Item 987654321/17476
67
0
0
全文
(2) 國立中央大學圖書館 碩博士論文授權書 本授權書所授權之論文全文與電子檔,為本人於國立中央大學,撰寫 之碩/博士學位論文。(以下請擇一勾選) ( V )同意 (立即開放) (. )同意 (一年後開放),原因是:. (. )同意 (二年後開放),原因是:. (. )不同意 ,原因是:. 授與國立中央大學圖書館,基於推動讀者間「資源共享、互惠合作」 之理念,於回饋社會與學術研究之目的,得不限地域、時間與次數, 以紙本、光碟、網路或其它各種方法收錄、重製、與發行,或再授權 他人以各種方法重製與利用。 研究生簽名: 論文名稱:. 林 祐 仲 經表面處理氮化鎵之特性研究. 指導教授姓名: 系所 : 學號:. 李 清 庭. 光電科學研究所. V. o博士. o碩士 班. 87246001. 日期:民國 90 年. 6. 月. 22. 日. 備註; 1. 本授權書親筆填寫後( 電子檔論文可用電腦打字) ,請影印裝訂於紙本論文 書名頁之次頁,未附本授權書,圖書館將不予驗收。 2. 上述同意與不同意之欄位若未勾選,本人同意視同授權立即開放。 2.
(3) 論文摘要. 在本次研究中,利用硫化銨 ((NH4) 2Sx) 溶液進行 N 型氮化鎵 (GaN) 表面處理,於蒸鍍鈦/鋁 (Ti/Al) 金屬後即形成特徵接觸電阻 值 5.0× 10-5 Ω-cm2 之歐姆接觸 (ohmic contact)。然後,再將它置於 高溫爐在氮氣環境下,以 300oC 進行熱處理 3 分鐘即可獲得低的特徵 接觸電阻值 3.0×10-6 Ω-cm2 。另外,對於氮化鎵硫化處理效應更以各 項實驗來加以驗證與推算,其包括:能夠完全去除氮化鎵表面原生氧 化層,並且改善氮化鎵表面特性及提高表面載子濃度,以致於能夠形 成非經熱處理之歐姆接觸、於氮化鎵表面形成鎵-硫(Ga-S) 鍵結防止 表面再氧化、能夠減少表面態密度及表面複合速度和增加光子激發光 光 譜 (photoluminescence) 之 強 度 、 能 獲 得 較 好 的 蕭 特 基 接 觸 (Schottky contact) 特性。. I.
(4) 目錄. 論文摘要--------------------------------------------------I 圖目------------------------------------------------------V 表目----------------------------------------------------VIII 一、前言 1. N 型氮化鎵之歐姆接觸--------------------------------1 2. 半導體的表面硫化處理-------------------------------3. 二、實驗方法與量測理論 1. X 光光電子能譜儀量測--------------------------------5 2. 反應離子蝕刻系統-----------------------------------6 3. 傳輸線模型-----------------------------------------7 4. 電容-電壓量測-------------------------------------8 5. 光子激發光光譜測量---------------------------------9. 三、 實驗步驟 1. 實驗 A---利用 XPS 觀測未使用與使用硫化銨溶液處理的氮化 鎵表面之差異----------------------------11. II.
(5) 2. 實驗 B---觀測未使用與使用硫化銨溶液處理的氮化鎵於蒸鍍 金屬電極後未經熱處理與經過熱處理之電特性分 析與比較--------------------------------12 3. 實驗 C---估算氮化鎵使用硫化銨溶液處理前後之表面態密度 變化------------------------------------13 4. 實驗 D---光子激發光光譜測量未硫化與經過硫化處理之氮 化鎵------------------------------------14. 四、 實驗結果與討論 1. 利用 XPS 觀測未使用與使用硫化銨溶液處理的氮化鎵表面 之差異-------------------------------------------16 2. 觀測未使用與使用硫化銨溶液處理的氮化鎵於蒸鍍金屬電極 後未經熱處理與經過熱處理之電特性分析與比較 -------------------------------------------------17 3. 估算氮化鎵使用硫化銨溶液處理前後之表面態密度變化 ------------------------------------------------23 4. 光 子 激 發 光 光 譜 測 量 未 硫 化 與 經 過 硫 化 處 理 之 氮 化 鎵 ------------------------------------------------27. III.
(6) 五、結論 ---------------------------------------------------------30 參考文獻 ---------------------------------------------------------32 本人發表之文章 ---------------------------------------------------------36. IV.
(7) 圖目. 圖 2-2-1 傳輸線模型完成之示意圖。 圖 2-2-2 傳輸線模型量測所得的電阻值為距離 L 之函數示意圖。 圖 3-3-1 蕭特基二極體結構圖。 圖 4-1-1 未硫化處理與經過硫化處理之 N 型氮化鎵 Ga2p3/2 的 XPS 能譜 圖。 圖 4-1-2 未硫化處理與經過硫化處理之 N 型氮化鎵 O1s 的 XPS 能譜圖。 圖 4-1-3 未硫化處理與經過硫化處理之 N 型氮化鎵 N1s 的 XPS 能譜圖。 圖 4-1-4 經過硫化處理之 N 型氮化鎵 S2p 的 XPS 能譜圖。 圖 4-2-1 四種不同表面處理 (10、20、30 分鐘(NH 4) 2Sx 溶液處理和 KOH 溶液處理) 之試片於蒸鍍 Ti(50 nm)/Al(150 nm)後的電流 電壓 (I-V) 特性曲線,其電極間距 25 微米。 圖 4-2-2 經過 20 分鐘(NH4) 2Sx 溶液處理試片之特徵接觸電阻值量測結 果。 圖 4-2-3 經過 30 分鐘(NH4) 2Sx 溶液處理試片之特徵接觸電阻值量測結 果。 圖 4-2-4 XPS 觀測未硫化處理和 10、20 和 30 分鐘硫化處理之氮化鎵 表面 S/N 原子濃度比。. V.
(8) 圖 4-2-5 XPS 觀測未硫化處理和 10、20 和 30 分鐘硫化處理之氮化鎵 表面 IGa-O/IGa-N 訊號峰強度比。 圖 4-2-6 經過 KOH 溶液處理之 N 型氮化鎵 O1s 的 XPS 能譜圖。 圖 4-2-7 經過 10、20 和 30 分鐘(NH4) 2Sx 溶液處理以及 KOH 溶液處理 之試片於氮氣環境下,以 300oC 進行熱處理,觀測特徵接觸 電阻值和熱處理時間之關係。 圖 4-2-8 未經熱處理 Ti/20min (NH 4) 2Sx-treated GaN 之 Ga2p3/2 的 XPS 能譜圖,並進行剖面分析。 圖 4-2-9 未經熱處理 Ti/20min (NH4) 2Sx-treated GaN 之 N1s 的 XPS 能譜圖,並進行剖面分析。 圖 4-2-10 未經熱處理 Ti/20min (NH 4) 2Sx-treated GaN 其界面之 S2p 的 XPS 能譜圖。 圖 4-2-11 20min (NH4) 2Sx-treated GaN 其表面之 Ga2p3/2 的 XPS 能譜 圖。 圖 4-2-12 未經熱處理 Ti/20min (NH 4) 2Sx-treated GaN其界面之 Ga2p3/2 的 XPS 能譜圖。 圖 4-2-13 Al/Ti/KOH-treated GaN 和 Al/Ti/(NH 4) 2Sx –treated GaN 的電特性。 圖 4-2-14 估算經過 20 分鐘硫化以後在氮化鎵近表面處所造成之電阻. VI.
(9) 的示意圖。(a) Al/Ti/KOH-treated GaN (b) Al/Ti/(NH4) 2. Sx –treated GaN。. 圖 4-3-1 利用 C-V 法量測未使用硫化處理和經過硫化處理的蕭特基二 極體之 A2/C2 和 V 的關係圖。 圖 4-3-2 未硫化與硫化之蕭特基二極體於順偏電壓時的 I-V 曲線圖。 圖 4-3-3 未硫化與硫化之蕭特基二極體於逆偏電壓時的 I-V 曲線圖。 圖 4-4-1 於 25K 觀測未硫化處理之氮化鎵試片的 PL。 圖 4-4-2 PL 於 25K 下觀測未硫化處理和 10、20 和 30 分鐘硫化處理 之氮化鎵試片的能帶邊緣輻射比例。 圖 4-4-3 PL 於 25K 觀測未硫化處理和 10、20 和 30 分鐘硫化處理之 氮化鎵試片。. VII.
(10) 表目. 表 4-1-1 XPS 觀測未硫化處理和 20 分鐘硫化處理之氮化鎵 Ga/N 原子 濃度比與距表面深度之關係。 表 4-3-1 C-V 法測得未硫化處理和 20 分鐘硫化處理之氮化鎵蕭特基二 極體的載子濃度和蕭特基位障。 表 4-3-2 C-V 法測得未硫化處理和 20 分鐘硫化處理之氮化鎵蕭特基二 極體的表面能帶彎曲值與表面態密度估計值。 表 4-3-3 I-V 法測得未硫化處理和 20 分鐘硫化處理之氮化鎵蕭特基二 極體的理想因子和蕭特基位障。. VIII.
