National Chiao Tung University PhD Thesis
國立交通大學博士學位論文
Characterization of PslB and PA3346, Two Bifunctional Enzymes in
Pseudomonas aeruginosa PAO1, and Analysis of the Galactose Stress on Gene
Expression of a Klebsiella pneumoniae CG43S3 ΔgalU Mutant
綠膿桿菌 PAO1 中兩個雙功能酵素 PslB 和 PA3346 功能特性之
研究與克雷白氏肺炎桿菌 CG43 galU 突變株在半乳糖壓力下其轉錄體之分析
生物科技學系
研究生:李蕙如
Student:Hui-Ju Lee
指導教授:張晃猷 博士
指導教授:
彭慧玲 博士
Advisor:Hwan-You Chang, Ph.D
Advisor:
Hwei-Ling Peng, Ph.D
中華民國一零三年一月
January, 2014
綠膿桿菌 PAO1 中兩個雙功能酵素 PslB 和 PA3346 功能特性之研究
與克雷白氏肺炎桿菌 CG43 galU 突變株在半乳糖壓力下其轉錄體之分析
Characterization of PslB and PA3346, Two Bifunctional Enzymes in
Pseudomonas aeruginosa PAO1, and Analysis of the Galactose Stress on
Gene Expression of a Klebsiella pneumoniae CG43S3 ΔgalU Mutant
研究生:李蕙如 Student:Hui-Ju Lee
指導教授:張晃猷 Advisor:Hwan-You Chang
指導教授:
彭慧玲
Advisor:
Hwei-Ling Peng
國立交通大學
生物科技學系
博士論文
National Chiao Tung University
Department of Biological Science and Technology
PhD Thesis
中華民國一零三年一月
January 2014
I
中文摘要
本論文共分為三個章節,第一章和第二章主要是探討綠膿桿菌 PAO1 中兩個雙功能 酵素 PslB 與 PA3346 之酵素特性,第三章則是分析克雷白氏肺炎桿菌 CG43 尿嘧啶雙磷 酸葡萄糖焦磷酸化酶(GalU)突變株在半乳糖存在下其基因轉錄之變化。 綠膿桿菌中 PslB 是具有磷酸甘露糖異構酶(PMI)與鳥苷雙磷酸甘露糖焦磷酸化酶 (GDP-man PPase)的雙功能酵素,分別參與鳥苷雙磷酸甘露糖(GDP-man)合成的第一步和 第三步催化反應。而目前為止,PslB 的酵素活性及參與催化反應的重要胺基酸位置尚未被報導。在第一章的研究裡,我們證實了 PslB 的確具有 PMI 與 GDP-man PPase 的酵
素功能。GDP-man PPase 酵素反應需要正二價鎂離子的幫助;PMI 酵素反應則是需要
正二價鈷離子,而且其活性會被 GDP-man 所抑制。另外,2, 3-丁二酮(2, 3-butanedione) 能將 PslB 的 PMI 活性去活化,顯示 PslB 上某個位置的精胺酸(Arg)可能在催化過程中 扮演重要的角色。我們進一步利用點突變的實驗技術,發現將 PslB 第 408 號位置的精 胺酸更改為賴胺酸(Lys)或者丙胺酸(Ala)後,其 PMI 的活性則是完全消失。經由圓二色 光譜的分析,野生株及定點突變株的 PslB 在蛋白質二級結構上未有改變。此結果證實 PslB Arg408 的確是參與催化反應的重要胺基酸,亦可做為未來了解 PMI 催化機制的初 步依據。 第二章主要研究綠膿桿菌另一雙功能酵素 PA3346。先前的研究發現,HptB 訊息傳
II
遞路徑在綠膿桿菌 PAO1 細菌群體移動與生物膜形成中扮演重要的調控角色。當細菌接
受到環境的刺激,感應激酶蛋白(PA1611, PA1976, PA2824)會自我磷酸化,其磷酸根再
經由 HptB 傳遞給下游的反應調控子 PA3346。PA3346 N 端磷酸化後,使得其絲胺酸蛋
白磷酸酶活性增加,並將 anti-sigma factor 拮抗子 PA3347 Ser56 上之磷酸根去除。然而,
能將 PA3347 Ser56 位置磷酸化的絲胺酸蛋白激酶仍然未知。經由蛋白質結構域分析,
我們發現 PA3346 C 端 (PA3346L408-A571
)的結構域與枯草桿菌的 SpoIIAB (絲胺酸蛋白激
酶/anti-sigma factor) 極為相似,因此推測 PA3346 L408-A571具有絲胺酸蛋白激酶的活性;
而且,PA3346-PA3347 可能與枯草桿菌 SpoIIE-SpoIIAB-SpoIIAA 的調控系統類似,形
成所謂的 partner-switching 調控模組。本論文建構 PA3346 L408-A571與 PA3347 重組蛋白質,
以進行試管內磷酸化反應。結果證實 PA3346L408-A571能將 PA3347 Ser56 位置磷酸化,其
絲胺酸蛋白激酶的活性需要 Mg2+
, Ca2+與 Mn2+等正二價金屬離子的幫助。在進行 GST
pull-down assay 與 GFP assembly assay 後,我們發現 PA3346L408-A571與 PA3347 蛋白質不
管是在試管內或是在細菌體內,都能夠互相結合。這些結果證實了 PA3346 L408-A571的確
具有絲胺酸蛋白激酶/anti-sigma factor 的功能。為了尋找參與 partner-switching 調控系統
的 sigma factor,我們亦進行了雙分子螢光互補實驗,而詳細的分子調控機制需要進一
步的探討研究。
本論文的第三章,主要對克雷白氏肺炎桿菌 galU 突變株進行研究。當使用 galU 突
III 甘油為主要碳源的 M9 培養液時,加入半乳糖會使得細菌生長緩慢並且有死亡的現象。 半乳糖對克雷白氏肺炎桿菌 galU 突變株的毒性,主要是因為尿嘧啶雙磷酸葡萄糖焦磷 酸化酶(UDP-glc PPase)缺損,導致了有毒的半乳糖代謝產物累積。然而,這些代謝產物 如何影響細菌基因表現及造成細菌死亡的原因仍然未知。因此,我們利用 RNA-seq 技 術研究在半乳糖存在的狀況下,克雷白氏肺炎桿菌 galU 突變株的基因表現變化。我們 發現在半乳糖的存在下,galETKM、galP、lacYZ 以及胺基酸合成相關的基因表現有上 升的趨勢;而甘油代謝(pduCDE)、鐵離子攝取系統(sitABCD、feoABC)與 12 個調控因子 (hns、csrA 與 10 個轉錄調控子)的基因表現則被抑制。廣泛型調控因子 H-NS 與 CsrA 控 制許多細菌的重要生理作用,如碳源代謝、毒性、移動與應激反應系統。我們推測本研 究使用的培養條件使得有毒的半乳糖代謝產物累積,並間接地抑制 H-NS、CsrA 與其它 轉錄調控子的基因表現,影響細菌的轉譯與轉錄作用,最後因為生理系統紊亂導致死 亡。
IV
Abstract
This thesis consists of three chapters. Chapter 1 and chapter 2 describe the functional
studies of two bifunctional enzymes, PslB and PA3346, in Pseudomonas aeruginosa PAO1.
Chapter 3 describes the results of transcriptome analysis of Klebsiella pneumoniae CG43S3
∆galU mutant under galactose stress.
Pseudomonas aeruginosa pslB gene encodes a bifunctional enzyme phosphomannose
isomerase/GDP-D-mannose pyrophosphorylase (PMI-GDP-man PPase). The enzyme
catalyzes the first and third steps in the GDP-D-mannose biosynthetic pathway, an important
precursor of many polysaccharides. So far, very little is known about PslB. In Chapter 1, we
demonstrate that Pseudomonas aeruginosa pslB encodes a protein with GDP-man PPase/PMI
dual activities. The GDP-man PPase activity is Mg2+-dependent, whereas the PMI activity is
Co2+-dependent and could be inhibited by GDP-mannose in a competitive manner.
Furthermore, the PMI activity could be inactivated by 2, 3-butanedione suggesting the
presence of a catalytic Arg residue. Site-specific mutations at R373, R472, R479, E410, H411,
N433 and E458 increase the KM approximately 8- to 20-fold. The PMI activity of PslB was
completely diminished with a R408K or R408A, reflecting the importance of this residue in
V
indicating that there is nearly no alterations of their secondary structures. Overall, these
results provide a basis for understanding the catalytic mechanism of PMI.
In chapter 2, we focus on the other bifunctional enzyme PA3346. We have previously
observed that the HptB-mediated phosphorelay pathway plays an important role in swarming
phenotype and biofilm formation in P. aeruginosa PAO1. Upon activation by an
environmental stress, sensor kinase (PA1611, PA1976 and PA2824) autophosphorylates itself
and then transfers a phosphoryl group to HptB, which then relays the signal to response
regulator PA3346. The phosphorylation on PA3346 N-terminal receiver domain results in an
increase in its Ser protein phosphatase activity leading to dephosphorylation of the putative
anti-sigma factor antagonist PA3347. While the target phosphorylation site on PA3347 has
been shown to be located at Ser-56, the corresponding serine protein kinase and anti-sigma
factor remain elusive. Protein domain analysis revealed that the C-terminal region of PA3346
(PA3346
L408-A571
) contains conserved domains similar to the Ser protein kinase/anti-sigma factor SpoIIAB in Bacillus subtilis. Thus, we proposed that PA3346-PA3347 forms a
partner-switching regulatory module as observed for SpoIIE-SpoIIAB-SpoIIAA in B. subtilis.
