含烯基及硫化合物電解液添加劑對於鋰離子電池石墨材料表面特性研究
張家欽1、賴志昀2、鍾怡芳2、詹勗忠3 1. 國立台南大學 環境與能源學系 2. 國立台南大學 材料科學系 3. 國立台南大學 綠色能源科技研究所 E-mail : [email protected]摘 要
本研究以碳酸亞乙烯酯(VC)和三硫代碳酸亞乙烯酯(VTC)作為電解液添加劑進行鋰離子電池測試。 並利用循環伏安法、半電池測試、電交流阻抗。由循環伏安發現隨著漸增至 3%濃度的電解液添加劑,VC 和 VTC 充分在石墨層上發展形成一層 SEI 膜。並以掃描式電子顯微鏡(SEM)用來觀察添加劑影響所形成 的 SEI 膜。透過半電池測試了解電解液添加劑對於電池效能所帶來影響。結果顯示,循環壽命性能在 3%濃 度時 VC 電解液添加劑優於 VTC 電解液添加劑。因此我們可以推測 VC 和 VTC 充放電的過程中,在石墨上 所形成的 SEI 膜對於循環壽命與參與反應的能力有所影響。1. 前 言
在商用鋰離子電池中,鋰離子在充電過程嵌入石墨層,放電時嵌出[1,2]。在鋰離子的遷移過程中,鋰 離子的導電率、遷移數量、電解質的穩定性等,絕大部分都取決於正極材料與電解液間的 SEI 膜。在正極 表面的 SEI 膜的性能與結構已經被廣泛地研究與討論[3-6]。這些反應基本上是不可逆的,然而,不可逆的 性能取決於電解液的種類[包括溶劑、添加劑和鹽類]。烷基碳酸鹽,例如:碳酸丙烯酯[PC],碳酸乙烯酯[EC], 碳酸二乙酯[DEC]和碳酸二甲酯[DMC], 為鋰離子電池最重要的電解液,因為它有具極性質子惰性和不易 揮發的特性。基底電解液 PC 會使石墨層剝落而且大幅分解游離釋放出有機的氣體在鋰-離子的電池當中。 [6] 碳材料已經被發現[7-10]具有好的循環特性並且在碳表面上形成好的 SEI 膜使得在 EC/DEC 電解液 中維持高潛能。Zaghib 等人[9] 發現形成 SEI 膜是不只是因為溶劑的分解而且在 EC/DEC 系統中也被鹽 的性質影響了。大體上,有還原能力的分子和 PF陰離子在 Li-石墨的負極上還原形成組成 ROCO2Li、
ROLi 和 Li2CO3 的表面薄膜。[11] 因此,在電解液添加劑上的研究主要專注於界面 graphitic 負極,電解 液添加劑參與 SEI 膜的形成程序[12],在 1990 年代中由 CO2 開始,許多電解液添加劑種類已被利用,像 是 N2O [13],Sx2- [13], SO2 [14,15], chloroethylene 碳酸鹽[16],乙烯基丙烯,vinylene 碳酸鹽 (VC) [6,18,19],乙烯基醋酸鹽[6],和乙烯亞硫酸鹽[6,21] 已經被評估。 實驗中我們計畫在 graphitic 材料中,將透過改良 SEI 的形成近一步改善鋰離子電池的性能表現。鋰 離子電池電解液添加劑相關文獻指出電池循環壽命及可逆電容量增加之添加劑, 一般將會改變 SEI 膜結 構, 如 venylene carbonate (VC)及 venylene trithiocarbonate (VTC)。此類添加劑結構中含乙烯基,其主要功 能是在充電過程中 Li+移往碳材表面行還原反應,並且與電解液中之 EC 在碳材表面反應產生 SEI 膜,乙烯 基於碳材表面受還原反應引發產生電聚合行為,參與 SEI 膜的形成反應;含氧或含硫官能基主要功能則在 與 Li+作遷移、電子供給、將 Li+還原成 Li 原子並遷移至石墨材料結構內部。目前之電解液添加劑研究均集 中於含氧官能基之探討較多 ,對於含硫官能基之探討較少。 本研究的目的為在電解液中添加入 VTC 或 VC,藉由乙烯基與 SEI 之形成反應特性,將與含硫或含氧 官能基之化合物修飾於碳材表面,並利用含硫或含氧官能基之陰電特性提高 Li+之吸附與轉移行為,預期將
改善 SEI 膜特性,利用循環伏安法與交流阻抗測試等電化學方法可以得到不同含乙烯基(Vinyl group)及 含硫官能基之化合物添加量對於負極碳材表面之薄膜特性作探討,並與目前已確定之含乙烯基(Vinylgroup) 及含氧官能基之 VC 化合物之行為作比對。
2. 實驗內容
本研究以 VC、VTC 作為電解液添加劑,添加於 EC 與 PC 兩種電解液中,將各種的電解液添加劑進行 基本材料測試,並進行硬幣型電池充放電測試、循環壽命測試。由結果顯示 VC、VTC 添加劑在適當濃度 下,的確有助於改善鋰電池鈍化膜的形成。 圖 1、SEI 膜的反應形成結構3. 結果與討論