(11) 一、 前言. 1. N 型 氮 化 鎵 之 歐 姆 接 觸 最近有非常多有關 III 族氮化物及其相關材料的研究與發展,並成 功地製造出電子與光電元件[1-3]。由於 III 族氮化物為寬能隙半導體, 要製作低電阻的歐姆接觸較不容易,因此截至目前有不少 III 族氮化 物和金屬之歐姆接觸的研究。早期的研究發現使用鋁和金 (Au) 蒸鍍 於氮化鎵 (GaN) 可獲得歐姆接觸,不過其特徵接觸電阻值(specific contact resistance,ρC) 仍相當高,約 10-3-10-4 Ωcm2。[4] Al 在蒸 鍍後顯示歐姆接觸,而 Au 在蒸鍍後形成整流特性,但在經過 575oC 熱 處理後得到歐姆接觸。另有研究 [5]指出蒸鍍 Ti /Al 於 N 型氮化鎵上, 經過高溫熱處理減少氮化鎵表面原生氧化層之後,便可形成歐姆接觸 得到較低之ρ C 值。 對於避免高溫熱處理所產生的缺點因此對於非熱合金化 (nonalloyed) 歐姆接觸的製程技術顯得十分重要。M. E. Lin 等人[5] 使用一個 10 周期的 InN(10 nm)/GaN(10 nm) 短周期超晶格結構 (short-period superlattice) 來達成氮化鎵非熱合金化歐姆接觸。 另外,J. D. Guo 等人[6]發現將 Ti/Ag (銀) 蒸鍍於高載子濃度的 N 型 氮化鎵 (1.7× 1019 cm -3),不需經過加熱處理即可得到歐姆接觸。K.V.. 1.
(12) Vassilevski 等人[7]也發現將 Ti/Ni (鎳) 蒸鍍於載子濃度 1.0× 1018 cm-3 的氮化鎵上,並於蒸鍍過程將基底加熱至 350oC,當蒸鍍完成後便可形 成歐姆接觸,但此歐姆接觸之熱穩定性很差。另一方面,J. K. Sheu 等人[8]也已經報導非熱合金化 Ti/Al 與氮化鎵的歐姆接觸,不過其特 徵接觸電阻值約 10-3-10-4 Ωcm2,對於元件的應用此特徵接觸電阻值仍 稍嫌偏高。N. A. Papanicolaou 等人[9]從實驗結果發現蒸鍍 Ti/Al 於 N 型氮化鎵 (載子濃度為 1.1×1018 cm-3) ,可得到約 10-3Ωcm2 之特徵 接觸電阻值,此值仍偏高。A. N. Bright 等人[10]也已經得到 3.6×10-5 Ωcm2 之特徵接觸電阻值,對於蒸鍍 Ti/Al 於載子濃度 4.5× 1018 cm-3 之 N 型氮化鎵上,不過再經過 335OC 的熱處理之後卻變差,反而形成整流 特性。 因此,在本次研究其目的便是欲製作其具有較低特徵接觸電阻值 的非熱合金化歐姆接觸於載子濃度低於 1018 cm-3 的 N 型氮化鎵,本論 文選用 Ti/Al 作為歐姆接觸之金屬電極。然而,要實現未經過熱處理 之歐姆接觸的首要條件便是要完全去除 N 型氮化鎵表面的原生氧化 層。H. Ishikawa 等人[11]曾報導其氮化鎵表面有一層厚約 2 nm 的氧化 鎵 (Ga 2O3) 和吸附的碳或碳氫化合物的污染物。L. L. Smith 等人[12] 也發現當氮化鎵在酸性溶液中處理過後只要於很短時間曝露在空氣 中,其表面便又會迅速地形成一層氧化層。總而言之,對於清除氮化. 2.
(13) 鎵表面的氧化層的方法,在這次研究中扮演重要的角色。. 2. 半 導 體 的 表 面 硫 化 處 理 在半導體及其元件的歷史,其表面/界面 (surface/interface) 的 控制是一個重要的問題。目前矽的界面的控制已經被解決,不過在其 它半導體方面 (砷化鎵、氮化鎵等)仍舊有待解決,甚至還未充分了解。 對於前人研究所報導表面硫化處理的種類有 H2S 、Na2S、(NH4) 2S、 (NH 4) 2Sx 和 CH3CSNH 2 等化學氣體或溶液。第一次在硫化處理的實驗是使 用 H2S 氣體。[13]不過,最早所報導有效濕式的硫化處理是 Na2S。 [14]在 文獻 (15) 和 (16) 也提出半導體在經過 Na2S 處理其氧化層被部份去 除,並改善雙極性電晶體之光子激發光光譜 (photoluminescence,簡 稱 PL) 或電流放大係數β,並且還指出減少表面複合速度 (surface recombination velocity)。 不過,B. J. Skromme 等人[17]卻發現這 Na2S 處理過的半導體表面在經過水洗後其改善的情形竟消失了。後 來 , M. S. Carpenter 等 人 [18] 實驗結果發現對於正-負接面 (p-n junction) 的改善其使用(NH4) 2S 處理比用 Na2S 有效。另一方面,使 用(NH4) 2S 和(NH4) 2Sx 溶液進行砷化鎵硫化處理仍十分有效。利用(NH4) 2. Sx 處理後會增強 PL 強度[19]、全部清除氧化層、蕭特基位障仰賴其接. 觸 金 屬 的 功 函 數. [20]. 以 及 改 善 金 屬 -絕 緣 體 -半導體. 3.
(14) (metal-insulator-semiconductor, 簡 稱 MIS) 結 構 之 電 容 -電 壓 (capacitance-voltag,簡稱 C-V) 特性[21,22]。P. S. Dutta 等人[23]則 認為在硫化處理所用的各種化學溶液中,以使用(NH4) 2Sx 處理其效果 最好。至於 InP 的 MIS 結構[24]以及其它 III-V 族化合物半導體材料[25] 使用(NH4) 2Sx 處理均是有效的。X. Zhang 等人[26]也報導使用 CH3CSNH2 溶液進行 III-V 族半導體表面鈍化 (passivated),其實驗結果顯示表 面形成穩定的硫鈍化層,並且降低表面非輻射複合和表面態(surface state)密度以及增加 PL 強度。 至於硫化處理應用於氮化鎵材料之研究則正處於發展的階段。X. A. Cao 等人[27]研究發現氮化鎵經(NH4) 2Sx 溶液處理之後會完全清除其表 面原生氧化層,而且因為在氮化鎵表面形成 Ga-S 的鍵結,於是可防止 再氧化的情形發生。還有,G. L. Martinez 等人 [28]也報導氮化鎵經(NH4) 2. Sx 溶液處過以後,減少表面複合速度和表面態密度,而且其觀測所得. 的 PL 強度增加為未經過表面處理的 4 至 6 倍,甚至將硫化過的氮化鎵 放置一段時間,其 PL 強度仍未衰減。 在這次的研究,為了能有效去除氮化鎵表面的原生氧化層並防止 再次氧化發生,因此在蒸鍍金屬電極前將氮化鎵進行(NH4) 2Sx (硫化銨) 處理,以便獲得良好的金屬半導體接面,達到非熱合金歐姆接觸。 並進行各項實驗,以便了解半導體於硫化處理後所造成的效應。. 4.
(15) 二、 實驗方法與量測理論. 1. X 光光電子能譜儀量測 (x-ray photoelectron spectroscopy,簡 稱 XPS) [29-31] X 光光電子能譜儀是藉著測量 X 光光電子能譜來分析材料的表面 特性,包括化學元素種類和化學鍵結型態,它也被稱為電子能譜化學 分 析 儀 (electron spectroscopy for chemical analyzer , 簡 稱 ESCA)。它的基本原理即是光電效應,利用 X 光照射到半導體的表面可 以激發游離光電子 (photoelectron)。藉由量測光電子的動能可以推 算出該光電子的束縛能 (binding energy),因此它可應用在研判發射 光電子的元素的種類以及其化學的鍵結情形。X 光光電子能譜儀可說 是目前化學表面分析非常靈敏一種量測技術。 被測定物試片的電子被特定深度量子化的能階束縛,對此類試片 照射特定能量的 X 光,於是被特定能階束縛的電子得以脫離,因此我 們能夠測得從此試片表面放出的各個特定動能形成光電子能譜圖。 X 光光電子能譜儀的能譜上之主要訊號來自於光電子,以光電子能 譜圖的能量值進行元素分析,以強度分析元素濃度,以化學位移分析 狀態。因此光電子訊號峰的位置及形狀與原子內的電子組態結構有密 切的關聯;由於電子自旋角動量與電子軌域角動量的藕合作用 (l -s. 5.
(16) coupling),對於 p、d、f 等電子軌域角動量大於零者會分裂成為高低 兩種能階。因此當光電子的發射不來自 s 副軌域時會產生兩條能譜 線,以 2p 電子而言,其符號分別為 2p1/2 和 2p3/2,兩者能量位置差異 即是這兩能階的位能差。當原子序很小時這能差小於 0.5 eV,而當原 子序越高則此能差增加。在此次的研究中對於硫原子的 2p 能譜線便必 須考慮 2p1/2、2p3/2 的組合分析。 X 光光電子能譜儀之裝置包含激發用 X 光源、電子能分析器、真空 系統和紀錄系統所組成。X 光光源常用 Mg Kα線或 Al Kα線。若假設 照射 X 光的量子能為 hυ,從表面放出的光電子動能為 Ek,測定電子 的束縛能量為 Eb,則它們之間關係為: Ebv = hυ - Ek – ( Φ SP – Φ S ) = EbF +Φ S Ebv 是從真空能階測定時的束縛能,ΦSP 和 Φ S 分別為分光裝置及試片 的功函數,EbF 是從費米能階測定的束縛能。 在本次研究中為了校正所觀測的 XPS 能譜圖,我們使用 Au 4f7/2 (峰 值位於 83.86 eV)和 Cu 2p3/2 (峰值位於 932.65 eV)。觀測的角度採用 45o。. 2. 反應離子蝕刻系統 (reactive ion etching,簡稱 RIE). [32]. 在本次研究,我們利用 RIE 來進行氮化鎵的蝕刻,例如製作 TLM. 6.
