen precluded N-glycosylation and prevented off-target reactions with activating reagents.62 At first, they chose thioglycosides B (Figure 4) as glycosyl donors using PST as a promoter that previously employed. However, a complex reaction mixture was obtained and the yield of the disaccharide was low. After that, they tried BSP/TTBP/Tf2O promoting system as previously reported by Crich et al. which enabled complete activation.
O AcO OAc SPhMe O NH O O AcO OAc SPhMe O N O Ac A B
Figure 4. N-Unsubstituted and N-acetyl oxazolidinone protected thioglycoside donors
Encouraged by this observation, they estimated the coupling of B with a varirty of acceptors (Table 4). Since Crich and co-workers formerly reported -glycosyl triflate intermediates during BSP/Tf2O activation of thioglycosides. A dynamic system where -glycosyl triflate is in equilibrium with its less stable but more reactive -glycosyl triflate was proposed.63,64 In addition, the molecular models of the - and -glycosyl triflate intermediates revealed that the N-acetyl moiety gave steric hindrance to -face on the -triflate which was attacked by hindered nucleophiles. As a result, they concluded that the stereoselectivity of glycosylation relied on the structure of the acceptors, N-substituent on the oxazolidinone ring, and promoters in glycosylations. These factors affect reaction rates and an equilibrium between - and -anomeric triflates, and finally induce the stereocontrol of the glycosidic bond.
Table 4. Glycosylation of oxazolidinone thioglycoside donor under BSP/Tf2O activation conditions O AcOAcOO NAc O SPhMe + R-OH BSP, Tf2O, TTBP CH2Cl2, -60oC O AcOAcOO NAc O OR Entry Acceptor (R-OH) Product t (h) Ratio (:) Yield (%) 1 H3C OH CbzHN COOMe O AcOAcOO NAc O O MeOOC NHCbz CH3 1 only 83 2 O HO OO O O O AcOAcOO NAc O O O O O OO 1 only 95 3 BnOBnO O HO O OCH3 O AcOAcOO NAc O O BnO BnO O O OCH3 4 only 90 4 HO O O OCH3 O O Ph AcOAcO O O NAc O O O O OCH3 O O Ph 12 1:20 82 5 BnOHO O MeOOC BnOOCH3 O AcOAcOO NAc O O O BnO MeOOC BnOOCH3 12 1:7 75 6 O O O NPhthOCH3 Ph HO O AcOAcOO NAc O O O O NPhthOCH3 Ph O 24 1:4.5 75 7 HOBnO O BMPO BnOOCH 3 O AcOAcOO NAc O O O BnO BMPO BnOOCH3 48 1.6:1 65 8 HOBzO O MeOOC BzO OCH3 O O BzO COOMe BzO OCH3 O AcOAcOO NAc O 48 2.8:1 77 9 HOBnO O NPhthOCH3
OBn AcOAcOO O
NAc
O BnOO O
NPhthOCH3 OBn
Later on, Kerns et al. reported another study for chemoselective manipulation of oxazolidinone derivatives of N-acetyl-D-glucosamine (Figure 5).65 They presented
specific methods that were useful for elaborating sugar glycosides of the glycoconjug- ates that containing variably substituted 2-amino-2-deoxy-D-hexopyranosides.
O O N O OR Ac PO OP O HO NH2 OR PO OP O HO NHAcOR PO OP O HO HN OR PO OP O R1O O HO HN OR PO OP O R2R1N
Figure 5. Chemoselective manipulation of N-acetyl protected oxazolidinone
Recently, Oscarson et al. extended their previous work and reported a more detailed investigation of their donor-promoter system and the reaction mechanism. Either - or -linked glucosamine-containing glycosides can be prepared by tuning the reaction conditions (Scheme 10).66 The stereochemical outcome could be initially
O SEt AcO AcO O N O Ac
NIS (2 equiv), AgOTf (0.1 equiv) CH2Cl2, rt, 10-20 min (method A) O AcO AcO O N O Ac O AcO AcO O N O Ac OR OR NIS (2 equiv), AgOTf (0.4 equiv)
CH2Cl2, rt, 25-45 min (method B) + ROH A O AcO AcO O N O Ac OR DMTST (2 equiv), DTBMP (2.7 equiv) CH2Cl2, 0 to 20oC, 30 min (method C)
formed -product in method A. To verify the assumption that the -product which anomerized to -glycoside in the presence of the oxazolidinone ring was most efficient under more acidic condition, the -product was treated with a catalytic amount of AgOTf in the NMR tube. An efficient and high-yielded anomerization was observed giving the -glycoside almost exclusively even an excess amount of methanol was added. Based on these results, they proposed that the mechanism is perhaps via an activation of the ring oxygen and consecutive endocyclic CO bond cleavage in preference to exocyclic cleavage (Scheme 11).