This thesis has cloned and purified PA3346
L408-A571
and PA3347-GST for in vitro phosphorylation and GST pull-down assays. The results clearly demonstrate that
PA3346
L408-A571
VI especially Mg 2+ , Ca 2+ and Mn 2+
dependent. The PA3347-S56A mutant protein could not
serve as a substrate for the kinase. In the GST pull-down assay, PA3346
L408-A571
could be co-eluted with PA3347-GST in the presence of ADP. As for the GFP fragment reassembly
assay, the bacterial cells harboring the plasmids of PA3346L408-A571-CGFP and
PA3347-NGFP display the green fluorescence signals. These data indicate that PA3346
L408-A571
is a serine protein kinase/anti-sigma factor playing a key role in the partner-switching regulatory module. In order to identify the specific sigma factor participated in this
partner-switching system, in vivo bimolecular fluorescence complementation assay are
performed and the detailed molecular regulation mechanism is under investigation.
Chapter 3 focuses on the response of K. pneumoniae CG43 ΔgalU mutant to the
galactose stress. K. pneumoniae ΔgalU mutant, which is defective in UDP-Glc PPase,
exhibited a growth-inhibition zone in the galactose disk diffusion assay. Galactose can
increase the mortality when the ΔgalU mutant was cultivated in M9 minimal medium with
2% glycerol as the sole carbon source. The toxic galatose metabolite affecting the gene
expression and causing the cell death remains unclear. Therefore, we investigate the gene
expression profiles of the K. pneumoniae ∆galU mutant under the galactose stress by
RNA-seq. The results indicate that galETKM, galP, lacYZ and genes responsible for
VII
expressions of glycerol metabolism (pduCDE), iron-acquisition systems (sitABCD and
feoABC) and 12 regulatory factors (hns, csrA and 10 transcriptional regulators) are repressed.
Notably, H-NS and CsrA are global regulatory proteins controlling a variety of physiological
processes. Besides, E. coli csrA gene was shown to be essential for bacterial growth in both
LB and minimal medium. The toxic galactose metabolites might indirectly repress the gene
transcription of H-NS, CsrA and several other transcriptional regulators and lead to the
VIII
Acknowledgement
博士班期間,受到了許多師長、同儕與朋友們的幫忙,才得以完成博士學業,要感 謝的人真的非常多。首先,最感謝的是我的指導教授張晃猷老師與彭慧玲老師。謝謝清 大分醫所張晃猷老師這幾年來的指導,讓我有機會跟不同領域的人合作,並參與許多研 究計畫。也謝謝張老師在百忙之中,抽空修改我的論文,使我的論文得以完善。謝謝彭 慧玲老師,在我最無助的時候,對我伸出援手,收我當研究生。老師總是設身處地為我 著想,替我做最好的安排,對於彭老師的感謝,真的不是言語能夠完全表達的。 謝謝楊昀良老師、梁美智老師與中國醫藥學大學林靖婷老師撥冗擔任我的博士班畢 業口試委員,老師們親切的提問,讓我回答問題時不那麼緊張,在口試時給了我許多建 議,而且非常仔細地看我的論文,幫我挑出很多小錯誤。謝謝林志生老師與中興大學鄧 文玲老師,在兩次的博士班資格考中擔任我的口試委員時,給我很多實驗設計與規劃的 建議,提醒我要加強報告與回答問題的技巧。最後,要感謝我的大學專題與碩士班指導 教授邱顯泰老師,在邱老師的實驗室裡,學到許多分生、生化實驗技術與實驗設計邏輯, 奠定了我做生物實驗的良好基礎。 感謝實驗室所有的成員們,尤其是同組的敏潔學姐,很懷念以前和學姐一起討論實 驗,一起出去吃飯,還有參加清大校園健走活動的日子;謝謝靜柔學姐,我們一起當了 微生物實驗課助教,而且,在兩次博士班資格考時,學姊幾乎一手包辦了大小瑣事,讓IX 我能夠專心準備資格考資料,真的非常謝謝學姊這些年來的幫忙與照顧;謝謝健誠學長 與小新學長,雖然與學長們相處的時間不多,但每次不管是聊實驗相關或是生活上的事 情,總是非常開心;要感謝的還有清大實驗室的貞儀學姊、韻如學姊、莉芳學姊、Poki 學長、錢小豬、婉禎、方瑜、宗葆、芊瑜、芝瑄、明融、幸珊、艾婷、吉姆、克萊兒、 柏融、Hubert、哈哈、小辣椒、Cain、楊不理、小蛙、毛毛、翼軒、小杜、慈芳、Maybe、
鄒宇、瑟玉、Maysam、Minh、Daniel Liu 與 Manish,大家在實驗室生活上的幫助、關
心與包容,讓我在清大的這幾年過得不孤單。
謝謝我的朋友們,偶爾與妳們聚餐聊天,是我攻讀博士班期間,最棒的生活點綴;
感謝我親愛的家人們,你們不求回報的付出與無條件的支持,對我而言,就是最大的鼓
勵,讓我能夠毫無後顧之憂的完成博士學業,我內心由衷的感謝,願將我所有的喜悅與
X
Abbreviations
AHL N-acylhomoserine lactones
ADP adenosine diphosphate
ATP adenosine triphosphate
ADP-Glc ADP-glucose
Ap ampicillin
C4-HSL N-butyryl-L-Homoserine lactone
CAS chrome azurol S
CD circular dichroism
DTT dithiothreitol
EDTA ethylenediaminetetraacetic acid
Fru-6-p fructose-6-phosphate
Gal galactose
Gal-1-p galactose-1-phosphate
GDP-Man GDP-mannose
GDP-Man PPase GDP-mannose pyrophosphorylase
Glc glucose
Glc-6-p glucose-6-phosphate
GFP green fluorescent protein
GST glutathione S-transferase
GTP guanosine triphosphate
HDTMA hexadecyltrimethylammonium bromide
HRP horseradish peroxidase
IPTG isopropyl thio--D-galactopyranoside
Km kanamycin
LB Luria-Bertani
Man-1-p mannose-1-phosphate
Man-6-p mannose-6-phosphate
NADH nicotinamide adenine dinucleotide
-NADP nicotinamide adenine dinucleotide phosphate
NB nutrient broth
PBS phosphate buffered saline
PIPES piperazine-N,N’-bis(2-ethanesulfonic acid)
XI
PVDF polyvinylidene fluoride
SDS sodium dodecyl sulfate
Sm streptomycin TDP-glc TDP-glucose 2CS two-component system UDP-Gal UDP-galactose UDP-Glc UDP-glucose UDP-GlcN UDP-glucosamine
XII
Table of Contents
中文摘要 ... I ABSTRACT ... IV ACKNOWLEDGEMENT ...VIII ABBREVIATIONS ... X TABLE OF CONTENTS ... XII LIST OF TABLES ... XV LIST OF FIGURES ... XVICHAPTER 1 ... 1
1.1ABSTRACT ... 2
1.2INTRODUCTION ... 2
1.3MATERIALS AND METHODS ... 4
1.3.1 Bacterial strains and growth conditions ... 4
1.3.2 Expression and purification of PslB ... 4
1.3.3 Determination of the enzyme activity ... 5
1.3.4 Inactivation of PslB by 2, 3-Butanedione ... 6
1.3.5 Amino acid sequence alignment and site-directed mutagenesis ... 6
1.3.6 Circular dichroism spectrum analysis ... 6
1.3.7 In vivo protein-protein interaction assay by GFP fragment reassembly ... 7
1.4RESULT AND DISCUSSION ... 7
1.4.1 PslB possesses PMI and GDP-Man PPase activity ... 7
1.4.2 Divalent cation requirement for PslB ... 8
1.4.3 The PMI activity of PslB is inhibited by GDP-Man ... 9
1.4.4 An Arg residue participates in the P. aeruginosa PMI activity ... 10
1.4.5 Arg408 substitutions abolished the PMI activity of PslB ... 11
1.4.6 The R408 mutations did not affect GDP-Man PPase activity of PslB ... 12
1.4.7 The GMP domain and the PMI domain influence the activities of each other ... 12
1.4.8 PslB cannot form protein complex with AlgC ... 14
1.5TABLES ... 16
1.6FIGURES ... 23
XIII
2.1ABSTRACT ... 33
2.2INTRODUCTION ... 34
2.3MATERIALS AND METHODS ... 36
2.3.1 Bacterial strains and growth conditions ... 36
2.3.2 Amino acid sequence alignments ... 36
2.3.3 Cloning, expression and purification of His-tag and GST-tag fusion protein ... 37
2.3.4 In vitro phosphorylation assay ... 38
2.3.5 Protein pull-down assays ... 38
2.3.6 Western blotting analysis ... 39
2.3.7 In vivo protein-protein interaction assay by GFP fragment reassembly ... 39
2.3.8 Chrome azurol S (CAS) plate assay ... 40
2.3.9 N-Acyl homoserine lactones (AHL) reporter plate bioassays ... 40
2.3.10 Congo red plate assay ... 41
2.4RESULT AND DISCUSSION ... 41
2.4.1 PA3346 L408-A571 contains the conserved N, G1 and G2 boxes ... 41
2.4.2 PA3346 L408-A571 possesses a Ser protein kinase activity toward PA3347 ... 42
2.4.3 PA2797 is not the substrate of PA3346L408-A571 ... 42
2.4.4 PA3346 L408-A571 interacts with PA3347 in the presence of ADP in vitro ... 43
2.4.5 PA3346 L408-A571 interacts with PA3347 in vivo ... 44
2.4.6 PA3346L408-A571 cannot interact with RpoN in the presence of ATP in vitro ... 45
2.4.7 ΔhptB mutant strain displays a higher production level of exopolysaccharides ... 46
2.4.8 No significant phenotype differences are found in CAS plate assay and AHL reporter assay ... 46
2.4.9 Is PA3346L408-A571 an anti-sigma factor? ... 47
2.5TABLES ... 51
2.6FIGURES ... 59
CHAPTER 3 ... 74
3.1ABSTRACT ... 75
3.2INTRODUCTION ... 75
3.3MATERIALS AND METHODS ... 77