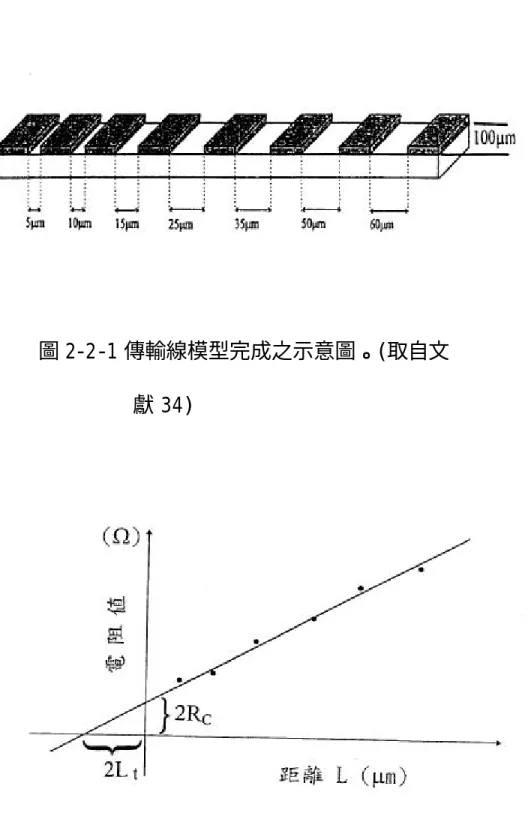
(17) 所需的高臺,還有製造蕭特基二極體中的圓柱狀蕭特基電極。 首先放試片於真空室 (chamber) ,然後將真空室抽真空,放置試 片的電極接射頻電源 (RF power 13.56 MHz) ,另一電極接地。然後 將選用的氣體導入真空室在兩電極間形成電漿,此時位在電漿中的試 片除了與到達試片表面的活性基 (reactive radical) 起化學反應 外,同時受到因電壓降而加速得離子的濺擊 (sputtering),因此包含 化學和物理蝕刻,其蝕刻速率快,其蝕刻機制具有非等向性 (anisotropic) 和高選擇性 (selectivity)。 在電漿蝕刻時,電漿中含有活性原子、活性離子及電子,當壓力 較低而且氣體兩端所加功率夠高時,活性離子便被迅速加速衝擊試片 表面,將其表面層的原子擊出,再與活性原子反應,而達到蝕刻的目 地,此即為 RIE 蝕刻原理。. 3. 傳輸線模型 (transmission line model,簡稱 TLM)[33,34] 在此次的研究,我們使用傳輸線模型來計算金屬半導體接面的特 徵接觸電阻 (specific contact resistance,ρC),藉由所得到的ρ C. 值來判斷其歐姆接觸 (ohmic contact) 特性的好壞。 從 TLM 得知:. ρ C = RC Lt W. (2-2-1). 7.
(18) 其中 R C 為接觸電阻值 (contact resistance,單位為歐姆),Lt 為轉 換長度 (transfer length,單位為微米),W 是電極之寬度 (單位為 微米) 。 圖 2-2-1 為此次研究所使用之 TLM 之圖樣,其每個電極的寬度 (W) 為 100 微米。我們設計兩兩電極間具有不同間距,其間距分別為 5、 10、15、25、35、50 和 60 微米,因此我們可測得七組不同間距的電 阻值,然後畫出電阻與間距 (R-L) 的關係圖,並將此七點做直線逼近 (linear fit),在 L=0 處之 R 軸截距就是 2RC,在 R=0 處之 L 軸截距 就是 2Lt (見圖 2-2-2)。然後,將此計算所得的 R C、Lt 代入 (2-2-1) 式便可得到ρ C 值。. 4. 電容-電壓 (capacitance-voltage,簡稱 C-V)法量測 對於蕭特基二極體的位障可以電容-電壓法來測得,其蕭特基位 障 (Schottky barrier height,簡稱 SBH)的電容-電壓關係如下 (1/C)2 = (2/εqNA2)( V i –V–kT/q). (2-3-1). 其中 V 為逆向偏壓之電壓值,A 為蕭特基電極的面積,ε為半導體的 介電常數 (氮化鎵的ε值為 9.5ε O[35])。 qΦ B = q (Vi +Vn). (2-3-2). 其中 qΦB 為 SBH,Vi 為內建電位(built-in potential),而 (2-3-2). 8.
(19) 式中的 Vn (導帶與費米能階的能量差) 如下 Vn = (kT/q)ln(NC/N). (2-3-3). 上式中的 NC 為半導體傳導帶之有效能階密度 (氮化鎵的 NC 值為 2.53× 1018 cm-3. [36]. )。. 於是,我們利用所測量所得的單位面積電容與電壓之關係後,利 用 (2-3-1)式得出雜值分佈。若 N 為定值則 A2/C2 對 V 作圖可得一條直 線,從此直線找出當 A2/C2 =0 的截距 (Vo,Vo = V i - kT/q) 即可得到 內建電位 Vi。然後將 N 代入 (2-3-3)式便可算出 Vn。最後將先後得到 的 Vi 和 Vn 代入 (2-3-2)式便可求出蕭特基位障。. 5. 光子激發光光譜 (photoluminescence,簡稱 PL) 測量 PL 量測系統是以氦鎘(He-Cd)雷射當光源,其原理為當雷射光入射 到真空室中的試片時,雷射所供給的能量必須大於半導體能隙的能 量,如此才能使位於價帶的電子獲得足夠能量躍升到導帶。換句話說, 也就是當光子入射到試片之價帶的位置,產生電子電洞對,並給予電 子大於能隙的能量,然後電子會再從導帶掉至價帶,此行為包含幾種 機制,分別是導帶的最低點到價帶的最高點的移動與結合,還有導帶 到受體 (acceptor)、施體(donor)到價帶,施體到受體的位置,都有 機會發生電子和電洞的結合與產生。也就是說,當電子從高能階掉至. 9.
(20) 低能階與電洞結合時便會放出光子。若電子從導帶直接掉到價帶,則 放出光的能量剛好為 hυ = EC – EV (eV) 也就是能隙之大,υ是光 的頻率,EC 是導帶的能量,EV 為價帶的能量。綜觀上述,不難理解利 用 PL 進行試片測量能夠得知材料的能隙,然後對發光元件所測出的波 長或頻率得以更深入探討元件內部結構與其缺陷 (defect)。. 10.
(21) 三、 實驗步驟. 1.實驗 A---利用 XPS 觀測未使用與使用硫化銨溶液處理的氮化鎵表面 之差異 <步驟> (a). 此 次 實 驗 所 選 用 氮 化 鎵 是 利 用 有 機 化 學 氣 相 沉 積 法 (metalorganic chemical vapor deposition,簡稱:MOCVD)長 於藍寶石基底上。先成長 650 nm 厚的緩衝層於藍寶石基底上, 其溫度為 520oC,再成長 1.2 微米厚的 Si 摻雜 N 型氮化鎵,其溫 度為 1050oC。磊晶成長源是高純度之三甲基鎵 (TMGa) 及氨氣 (NH 3), 其 Si 摻雜源為 SiH4 。 此 N 型氮化鎵之載子濃度為 6.2×1017cm-3,遷移率為 350 cm2/V-s。 (b).將兩片相同氮化鎵試片分別以下列化學藥品各處理五分鐘:三氯 乙烯(TCE)、丙酮(ACE)和甲醇(methonal)等溶液。 (c).將試片以去離子水(DI water)沖洗五分鐘並用氮氣槍吹乾。 (d).取其中一片試片放入 60oC 的黃色硫化銨溶液 (含硫量為 6%) 中持 續加熱 20 分鐘。然後浸於去離子水中再用氮氣槍吹乾。 (e).利用 XPS 觀測此兩片氮化鎵試片。. 11.
(22) 2. 實驗 B---觀測未使用與使用硫化銨溶液處理的氮化鎵於蒸鍍金屬 電極後未經熱處理與經過熱處理之電特性分析與比較 <步驟> (a).此次實驗所選用氮化鎵是利用有機化學氣相沉積法長於藍寶石 基底上。先成長 650 nm 厚的緩衝層於藍寶石基底上,其溫度為 520 oC,再成長 1.2 微米厚的 Si 摻雜 N 型氮化鎵,其溫度為 1050 oC。磊晶成長源是高純度之三甲基鎵 (TMGa) 及氨氣 (NH3), 其 Si 摻雜源為 SiH4。此 N 型氮化鎵之載子濃度為 6.2× 1017cm-3, 遷移率為 350 cm2/V-s。 (b).製作 TLM 之矩形高台於氮化鎵表面:先蒸鍍 Ni/Au(50/600 nm) 於 氮化鎵表面製作高台形狀的金屬面罩,然後利用反應離子蝕刻系 統(RIE)來進行氮化鎵的乾式蝕刻。然後將試片浸泡於王水溶液 中,以便移除 Ni/Au 金屬面罩。 (c).將氮化鎵試片切割成四片,分別以下列化學藥品各處理五分鐘: 三氯乙烯、丙酮和甲醇等溶液。 (d).將四片試片以去離子水沖洗五分鐘並用氮氣槍吹乾。 (e).取其中三片試片放入 60oC 的黃色硫化銨溶液 (含硫量為 6%) 中分 別持續加熱 10、20 和 30 分鐘。然後浸於去離子水中再用氮氣槍 吹乾。. 12.