O AcO AcO O NAC O O O BnO OBn OBn OMe AcO O AcO O NAC O O O BnO OBn OBn OMe E O AcO AcO O N O O O BnO OBn OBn OMe E Ac O AcO AcO O N O O O BnO OBn OBn OMe E Ac O AcO AcO O N O O O BnO OBn OBn OMe Ac E+ -E+
Scmeme 11. Proposed mechanism for the anomerization via endocyclic cleavage
To investigate the relationship between the stereochemistry outcomes of glycosylation and pre-activation protocol, Ye and et al. chose the known 2,3- oxazolidinone protected thioglycoside A (Scheme 12) as the glycosyl donor.67 Donor
A was preactivated at 73oC in anhydrous dichloromethane using benzenesulfinyl morpholine (BSM) and Tf2O in the presence of TTBP.68 After disappearance of donor
O STol AcO AcO O N O Ac BSM, Tf2O CH2Cl2, -73oC BSM, TTBP, Tf2O CH2Cl2, -73oC ROH -73oC to rt ROH -73oC to rt O AcO AcO O N O Ac O AcO AcO O N O Ac A OR OR
Scmeme 12. Stereoselectivity-controllable glycosylation of the N-acetyl oxazolidinone donor
A, the acceptor was added to the reaction mixture to furnish the glycosidic bond
formation. The yields were high and the glycosylations proceeded with excellent -selectivity. They attributed the -selectivity to a SN2-like process via the -glycosyl triflateintermediate based on the preactivation protocol.64 Since the use of hindered base (TTBP) is not necessary in their previous pre-activation protocol, they withdrew the base using BSM-Tf2O promoter system in glycosylations.68,69 Surprisingly, the yields are high and the glycosylations proceeded with excellent -selectivity. The reversal of the stereoselectivity in the absence of TTBP probably resulted from in situ anomerization of the -glycoside under acidic conditions. As a result, either - or -linked glucosamine-containing glycosides were prepared from the BSM-Tf2O promoter system in the presence of TTBP or not. Next, they applied the protocol to a variety of oxazolidinone protected glucosamine thioglycosides (A-F) with the same acceptor G (Table 5).70 The different stereoselectivities might arise from three major types of intermediates: oxocarbenium ion, -triflate, -triflate; the trend of the stereoselectivity remained inconsistent. In terms of the glycosylation results, the coupling between donor B and acceptor G showed the excellent -stereoselectivity. Then they used this pair of donor and acceptor to investigate the influence on the stereoselectivity resulting from different glycosylation conditions such as solvent, te-
Table 5. Couplings of various oxazolidinone protected donors O O R1RO 1O N O R 2STol BSM, Tf2O CH2Cl2, -73oC O O O Ph HO BnO OMe O O R1RO 1O N O R 2 O O O Ph O BnOOMe G A-F AG, BG, DG, FG Entry Donor T
(oC) Product Yield (%) Ratio (:)
1 AcOO O AcO NH O STol A 73 AcOO O AcO NH O O O O Ph O BnOOMe AG 87 1:2 2 BnOO O BnO N O Bn STol B 73 BnOO O BnO N O Bn O O O Ph O BnOOMe BG 85 only 3 BnOO O BnO NH O STol C 73 no coupling --- --- 4 BnOO O BnO N O Ac STol D 60 BnOO O BnO N O Ac O O O Ph O BnOOMe DG 84 1:1 5 O O N O Bn STol O O Ph E --- no activation --- --- 6 AcOO O AcO N O Bn STol F 50 AcOO O AcO N O Bn O O O Ph O BnOOMe FG 94 3:1
mperature, and promoter system (Table 6).70 According to Ye’s study, the stereo- chemistry outcomes of glycosylations are strongly influenced by the protecting groups in oxazolidinone protected glycosyl donors and the activation manner of donors; in addition, the properties of glycosyl acceptors also affect the stereoselectivity.
Table 6. The coupling of donor B and acceptor G (both in Table 5) under different conditions Entry Promoter system Solvent T (oC) Yield (%) Ratio (:) 1 BSM, TTBP, Tf2O CH2Cl2 73 88 only
2 BSP (or Ph2SO), Tf2O CH2Cl2 73 8587 only
3 N-(phenylthio)--caprolactam, Tf2O CH2Cl2 15 86 1:1.5a
4 N-(phenylthio)--caprolactam, Tf2O CH2Cl2 45 75 1:1a
5 N-(phenylthio)--caprolactam, Tf2O toluene 15 87 1:1a
6 NIS, AgOTf CH2Cl2 10 86 1:2.5a
a Anomeric ratio was determined by the integration of 1H NMR spectrum of the anomeric mixture.