3.3.1 Bacteria strains and growth medium ... 77
3.3.2 Bacterial growth curve ... 77
3.3.3 Disk diffusion assay ... 78
3.3.4 LIVE/DEAD® BacLight™ Bacterial Viability assay ... 78
3.3.5 RNA purification ... 78
3.3.6 RNA sequencing ... 80
XIV
3.4.1 A growth-inhibition zone is observed in K. pneumoniae ΔgalU mutant strain in galactose disk
diffusion assay ... 80
3.4.2 Galactose inhibits growth of K. pneumoniae ΔgalU mutant ... 81
3.4.3 Galactose induces death of K. pneumoniae ΔgalU mutant... 82
3.4.4 Transcriptome analysis of K. pneumoniae ΔgalU gene expression profiles under galactose stress ... 82
3.5TABLES ... 88 3.6FIGURES ... 96 REFERENCES ... 99 APPENDIX I ... 112 APPENDIX II ... 120 CURRICULUM VITAE ... 122
XV
List of Tables
Table 1.5.1 Bacterial strains and plasmids used in this study ... 16
Table 1.5.2 Primers used in this study ... 18
Table 1.5.3 Divalent cation effects on PslB ... 20
Table 1.5.4 Effects of potential inhibitors on the PMI activity ... 21
Table 1.5.5 PMI kinetic parameters of the wild type or mutated PslB ... 22
Table 2.5.1 Bacterial strains and plasmids used in this study ... 51
Table 2.5.2 Primers used in this study ... 54
Table 2.5.3 Genes used in the GFP assembly assay ... 58
Table 3.5.1 Upregulated genes in CG43S3 ΔgalU (+Gal) comparing with CG43S3 ΔgalU ... 88
Table 3.5.2 Downregulated genes in CG43S3 ΔgalU (+Gal) comparing with CG43S3 ΔgalU ... 90
Table 3.5.3 Predicted CRP binding site in the upstream region of galactose regulated genes ... 93
Table 3.5.4 Predicted H-NS binding site in the upstream region of the galactose regulated genes ... 94
XVI
List of Figures
Fig. 1.6.1 Protein purity of wild type and mutant PslB ... 23
Fig. 1.6.2 Effects of various concentrations of cobalt ion on the PMI activity ... 24
Fig. 1.6.3 Effects of Mg2+ on the PMI activity of PslB... 25
Fig. 1.6.4 Double reciprocal plot at various GDP-Man concentrations ... 26
Fig. 1.6.5 Inactivation of PslB by 2, 3-butanedione ... 27
Fig. 1.6.6 Amino acid sequence alignment of GDP-Man PPase /PMIs ... 28
Fig. 1.6.7 Circular dichroism spectra of wild type and mutant PslB ... 29
Fig. 1.6.8 Time dependent profiles of the GDP-man PPase activity assay ... 30
Fig. 1.6.9 Time dependent profile of PMI activity assay ... 31
Fig. 2.6.1 Multiple sequence alignment of PA3346L408-A571 with other Ser protein kinases. ... 59
Fig. 2.6.2 Time course analysis of PA3347 in vitro phosphorylation. ... 60
Fig. 2.6.3 In vitro phosphorylation assay of purified PA3347 and PA3347S56A mutant protein ... 61
Fig. 2.6.4 Divalent cation dependence on phosphorylation reaction. ... 62
Fig. 2.6.5 Phosphorylation level of PA3347 analysis by Pro-Q Diamond staining ... 63
Fig. 2.6.6 Multiple sequence alignment of PA2797 with other anti-sigma factor antagonist. ... 64
Fig. 2.6.7 In vitro phosphorylation of purified GST, GST-PA2797 and GST-PA3347 proteins ... 65
Fig. 2.6.8 Analysis of PA3346L408A-A571 and PA3347 protein-protein interaction by using GST pull down assay.66 Fig. 2.6.9 Protein interaction of PA3347 and PA3346L408-A571 examined using the GFP fragment reassembly assay ... 67
Fig. 2.6.10 Analysis of PA3346 and RpoN protein-protein interaction using GST pull down assay. ... 68
Fig. 2.6.11 Colony morphology of P. aeruginosa PAO1 and mutants on Congo red plate ... 69
Fig. 2.6.12 The CAS plate assay ... 70
Fig. 2.6.13 The N-Acyl homoserine lactones (AHL) reporter plate bioassays ... 71
Fig. 2.6.14 A hypothetical model of the combination of the HptB-mediated phosphorelay and partner switching regulatory system. ... 72
XVII
Fig. 3.6.1 Effects of glucose or galactose on K. pneumoniae CG43 S3 and ΔgalU mutant using the disk diffusion assay ... 96 Fig. 3.6.2 Growth curve of K. pneumoniae CG43 S3 and the ΔgalU mutant ... 97 Fig. 3.6.3 The growth curve and Live/Dead cell staining of the K. pneumoniae CG43 ΔgalU mutant ... 98
1
Chapter 1
Identification of amino acid residues important for the
phosphomannose isomerase activity of PslB in Pseudomonas
2
1.1 Abstract
Phosphomannose isomerase (PMI) plays a pivotal role in biosynthesis of GDP-mannose,
an important precursor of many polysaccharides. We demonstrate in this study that
Pseudomonas aeruginosa pslB encodes a protein with GDP-mannose
pyrophosphorylase/PMI dual activities. The GDP-man PPase activity is Mg2+-dependent,
whereas the PMI activity is Co2+-dependent and could be inhibited by GDP-mannose in a
competitive manner. Furthermore, the PMI activity could be inactivated by 2, 3-butanedione
suggesting the presence of a catalytic Arg residue. Site-specific mutations at R373, R472,
R479, E410, H411, N433 and E458 increase the KM approximately 8- to 20-fold. The PMI
activity of PslB was completely diminished with a R408K or R408A, reflecting the
importance of this residue in catalysis. The CD spectra of R408A, R408K and wild type PslB
are nearly identical, indicating that there is nearly no alterations of their secondary structures.
Overall, these results provide a basis for understanding the catalytic mechanism of PMI.
1.2 Introduction
Phosphomannose isomerase (PMI) catalyzes the reversible interconversion of
fructose-6-phosphate (Fru-6-p) and mannose-6-phosphate (Man-6-p) (1). This reaction links
Man-6-p into the mannose metabolism pathway resulting in the generation of GDP-mannose
3
GDP-fucose, and for mannosylation of various bacterial structural components such as
lipopolysaccharides and glycoproteins (2,3). PMI also plays an essential role in yeasts and
therefore can be regarded as an appropriate target to combat both bacterial and mycotic
infections (4).
PMI can be classified into three major types based on sequence similarity and domain
organization (5,6). Type I PMIs are found in all eukaryotes including yeast Candida albicans,
and certain bacteria including Escherichia coli. Type II PMIs are bifunctional enzymes
possessing GDP-mannose pyrophosphorylase (GDP-Man PPase)/PMI dual activities and are
primarily found in bacteria. Type III PMIs, first identified in Rhizobium meliloti, are
evolutionally more distinct from the other two PMI types. The structural basis on how these
seemingly unrelated proteins can catalyze an identical reaction is not clear.
Pseudomonas aeruginosa PAO1 contains three genes encoding a product homologous
with the Type II PMI. These genes, designated algA, wbpW and pslB, are located individually
within three distinct polysaccharide biosynthesis gene clusters (3). AlgA and WbpW have
been shown to participate in the production of alginate and A-band lipopolysaccharide,
respectively (3,7). The gene pslB is located in a 15-gene psl operon required for
exopolysaccharide synthesis and biofilm formation (8,9). Whether PslB is indeed a
4
The major aim of this study is to address the structural-functional relationship of type II
PMIs that is been largely unexplored. We have cloned, overexpressed, and characterized the
PMI activity of PslB. This was followed by site-directed mutagenesis of conserved amino
acid to identify important residues participating in the PMI activity.