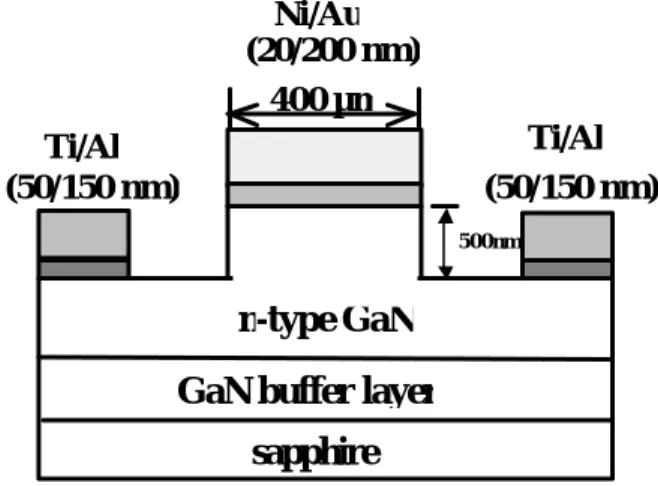
(23) (f).取其中一片試片放入 70oC 1M KOH 溶液中持續加熱 30 分鐘。然後 浸於去離子水中再用氮氣槍吹乾。 (g).將四片試片利用曝光顯影技術完成欲蒸鍍金屬電極的圖案,接著 再以電子槍蒸鍍機(E-gun evaporator)蒸鍍鈦/鋁(50/150 nm)於 試片上。 (h).利用 HP4145B 半導體元件參數分析儀量測此四組試片的電特性。 (i).將所有製作完成之試片放入高溫爐於氮氣環境,以 300oC 進行熱 處理並用 HP4145B 分別量測其電特性。. 3. 實驗 C---估算氮化鎵使用硫化銨溶液處理前後之表面態密度變化 <步驟> (a).此次實驗製作氮化鎵蕭特基二極體,其所選用氮化鎵是利用有機 化學氣相沉積法長於藍寶石基底上。先成長 650 nm 厚的緩衝層 於藍寶石基底上,其溫度為 520oC,再成長 1.2 微米厚的 Si 摻雜 N 型氮化鎵,其溫度為 1050oC。磊晶成長源是高純度之三甲基鎵 (TMGa) 及氨氣 (NH3),其 Si 摻雜源為 SiH4。此 N 型氮化鎵之載 子濃度為 6.2×1017cm-3,遷移率為 350 cm2/V-s。 (b).製作蕭特基電極圓柱狀之高台於氮化鎵表面 (圓形接面的直徑為 400 微米):先蒸鍍 Ni/Au(50/600 nm) 於氮化鎵表面製作圓柱高. 13.
(24) 台形狀的金屬面罩,然後利用反應離子蝕刻系統 (RIE) 來進行 氮化鎵的乾式蝕刻。蝕刻深度估計約為 500 nm。 (c).將兩片相同氮化鎵試片分別以下列化學藥品各處理五分鐘:三氯 乙烯、丙酮和甲醇等溶液。 (d).將兩片試片以去離子水沖洗五分鐘並用氮氣槍吹乾。 (e).將兩片試片放入 60oC 的黃色硫化銨溶液 (含硫量為 6%) 中持續加 熱 20 分鐘,再以去離子水沖洗並用氮氣槍吹乾。 (f).製作蕭特基二極體的歐姆電極: 將兩片試片利用曝光顯影技術完 成欲蒸鍍歐姆金屬電極的圖案,再以電子槍蒸鍍機蒸鍍鈦/鋁 (50/150 nm)於試片上,然後於氮氣中以 300oC 進行熱處理 4 分鐘。 (g).將其中一片試片放入 60oC 的黃色硫化銨溶液 (含硫量為 6%) 中持 續加熱 20 分鐘,再以去離子水沖洗並用氮氣槍吹乾。 (h).然後將兩片試片浸泡於王水溶液中,以便移除 Ni/Au 金屬面罩。 (i).製作蕭特基二極體的蕭特基電極:將兩片試片利用曝光顯影技術 完成欲蒸鍍蕭特基金屬電極的圖案,再以電子槍蒸鍍機蒸鍍鎳/ 金(20/200 nm)於試片上。此圓形蕭特基電極的直徑為 400 微米。 (j).圖 3-3-1 為完成之蕭特基二極體結構圖。. 4. 實驗 D--光子激發光光譜測量未硫化與經過硫化處理之氮化鎵. 14.
(25) <步驟> (a).此次實驗所選用氮化鎵是利用有機化學氣相沉積法長於藍寶石 基底上。先成長 650 nm 厚的緩衝層於藍寶石基底上,其溫度為 520 oC,再成長 1.2 微米厚的 Si 摻雜 N 型氮化鎵,其溫度為 1050 oC。磊晶成長源是高純度之三甲基鎵 (TMGa) 及氨氣 (NH3), 其 Si 摻雜源為 SiH4。此 N 型氮化鎵之載子濃度為 6.2× 1017cm-3, 遷移率為 350 cm2/V-s。 (b).將氮化鎵試片切割成四片,分別以下列化學藥品各處理五分鐘: 三氯乙烯、丙酮和甲醇等溶液。 (c).將四片試片以去離子水沖洗五分鐘並用氮氣槍吹乾。 (d).取其中三片試片放入 60oC 的黃色硫化銨溶液 (含硫量為 6%) 中分 別持續加熱 10、20 和 30 分鐘。然後浸於去離子水中再用氮氣槍 吹乾。 (e).將此四片經過表面處理之氮化鎵試片利用 PL 於室溫和 25 K 下進 行測量。. 15.
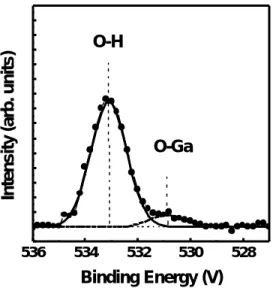
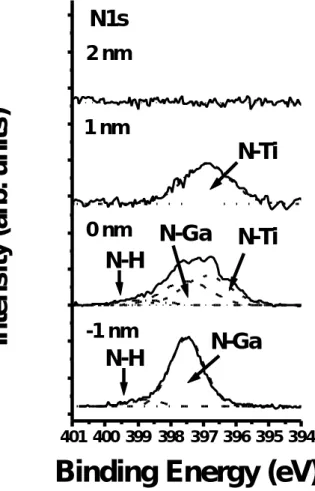
(26) 四、 實驗結果與討論. 1. 利用 XPS 觀測未使用與使用硫化銨溶液處理的氮化鎵表面之差異. 圖 4-1-1 顯示 Ga2p3/2 XPS 的能譜圖,從圖中我們發現對於未經過 硫化銨溶液處理的氮化鎵表面有 Ga-N 的鍵結 (位在 1117.1 eV[37]) 以 及 Ga-O 的鍵結 (位在 1118.9 eV[38] )。然而對於經過硫化處理的試片 除了有 Ga-N 的鍵結(位在 1117.1 eV)、Ga-S 的鍵結 (位在 1118.6 eV[38] ) 外並未發現 Ga-O 的鍵結。從上述結果可知,氮化鎵經過硫化銨溶液處 理之後其表面的原生氧化層可以被去除並且因為形成 GaSx 而使表面鈍 化了。 圖 4-1-2 顯示 O1s XPS 的能譜圖,對於未硫化的試片其表面發現 O-Ga 的鍵結 (位在 530.8 eV [38]) 和 O-H 的鍵結 (位在 533.1 eV [39]), 不過經過硫化銨溶液處理之後我們只發現 O-H 的鍵結,至於 O-Ga 的鍵 結則完全被去除了。 圖 4-1-3 顯示 N1s XPS 的能譜圖,從圖中我們發現氮化鎵在硫化 處理前後,其 N1s XPS 的能譜圖並未有什麼不同,同樣顯示 N-Ga 鍵結 (397.5 eV[37] )、N-H 鍵結 (398.8 eV[40])。對於 N-H 鍵結中的氫推測來 自磊晶成長源,即是高純度之三甲基鎵 (TMGa) 及氨氣 (NH3)。 [41] 另. 16.
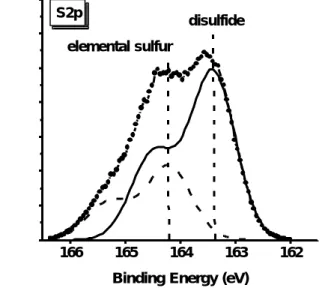
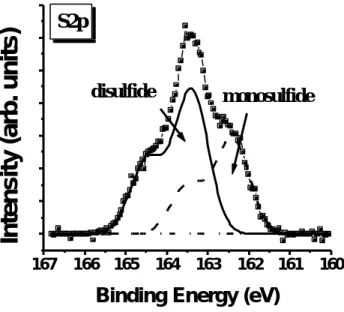
(27) 外,我們並未發現氮與氧或氮與硫所形成的鍵結。 圖 4-1-4 為 S2p XPS 能譜圖,其顯示硫化後的氮化鎵表面硫的鍵 結情形。基於考慮由於電子自旋角動量與電子軌域角動量的藕合作 用,S2p1/2:S2p 3/2=1:1.95[42],兩者 (S2p 1/2、S2p 3/2) 分隔的能量差為 1.08 eV 以及兩者半高寬為 0.96 eV[43],我們分析得到兩種硫的鍵結情 形。一種屬於元素硫(elemental sulfur)鍵結的 S2p3/2 的峰值位在 164.2 eV[44],另一種屬於雙硫 (disulfide,Ga-S-S-Ga) 鍵結的 S2p3/2 的峰值位在 163.4 eV,此兩種鍵結的束縛能差值約在-0.3到-0.9eV [45]。 表 4-1-1 顯示在經過硫化處理的氮化鎵表面的 Ga/N 原子比例為 0.38 比未經過硫化處理的氮化鎵表面的值 (Ga/N = 0.68) 要低。這 說明氮化鎵在經過硫化銨溶液進行表面處理之後會移去其表面的氧化 層並形成 N-rich 的表面 (或說是形成相當數量的鎵空位)。 J. K. Kim 等人也發現當氮化鎵表面去除這原生氧化層 (GaO x) 之後其表面會存 在鎵空位。 [46]. 2. 觀測未使用與使用硫化銨溶液處理的氮化鎵於蒸鍍金屬電極後未 經熱處理與經過熱處理之電特性分析與比較. 圖 4-2-1 顯示四種不同表面處理 (10、20、30 分鐘硫化處理和 KOH. 17.