1.3.3.3 N-Benzyl oxazolidinone
As mentioned before, although Kerns et al. made use of 2,3-N,O-carbamate protected glucosamine donor which gives high -selectivity in glycosylation, this glycosyl donor has several disadvantages: (1) side reactions include N-sulfenylation and N-glycosylation, and (2) requirement of excess amount of promoter. To avoid these disadvantages, Kerns and Oscarson both protected the oxazolidinone nitrogen with acetyl group, but the -selectivity depended on the activation conditions (e.g. lewis acid, reaction time, etc.) and the properties of donors and acceptors (e.g. reactivity, conformation, etc.). During this period, the N-substituent on the oxazolidinone ring except for acetyl group was not developed until Ito et al. reported the use of N-benzyl-2,3-trans-oxazolidinone novel glycosyl donors (E and F) for the 1,2-cis--glycosylation for 2-amino-2-deoxy sugars (Scheme 13).71 The direct synthesis of E and F starts with the one-step conversion of trichloroethyl carbamate protected GlcNAc derivative A to oxazolidine derivative B. Reductive benzylidene acetal ring opening under typical conditions using Et3SiHBF3·OEt2 gave C (72%)
O O O Ph HO NHTroc SPh O O O Ph O N SPh O Bn O BnO HO O N SPh O Bn + O BnO HO O N O BnSPh O BnO ClAcO O N SPh O Bn O O N SPh O Bn OBn AcO (a) (c) (d) A B C D E F (b)
Scmeme 13. Synthesis of N-benzyl oxazolidinone protected donors and acceptors
Reagents and cinditions: (a) BnBr, NaH, DMF, 96%. (b) Et3SiH, BF3·OEt2, CH2Cl2, C, 72%, D, 11%
(c) chloroacetic anhydride, pyridine, CH2Cl2, 99%. (d) (i) Tf2O (2 equiv), pyridine (4 equiv), CH2Cl2,
-40 to -20 oC; (ii) NaOAc, DMF; 73% (2 steps).
and D (11%) with a free hydroxyl group at the 4-position.72 The anomerization result was similar to previous reports proposed from Crich and Oscarson. Then C was converted to glucosamine donor E and galactosamine donor F. After a series of experiments, they selected PhSOTf/DTBMP promoting system in a solvent system of toluene/1,4-dioxane (3:1) as their optimized condition.73 Then they used E and F in a one-pot operation to synthesize a trisaccharide which is a component of the immune system stimulating O-specific polysaccharide from Proteus mirabilis O48 (Scheme 14). The trisaccharide was obtained with complete -selectivity, although Kerns reported the difficulty of activation of the disaccharide having 2,3-trans- oxazolidinone ring. The complete activation repated by Ito may be attributed to the protection of the N atom on oxazolidinone ring avoiding side reactions such as N-sulfenylation and N-glycosylation.
O ClAcO O N SPh O Bn O O N SPh O Bn OBn AcO E F OBn O ClAcO O N O Bn OBn Br O O N SPh O Bn OBn HO O ClAcO O N O Bn OBn O O N SPh O Bn OBn O O AcO O NPhth OMP OBn O ClAcO O N O Bn OBn O O N O Bn OBn O O AcO NPhth OMP OBn HO (a) (b) (c) (d)
Scmeme 14. One-pot synthesis of trisaccharides via oxazolidinone glycosides
Reagents and conditions: (a) Br2, CH2Cl2, 91%. (b) NaOMe, MeOH/1,4-dioxane, 99% (c) AgOTf, MS
4A, di-tert-butylmethylpyridine, toluene/1,4-dioxane (3:1). (d) AgOTf, PhSCl, 81% (2 steps)
Based on this successful result, the same group applied the N-benzyl-2,3-trans- oxazolidinone to the synthesis of anti-Helicobacter pylori oligosaccharides.74,75 The hexasaccharide C was synthesized after a series of stereoselective glycosylations and deprotections (Scheme 15). This is the first total synthesis of an anti-Helicobacter pylori oligosaccharide which is completed with high overall efficacy by employing a N-benzyl-2,3-trans-oxazolidinone donor.
O ClAcO O N O Bn OBn O AcO OAc OBn O O O OBn PhthN BnO O O ClAcO O N O Bn OBn O AcO OAc OBn O O N3 O BnO OMe O HO O N O Bn OBn O HO OH OBn O O O OBn H2N BnO O O HO O N O Bn OBn O HO OH OBn O O N3 O BnO OMe O HO HO AcHN OH O HO OH OH O O O OH AcHN HO O O HO HO AcHN OH O HO OH OH O O AcHN O HO OMe (a) (b)-(d) A C B
Scmeme 15. The deprotection sequence of an anti-Helicobacter pyroli oligosaccharides
Reagents and conditions: (a) N, N’-ethylenediamine, BuOH. (b) 1M NaOH, dioxane. (c) H2, 20%
1.4 Motivation
Based on the literatures described before, we are attracted by the anomeric activity and its related stereoselectivity of N-benzyl oxazolidinone protected thioglycoside because these GlcNAc building blocks are relatively easy to prepare. Meanwhile, Kerns’ study for chemoselective manipulation of N-acetyl oxazolidinone derivatives inspires us to consider the synthesis of novel donors via N-benzyl oxazolidinones for 1,2-trans--glycosylations (Figure 5). We herein report the investigations and findings based on our observations mentioned above.