1.3 Materials and Methods
1.3.1 Bacterial strains and growth conditions
E. coli XL-1 Blue was used for plasmid preparation and recombinant DNA manipulation.
E. coli Nova Blue was used for protein expression. All bacteria used in this study were grown
in Luria-Bertain medium containing ampicillin (100 μg/ml) or kanamycin (50 μg/ml), with
150 rpm shaking at 37oC. Bacteria strains and plasmids used in this study were listed in Table
1.5.1.
1.3.2 Expression and purification of PslB
E. coli Nova Blue harboring the pslB overexpression plasmid was grown in 200 ml
Luria-Bertani broth supplemented with 50 g/ml kanamycin and 100M isopropyl thio--D-galactopyranoside (IPTG) at 30o
C with vigorous shaking. After 10 h, cells were
collected by centrifugation at 3100 rpm for 15 min and washed with 20 mM Tris-HCl buffer
pH 7.5. The pellets were resuspended in 20 mM Tris-HCl buffer and the bacteria were
5
centrifugation at 14000 g for 20 min at 4oC. PslB was purified by nickel-charged affinity
chromatography following the standard purification protocol (Novagen). The eluted protein
was dialyzed against 20 mM Tris-HCl buffer (pH 7.5) to remove imidazole and the protein
concentration was determined by the Bradford method with bovine serum albumin as a
standard.
1.3.3 Determination of the enzyme activity
The PMI activity was determined in a 1-ml reaction mixture containing 50 mM Tris-HCl
pH 7.0, 2 mM CoCl2, 1 mM Man-6-p, 1 mM -NADP, 5 U phosphoglucose isomerase and 5
U Glc-6-p dehydrogenase. The reduction of -NADP was measured at 340 nm at 25o
C using
a spectrophotometer. The GDP-Man PPase activity was assayed in a reaction mixture
containing GDP-Man, 5 mM sodium pyrophosphate, 2 mM MgCl2, 2 mM
3-phosphoglycerate, 20 U glyceraldehyde-3-phosphate dehydrogenase and 10 U
phosphoglycerate kinase. The decrease in NADH absorption at 340 nm was monitored by a
spectrophotometer (10). Where indicated, 0.5 mM of Man-1-p, GDP-Man, GTP, UDP-Glc,
UDP-Gal, ADP-Glc, TDP-Glc, UDP-GlcN, or sodium pyrophosphate was included in the
PMI reaction to determine the inhibition activity of these compounds. For determination of
the effects of divalent cation, PslB was dialyzed extensively in 20 mM Tris HCl pH 7.5, 140
6
chloride form divalent cation including CaCl2, MnCl2, MgCl2, CoCl2, NiCl2 and ZnCl2 (2,11).
1.3.4 Inactivation of PslB by 2, 3-Butanedione
PslB (3.2 mg/ml) was incubated with different concentrations of 2, 3-butanedione (150
mM borate buffer, pH 9.0) at 22oC in the dark for 0 to 60 min (12,13), then the excess
2,3-butanedione was removed by passing the reaction mixture through a Sephadex G-25
column (GE heahthcare). The PMI activity was measured as described above.
1.3.5 Amino acid sequence alignment and site-directed mutagenesis
The multiple sequences alignment of several type II PMIs, including Xanthomonas
campestris XanB (14), Helicobacter pyroli HP0043 (2), Gluconacetobacter xylinus AceF
(15), and Vibrio vulnificus YJ016 VV0352, were conducted by using the Vector NTI
(Invitrogen) and the result is shown in Fig. 1.6.6. Site-specific mutations were performed
using the QuikChange site-directed mutagenesis kit purchased from Stratagene.
Oligonucleotide primer sequences are provided in Table 1.5.2.
1.3.6 Circular dichroism spectrum analysis
The CD spectra of wild type and mutant PslB were recorded by using a CD
spectrophotometer (AVIV 62A PS) with 1-mm path length cell, 0.5 nm wavelength step, and
7
measurement. The CD spectra signals were collected from 190 nm to 260 nm in 20 mM
Tris-HCl buffer at 25oC and averaged over three scans (16).
1.3.7 In vivo protein-protein interaction assay by GFP fragment reassembly
The vectors used in this study were provided by Dr. Lynne Regan of Yale University as a
gift (17). Gene algC was cloned into pMRBAD-link-CGPF. Three GMP-PMIs (algA, pslB
and wbpW) were cloned into pET11a-link-NGFP. The two plasmids were co-transformed into
E. coli BL21 (DE3) and cultured in LB medium containing 100 μM ampicillin and 35 μM
kanamycin at 37oC for 12 to 16 h. To perform the GFP reassembly, ten microliters of the
overnight cultures were applied to the LB agar plates containing the same antibiotics, 100 μM
IPTG and 0.05% arabinose. Then, the plates were statically incubated at 20°C for 2 to 3 days.
The bacteria cells grown on the plates were re-suspended with PBS buffer and examined by
fluorescence microscope. All images were captured with an upright microscope (BX-51;
Olympus) connected with a CCD camera. The data were analyzed by using the SPOT
Advanced Plus Imaging software (Sterling Heights, MI).
1.4 Result and Discussion
1.4.1 PslB possesses PMI and GDP-Man PPase activity
Based on the domain analysis result of InterProScan, the N-terminal domain of AlgA,
8
domain. Among the three PMI-GMPs in P. aeruginosa PAO1, the GDP-Man PPase/PMI
activities of AlgA have been proved (7), whereas that of PslB has not been reported before.
Therefore, we first examined whether PslB is indeed a GDP-Man PPase/PMI bifunctional
enzyme. Recombinant PslB was purified from E. coli by nickel affinity chromatography and
its purity was confirmed on a SDS-polyacrylamide gel (Fig. 1.6.1). Under the standard assay
conditions, both GDP-Man PPase and PMI activities could clearly be detected for PslB. The
KM value of Man-6-p for the PMI activity was 1.18 mM, and the KM of GDP-Man for
GDP-Man PPase was 0.11 mM, both values are in good agreement with that reported for
AlgA in P. aeruginosa 8822 (7). Bacteria with a large genome commonly harbor duplicated
genes encoding functionally identical enzymes. For example, there are at least two UDP-Glc
dehydrogenases in P. aeruginosa PAO1. The enzyme kinetic parameters and expression
profiles are somewhat different for these UDP-Glc dehydrogenases that ensure the bacterium
can adapt to broader environments (18). A similar mode of regulation may also occur for PMI.
It has been demonstrated previously that a P. aeruginosa strain deficient in WbpW could not
produce normal amounts of A-band lipopolysaccharide despite the presence of functional
algA and pslB (3), suggesting that WbpW has distinct expression profiles or kinetic
parameters from PslB and AlgA.
9
We measured the PMI and GDP-Man PPase activities of PslB at a fixed concentration (2
mM) of different divalent ions to evaluate cation dependence of the protein. The GDP-Man
PPase of PslB could utilize different divalent metal ion with the highest activity in the
presence of Mg2+, followed by Co2+, then Mn2+ (Table 1.5.3). The PMI activity of PslB
showed high specificity to Co2+, yielding about 3-fold higher activity than that observed with
Mn2+ (Table 1.5.3). Other divalent cations, including Mg2+, Zn2+, Ca2+, and Ni2+, could not
serve effectively as a cofactor for the PMI activity of PslB, consistent with several previous
reports (2,7,19). The optimal Co2+ concentration for the PMI was 0.2 mM, which yielded an
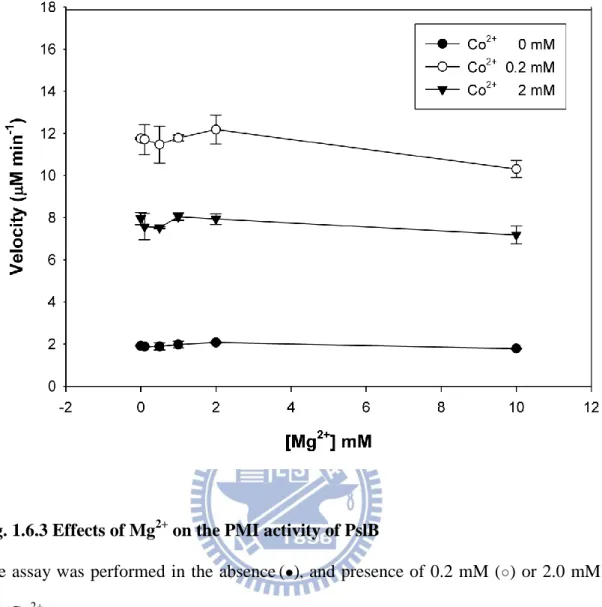
activity approximately 1.5 fold higher than that of 2.0 mM (Fig. 1.6.2). Mg2+ at 2.0 mM
showed little interference on the PMI activity in the presence of either 0.2 mM or 2.0 mM
Co2+ (Fig. 1.6.3), indicating that Co2+ is capable of functioning as a cofactor for the PMI in
bacterial cells which commonly contain millimolar Mg2+ (20). Interestingly, both PMI and
GDP-Man PPase activities in PslB could not be activated in ZnCl2 despite the fact that the
protein contains a zinc-binding sequence Q403XH405 (X represents any amino acid), a feature
that is also present in type I PMIs (6).