3.1 半電池性能測試 實驗中為了要了解電解液添加劑對於循環性能之貢獻,我們分別添加 3% 重量比的 VC 和 VTC 在 PC/DEC (1:2,w/w)以及 EC/DEC (1:2,w/w)之中 ,且製作成硬幣型的電池進行測試。同時也 針對未含添加劑的電解液進行硬幣電池的測試。測試的結果在圖 2 中展示。 在 NG 的負極材料中, VC 添加劑效果超過 VTC ,添加劑參與反應的效果,使得 PC 電解液並未產生共崁入的現象,天然石墨層並未 剝落。由於負極 SEI 膜形成良好,沒有添加劑的 EC 還原在碳表面[3-6] 上也展現出良好的循環性能表現。 我們比較半電池性能測試中,以 0.1C 作為充放電電流時,充電與放電之間的比值,其數值整理在表一 之中,由表一我們可觀察出,使用 VC 作為電解液添加劑擁有最低的不可逆電容量。在電池活化期間。不 可逆的電容量主要是因為在石墨電極表面形成固液界面膜 (SEI)所引起的。 在這半電池性能測試中,在 3% VC-EC/DEC 不可逆電容量最低,,其次為 3% VC-PC/DEC。基於這些現象,我們能假設 VC 的 SEI 膜的形成較 VTC 少量。 依照上述的資料,它指出,在以 EC 和 PC 電解液為基礎 的 3 wt % VC 添 加劑 為鋰離子半電池提高充放電的效率。表.1 以 DC/DEC (1:2 by w/w)加入 1 mol dm-3
LiPF6或 PC/DEC (1:2 by w/w) 1 mol dm-3
LiPF6為電解液,添加 VC、VTC 或無添加劑的可逆電容量比。 First charge capacity Reversible capacity ratio% EC-BLANK 351.5 mAh/g 87.2% EC-VC- 3% 332.4 mAh/g 89.2% EC-VTC- 3% 67.8 mAh/g 41.4% PC-VC- 3% 318.7 mAh/g 88.1% PC-VTC- 3% 242.8mAh/g 72.7% --●--PC-VC --▲--PC-VTC --■--EC-VTC --■--EC-VC --●--EC-BLANK -- X --PC-BLANK 0 100 200 300 400 0 10 20 30 40 50 Cycle number Capacity (mA h /g ) 圖 2. 以 DC/DEC (1:2 by w/w)加入 1 mol dm-3
LiPF6或 PC/DEC (1:2 by w/w) 1 mol dm-3
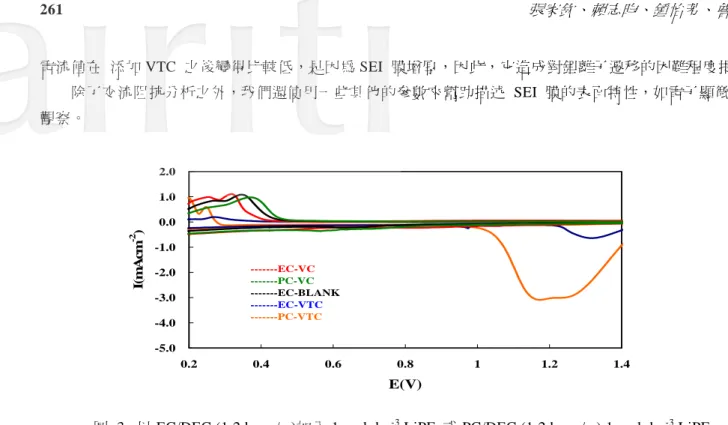
LiPF6為電解液,添加 VC、VTC 或無添加劑的循環壽命特性。 3.2 電化學測試 依照上述的半電池結果,我們能推論 VC 和 VTC 電解液添加劑參與 SEI 膜的形成。 為了要確定這 一項假定,我們使用循環伏安法來確認其反應。在 1 mol dm-3 LiPF6 的石墨電極的循環伏安法中以 EC/ DEC 或 PC/DEC(1:2 w/w)並分別添加 VC 或 VTC 添加劑. 對於圖 3 的所有圖示中,大約在 0.5 ~0.6 V,有一個明顯的氧化波峰,推測此為鋰離子嵌入碳層時與電解液反應之結果,此外,我們能在 0.75 V 和 1.25V 觀察另外兩個氧化波峰分別為 VC 和 VTC。此為添加劑參與 SEI 膜反應之現象。在 VTC 的樣品中, 波峰電位較高,因此 VTC 參與 SEI 膜的反應最為優先,而形成的 SEI 膜將是影響鋰離子進出阻抗的重要 因素之一,我們可透過 AC 交流阻抗分析來審視鋰離子在穿越 SEI 膜時的阻抗大小。 因此在循環伏安法 掃描之後,我們立刻在極片上進行了交流阻抗分析。 其阻抗頻譜我們整理在圖 4 。添加 VC 的負極極片 顯示擁有較低的阻抗值。 低阻抗 SEI 膜的形成不僅改善電池循環性能表現,仍可提升電池的充放電效率, 因為 VC 減少了電解液還原的可能。VTC 的阻抗值較高,這可能成為是循環測試時電容量快速衰退的因素。 我們推估 SEI 膜形成量愈少,石墨層能被用來儲藏鋰的效用也愈增強。 然而,圖 3 中,它能在還原波峰
電流值在 添加 VTC 之後變得比較低,是因為 SEI 膜增厚,因此,它造成對鋰離子遷移的困難程度提高。 除了交流阻抗分析之外,我們還使用一些其他的參數來幫助描述 SEI 膜的表面特性,如電子顯微鏡的 觀察。 -5.0 -4.0 -3.0 -2.0 -1.0 0.0 1.0 2.0 0.2 0.4 0.6 0.8 1 1.2 1.4 E(V) I( m A cm -2 ) ---EC-VC ---PC-VC ---EC-BLANK ---EC-VTC ---PC-VTC 圖 3. 以 EC/DEC (1:2 by w/w)加入 1 mol dm-3