2 Results and discussion
2.1 Anomerization phenomenon of oxazolidinone protected thioglycoside 4
The amino protecting group that routinely used in our laboratory is the N-2,2,2-trichloroethoxycarbonyl group (N-Troc), and the transformation from the N-Troc to the N-benzyl oxazolidinone protecting group only needs one step. For an easier preparation, we seleted the N-benzyl oxazolidinone instead of the N-acetyl one. To investigate the chemical properties of the N-benzyl oxazolidinone derivative, the oxazolidinone glucosamine donor 5 was first prepared (Scheme 16).
O HO OH HO NTroc STol O O O Ph HO NTroc STol O O O Ph O NBn STol O O HOO NBn STol O OBn O AcOO NBn STol O OBn 1 2 4 5 3 (a) (b) (c) (d)
Scmeme 16. Synthesis of N-benzyl oxazolidinone protected glucosamine donor 5
Reagents and conditions: (a) C6H5CH(OMe)2, cat. TsOH, CH3CN, rt, 92%. (b) BnBr, NaH, DMF, 0
.
oCrt, 80%. (c) Et
3SiH, CF3CO2H, 4A MS, CH2Cl2, -20 oC, 80%. (d) Ac2O, pyridine, rt, 95%
The N-Troc-protected thioglycoside 1 was converted to 4,6-O-benzylidene-2-N- benzyl-2,3-N,O-carbonyl-2-deoxy thioglucopyranoside 3 via benzylidene acetal intermediate 2.70,76 Reductive opening of the benzylidene acetal 3 afforded 4-hydroxy thioglycoside 4, which was further acetylated to furnish the oxazolidinone glucosamine donor 5.77 In the selective opening of the benzylidene ring step, Crich reported that the → anomerization was observed when they reductively opened the benzylidene acetal using sodium cyanoborohydridehydrogen chloride
(NaBH3CNHCl)59,60 and Ito also mentioned this phenomenon using triethylsilane boron trifluoride etherate (Et3SiHBF3·OEt2)72. Both of them proposed the / mixture was from the → anomerization through endo-cleavage of C1O5 bond under acidic conditions, while physical evidence for this phenomenon was given. This work was completed by Ito, who trapped the intermediate in the endo-cleavage process.78 They obtained the reductive product of the pyranosides having 2,3-trans- carbamate and –carbonate during the reductive opening of the benzyldene ring and proposed the mechanism based on quantum mechanical computations and other related experimental data (Scheme 17). They found that acids, temperature, reaction time, solvents, and the conformation of fused ring of pyranosides affect the extent of → anomerization.78-80 O O X O PO YR endo-cleavage O O X O PO YR O O X O PO YR O O X O PO YR cyclization rotation reduction OH O X O PO YR X = N-R' or O Y = S or O
Scmeme 17. Proposed mechanism for the anomerization of cyclic carbonate or -carbamate pyranosides
temperature and addition of molecular sieves, the formation of -anomer was ultimately eliminated when we used triethylsilanetrifluoroacetic acid (TES/TFA) combined with low reaction temperature (Table 7).77
Table 7. Optimizations of the reductive benzylidene ring opening of thioglycoside 3
O O O Ph O NBn STol O Et3SiH, Acid, 4A MS, CH2Cl2, T oC, yield% O HOO NBn O OBn R1 3 4: R 1 = STol, R2 = H 6: R1 = H, R2 = STol R2
Entry Acid (equiv) Et3SiH (equiv) T Yield (%)a Ratio (:)
1 TFA (6) 5 25 35 1:1
2 TFA (6) 5 0 57 1:10
3 TFA (6) 5 20 80 only
4 BF3·OEt2(2) 12 20 65 1:6b
a Total yield of 4 and 6 after chromatography purification. b The method was referred to ref 71.
To our delight, exclusive oxazolidinone protected -thioglucopyranoside 4 was formed in high (80%) yield at 20 oC (Table 7, entry 3). The -anomeric configuration of 4 was supported by the 13C chemical shift at 86.7 ppm and 1JCH coupling constant of 161 Hz.81 Nevertheless, the anomerization of -anomer 4 to -anomer 6 and trace amount of complete deacetalation product were observed at higher temperatures (Table 7, entries 1 and 2). Interestingly, the use of literature procedure resulted in a 1:6 /-amomeric mixture at 20 oC, which was consistent with literature finding (Table 7, entry 4).71 We speculate that the weak coordinateon affinity of TFA with ring oxygen atom at low reaction temperature compared with BF3·OEt2 may be the main reason for the reduction of anomerization.