(28) 溶液處理) 之試片其電極間距 25 微米的電流電壓 (I-V) 特性曲線。 我們發現經過20分鐘和 30分鐘硫化處理的氮化鎵試片於蒸鍍鈦/鋁金 屬之後,其 I-V 的曲線為直線顯示有較佳的歐姆特性,經過 TLM 觀測 計算所得到的ρC 值為 5.0× 10-5 Ω-cm2 和 7.8×10-5 Ω-cm2 (見圖 4-2-2 和圖 4-2-3)。至於經過 10 分鐘硫化處理和 KOH 溶液處理之試片的 I-V 曲線為非線性,呈現整流特性。從 XPS 資料分析結果再加上 I-V 特性 觀測,清楚地說明氮化鎵經過 20 分鐘硫化銨溶液處理之後,確實能夠 移除其表面原生氧化層並且因為 Ga-S 鍵結形成表面鈍化,因此於蒸鍍 金屬後便容易形成歐姆接觸。對於 30 分鐘硫化處理也能形成歐姆接觸 之原因在於雖已完全去除表面原生氧化層,但過度硫化使氮化鎵表面 吸附更多硫原子因而輕微影響電特性。然而 10 分鐘硫化處理卻無法形 成歐姆接觸在於因硫化處理時間稍短,並未完全清除其表面原生氧化 層,因此顯示出整流的特性。從圖 4-2-4 和圖 4-2-5 便可清楚了解上 述對 10、20 和 30 分鐘硫化處理所做的電特性分析。圖 4-2-4 顯示經 由 XPS 觀測 10、20 和 30 分鐘硫化處理之氮化鎵表面 S/N 原子濃度比 的情形,對於 30 分鐘硫化處理之試片有最高的比例值。圖 4-2-5 顯示 經由 XPS 觀測未硫化處理和 10、20 和 30 分鐘硫化處理之氮化鎵表面 Ga-O 和 Ga-N 鍵結之訊號峰強度比的情形,對於未經硫化處理之試片 有最高的比例值,其次為 10 分鐘硫化處理之試片。圖 4-2-4 選用 N. 18.
(29) 當作一標準值之理由是氮化鎵於硫化處理前後,N1s 的 XPS 訊號峰變 化不大。除此之外,對於經過 KOH 溶液處理的試片也形成整流特性, 根據文獻 (47) 得知其氮化鎵表面之氧化層 (Ga2O3)也可以被 KOH溶液 清除,不過從 XPS 觀測經過 KOH 溶液處理之氮化鎵表面發現其原生氧 化層被去除但表面卻吸附太多 OH 離子,因而影響其電特性無法形成歐 姆接觸 (見圖 4-2-6)。 圖 4-2-7 顯示將經過 10、20 和 30 分鐘硫化處理以及 KOH 溶液處 理之試片置入高溫爐中於氮氣環境下,以 300oC 進行熱處理。對於 20 和 30 分鐘硫化處理試片分別於熱處理 3 分鐘和 4 分鐘可獲得低的特徵 接觸電阻值 3.0×10-6 Ω-cm2。因為 20 分鐘以上的硫化處理足以完全去 除其表面原生氧化層,因此在 300oC 低溫熱處理時 Ti 和 N 會反應形成 TiN,另外 Al 穿透 Ti 層形成具較低功函數之 Al -Ti 合金[48],於是便 可得到低的特徵接觸電阻值。於 300oC 熱處理形成 Al-Ti 合金也已經 被報導了。 [49] 對於硫化 10 分鐘處理之試片再經過熱處理 8 分鐘得到 最低之特徵接觸電阻值 2.3× 10-5 Ω-cm2。另外,KOH 溶液處理之試片 2 再經過 5 分鐘熱處理後可獲得最低之特徵接觸電阻值 1.3× 10-5 Ω-cm。. 另一方面,S. C. Binari 等人已經報導了當鈦蒸鍍在 N 型氮化鎵 後觀測發現其蕭特基位障為 0.58 eV。[50] 我們認為此次工作之所以能 夠得到良好的非經過熱處理之歐姆接觸可歸納為三個因素。第一,其. 19.
(30) 氮化鎵表面之原生氧化層能夠被硫化銨溶液完全去除,並形成 Ga-S 鍵結防止其表面再度氧化。第二、可以從表 4-1-1 得知,因為硫化後 的氮化鎵為 N-rich 的表面因此在蒸鍍鈦時較易形成 TiN,此具金屬性 的 TiN 不僅有較低的電阻係數 (~20μΩ cm) [51] 還有較低的金屬功函 數 (3.74 eV)[52],因此能形成歐姆接觸。C. T. Wu 和 A. Kahn 也提出 在室溫下對於 Ti/GaN 界面出現 Ti 與 N 反應的現象。[53] S. Ruvimov 等人也利用電子繞射觀測剛蒸鍍 Ti/GaN 界面發現已有 TiN 形成。[54] 為 了證明此一推斷,於此次研究也進行未經過熱處理 Ti(8nm)/20min (NH 4) 2Sx-treated GaN 之 XPS 剖面觀測。從圖 4-2-8 和圖 4-2-9 可以 發現位於 Ti/GaN 界面的氮原子有些微地向外擴散並形成 TiN,這也可 能伴隨產生氮空位進而提高表面載子濃度[8]。此外,我們並未觀測出 鎵原子向外擴散的跡象,其理由是鎵與硫產生鍵結而氮並未與硫形成 鍵結所致,且硫原子於蒸鍍金屬後仍處於界面,文獻 (26) 也提出當 金屬蒸鍍於硫化過的半導體時,其因硫化所產生的硫原子仍留在金半 界面處。另一方面,圖 4-2-10 顯示此位於 Ti/GaN 界面的硫原子之 S2p XPS 能譜圖有向低束縛能移動的趨勢,這和圖 4-1-4 比較,發現其元 素硫之訊號峰於蒸鍍 Ti 後即消失,並產生單硫 (monosulfid) 鍵結的 訊號峰,這可以被推測當氮原子向外擴散時所形成鎵的懸浮鍵 (dangling bonds) 將和多餘的硫原子產生鍵結,從圖 (4-2-11) 和圖. 20.
(31) (4-2-12) 比較發現便可得知,蒸鍍金屬 Ti 前後其 Ga2p3/2 的 XPS 能譜 圖中的 Ga-S 鍵結之訊號峰強度產生變化,其 Ga-S 鍵結之訊號峰強度 和 Ga-N 鍵結之訊號峰強度的比值在蒸鍍 Ti 後明顯變大。文獻 (26) 也 發現此硫之 XPS 能譜圖之訊號峰向低結合能位移的情形。第三,經過 (NH 4) 2Sx 溶液處理後的氮化鎵表面載子濃度被提高,因此於蒸鍍 Ti/Al 後便易於形成歐姆接觸。根據先前 XPS 的剖面資料 (見表 4-1-1) 發 現,在氮化鎵近表面約 1 nm 範圍受到硫化效應影響,因此其硫化所改 善的表面層及被提高載子濃度層推測約在近表面 1 nm 處。 為了估算此提高之表面載子濃度,本研究選用 TLM 之 Al/Ti/20 min (NH4) 2Sx –treated GaN 和 Al/Ti/KOH-treated GaN 兩組試片, 並於氮氣中以 300oC 進行 1 分鐘熱處理,兩組試片均形成歐姆接觸 (見 圖 4-2-13)。因為氮化鎵經過 20 分鐘硫化處理可以全部去除原生氧化 層,另外根據文獻 (47) 得知其氮化鎵表面之氧化層 (Ga2O3)也可以被 KOH 溶液清除,如此一來便可用兩組所獲得之實驗資料來推算因硫化 所提高之表面載子濃度值。首先觀測兩試片各間距之電阻,並利用圖 4-2-14 之簡易模式和公式 (4-1-1) 和公式 (4-1-2) 推算其經過 20 分鐘硫化以後在近表面處所造成之電阻 RSH3。 R= 2RC + RSH. (4-1-1). RSH2 = RSH1∥RSH3. (4-1-2). 21.
(32) 上式中,對於 KOH 處理的試片其 RSH= RSH1,至於(NH4) 2Sx 處理的試片其 RSH= RSH2。另外,R 是用 HP4145B 所量測各間距之電阻。假設測得兩組 不同間距 Al/Ti/KOH-trerated GaN 的 R 值便可消去 RC (接觸電阻) , 並求得 RSH1 的項。同樣地,依照相同的方法也可求出 Al/Ti/20 min (NH4) 2. Sx –treated GaN 的 RSH2,然後從公式 (4-1-2) 算出 RSH3 。接著代入. 公式 (4-1-3) 估算硫化後氮化鎵近表面的電阻率ρL,此ρL 經由公式 (4-1-4) 即可求得硫化後氮化鎵近表面的載子濃度值 n。 RSH3 = ρ L ρL =. L A. (4-1-3). 1 nqµ. (4-1-4). 其中 L 為方形電極之間距,μ為遷移率,q 是基本電荷量,A 是氮化鎵 近表面處之傳遞電流的截面積 (A = 0.001×100 μm2)。 經由上述簡易模式所估算氮化鎵經過 20 分鐘硫化以後其近表面 (~1 nm)處之提高載子濃度值約為 ~1019 cm-3。因為氮化鎵表面載子濃 度的增加,於蒸鍍 Ti/Al 之後並形成良好之歐姆接觸,其特徵接觸電 阻值低至 ~10-5 Ωcm2,此結果可從文獻 (6) 所報導之結論獲得驗證。 在文獻 (6) 提到蒸鍍 Ti/Ag 於載子濃度值為 ~1019 cm-3 之氮化鎵上, 不需經過熱處理即可獲得~10-5 Ωcm2 之特徵接觸電阻值。在這次簡易 模式所估算經過硫化處理的氮化鎵表面載子濃度戲劇性的提高十幾倍 之多,這可能由於因為硫化處理後完全清除表面氧化層並且改善其表 22.