LiPF6或 PC/DEC (1:2 by w/w) 1 mol dm-3 LiPF6
為電解液,添加 VC、VTC 或無添加劑的第一圈循環伏安特性﹙0.1 mV s-1﹚。 0 50 100 150 200 250 0 200 400 600 800 1000 Z'/ohm Z ''/o h m ---EC-VTC ---EC-VC 圖 4. 交流阻抗分析 在 1 mol dm-3 LiPF 6 EC/DEC 電解液(1:2 by w/w) 中加入 VC 或 VTC 電解液添加劑並進行測試,且控制電壓固定在 0.03V 圖 4. 石墨電極的 5000 倍 SEM 影像:原始石墨。
圖 5. 石墨電極的 5000 倍 SEM 顯微像:(a)PC/DEC + 3% VC;(b)PC/DEC + 3% VTC; (c)EC/DEC + 3% VC;(d)EC/DEC + 3% VTC。每張照片中的比例尺為 5μm
3.3 表面結構
藉由掃描電子顯微鏡使用 (SEM)觀察極片表面的型態學,我們可進一步了解 SEI 膜的表面型態,圖 4 所顯示的是 EC 電解液未添加任何添加劑的樣品,如圖中 SEM 所顯示的,當使用包含添加劑的電解液時, 所有的負極極片上完全地佈滿 SEI 膜。 圖 5(a)- (d)成功拍攝出天然石墨上所形成的 SEI 膜表面。 相當有趣地,由形態學的觀察,在 添加 VC 的樣品中,圖 5(a)為以 PC 為基底的電解液樣品, 圖 5(c) 為以 EC 為基底的電解液樣品,兩者的 SEI 膜非常相似. 這顯示在充放電活化的過程中,在天然石墨表面所 形成的 SEI 膜,不管是以 PC 為基礎的電解液 或者 以 EC 為主的電解液, VC 電解液添加劑參與 SEI 膜 反應後,其形態學一致且對於電池的性能展現有相當的助益。 圖 5(b)、 (d)是以 VTC 作為電解液添加 劑 拍攝在天然石墨上所形成的 SEI 膜形態。 很明顯的是,天然石墨表面徹底地佈滿果肉狀膜層,不同於 圖 5(a)、(c)添加 VC 電解液添加劑的形態學,在天然石墨表面所形成的 SEI 膜,不管是以 PC 為基礎的 電解液 或者 以 EC 為主的電解液, VTC 電解液添加劑參與 SEI 膜反應後,形成一層較厚的 SEI 膜。一 層緻密的 SEI 膜能良好的阻隔石墨層與電解液發生反應。然而,SEI 膜過厚也容易惡化鋰離子電池的性能表 現,因為它可能妨礙鋰離子的進出。 舉例來說,在天然石墨表面,因為 VTC 還原後所形成的果肉狀 SEI 膜與其他的添加劑比較後是最厚的,也影響了鋰離子傳遞的能力,如果比較圖 4 中 SEI 膜的阻抗值,我們 發現,添加 VTC 添加劑的阻抗值較高,表示鋰離子的傳遞能力受到 VTC 所形成的 SEI 膜影響。而另外 一部分, EC 和 VC 所形成的 SEI 膜展現較低阻抗和較薄的厚度,結果包含這些有機溶劑的硬幣電池展 現出非常好的循環性能。因此, SEI 膜的厚度對碳負極的循環性能表現佔有一定的比例。 此外, SEI 膜 的成份也可以在電化學反應中擔任一個非常重要的角色。
4. 結 論
藉由半電池測試、循環伏安法、電交流阻抗分析研究添加有 VC 或 VTC 的電解液在石墨陽極的影響。 電解液中的 VC 和 VTC 顯著地提高鋰離子電池的循環性能與效率周期表現和效率。從半電池測試的結果來 看,添加 3 wt % VC 的半電池有較高的可逆能力比。根據循環伏安測試,發現 SEI 膜的形成電位,有 VC 和 VTC 電解液添加劑比無添加有更高的還原電位。由 SEM 看的出來,在含 VC 和 VTC 添加劑電解液中 SEI 膜有較平滑且均勻的結構。 在負極的 SEI 膜中觀察得知 VC 和 VTC 添加劑能有效影響負極特性,像是循環性能,周期壽命和負 極的阻抗等相當重要之因素。添加劑在電極上的表現能清楚地歸因於他們對於石墨陽極的表面化學的影響。參考文獻
[1] M. Winter, P. Novak, A. Monnier, J. Electrochem. Soc. 145 (1998) 428. [2] T. Nakajima, J. Fluorine Chem. 105 (2000) 229.
[3] D. Aurbach, A. Zaban, Y. Ein-Eli, I. Weissman, O. Chusid, B. Markovsky, M. Levi, E. Levi, A. Schechter, E. Granot, J. Power Sources 68 (1997) 91.