2.2 Glycosylation studies of the thioglycoside donor 5
After the preparation of oxazolidinone thioglycoside 5, we focused on the electrophilic activation of this donor and tried to use promoter systems which have not been reported. The activation of this donor was investigated using primary alcohol
7 as the acceptor, and the result was shown in Table 8. The thioglycoside 5 remained
intact upon treatment with the N-iodosuccinimide/trimethylsilyl trifluoromethane- sulfonate combination (NIS/TMSOTf) at temperature range between 78 to 20 oC (Table 8, entry 1),82 dimethyl(thiomethyl) sulfonium trifluoromethane sulfonate/ tetrabutyl ammonium bromide combination (DMTST/Bu4NBr) at the temperature range of 78 to 0 oC (Table 8, entry 3)83, N-iodosuccinimide/silver trifluoromethane- sulfonate combination (NIS/AgOTf) at temperature range between 78 to 0 oC (Table 8, entry 5), benzenesulfinyl piperidine/triflic anhydride/2,6-di-tert-butyl-4-methyl pyridine combination (BSP/Tf2O/DTBMP) in CH2Cl2 at temperature range between 78 to 20 oC (Table 8, entry 7),50 and BSP/Tf
2O/DTBMP in 1,4-dioxane/toluene (1:3) solvent mixtures at the temperature range of 78 to 25 oC (Table 8, entry 8).50,73 When the temperature was raised, the thioglycoside 5 was activated but no glycosylation product was obtained (Table 8, entries 2, 4, and 6). From Table 8, we found that the activation temperature of the thioglycoside 5 was between 10 oC to room temperature among the promoter systems we selected.
Table 8. Glycosylation studies of N-benzyl oxazolidinone protected donor 5 and acceptor 7 O AcO OBn O STol NBn O + 5 7 Promoters solvent, T Results HO(CH2)6Cl
Entry Promoters (equiv) Solvent T (oC) Results
1 NIS (1.1), TMSOTf (0.3) CH2Cl2 78 to 20 no activation of 5
2 NIS (1.1), TMSOTf (0.3) CH2Cl2 10 decomposition of 5
3 Bu4NBr (3), DMTST (3) CH2Cl2 78 to no activation of 5
4 Bu4NBr (3), DMTST (3) CH2Cl2 decomposition of 5
5 NIS (1.5), AgOTf (0.3) CH2Cl2 78 to no activation of 5
6 NIS (1.5), AgOTf (0.3) CH2Cl2 to decomposition of 5
7 BSP (1.3), TfDTBMP (2.6) 2O (1.3), CH2Cl2 78 to 20 no activation of 5
2.3 Reactivity based chemoselective glycosylations of thioglycoside 4
The reactivity of the thiotolyl leaving group in oxazolidinone protected donor 5 was poor in NIS/TMSOTf promoter system. Perhaps, we could use the oxazolidinone protected thioglycosides as glycosyl acceptors in reactivity-based chemoselective glycosylations.46,84 To test our hypothesis, a series of glycosylations were studied using the NIS/TMSOTf promoter system and the results were shown in Table 9. In first instance, thiogalactopyranoside 8 85 was selected as a donor to glycosylate with the thioglycoside acceptor 4. As we expected, the Gal--(1→4)-GlcNAc disaccharide
13 was obtained in acceptable yield and complete -selectivity was observed (Table 9, entry 1). This result encouraged us to further investigate the glycosylation with thiogalactopyranoside with an ester groups at C-2 position. Glycosylations of 911 84 with 4 produced Gal--(1→4)-GlcNAc (type 2 LacNAc) disaccharides 1416 with moderate to high (5080%) yields (Table 9, entries 24). When the C-2 position of the thiogalactopyranoside was protected by the benzoyl group, the yield was high in comparison with the acetyl group which may induce side reactions such as acyl migrations to lower the yield of glycosylation. Intriguingly, the glycosylation of thiofucopyranoside 12 86 with 4 furnished Fuc--(1→4)-GlcNAC disaccharide 17, but the thiotolyl leaving group in the disaccharide underwent → anomerization forming an inseparable 1:3.5 /-anomeric mixture at temperature of -60 oC (Table 9, entry 5). This anomerization could be explained by C1O5 endocyclic bond cleavage as described before. Due to the deactivation of the oxazolidinone, self-condensation of 4 was not observed under the present reaction conditions. The results implicates that the reactivity-based chemoselective glycosylation is feasible and oxazolidinone GlcNAc acceptor can be used as an acceptor with low donor reactivity.