(33) 面狀況、減少表面態密度有關,此外六族的硫原子將會佔據五族氮的 空位形成施體所致(見實驗 D)。. 3. 估 算 氮 化 鎵 使 用 硫 化 銨 溶 液 處 理 前 後 之 表 面 態 密 度 變 化. 圖 4-3-1 是經過硫化處理和未使用硫化處理的蕭特基二極體利用 C-V 法所量測當 A2/C2=0 時的 Vo 值分別為 1.041 eV 和 0.793 eV,圖中 經由直線斜率關係可計算得到經過硫化處理和未使用硫化處理之 GaN 的載子濃度為 7.4×1017 和 6.2×1017cm-3,其顯示經過硫化處理之蕭特基 二極體除了有較大的 Vo 值外,其載子濃度也些微增加。文獻 (28) 也 曾提出氮化鎵經硫化處理後其載子濃度增加的現象。另外,文獻 (55) 指出這是因為硫化後形成 Ga-S 鍵結所致。不過,利用 C-V 法測得的載 子濃度比先前運用簡易模式所估算表面載子濃度值要低,其原因是因 為硫化改善層位於近表面 1 nm 處,此層厚度極薄無法用 C-V 法測量求 得。 表 4-3-1 列出經過硫化處理和未使用硫化處理的蕭特基二極體所 量測的載子濃度值和蕭特基位障。這經過硫化處理的蕭特基二極體其 蕭特基位障值為 1.099 eV,此值非常近似以鎳蒸鍍於 N 型氮化鎵上所 形成的蕭特基位障 (鎳的金屬功函數 (work function) 為 5.2eV[56],. 23.
(34) 而氮化鎵的電子親和力 (electron affinity) 為 4.1eV[57] )。另一方 面,未經過硫化處理的蕭特基二極體其蕭特基位障值 0.856eV,推測 這是因為未經硫化處理之蕭特基二極體其界面有較多表面態密度,並 引發更強的表面能帶彎曲效應。根據 S. J. Pearton 等人[58]指出在已 發表之文獻記載,對於 Ni 於 N 型氮化鎵上所形成的蕭特基位障十分不 一致,其分布的範圍從 0.66eV [59]至 0.99eV [60]。為了進一步探討,我們 考慮表面能帶彎曲項 ( q V S ),希望來調整蕭特基位障與半導體電子 親和力之間的關係。對於一個 N型氮化鎵的表面功函數 (surface work function,qΦ S) 可表示如下: qΦ S = q Vn + χ +. (4-3-1). qVS. 其中χ= 4.1eV。另外,蕭特基位障 (qΦ B) 公式也可顯示如下: qΦ B= qΦ m - qΦ S + q Vn. (4-3-2). 其中鎳的金屬功函數 qΦ m = 5.2eV。 將 (4-3-1)式 代入 (4-3-2)式可得到: qΦ B= qΦ m – χ – q V S. = 5.2 – 4.1– q VS. = 1.1 – q V S (4-3-3). 因此,我們可以利用 (4-3-3)式計算求得表面能帶彎曲項的值,從表 4-3-2 得知未使用硫化處理和使用硫化處理的蕭特基二極體所計算之 表面能帶彎曲值分別為 0.244 eV 和 0.001 eV 。從此結果得知,氮化. 24.
(35) 鎵經過硫化處理所製成的蕭特基二極體具有較小的表面能帶彎曲值。 然後再經由 (4-3-4)式 [61] 的估算,便可得到表面態密度 (surface state density) 的值。. (qN S ) 2 q VS = 2εN sc kT. (4-3-4). 其中 NS 便是表面態密度,而 NSC 為半導體的載子濃度。對於未經過硫化 處理和硫化處理過的蕭特基二極體其 NSC則代入 C-V 所量測到的載子濃 度來計算表面態密度的值。對於鎳蒸鍍於 N 型氮化鎵形成蕭特基位障 後於半導體接面會產生空乏層,經計算得知當電壓為零時此空乏層之 寬度為 39 nm,對於先前用簡易模式所估算 20 分鐘硫化後之氮化鎵近 表面 1 nm 處的表面載子濃度並不適用。 表 4-3-2 顯示經由計算所得的表面態密度,對於未經過硫化處理 的蕭特基二極體其表面態密度值為 2.0× 1011 cm -2,至於經過硫化處理 的蕭特基二極體其表面態密度值為 1.3× 1010 cm-2。這表面態密度值在 歷經硫化銨溶液處理後被降低,因此能夠得到高的蕭特基位障值。H. Hasegawa 等人曾說明了當蕭特基位障不那麼仰賴於金屬功函數的情 況將導至所謂的費米能階被釘住 (Fermi level pinning),[62]不過此 次研究發現並未有非常嚴重的費米能階被釘住的現象。此外,這次研 究所估算出氮化鎵的表面態密度值與前人研究所報導的數量十分近 似。 [63,64] 25.
(36) 對於經過 20 分鐘硫化處理後其表面態密度被減少可能與完全清除 表面氧化層有關,M. A. Reshchikov 等人[65]的研究報導指出,氮化鎵 的表面氧化層會對其表面態進行充電效應,因而導致能帶彎曲。然而, 本次研究在經過 20 分鐘硫化處理後,可以除去此氧所引發的表面態分 布,減少表面能帶彎曲,所以經過硫化處理能夠降低表面態密度的值。 另一方面,對於氮化鎵經過硫化處理所製成的蕭特基二極體可以 得到較高的蕭特基位障除了表面態密度被降低之外,根據文獻 (59) 報導當 Ni 蒸鍍於 N 型氮化鎵時,因為形成鎵鎳合金 (Ga4Ni3) 而影響 其蕭特基位障。不過,於實驗 A 之研究發現,氮化鎵經過硫化處理以 後,會在其表面形成 Ga-S 鍵結,因此推斷此 Ga-S 鍵結將會阻礙 Ga4Ni3 之形成,因而實現 Au/Ni/GaN 高的蕭特基位障。此外,Ga-S 鍵結將會 減少表面鎵原子的懸浮鍵 (dangling bond),進而降低未佔據之表面 態 (unoccupied surface states),因此可以判定試片經過硫化處理 以後其表面態密度之所以會減少,也是因為 Ga-S 鍵結的緣故。總而言 之,Ga-S 鍵結對於達成 Au/Ni/GaN高的蕭特基接觸也是重要因素之一。 另外,在本次研究也利用電流電壓 (I-V) 法,進行蕭特基位障高 度的量測。根據熱游子放射 (thermionic emission) 理論得知 J = Js exp[(qV/ηkT)-1]. (4-3-5). Js = A**T2 exp(-qΦ B/kT). (4-3-6). 26.
(37) 其中為 J 電流密度, Js 為飽和電流密度,η是理想因子 (ideality factor),A**為理論理查遜常數 (theoretical Richardson constant),對 N 型 GaN 而言,此常數為 26.4 A cm-2 K-2。 [66] 圖 4-3-2 和圖 4-3-3 分別顯示未經硫化與經過硫化處理之蕭特基二 極體所觀測順偏和逆偏的 I-V 曲線圖。對於經硫化處理的蕭特基二極 體具有較小的逆偏電流。另一方面,經由公式 4-3-5 和 4-3-6 可計算得 到理想因子和蕭特基位障 (列於表 4-3-3)。經過硫化處理的蕭特基二 極體具有較大的蕭特基位障 (0.92 eV) 和較接近 1 的理想因子 (η =1.08)。不過,利用 I-V 法所觀測的蕭特基位障仍較 C-V 法所觀測的 結果來的低,這現象在前人的研究中也已經被報導了。 [66-68]. 4. 光 子 激 發 光 光 譜 測 量 未 硫 化 與 經 過 硫 化 處 理 之 氮 化 鎵. 圖 4-4-1 顯示於 25K 下其 PL 觀測未硫化處理的氮化鎵試片,從圖 中 可 以 清 楚 地 看 到 有 三 個 區 域 , 分 別 為 黃 光 區 (yellow luminescence,簡稱 YL)、施體-受體帶 (Donor-Acceptor band,簡稱 D-A band)以及能帶邊緣 (Band-edge) 輻射。 圖 4-4-2 顯示於 25K 下其 PL 觀測未硫化處理和 10、20 和 30 分鐘 硫化處理之氮化鎵試片的能帶邊緣輻射 (位於 ~3.4 eV) 相關強度變 化。圖中將未經硫化處理試片之 PL 的強度設定為一,再分別標示出 27.
(38) 10、20 和 30 分鐘硫化處理之氮化鎵試片以此為標準值之 PL 相對值。 圖 4-4-2 很清楚地看出,以 20 分鐘硫化處理之試片的相對強度最高, 這和 XPS 觀測的資料相吻合,其原因說明 20 分鐘硫化處理能夠將表面 氧化層完全地去除,並形成 Ga-S 鍵結以致於有效地鈍化其表面,減少 表面態密度和表面複合速度,達到較高的能帶邊緣輻射強度。這結果 和文獻 (28) 所指出其 N 型氮化鎵經過硫化處理後其 PL 強度增強並降 低表面複合速度是一致的。 另外,圖 4-4-2 中對於 10 分鐘硫化處理試片其能帶邊緣輻射強度 比未經硫化處理要低,此乃因為硫化進行去除氮化鎵表面氧化層之蝕 刻效應導致非輻射複合中心產生,因此觀測到較低之能帶邊緣輻射 值。另外,30 分鐘硫化處理試片其能帶邊緣輻射強度比 20 分鐘硫化 處理稍低,其原因是此硫化處理形成之表面鈍化層的漸變效應 [23],這 可以從圖 4-2-4發現 30分鐘硫化處理試片之表面所吸附的硫原子明顯 增加。 圖 4-4-3 為 25K 下對於四組試片之 PL 觀測 2.9eV 到 3.32eV 的情 形。根據 E. Silkowski 等人[69]的研究指出,發生在 3.27eV 的 PL 稱為 施體-受體帶 (D-A band),並且進一步發現當氮化鎵的氮空位增加 時,此 D-A band 之強度也跟著增強。從圖 4-4-3 中可以發現硫化 10 分鐘之試片的強度比未經過硫化處理之試片還高,而硫化 20 和 30 分. 28.
(39) 鐘之試片其強度最弱。這說明了硫化 20 和 30 分鐘將會因為 Ga-S 鍵結 形成和硫原子佔據氮的空位,因而使得 D-A band 的強度減弱,至於硫 化 10 分鐘的試片尚未完全清除表面氧化層且因侵蝕效應導致輻射性 缺陷產生,因此增加 D-A band 的強度。在實驗 C 中所估算的表面態密 度在 20 分鐘硫化處理後被降低了,其原因除了清除表面氧化層以及 Ga-S 鍵結形成外,也可能與硫佔據氮空位有關。. 29.