[4] O. Chusid, Y. Ein-Eli, D. Aurbach, M. Babai, Y. Carmeli, J. Power Sources, 43/44 (1993) 47. [5] D. Aurbach, M. Levi, E. Levi, A. Schechter, J. Phys. Chem. B. 101 (1997) 2195.
[6] K. Abe, H. Yoshitake, T. Kitakura, T. Hattori, H. Wang, M. Yoshio, Electrochimica Acta 49 (2004) 4613–4622 [7] K. Tatsumi, K. Zaghib, H. Abe, S. Higuch, T. Ohsaki, Y. Sanade, J. Power Sources 54, (1995) 425.
[8] H. Abe, K. Zaghib, K.Tatsumi, S.Higuchi, J. Power Sources 54 (1995) 236.
[9] K. Zaghib, K. Tatsumi, H. Abe, T. Ohsaki, Y.Sawada, S. Higuchi, J. Power Sources 54 (1995) 435. [10] G. Li, R. Xue, L. Chen, Y. Huang, J. Power Sources 54 (1995) 271.
[11] E. Peled, D. Golodnitsky, G. Adriel, J. Electrochem. Soc. 144 (1997) 823.
[12] D. Aurbach, E. Zingigrad, Y. Cohen, H. Teller, Solid State Ionics 148 (2002) 405-416.
[13] J. O. Besenhard, M. W. Wagner, M. Winter, A. D. Jannakoudakis, P. D. Jannakoudakis, E. Theodoridou, J. Power Sources 43 – 44 (1993) 413.
[14] J. D. Besenhand, M. Winter, J. Yang, W. Biberacher, J. Power Sources 54 (1995) 228. [15] Y. Ein - Eli, S. R. Thomas, V. R. Koch, J. Electrochem. Soc. 142 (1997) 1159. [16] Z. X. Shu, R. S. McMillan, J. J. Murray, J. Electrochem. Soc. 142 (1995) L161.
[17] K. Abe, H. Yoshitake, T. Kitakura, T. Hattori, H. Wang, M. Yoshio, Electrochim. Acta 49 (2004) 4613. [18] G. H. Wrodnigg, J. O. Besenhard, M. Winter, J. Electrochem. Soc. 146 (1999) 470.
[19] H.-C. Wu, C.-Y. Su, D.-T. Shieh, M.-H. Yang, N.-L. Wu, Electrochemical and Solid-State Letters, 9 (2006) A537-A541. [20] D. Aurbach, J. S. Gnanaraj, W. Geissler, M. Schmidt, J. Electrochem. Soc. 151 (2004) A23-A30.
[20] T. Hamamoto, A. Hitaka, Y. Nakada, K. Abe, Patent US 6033809. [21] K. Tomishige, K. Kunimori, Appl. Catal. A: Gen. 237 (2002) 103.
[22] M. Yoshio, H. Wang, K. Fukuda, Y. Hara, Y. Adachi, J. Electrochem. Soc. 147 (2000) 1245. [23] D. Aurbach, M. L. Daroux, P. W. Faguy, E. Yeager, J. Electrochem. Soc. 134 (1987) 1611.