Table 9. Glycosylation studies of N-benzyl oxazolidinone acceptor 4
thioglycoside donor glucosamine acceptor disaccharide +
8, 9, 10, 11, or 12 4 13, 14, 15,16, or 17 NIS, cat. TMSOTf, 4A MS
CH2Cl2, T
Entry Thioglycoside donor
T
(oC) Disaccharide product Yield (%)
1 O BnO OLev BnO OBnSTol 8 70 O O OBn O NBn STol O O BnO OLev BnO BnO 13 70 2 O BnO OBn BnO OAcSTol 9 65 O BnO OBn BnO OAc O O OBn O NBn STol O 14 50 3 O BnO OBn BnO OLevSTol 10 65 O BnO OBn BnO OLev O O OBn O NBn STol O 15 65 4 O BnO OBn BnO OBzSTol 11 70 O BnO OBn BnO OBz O O OBn O NBn STol O 16 80 5 12 O STol
BnOOBn OBn 60
O BnOOBn OBn O O OBn O NBn STol O 17 (: = 1:3.5) 85
2.4 Retrosynthetic analysis of tetrasaccharide Lewis Y
On a close look at the structure of Lewis Y, empirically it could be synthesized from disaccharide 16 (Table 9, entry 4) which had the backbone structure of Gal--(1 →4)-GlcNAc. If we can further elaborate disaccharide 16, the synthetic route would be attainable to get tetrasaccharide which is part of Lewis Y oligosaccharide (Scheme 18). After removal of the oxazolidinone in 16, the exposed secondary amine should be protected by a suitable protecting group to avoid N-glycosylation. This prompted us to design a desymmetry tertiary amino protecting function as illustrated by disaccharide
18. O HO OH HO O O OH O NHAc OR O O HOOH OH O OH OH OH O P'gO OP'g P'gO O O OP'g HO NP''gP'''g LG OH O P'gOOP'g OP'g LG O BnO OBn BnO O O OBn HO N(P''g)Bn STol OH O BnOOBn OBn STol O BnOOBn OBn STol O BnO OBn BnO OBz O O OBn O NBn STol O 16 12 + + + + + + 18 HOR HOMe HOMe synthetic target 12
2.5 Removal of oxazolidinone ring in monosaccharide
However, the reaction conditions for the removal of oxazolidinone ring should be carefully studied prior to the design of the tertiary amine. To start our study, glucosamine derivative 1 was transformed to oxazolidinone glucosamine 19 with only one step using the known literature procedure.70 After preparation of thioglycoside 19, it was used as a model and subjected to a range of reaction conditions to the suitable method for the removal of oxazolidinone rings (Table 10). Substrate 19 was remained when it was treating with p-toluenesulfonic acid (PTSA) in methanol,87 and obtained in low yield with methyl magnesium chloride (CH3MgCl) in THF (Table 10, entries 1 and 2).88 In the event, the literature procedures were the suitable methods for the removal of the oxazolidinone ring. Substrate 19 under basic condition (1,4-dioxane/
Table 10. Removal studies of the N-benzyl oxazolidinone ring in monosaccharide 19
O BnO OBn O NBn STol O O BnO OBn HO NHBn STol Reagents solvent, T, t 19 20
Entry Reagents (equiv) Solvent T (oC) t Yield (%)
1 CH MgCl (10) 3 THF 25 10 min 42
2 PTSA ( ) MeOH 10 12h no reaction
3 NaOHa 1,4-dioxane 25 12h no reaction
4 NaOHa 1,4-dioxane 60 12h 73
DMSO 25 30 min 75
1
a
1M NaOH, v/v = 1:1) was deprotecd at 60 C, but is was intact at room temperature (Table 10, entries 3 and 4). Also, substrate 19 was deprotected under basic condition (t-BuOK/DMSO) at room temperature (Table 10, entry 5). We finally selected the t-BuOK/DMSO basic condition (Table 10, entry 5) to remove the oxazolidinone ring because of the highest deprotection yield and shorter reaction time.
2.6 Synthesis and characterization of desymmetric aminoprotecting groups
With glucosamine derivative 20 in hand, our attention was focused on the selection of a protecting group which can induce 1,2-trans--glycosylation and also can be removed under mild conditions. At first, acetyl group was our choice on account of the natural N-acetyl function, and it would simplify the subsequent deprotection. Although a number of conditions we had tried, the yield of glucosamine derivative 21 was not improved (Scheme 19). Hence, we changed to use tert-butoxyc-
1 M aqueous NaOH solution
o 71 70 O BnO OBn HO NHBn STol BnO O OBn HO N(Boc)Bn STol O BnO OBn HO N(Ac)Bn STol O BnO OBn HO N(Cbz)Bn STol 20 25 22 21 O BnO OBn HO N(R1)Bn OR2 26, R1 = Cbz R2 = (CH2)6Cl 27, R1 = H R2 = (CH2)6Cl O BnO OBn RO BnN O O 23, R = H 24, R = Ac (a) (b) (e) (c) (f) (g) (d)
Scmeme 19. Investigations of desymmetric glucosamine acceptors
Reagents and conditions: (a) Ac2O, NaHCO3, MeOH, rt, 30%. (b) Boc2O, NaHCO3, MeOH, rt, 76%.