(40) 五、 結論. 在這次研究發現 N 型氮化鎵置於 60oC (NH4) 2Sx 溶液進行 20 分鐘 表面處理,於蒸鍍鈦/鋁金屬後即形成特徵接觸電阻值 5.0×10-5 Ω-cm2 之歐姆接觸。然後,於氮氣環境下,以 300oC 進行熱處理 3 分鐘即可 獲得低的特徵接觸電阻值 3.0× 10-6 Ω-cm2 。如果硫化時間低於 20 分 鐘其表面原生氧化層無法完全去除,便不能獲得非熱合金化之歐姆接 觸。如果硫化時間高於 20 分鐘其表面原生氧化層雖已完全去除,但因 過多的硫原子吸附在表面,反而影響非熱合金化歐姆接觸之電特性。 至於 KOH 溶液處理過之氮化鎵試片也未能得到非熱合金化歐姆接觸, 其原因是吸附的 OH 離子影響了金屬半導體接面。 最後,將氮化鎵硫化處理所產生的效應歸納如下: 第一、從 XPS 觀測發現 20 分鐘硫化處理能夠有效去除 N 型氮化鎵表 面原生氧化層,並於氮化鎵表面形成 Ga-S 鍵結防止表面再氧 化,有助於形成非經熱處理之歐姆接觸。 第二、硫化處理改善氮化鎵表面以及提高表面載子濃度。其表面載子 濃度從原先的 6.2× 1017 cm-3 提高至~1019 cm-3。推斷此為形成氮 化鎵非經熱處理之歐姆接觸之另一項因素。 第三、硫化處理可降低氮化鎵表面態密度,從原先的 2× 1011 cm-2 減少. 30.
(41) 為 1.3× 1010 cm-2,這和表面氧化層完全被清除、形成 Ga-S 鍵結 以及硫原子佔據氮空位有關。另外,於製程中經過硫化處理的 蕭特基二極體能獲得較高的蕭特基位障。 第四、進行 20 分鐘硫化處理能增強光子激發光光譜 (PL) 之強度, 並減少表面複合速度。此外,從 D-A band 的強度變化推斷在 20 和 30 分鐘硫化處理過後,其硫原子會佔據氮空位。. 31.
(42) 參考文獻. 1. M. A. Khan, A. Bhattarai, J. N. Kuznia, and D. T. Olson, Appl. Phys. Lett. 63, 1214 (1993). 2. S. Yoshida and J. Suzuki, J. Appl. Phys. 85, 7931 (1999). 3. S. Nakamura, M. Senoh, S. Nagahama, N. Iwasa, T. Yamada, T. Matsushita, H. Kiyoku, Y. Sugimoto, T. Kozaki, H. Umemoto, M. Sano, and K. Chocho, Appl. Phys. Lett. 72, 2014 (1998). 4. J. S. Foresi, T. D. Moustakas, Appl. Phys. Lett. 62, 2859 (1993). 5. M. E. Lin, Z. Ma, F. Y. Huang, Z. Fan, L. H. Allen, and H. Morkoc, Appl. Phys. Lett. 64, 1003 (1994). 6. J. D. Guo, C. I. Lin, M. S. Feng, F. M. Pan, G. C. Chi, and C. T. Lee, Appl. Phys. Lett. 68, 235 (1996). 7. K. V. Vassilevski, M. G. Rastegaeva, A. I. Babanin, I. P. Nikitina, and V. A. Dmitriev, Mater. Sci. Engineering B 43, 292 (1997). 8. J. K. Sheu, Y. K. Su, G. C. Chi, M. J. Jou, C. C. Liu, C. M. Chang, W. C. Hung, J. S. Bow, and Y. C. Yu, J. Vac. Sci. Technol. B 18, 729 (2000). 9. N. A. Papanicolaou, M. V. Rao, J. Mittereder, and W. T. Anderson,J. Vac. Sci. B 19, 261 (2001). 10. A. N. Bright, D. M. Tricker, C. J. Humphreys, and R. Davies, J. Electron. Mater. 30, L13 (2001). 11. H. Ishikawa, S. Kobayashi, Y. Koide, S. Yamasaki, S. Nagai, J. Umezaki, M. Murakami, J. Appl. Phys. 81, 1315 (1997). 12. L. L. Smith, S. W. King, R. J. Nemanich, and R. F. Davis. J. Electron. Mater. 25, 805 (1996). 13. J. Massies, J. Chaplart, M. Laviron, and N. T. Linh, Appl. Phys. Lett. 38, 693 (1981). 14. C. J. Sandroff, R. N. Nottenburg, J. C. Bischoff, and R. Bhat, Appl. Phys. Lett. 51, 33 (1987). 15. Y. Nannichi, J. Fan, H. Oigawa, and A. Koma, Jpn. J. Appl. Phys. 27, L2367 (1988). 16. Y. Nannichi and H. Oigawa, Extended Abstracts, 22nd Conf. Solid State Devices & Materials, Sendai 1990, 453 (Business Center for Academic Societies, Tokyo). 17. B. J. Skromme, C. J. Sandroff, E. Yablonovitch, and T. Gmitter, Appl. Phys. Lett. 51, 2022 (1987). 32.
(43) 18. M. S. Carpenter, M. R. Melloch, M. S. Lundstrom, and S. P. Tobin, Appl. Phys. Lett. 52, 2157 (1988). 19. H. Oigawa, J. Fan, Y. Nannichi, K. Ando, K. Saiki, and A. Koma, Extended Abstracts, 20th Conf. Solid State Devices & Materials, Sendai 1988, (Business Center for Academic Societies Japan, Tokyo, 1988). p. 263. 20. J. Fan, H. Oigawa, and Y. Nannichi, Jpn. J. Appl. Phys. 27, L2125 (1988). 21. J. Fan, H. Oigawa, and Y. Nannichi, Jpn. J. Appl. Phys. 27, L1331 (1988). 22. J. Fan, Y. Kurata, and Y. Nannichi, Jpn. J. Appl. Phys. 28, L2255 (1989). 23. P. S. Dutta, K. S. Sangunni, H. L. Bhat, and Vikram Kumar, Appl. Phys. Lett. 65, 1695 (1994). 24. H. Ishimura, K. Sasaki, and H. Tokuda, Int. Symp. GaAs and Related Compounds, Karuizawa, 1989, p. 405. 25. H. Oigawa, Y. Kurata, J. Fan, and Y. Nannichi, Extended Abstracts of the 37th Spring Meeting, 1990 (The Japan Society of Applied Physics and Related Societies, Chiba, 1990), paper 30a-M9. 26. X. Zhang, F. Zhang, E. Lu, and P. Xu, Vacuum, 57, 145 (2000). 27. X. A. Cao, S. J. Pearton, G. Dang, A. P. Zhang. F. Ren, and J. M. Van Hove, Appl. Phys. Lett. 75, 4130 (1999). 28. G. L. Martizen, M. R. Curiel, B. J. Skromme, and R. J. Molnar, J. Electron. Mater. 29, 325 (2000). 29. 汪建民,材料分析,中國材料科學學會 (1998). 30. 王志方,材料表面測定技術,復漢出版社 (1999). 31. 黃振昌, “X 光光電子能譜儀”, 收錄於儀器總覽 6-表面分析儀器 p.5, 行政院國家科學委員會精密儀器發展中心出版 (1998). 32. 張勁燕, “電子材料”, 五南出版社 (2000). 33. G. K. Reeves and H. B. Harrison, IEEE Electron Device Lett. EDL 3, 111 (1982). 34. 高孝維, “N-型氮化鎵高熱穩定性歐姆接觸之研究”, 國立中央大學. 33.
(44) 光電科學研究所碩士論文 (1999). 35. Semiconductors Group IV Elements and III-V Compounds, edited by O. Madelung (Springer, Berlin, 1991), p. 89. 36. P. Hacke, T. Detchprohm, K. Hiramatsu, and Sawaki, Appl. Phys. Lett. 63, 2676 (1993). 37. K. Kuriyama, T. Tsunoda, N. Hayashi, and Y. Takahashi, Phys. Research B148, 432 (1999). 38. C. D. Tsai and C. T. Lee, J. Appl. Phys. 87, 4230, (2000). 39. J. D. Choi and L. T. Thompson, Appl. Surface Sci. 93, 143 (1996). 40. Y. Inoue, M. Nomiya, and O. Takai, Vacuum, 51, 673 (1998). 41. J. P. Zhang, D. Z. Sun, X. B. Li, X. L. Wang, M. Y. Kong, Y. P. Zeng, J. M. Li, and L.Y. Lin, J. Crystal Growth, 201/202, 429 (1999). 42. I. M. Band, Y. I. Kharitonov, and M. B. Trzhaskovskaya, At. Data Nucl. Data Table, 23, 443 (1979). 43. Y. Fukuda, Y. Suzuki, and N. Sanada, J. Appl. Phys. 76, 3059 (1994). 44. M. Sakata and K. Ikoma, Jpn. J. Appl. Phys. 33, 3813 (1994). 45. M. M. Hyland and G. M. Bancroft, Geochim. Cosmochin. Acta 53, 367 (1989). 46. J. K. Kim, J. L. Lee, J. W. Lee, Y. J. Park, and T. Kim, J. Vac. Sci. Technol. B17, 497 (1999). 47. 廖清賢, “氮化鎵之光電化學反應與應用”, 國立台灣大學光電工程 學研究所碩士論文 (2000). 48. B. P. Luther, S. E. Mohney, T. N. Jackson, M. Asif Khan, Q. Chen, and J. W. Yang, Appl. Phys. Lett. 70, 57 (1997). 49. M. Ben-Tzur, M. Eizenberg, and J. Greenblatt, J. Appl. Phys. 69, 3907 (1991). 50. S. C. Binari, H. B. Dietrich, G. Kelner, L. B. Rowland, K. Doverspike, and D. K. Gaskill, Electron. Lett. 30, 909 (1994). 51. M. Wittmer, J. Vac. Technol. A3, 1797 (1985). 52. L. L. Smith, R. F. Davis, R-J. Liu, M. J. Kim, and R. W. Carpenter, J. Mater. Res. 14, 1032 (1999). 53. C. T. Wu and A. Kahn, J. Vac. Sci. Technol. B16, 2218 (1998). 54. S. Ruvimov, Z. Liliental-Weber, J. Washburn, K. J. Duxstad, E. E. Haller, Z. F. Fan, S. N. Mohammad, W. Kim, A. E. Botchkarev, and H. Morkoc, Appl. Phys. Lett. 69, 1556 (1996). 34.