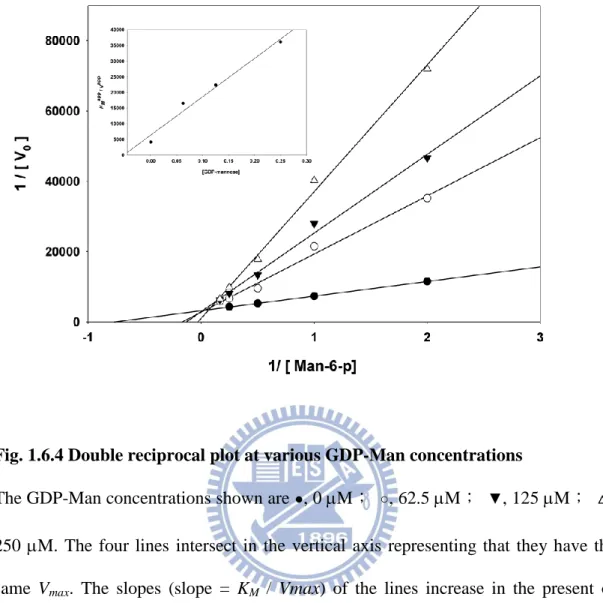
1.4.3 The PMI activity of PslB is inhibited by GDP-Man
Several molecules participating in GDP-Man biosynthesis and six nucleotide sugars
10
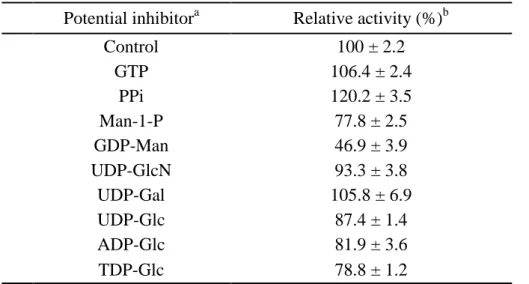
mM, only GDP-Man significantly inhibited the PMI activity, which decreased to
approximately 47% than that observed for the control (Table 1.5.4). Further investigation on
the GDP-Man inhibition type demonstrated a competitive inhibition (Fig. 1.6.4) indicating
that the GDP-Man biosynthesis pathway is well controlled by the end product, GDP-Man, as
also is found for Helicobacter pylori PMI (2). A slight deviation of the 250 M GDP-Man
lines from the other three lower concentration lines was observed suggesting that the
inhibition is attenuated in higher concentrations of the inhibitor (GDP-man). The results
suggest that the mannose group in GDP-Man competes with Man-6-p at the PMI active site
to regulate the mannose utilization in P. aeruginosa PAO1.
1.4.4 An Arg residue participates in the P. aeruginosa PMI activity
To identify if PslB is a useful target to combat P. aeruginosa infections, it is essential to
locate the active site participating in the PMI catalytic activity. It has been shown previously
that R304 is an active site residue in a type I PMI of C. albicans (21). Although type I and
type II PMI share low amino acid sequence homology, their catalytic mechanisms may be
similar. To determine whether an Arg is involved in the PMI activity of PslB, this study
utilized 2, 3-butanedione to modify Arg in the protein and the PMI activity was determined
(22). Figure 1.6.5 shows that the PMI activity decreased with increasing 2, 3-butanedione
11 involved in PMI catalysis.
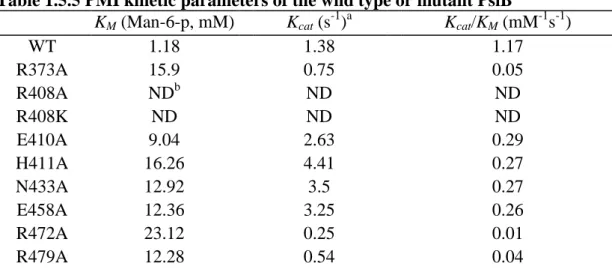
1.4.5 Arg408 substitutions abolished the PMI activity of PslB
Based on sequence alignment with other type II PMI, R373, R408, R472 and R479 were
chosen and changed to Ala by site-directed mutagenesis. In addition, this study adopted the
molecular structure information of Pyrobaculum aerophilum bifunctional enzyme
phosphoglucose/phosphomannose isomerase (PaPGI/PMI) (23), and selected E410, H411,
N433 and E458 for site-directed mutagenesis. The kinetic constants of the wild type and
mutant PslB are shown in Table 1.5.5. Substitution of Arg at position 408 to either a Lys or
an Ala abolished nearly all of the PMI activity. Other mutant proteins still retained PMI
activities and the KM values were 8- to 20- fold larger compared to those values observed for
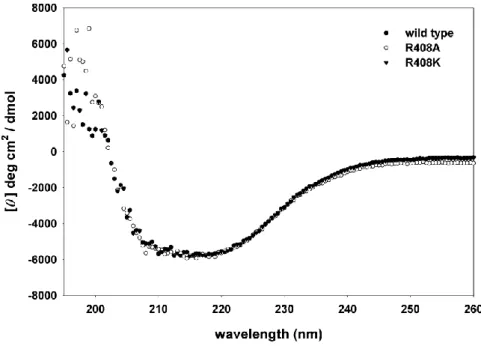
wild type. The CD spectra (Fig. 1.6.7) of R408A, R408K and wild type PslB are nearly
identical, all showing a minimum point at 208 nm and 222 nm, a typical spectrum of high -helix content protein. The data indicate that there is no major alteration of the secondary
structures of the mutant proteins.
Overall, the results indicate that R408, which lies within the well-conserved motif
shared by most, if not all, type II PMI is an important residue for PMI catalysis. The residue
is likely to participate in the interconversion of sugar moieties by providing the binding of the
12 binding between substrates and enzymes.
1.4.6 The R408 mutations did not affect GDP-Man PPase activity of PslB
The fusion of the GDP-Man PPase and PMI domains in the type II PMI implies these two
activities may interact with each other. The GDP-Man PPase activity of the PslB deficient in
PMI activity was determined and no difference was observed. The KM values of wild type,
R408A and R408K were 0.068 mM, 0.072 mM and 0.078 mM, respectively. The similar KM
values of wild type and mutant PslB provide strong evidence that PMI and GDP-Man PPase
are actually two independent domains.
1.4.7 The GMP domain and the PMI domain influence the activities of each other
Based on the domain analysis result of InterProScan, the N-terminal domain (9-297 a.a.)
of PslB is GMP domain and the C-terminal domain (325-475 a.a.) of PslB is the PMI domain.
Although previous literatures also reported that Type II PMIs own two independent domains
(2,6,14,24,25), no article has reported if the PMI and GDP-Man PPase activities would be
influenced when Type II PMIs lacked the PMI domain or the GMP domain. To further
understand if the separate domains influence the activities of each other, the GMP domain
and the PMI domain were cloned into the protein expression vector individually. According
to the amino acid sequence alignment result reported by Dr. Leitao J. H. and colleagues (25),
13
to constructed a C-terminal His tag fusion protein. To increase the protein solubility, the pslB
DNA fragment of 930-1464 (PslB310-488) was cloned into pGEX-5X-1 to constructed an
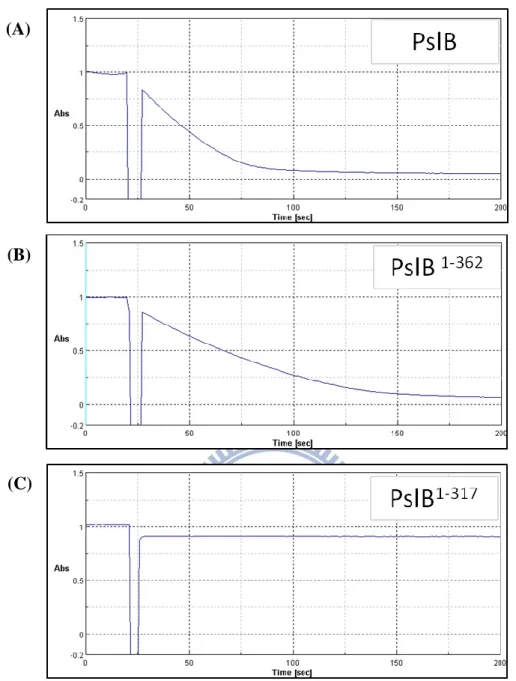
N-terminal GST tag fusion protein. As shown in Fig. 1.6.8, PslB and PslB1-362 display the
GDP-man PPase activities, but PslB317 donot. The KM values (GDP-man) of PslB and
PslB1-362 were 0.089 mM and 0.078 mM, respectively. The similar KM values of PslB and
PslB1-362 indicated that the GDP-man PPase activity would not be influenced when lacking
the C-terminal domain from 363-488. Interestingly, GDP-man PPase activity was lost when
lacking the fragment residues from 317-362 predicted to be belonged to the PMI domain (25).
Taking those results together, the complete GDP-man PPase activity still required the
existence of partial PMI domain. In the case of PMI domain, the time dependent profile (Fig.
1.6.9) displayed that the PMI activity of PslB310-488 were relatively poor than that of PslB.
The KM (Man-6-p) values of PslB and PslB310-488 were 0.92 mM and 2.89 mM, respectively.
The KM value of PslB310-488 decreased 3 folds, indicating that the N-terminal domain is also
important for the complete PMI activity. PslB1-362 had no PMI activity, revealing that the
C-terminal domain was important for PMI activity. May T. B et al. made the similar
conclusion using chymotrypsin digestion to produce a 1-kDa C-terminal deletion of AlgA.