(c) HO(CH2)6Cl 7, NIS, cat. TMSOTf, 4A MS, -40 oC; (d) Ac2O, pyr., rt; 50% (2 steps). (e) CbzCl,
Et3SiH, Et3N, CH2Cl2, rt, 45%.
89
90
89
arbonyl group for its versatile deprotection methods. Treatment of 20 with NaHCO3 in MeOH, followed by di-tert-butyl dicarbonate gave desymmetric N-benzyl-N-tert- butoxy protected glucosamine thioglycoside 22 in 76% yield. Unfortunately, subsequent glycosylation of 22 with acceptor 7 was characterized by the cleavage of the N-substituent, and the main compound recovered was the amine 23. The amine 23 was further acylated to 24 for characterizations. After searching literatures, we found that the experimental result was similar with Boullanger’s work, and the formation of
23 could be due to removal of one of the carbamate protons from the intermediate A
(Scheme 20) to form isobutylene together with 23. Boullanger et al. conducted a study on the glycosylation of simple acceptor alcohols with various N-alkoxycarbonyl derivatives of glucosamine. Their investigation inspired us to adopt the benzyloxycarbonyl group to protect the secondary amine in glucosamine derivative 20. Again, treatment of 20 with NaHCO3 in MeOH, followed by benzyloxycarbonyl chloroformate gave desymmetric N-benzyl-N-benzyloxycarbonyl (N-BnCbz) protected glucosamine thioglycoside 25 in high yield of 82%. Subsequent glycosylation of 25 with primary alcohol 7 gave the -glycoside 26 in 88% yield (Scheme 19). Although the yields of 25 and 26 were satisfactory and their preliminary
O BnO HO OBn N Bn O O S O BnO HO OBn N Bn O O Nu H O BnO HO OBn N Bn O O intermediate A S Tol 23
identifications were evidenced by HRMS, the assignments of NMR spectra were difficult due to the peak broadening of the resonance signals. After searching literatures, we found that the peak broadening phenomenon was quite similar with Lafont’s research.91 The 1H NMR spectrum of N-acetyl-N-allyloxycarbonyl protected glucosamine derivative A (Figure 6) revealed the presence of two conformers (45:55). They pointed that this observation could be due to the restricted rotations around the amide bond, and the coalescence of signals appeared when the temperature was increased. Compared to our study, the 1H NMR spectrum was unclear at room temperature. When the temperature was raised, the broadened peaks became sharp at 100 oC as shown in Figure 7 (ca VT-NMR from 30 to 100 oC in deuterated DMSO solvent). This phenomenon could also be attributed to the restricted rotations around the amide bond based on our VT-NMR spectra and Lafont’s research. Although we could get clear spectrum at elevated temperature of 100 oC, it was unpractical since the operation of NMR machine at such elevated temperature were quite time-spending and laborious. We predicted that the broadening of resonance signal was resulted from the presence of the carbamate function. In order to prove our predictions, the Cbz protecting group was removed under mild conditions (Scheme 19).92 Consistent with our reasoning, the broadening phenomenon was disappeared, and the clear NMR spectrum of -glycoside 27 (data were shown in experimental section) could be obtained at room temperature.
O AcO OAc AcO N OAc Ac Alloc compound A
Figure 6. 1H NMR spectra of compound A at different temperatures
O BnO OBn HO N(Cbz)Bn OR 26, R = (CH2)6Cl
2.7 Glycosylation studies of the glucosamine acceptor 25
After characterizations of the desymmetric N-BnCbz protected glucosamine 25, the next step was glycosylation studies of the acceptor 26 with thioglycosides 11, 12,
28, and 29 (Table 11). Glycosylations of 26 with thioglactopyranosides 11, 28, and 29
furnished the expected Gal--(1→3)-GlcNAc (type 2 LacNAc) disaccharides 3032 in high (73-80%) yields (Table 11, entries 13). The coupling of thiofucopyranoside
Table 11. Glycosylation studies of the desymmetric glucosamine acceptor 26
thioglycoside donor glucosamine acceptor disaccharide +
11, 12, 28, or 29 26 30, 31, 32, or 33 NIS, cat. TMSOTf, 4A MS
CH2Cl2, T
Entry Thioglycoside donor T
(oC) Disaccharide product Yield (%)
1 O BnO OBn BnO OBzSTol 11 65 O BnO OBn O N(Cbz)BnOR O BnOBnO BnO OBz 30, R = (CH2)6Cl 80 2 O BnO OBn AllO OBzSTol 28 65 O BnO OBn O N(Cbz)BnOR O BnOBnO AllO OBz 31, R = (CH2)6Cl 73 3 O BnO OBn PMBO OBzSTol 29 65 O BnO OBn O N(Cbz)BnOR O BnO BnO PMBO OBz 32, R = (CH2)6Cl 70 4 12 O STol BnOOBn OBn 70 O BnO OBn O N(Cbz)BnOR 33, R = (CH2)6Cl O BnOOBn OBn 93
12 and acceptor 26 led to the Fuc--(1→3)-GlcNAc disaccharide 33 which is the backbone of type 1 Lewis antigens in high yield of 93% (Table 11, entry 4). All the glycosylations proceeded smoothly and the yields were satisfactory. The peak broadening was also occurred in the NMR spectroscopy of disaccharides 3033. We tried in vain to remove the Cbz protecting group in disaccharide 30 using the same condition which was feasible for monosaccharide 26 (Scheme 21). The reason is not clear at this stage.
PdCl2, Et3SiH, NEt3 CH2Cl2, rt, 45% O BnO OBn HO N(Cbz)Bn OR 26, R = (CH2)6Cl O BnO OBn HO NHBn OR 27, R = (CH2)6Cl O BnO OBn O N(Cbz)Bn OR O BnO OBn BnO OBz 30, R = (CH2)6Cl PdCl2, Et3SiH, NEt3 CH2Cl2, rt O BnO OBn O NHBn OR O BnO OBn BnO OBz R = (CH2)6Cl
2.8 Hydrogenolysis of oxazolidinone protected disaccharides
Hence, the Pd-catalyzed hydrogenolysis method may be another choice for the removal of Cbz protecting groups. At the same time, O-benzyl ethers and N-benzyl groups could be removed simultaneously during hydrogenolysis reaction. Consequently, the conditions for the total debenzylations in the presence of Cbz protecting groups were optimized. We used glucosamine derivative 26 as a model to study hydrogenolysis reactions and the results were shown in Table 12.
Table 12. Deprotection of desymmetric glucosamine acceptor 26
catalyst, H2 (1 atm) solvent, T, 12h O BnO OBn HO N(Cbz)Bn OR 26, R = (CH2)6Cl O AcO OAc AcO NHAc OR 34, R = (CH2)6Cl
Entry Catalyst Solvent T (oC) Results
1 Pd MeOH:AcOH = 10:1 25 26a
2 Pd MeOH:HCOOH = 10:1 25 not fully deprotecteda
3 Pd EtOH:HCOOH = 10:1 25 not fully deprotecteda
4 Pd THF:H2O = 4:1 25 26a
5 Pd THF:H2O:AcOH = 8:2:1 25 26a
6 Pd THF:H2O:HCOOH = 8:2:1 25 not fully deprotecteda
7 Pd(OH)2 AcOH 25 26a
8 Pd(OH)2 MeOH:AcOH = 2:1 25 26a
9 Pd(OH)2 EtOAc:H2O:AcOH = 2:1:4 25 26a
10 Pd(OH)2 EtOAc:H2O:AcOH = 2:1:4 60 34 (70%)b a Judged from TLC plates. b After acetylation, we got the low mass data.
At first, hydrogenolysis of monosaccharide 26 did notproceed over 10% Pd/C in solvent mixtures of MeOH/AcOH (10:1), THF/H2O (4:1), and THF/H2O/AcOH (8:2:1) at room temperature (Table 12, entries 1, 4 and 5).70 Incomplete deprotections were observed and a series of spots were noted on the TLC plate, and changing the solvent mixture to MeOH/HCOOH (10:1), EtOH/HCOOH (10:1), and THF/H2O/HCOOH (8:2:1) did not give the desired product either. After these failures, we took a step back to do a search hoping for finding the solutions to overcome this problem. We found that 20 % Pd(OH)2 catalyst was used for the removal of N-benzyl groups, maybe it would fulfill all our requirements for total debenzylations along with the removal of Cbz protecting group. Hydrogenolysis reactions of monosaccharide 26 were not successful even over 20 % Pd(OH)2 in solvent system of AcOH, MeOH/AcOH (10:1), EtOAc/H2O/AcOH (2:1:4) at room temperature (Table 12, entries 79).93,94 Eventually, the hydrogenolysis temperature was best performed at 60 oC.74 After raising the temperature to 60 oC, the deprotection was achieved (Table 12,
entry 10). To our delight, we applied the optimized condition to disaccharides 30 and
33. As shown in Scheme 22, both of their hydrogenolysis reactions worked well in
solvent mixture of EtOAc/H2O/AcOH (the ratio was based on the solubility of disaccharide) over 20% Pd(OH)2 under 1 atm H2 at 60 oC. For NMR characterization, the resulting debenzylated products were further acetylated to produce the type 1 LacNAc glycoside 35 and the peracetyl Fuc--(1→3)-GlcNAc disaccharide 36 which was similar to part of Lewis Y structure.