The chymotrypsin digestion of AlgA retained 81% GMP activity but lost 92% PMI activity
14
the activities of each other. This result may explain why the two domains fused together.
1.4.8 PslB cannot form protein complex with AlgC
Many of the bifunctional enzymes have been discovered that they catalyze the
continuous reactions in the biosynthesis pathway. For example, tryptophan synthase which is
commonly found in bacteria, fungi and plants is a bifunctional enzyme that catalyzes the final
two steps in the biosynthesis of tryptophan (26). Some bifunctional enzyme, such as
PMI-GMPs, catalyzes the noncontinuous reactions. PMI-GMPs participates the first and third steps of the GDP-mannose biosynthesis pathway (Fru-6-p PslB (PMI) Man-6-p AlgC
(PMM) Man-1-p + GTP PslB (GMP) GDP-man + ppi). It is reasonable to speculate that
PMI-GMPs are associated with PMM (AlgC) by protein-protein interaction to form the
channeling of Fru-6-p to GDM-man (7,27). If PMI-GMP from a protein complex with PMM,
it could increase the reaction efficiency by avoiding the loss of any reaction intermediates. In
order to clarify this speculation, three PMI-GMPs (algA, pslB and wbpW) and algC in P.
aeruginosa PAO1 were cloned into pET11a-link-NGFP and pMRBAD-link-CGFP
respectively for performing the GFP fragment reassembly assay. The bacteria cells harboring
the two plasmids were examined by a fluorescence microscope. Only the bacteria harboring
pET11a-Z-NGFP and pMRBAD-Z-CGPF displayed the green fluorescence emission (28).
15
fluorescence signals. Our result does not prove if the PMI-GMPs form protein complex with
16
1.5 Tables
Table 1.5.1 Bacterial strains and plasmids used in this study
Strains or Plasmids Descriptions Refs. or source Strains
E. coli
BL21 (DE3) F– ompT hsdSB(rB–, mB–) gal dcm (DE3) Invitrogen
Nova Blue endA1 hsdR17(rK12 –
mK12+) supE44 thi-1 recA1 gyrA96 relA1 lac F’[proA+B+lacIqZΔM15::Tn10] (TetR) Novagen XL-1 Blue recA1 endA1 gyrA96 thi-1 hsdR17 supE44 relA1 lac [F´ proAB lacIq Z∆M15 Tn10 (Tetr)] Stratagene
Plasmids
pET28a Kmr, His tag protein expression vector Novagen
pET30a Kmr, His tag protein expression vector Novagen
pGEX-5X-1 Apr, GST tag protein expression vector GE Healthcare
pET11a-Z-NGFP Apr, plasmid vector that expresses a fusion of an antiparallel leucine zipper peptide to NGFP (29) pET11a-link-NGFP Apr, plasmid vector designed for fusion of a target protein to the N-terminal fragment of GFP (1-157) (29) pMRBAD-link-CGFP Kmr, plasmid vector designed for fusion of a target protein to the C-terminal fragment of GFP (158-238) (29) pMRBAD-Z-CGFP Kmr, plasmid vector that expresses a fusion of an antiparallel leucine zipper peptide to CGFP (29) pETPslB Kmr, a fragment containing entire pslB (PA2232) coding region cloned into pET30a (NdeI&EcoRI) this study
pHL17 Kmr, pslB with the mutation E410A cloned into pET30a this study
pHL18 Kmr, pslB with the mutation E411A cloned into pET30a this study
pHL19 Kmr, pslB with the mutation E433A cloned into pET30a this study
pHL20 Kmr, pslB with the mutation E458A cloned into pET30a this study
pHL25 Kmr, pslB with the mutation E373A cloned into pET30a this study
pHL26 Kmr, pslB with the mutation E472A cloned into pET30a this study
17
pHL30 Kmr, pslB with the mutation E408A cloned into pET30a this study
pHL31 Kmr, pslB with the mutation E408K cloned into pET30a this study
pHL24 Kmr, a fragment containing entire pslB (PA2232) coding region cloned into pET30a (NdeI&HindIII) this study pHL29 Kmr, a fragment containing residues 280-488 of PslB coding region cloned into pET30a this study pHL75 Kmr, a fragment containing residues 1-362 of PslB coding region cloned into pET30a this study pHL76 Kmr, a fragment containing residues 1-317 of PslB coding region cloned into pET30a this study pHL77 Kmr, a fragment containing residues 310-488 of PslB coding region cloned into pET30a this study pHL78 Kmr, a fragment containing residues 1-362 of PslB coding region cloned into pET28a this study pHL79 Kmr, a fragment containing residues 1-317 of PslB coding region cloned into pET28a this study pHL80 Kmr, a fragment containing residues 310-488 of PslB coding region cloned into pET28a this study pHL81 Apr, a fragment containing residues 310-488 of PslB coding region cloned into pGEX-5X-1 this study pHL87 Kmr, a fragment containing entire algA (PA3551) coding region cloned into pET30a this study pHL88 Kmr, a fragment containing entire wbpW (PA5452) coding region cloned into pET30a this study pHL89 Kmr, a fragment containing entire algC (PA5322) coding region cloned into pET30a this study pHL90 Apr, a fragment containing entire algC (PA5322) coding region cloned into pGEX-5X-1 this study pHL91 Kmr, a fragment containing entire algC (PA5322) coding region cloned into pMRBAD-link-CGFP this study pHL92 Apr, a fragment containing entire algA (PA3551) coding region cloned into pET11a-link-NGPF this study pHL93 Apr, a fragment containing entire pslB (PA2232) coding region cloned into pET11a-link-NGPF this study pHL94 Apr, a fragment containing entire wbpW (PA5452) coding region cloned into pET11a-link-NGPF this study
18
Table 1.5.2 Primers used in this study
Primer name Primer sequence (5'3') Descriptions
Or488-F1 CATATGAACGCCGTCGCCCCGCTG clone pslBinto pET30a
Or488-R1 AAGCTTGGCTTTCTTCTCGTCGCTGG clone pslB and PslB280-488 into pET30a
R373A-F2 CACCGCACGGTCAGCGCGCCCTGG pslB- pET30a-R373A mutant
R373A-R2 CCAGGGCGCGCTGACCGTGCGGTG pslB- pET30a-R373A mutant
R408AF1 TGCACCACCATGCCAGCGAGCACTGGAT pslB- pET30a-R408A mutant R408AR1 ATCCAGTGCTCGCTGGCATGGTGGTGCA pslB- pET30a-R408A mutant R408KF1 ATGCACCACCATAAGAGCGAGCACTGGAT pslB- pET30a-R408K mutant R408KR1 ATCCAGTGCTCGCTCTTATGGTGGTGCAT pslB- pET30a-R408K mutant IsoE410A-F1 CATCGCAGCGCGCACTGGATCGTGGTC pslB- pET30a-E410A mutant IsoE410A-R1 GACCACGATCCAGTGCGCGCTGCGATG pslB- pET30a-E410A mutant IsoH411A-F1 CATCGCAGCGAGGCCTGGATCGTGGTC pslB- pET30a-H411A mutant IsoH411A-R1 GACCACGATCCAGGCCTCGCTGCGATG pslB- pET30a-H411A mutant IsoN433A-F1 CTCCTCAACACCGCGGAATCCACCTTCATCC pslB- pET30a-N433A mutant IsoN433A-R1 GGATGAAGGTGGATTCCGCGGTGTTGAGGAG pslB- pET30a-N433A mutant IsoE458A-F1 GGTGATGATCGCGGTACAGAGCGGCGAG pslB- pET30a-E458A mutant IsoE458A-R1 CTCGCCGCTCTGTACCGCGATCATCACC pslB- pET30a-E458A mutant R472A-F1 GGACGACATCGTCGCGTTCAACGACATC pslB- pET30a-R472A mutant R472A-R1 GATGTCGTTGAACGCGACGATGTCGTCC pslB- pET30a-R472A mutant R479A-F1 CGACATCTACGGTGCCGCCCCCGCCAGC pslB- pET30a-R479A mutant R479A-R1 GCTGGCGGGGGCGGCACCGTAGATGTCG pslB- pET30a-R479A mutant PMI-f3 CATATGAGCGACATCGGCTCCTGGCA clone PslB280-488 into pET30a
19
GMP317NR ATTCTCGAGTCACGAATCGATGTAGCAGTTGC clone PslB1-317 into pET-28a GMP317CR AATCTCGAGCGAATCGATGTAGCAGTTGCTG clone PslB1-317 into pET-30a GMP362NR AATCTCGAGTCAGTGGCCGCGACGCTTG clone PslB1-362 into pET-28a GMP362CR AATCTCGAGGTGGCCGCGACGCTTGAG clone PslB1-362 into pET-30a
PMI-F ATTCATATGGTCAGCAACTGCTACATCGATTCG clone PslB310-488 into pET vector and pGEX-5X-1 PMI-NR AATGCGGCCGCTCAGGCTTTCTTCTCGTC clone PslB310-488 into pET-28a
PMI-CR AATGCGGCCGCGGCTTTCTTCTCGTC clone PslB310-488 into pET-30a PMI GST F GCGGATCCACGTCAGCAACTGCTACATCG clone PslB310-488 into pGEX-5X-1
3551 F GCCATATGATCCCAGTAATCCTTTC clone algA into pET30a
3551 R AATCTCGAGGCGGCTGCCGGCGACC clone algA into pET30a
5452 F AATCATATGCTGATTCCCGTGGTG clone wbpW into pET30a
5452 R AATGCGGCCGCGACCACCCTGCCGTACTG clone wbpW into pET30a
PMM-F AACATATGAGCACTGCAAAAGCACCGACG clone algC into pET30a and pGEX-5X-1 pMM-R AAGCGGCCGCGAAGGGCACGGGCAGCGAGG clone algC into pET30a
pMMgst-F AAGGATCCCCATGAGCACTGCAAAAGCACCGACG clone algC into pGEX-5X-1 AN-F AACTCGAGCATGATCCCAGTAATCCTTTC clone algA into pET11a-link-NGFP
AN-R ATGGATCCTCAGCGGCTGCCGGC clone algA into pET11a-link-NGFP
BN-F GCATGAACGCCGTCGCCC clone pslB into pET11a-link-NGFP
BN-R ATGGATCCTCAGGCTTTCTTCTCGTC clone pslB into pET11a-link-NGFP
WN-F GCATGCTGATTCCCGTGGTGCT clone wbpW into pET11a-link-NGFP
WN-R ATGGATCCTCAGACCACCCTGCC clone wbpW into pET11a-link-NGFP
MC-F AATCATGACTATGAGCACTGCAAAAGCACCG clone algC into pMRBAD-link-CGFP
20
Table 1.5.3 Divalent cation effects on PslB
Divalent cation Relative activity (%)
b
PMI GDP-Man PPase
Controla 9.4 ± 3.6 NDc MgCl2 10.1 ± 0.3 100 CaCl2 6.1 ± 2.7 38.8 ± 0.1 MnCl2 31.7 ± 1.8 38.6 ± 2.0 CoCl2 100 86.5 ± 1.3 NiCl2 5.5 ± 0.6 9.9 ± 0.2 ZnCl2 2.6 ± 0.4 4.7 ± 0.2 a
Control reaction was assayed without adding any divalent cation.
b
The relative activity was compared to the optimal activity of the divalent cation set as 100% for Co2+, in case of PMI and Mg2+, in case of GDP-Man PPase.
c
21
Table 1.5.4 Effects of potential inhibitors on the PMI activity
Potential inhibitora Relative activity (%)b
Control 100 ± 2.2 GTP 106.4 ± 2.4 PPi 120.2 ± 3.5 Man-1-P 77.8 ± 2.5 GDP-Man 46.9 ± 3.9 UDP-GlcN 93.3 ± 3.8 UDP-Gal 105.8 ± 6.9 UDP-Glc 87.4 ± 1.4 ADP-Glc 81.9 ± 3.6 TDP-Glc 78.8 ± 1.2 a
The PMI activity was analyzed in the presence or absence of 0.5 mM potential inhibitors.
b
The relative activity was calculated by comparing with control, whose activity was set as 100%
22
Table 1.5.5 PMI kinetic parameters of the wild type or mutant PslB
KM (Man-6-p, mM) Kcat (s-1)a Kcat/KM (mM-1s-1)
WT 1.18 1.38 1.17 R373A 15.9 0.75 0.05 R408A NDb ND ND R408K ND ND ND E410A 9.04 2.63 0.29 H411A 16.26 4.41 0.27 N433A 12.92 3.5 0.27 E458A 12.36 3.25 0.26 R472A 23.12 0.25 0.01 R479A 12.28 0.54 0.04 a
Kcat was calculated from the equation Vmax=Kcat × [Eo], where Eo is the molar concentration of PslB. The results are the average of three independent experiments.
b
23
1.6 Figures
Fig. 1.6.1 Protein purity of wild type and mutant PslB
The samples were resolved on a 0.5% sodium dodecyl sulfate-12.5% polyacrylamide gel. Lanes 1, wild type; 2, R373A; 3, R408A; 4, R408K; 5, E410A; 6, H411A; 7, N433A; 8, E458A; 9, R472A; 10, R479A.
24
Fig. 1.6.2 Effects of various concentrations of cobalt ion on the PMI activity
The concentrations used in this study were: 0 mM, 0.002 mM, 0.01 mM, 0.05 mM, 0.1 mM, 0.2 mM, 0.5 mM, 1 mM, 2 mM and 4 mM (the X axis). The Y axis is the formation velocity (M min-1) of -NADPH representing the PMI activity.
25
Fig. 1.6.3 Effects of Mg2+ on the PMI activity of PslB
The assay was performed in the absence(●), and presence of 0.2 mM (○) or 2.0 mM (▼) Co2+.
26
Fig. 1.6.4 Double reciprocal plot at various GDP-Man concentrations
The GDP-Man concentrations shown are ●, 0 M; ○, 62.5 M; ▼, 125 M; △, 250 M. The four lines intersect in the vertical axis representing that they have the same Vmax. The slopes (slope = KM / Vmax) of the lines increase in the present of higher concentration of GDP-Man, indicating that their KM values increase. Overall, this plot shows a competitive inhibition of GDP-Man for PMI activity. The inset illustrates the reciprocal slopes from Fig. 1.6.4 versus GDP-Man concentrations.
27
Fig. 1.6.5 Inactivation of PslB by 2, 3-butanedione
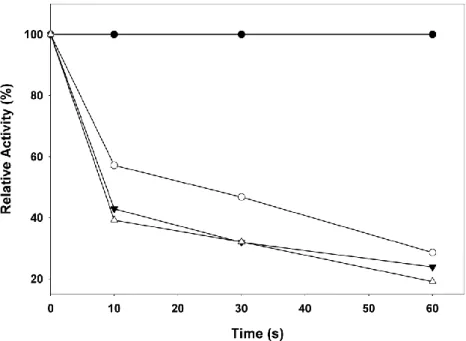
Relative activity (%) was calculated against the activity without 2, 3-butanedione. The final concentration of 2, 3-butanedione were ●, 0 mM; ○, 20 mM; ▼, 40 mM; ▽, 80 mM.
28
Fig. 1.6.6 Amino acid sequence alignment of GDP-Man PPase /PMIs
Only the C-terminal PMI comprising regions of the proteins are shown. The GenBank accession numbers for these proteins are: P. aeruginosa PAO1 PslB, NP_250922; X.
campestris XanB, YP_933589; Helicobacter pylori 26695 HP0043, NP_206844;
Gluconacetobacter xylinus AceF, CAA72316; Vibrio vulnificus YJ016 VV0352,
29
Fig. 1.6.7 Circular dichroism spectra of wild type and mutant PslB
30
Fig. 1.6.8 Time dependent profiles of the GDP-man PPase activity assay
The profiles exhibited the GDP-Man PPase activity of (A) full length of PslB (B) the N-terminal GMP region including partial of Man-6-p isomerase domain (C) the N-terminal GMP region of PslB. The Y axis is the absorption values of -NADH representing the GDP-man PPase activity.
(A)
(B)
31
Fig. 1.6.9 Time dependent profile of PMI activity assay
This profile showed the PMI activity of full length PslB was better than that of the PMI domain only. The Y axis is the absorption values of -NADPH representing the PMI activity.
32
Chapter2
Characterization of PA3346, a response regulator with dual
Ser protein phosphatase and kinase activities in P.
33
2.1 Abstract
Our laboratory has previously observed that the HptB-mediated phosphorelay pathway plays an important role in swarming phenotype and biofilm formation in P.
aeruginosa PAO1. Upon activation by an environmental stress, sensor kinase
(PA1611, PA1976 and PA2824) autophosphorylates itself and then transfers a phosphoryl group to HptB, which then relays the signal to response regulator PA3346. The phosphorylation on PA3346 N-terminal receiver domain results in an increase in its Ser protein phosphatase activity leading to dephosphorylation of the putative anti-sigma factor antagonist PA3347. While the target phosphorylation site on PA3347 has been shown to be located at Ser-56, the corresponding Ser protein kinase and anti-sigma factor remain elusive. Protein domain analysis revealed that the C-terminal region of PA3346 (PA3346
L408-A571
) contains conserved domains similar to the serine protein kinase/anti-sigma factor SpoIIAB in Bacillus subtilis. Thus, we proposed that PA3346-PA3347 forms a partner-switching regulatory module as observed for SpoIIE-SpoIIAB-SpoIIAA in B. subtilis. Using standard recombinant DNA techniques and protein purification procedures, we have cloned and purified PA3346L408-A571 and PA3347-GST for in vitro phosphorylation and GST pull-down assays. The results of in vitro phosphorylation assay clearly demonstrated that PA3346
L408-A571
possesses a kinase activity toward PA3347 and the activity is divalent cation, especially Mg 2+ , Ca 2+ and Mn 2+
, dependent. The PA3347-S56A mutant protein could not serve as a substrate for the kinase. In the GST pull-down assay, PA3346
L408-A571
could be co-eluted with PA3347-GST in the presence of ADP. As for the GFP fragment reassembly assay, the bacterial cells harboring the plasmids of PA3346L408-A571-CGFP and PA3347-NGFP display the green fluorescence signals.
34
These data indicate that PA3346 L408-A571 is a serine protein kinase/anti-sigma factor playing a key role in the partner-switching regulatory module. In order to identify the specific sigma factor participated in this partner-switching system, in vivo bimolecular fluorescence complementation assays were performed and the detail molecular regulation mechanism was reported in this study.
2.2 Introduction
Bacteria mainly use two-component systems (2CS) to regulate gene expressions for rapid adaptation to different environmental challenges (30). A 2CS is usually composed of two modules, a sensor kinase and a response regulator. The sensor kinase can autophosphorylate itself when environmental cue stimulates, and the signal is transmitted through a simple HisAsp pathway or a more complicated HisAspHisAsp pathway to activate the cognate response regulator, which mostly functions as a transcription regulator (31). For example, response regulator BvgA in Bordetella bronchiseptica is activated by sensor kinase BvgS. Then, the C-terminal DNA-binding domain of BvgA-P binds to frl promoter and represses the gene expressions required for flagella biosynthesis, causing a defect in motility (32). Oppositely, MrpAB in Myxococcus xanthus positively regulates its own promoter and the expression of mrpC (33).
The response regulator PA3346 in the HptB mediated signal transduction pathway in P. aeruginosa PAO1 is totally different from transcriptional regulators. The phosphorylation on PA3346 N-terminal receiver domain results in an increase in its Ser protein phosphatase activity and leads to dephosphorylation at Ser-56 of the putative anti-sigma factor antagonist PA3347 (34). More interestingly, the gene organization of PA3346-(unknown Ser protein kinase/anti-sigma factor)-PA3347 is
35
similar to SpoIIE-SpoIIAB-SpoIIAA and RsbU-RsbW-RsbV in B. subtilis which are so-called partner switcher system (35). In partner switcher regulatory system, Ser protein kinase /phosphatase modulate the activities of anti-sigma factor antagonist by state conversion between phosphorylated or dephosphorylated form. SpoIIE-SpoIIAB-SpoIIAA regulates the sporulation-related gene expression by phosphorylation or dephosphorylation at Ser-58 of the anti-sigma factor antagonist SpoIIAA (36). The phosphorylated state SpoIIAA was an inactive form and the anti-sigma factor SpoIIAB was released to form a SpoIIAB ‧F complex for repressing gene transcriptions. In a Bordetella spesies, BtrU-BtrW-BtrV is also a partner switcher system and is required for the production of type III secretion system through the regulation of stress response B
(37). However, in the HptB-PA3346- PA3347 system, the Ser protein kinase, anti-sigma factor and specific sigma factor remain unknown. Therefore, the major goal of this study is to search for the Ser protein kinase, which regulates the activity of anti-σ antagonist (36). We recently noted that, by using the InterProScan analysis (http://www.ebi.ac.uk/Tools/pfa/iprscan/), the C-terminal region of PA3346 possesses a putative HATPase domain. Hence, our first aim is to determine biochemically whether PA3346 is indeed a Ser protein kinase. It is somewhat unusual if the hypothesis turns out positive, because the Ser protein kinase and phosphatase of partner switcher systems are in two separate protein identities in B.
subtilis and Bordetella species. How the two opposite enzyme activities are regulated
in a single protein molecule would be an interesting question to address. Although the enzyme functions of two Ser/Thr protein kinases PpkA and Stk1 in P. aeruginosa PAO1 have been proved (38-40), the biochemical and biophysical properties concerning prokaryotic Ser protein kinases are still rarely defined. Therefore, this thesis investigates kinase activity and characterizes the metal ion dependence of the
36
enyzme by in vitro phosphorylation assay. The result of this enzymatic study will help us to understand the regulation mechanism of this system further.
2.3 Materials and Methods
2.3.1 Bacterial strains and growth conditions
E. coli XL-1 Blue was used for plasmid preparation and recombinant DNA
manipulation. E. coli BL21 (DE3) was used for protein expression. All bacteria used in this study were grown in Luria-Bertain (LB) medium containing ampicillin (100 μg/ml) or kanamycin (25 μg/ml), with 150 rpm shaking at 37o
C. Nutrient broth (NB) contains nutrient broth powder (DifcoTM) and 0.5% glucose (41). M8 minimal (M8) medium contains 64 g/L Na2HPO4.7 H2O, 15 g/L KH2PO4, 2.5 g/L NaCl, 2 mM
MgSO4, 0.1 mM CaCl2, 0.5% Casamino acid, 0.4% glucose and 10 mM L-glutamate
(42,43). Bacteria strains and plasmids used in this study were listed in Table 2.5.1.
2.3.2 Amino acid sequence alignments
The HATPase region of PA3346, ranging from amino acid position 408 to 571, was used for searching the homologous protein by protein BLAST in the NCBI database, and four proteins predicted as a Ser protein kinase in Rhodopseudomonas
palustris BisA53 (YP_780133), Algoriphagus sp. PR1 (ZP_01718475) Ser-protein
kinase RsbW, Bacillus selenitireducens MLS10 (ZP_02169479) and Chloroflexus
aggregans DSM 9485 (YP_002463389) were found. To analyze the conserved region
in these Ser protein kinases, four additional proteins, Geobacillus stearothermophilus SpoIIAB (AAB81193) (44), B. subtilis SpoIIAB (NP_390227) (45) B. subtilis RsbW (NP_388353) (46), Bordetella bronchiseptica BtrW (NP_888190) (47), were also used in the sequence alignment. The amino acid sequences were analyzed by VectorNTI and the result was shown in Figure 2.6.1.
37
2.3.3 Cloning, expression and purification of His-tag and GST-tag fusion protein
DNA regions encoding PA3347 and the C-terminal region of PA3346 (1222-1713) were amplified by polymerase chain reaction and cloned into pGEX-5X-1 and pET30a, respectively. Primers used in this study are listed in Table 2.5.2. E. coli BL21 (DE3) containing the genes of interest were refreshed in LB medium supplemented with kanamycin or ampicillin with shaking at 37°C until optical density (OD595) reached 0.4 to 0.6. Protein expression was induced by adding
isopropyl-1-thio-β-D-galactopyranoside (IPTG) to a final concentration of 100 μM, and then the bacteria were propagated at 20°C with 150 rpm shaking for 20 h. The bacterial cells were collected by centrifugation at 5000 rpm for 10 min at 4°C. Cell pellets were washed and resuspended in cold suspension buffer (20 mM Tris-HCl, 140 mM NaCl pH 8.0). The cells were broken by sonication and the cell pellets were removed by centrifugation at 14000 rpm for 20 min at 4°C. His-tagged fusion proteins were purified by Nickel-charged resin (Ni sepharose
TM
High Performance, GE Healthcare). The unbinding proteins were washed by wash buffer (20 mM imidazole, 20 mM Tris-HCl, 140 mM NaCl pH 8.0) and the His-tag fusion proteins were eluted by 300 mM imidazole, 20 mM Tris-HCl, and 140 mM NaCl, pH 8.0. The method of induction, protein expression and collection of E. coli BL21 (DE3) containing PA3347-GST-tagged fusion plasmid was the same as His-tagged fusion protein described above. The GST-tagged protein were purified via glutathione sepharose
TM
4 fast flow resin (GE Healthcare), washed by PBS buffer and eluted by 50 mM Tris-HCl buffer containing 10 mM reduced form glutathione pH 8.0 (48). Finally, those purified proteins were dialyzed against the dialysis buffer (50 mM Tris-HCl, 200 mM KCl, 10 mM MgCl
2, 1 mM EDTA, 1 mM DTT and 50% glycerol) and
38
2.3.4 In vitro phosphorylation assay
To detect the Ser protein kinase activity, PA3346
L408-A571
and PA3347 were incubated in buffer containing 100 mM Tris-HCl pH 7.5, 50 mM KCl, 1.5 mM MgCl
2,
1 μCi [γ-32P] ATP or 2 mM ATP at 37°C for 1 h. The protein ratio of PA3346
L408-A571
and PA3347 was 1:1; as for in vitro phosphorylation assay of another putative anti-sigma antagonist protein PA2797, GST and GST-PA3347 proteins were used as negative control and positive control, respectively. GST-PA2797 (2 g) was incubated with PA3346L407-A571 (1 g) and 1 mM ATP at 37oC for 10, 30 and 60 min. The reactions were quenched by adding the same volume of SDS-PAGE loading buffer (50 mM Tris-HCl pH 6.8, 100 mM DTT, 2% sodium dodecyl sulfate, 0.1% bromophenol blue, 10% glycerol, and 10% β-mercaptoethanol) and heated at 95oC for 10 min. The phosphorylation pattern was detected by autoradiography or Pro-Q® Diamond phosphoprotein gel stain (Molecular Probes, USA). Pro-Q® Diamond phosphoprotein gel stain is an in-gel detection method of protein phosphorylation. This fluorencence dye could detecte the phosphate group attached on Ser, Thr or Tyr residues (49,50) and the signals could be visualized by using a Typhoon 9200 Imager (GE Healthcare) or a UV Box.
2.3.5 Protein pull-down assays
Glutathione agarose beads were suspended with the equal volume of PBS buffer (pH 7.4). Equal quantity of PA3347-GST (27.4 g) and PA3346L408-A571-His
6 were
incubated with 500 μl of 50% slurry of glutathione agarose beads and 2 mM MgCl2 in
the presence or absence of 2 mM ADP. After end-over-end mixing at 4oC for 2 h, the protein complexes were washed by 1 ml PBS buffer, eluted with 20 mM glutathione