(45) 55. P. Moriarty, B. Murphy, L. Roberts, A. A. Caffola. G. Hughes, L. Koenders, and P. Bailey, Phys. Rev. B50, 14237 (1994). 56. H. B. Michaelson, J. Res. Dev. 22,72 (1978). 57. S. Arulkumaran, T. Egawa, H. Ishikawa, T. Jimbo, and M. Umeno, Appl. Phys. Lett. 73, 809 (1998). 58. S. J. Pearton, J. C. Zolper, R. J. Shul, and F. Ren, J. Appl. Phys.86, 1 (1999). 59. J. D. Guo, F. M. Pan. M. S. Feng, R. J. Guo, P. F. Chou, and C. Y. Chang, J. Appl. Phys. 80, 1623 (1996). 60. A. C. Schmitz, A. T. Ping, M. A. Khan, Q. Chen, J. W. Yang, and I. Adesida,Semicond. Sci. Technol. 11, 1464 (1996). 61. Semiconductor Surface and Interface, Ed. W. Mönch (Springer, Berlin, 1995). 62. H. Hasegawa, Y. Koyama, and T. Hashizune, Jpn. J. Appl. Phys. 38, 2634 (1999). 63. I. Shalish, L. Kronik, G. Segal, Y. Rosenwaks, Yoram Shapira, U. Tisch, and J. Salzman, Phys. Rev. B59, 9748 (1999). 64. P. M. Bridger, Z. Z. Bandic, E. C. Piquette, and C. T. McGill, Appl. Phys. Lett. 74, 3522 (1999). 65. M. A. Reshchikov, P. Visconti, and H. Morkoç, Appl. Phys. Lett. 78, 177 (2001). 66. M. Sawada, T. Sawada, Y. Yamagata, K. Imai, H. Kimura, M. Yoshino, K. Iizuka, and H. Tomozawa, J. Crystal Growth, 189/190, 706 (1998). 67. P. Hacke, T. Detchprohm, K. Hiramatsu, N. Sawaki, Appl. Phys. Lett. 63, 2676 (1993). 68. A. T. Ping, A. C. Schmitz, M. Asif, I. Adesida, Electron. Lett. 32, 68 (1996). 69. E. Silkowski, G. S. Pomrenke, Y. K. Yeo, and R. L. Hengehold, Physica Scripta, T69, 276 (1997).. 35.
(46) 本人發表之文章 1. C. D. Tsai, C. H. Fu, Y. J. Lin, and C. T. Lee, Solid-State Electron. 43, 665 (1999). 2. Y. T. Lyu, K. L. Jaw, C. T. Lee, C. D. Tsai, Y. J. Lin, and Y. T. Cherng, Mater. Chem. Phys. 63, 122 (2000). 3. Y. J. Lin, C. D. Tsai, Y. T. Lyu, and C. T. Lee, Appl. Phys. Lett. 77, 687 (2000). 4. Y. J. Lin and C. T. Lee, Appl. Phys. Lett. 77, 3986 (2000). 5. C. D. Tsai, Y. J. Lin, D. S. Liu, and C. T. Lee, The International Society for Optical Engineering, In Optoelectronic Materials and Devices II, pp.725 (2000). 6. Y. J. Lin and C. T. Lee, 2000 International Electron Devices and Materials Symposia, pp.300 (2000). 7. Y. J. Lin, H. Y. Lee, F. T. Hwang, and C. T. Lee, J. Electron. Mater. 30, 532 (2001). 8. Y. J. Lin and C. T. Lee, J. Vac. Sci. Technol. B, accepted (2001).. 36.
(47) 圖 2-2-1 傳輸線模型完成之示意圖。(取自文 獻 34). 圖 2-2-2 傳輸線模型量測所得的電阻值為距 離 L 之函數示意圖。(取自文獻 34). 37.
(48) Ni/Au (20/200 nm) 400 µm Ti/Al (50/150 nm). Ti/Al (50/150 nm). 500nm. n-type GaN GaN buffer layer sapphire. 圖 3-3-1 蕭特基二極體結構圖。. 38.
(49) Ga2p3/2. Ga-N. as-cleaned GaN. Intensity (arb. units). Ga-O. (NH4)2Sx-treated GaN. Ga-S. 1122. 1120. 1118. 1116. 1114. Binding Energy (eV). 圖 4-1-1 未硫化處理與經過硫化處理之 N 型 氮化鎵 Ga2p3/2 的 XPS 能譜圖。. 39.
(50) as-cleaned GaN. O1s. Intensity (arb. units). O-Ga. O-H. (NH4)2Sx-treated GaN. 536. 534. 532. 530. 528. Binding Energy (eV). 圖 4-1-2 未硫化處理與經過硫化處理之 N 型 氮化鎵 O1s 的 XPS 能譜圖。. 40.
(51) as-cleaned GaN. N1s. Intensity (arb. units). N-Ga. N-H. (NH4)2Sx-treated GaN. 401 400 399. 398 397 396. 395. Binding Energy (eV). 圖 4-1-3 未硫化處理與經過硫化處理之 N 型 氮化鎵 N1s 的 XPS 能譜圖。. 41.
(52) Intensity (arb. units). S2p. disulfide. elemental sulfur. 166. 165. 164. 163. 162. Binding Energy (eV). 圖 4-1-4 經過硫化處理之 N 型氮化鎵 S2p 的 XPS 能譜圖。. 42.
(53) current (mA). 25 20 15 10 5 0 -5 -10 -15 -20 -25. -6. -4. -2. 0. 2. 4. 6. Voltage (V) 10min (NH2)4Sx treatment 20min (NH2)4Sx treatment 30min (NH2)4Sx treatment KOH treatment. 圖 4-2-1 四種不同表面處理 (10、20、30 分鐘(NH4) 2Sx 溶液處理和 KOH 溶液處理) 之試片於蒸鍍 Ti(50 nm)/Al(150 nm)後的電流電壓 (I-V) 特性曲線,其電極間距 25 微米。. 43.
(54) 100. resistance (Ω ). 80. 60. 40. Al/Ti/n-GaN -5 2 ρc= 5.0 x 10 Ω cm. 20. 0. 10. 20. 30. 40. 50. 60. space ( µ m). 圖 4-2-2 經過 20 分鐘(NH4) 2Sx 溶液處理試片之 特徵接觸電阻值量測結果。 100. resistance (Ω). 80. 60. 40. Al/Ti/n-GaN -5 2 ρc = 7.8 x 10 Ωcm. 20. 0. 10. 20. 30. 40. 50. 60. space ( µ m). 圖 4-2-3 經過 30 分鐘(NH4) 2Sx 溶液處理試片之 特徵接觸電阻值量測結果。 44.
(55) 0.16. S/N. 0.12 0.08 0.04 0.00 as-cleaned. (NH4 )2Sx (NH4)2 Sx (NH4)2Sx treatment treatment treatment for 10 min for 20 min for 30 min. 圖 4-2-4 XPS 觀測未硫化處理和 10、20 和 30 分鐘 硫化處理之氮化鎵表面 S/N 原子濃度比。. 0.30. IGa-O / IGa-N. 0.25 0.20 0.15 0.10 0.05 0.00 as-cleaned. (NH4 )2Sx (NH4) 2Sx treatment treatment for 10 min for 20 min. (NH 4)2 Sx treatment for 30 min. 圖 4-2-5 XPS 觀測未硫化處理和 10、20 和 30 分鐘 硫化處理之氮化鎵表面 IGa-O/IGa-N 訊號峰強 度比。 45.
(56) Intensity (arb. units). O-H. O-Ga. 536. 534. 532. 530. 528. Binding Energy (V). 圖 4-2-6 經過 KOH 溶液處理之 N 型氮化鎵 O1s 的 XPS 能譜圖。. Specific Contact Resistance -6 2 x10 ( Ω cm ). 180 150. (NH4 )2S x-treated GaN 10 min 20 min 30 min KOH-treated GaN. 120 90 60 30 0 1 0 2 4 6 8 10 12 14 16 18 20. Annealing Time (min). 圖 4-2-7 經過 10、20 和 30 分鐘(NH4) 2Sx 溶液處理以及 KOH 溶液處理之試片於氮氣環境下,以 300oC 進行熱處理,觀測特徵接觸電阻值和熱處理 時間之關係。 46.
(57) Ga2p3/2 2 nm. Intensity (arb. units). 1 nm. 0 nm. Ga-N. Ga-S. Ga-N. -1 nm. 1121 1120 1119 1118 1117 1116 1115. Binding Energy (eV). 圖 4-2-8 未經熱處理 Ti/20min (NH4) 2Sx-treated GaN 之 Ga2p3/2 的 XPS 能譜圖,並進行剖面分析。 (正方向為蒸鍍之金屬,負方向為半導體). 47.
(58) N1s. Intensity (arb. units). 2 nm. 1 nm. N-Ti. 0 nm. N-Ga. N-Ti. N-H -1 nm. N-Ga. N-H. 401 400 399 398 397 396 395 394. Binding Energy (eV). 圖 4-2-9 未經熱處理 Ti/20min (NH4) 2Sx-treated GaN 之 N1s 的 XPS 能譜圖,並進行剖面分析。(正 方向為蒸鍍之金屬,負方向為半導體). 48.
(59) Intensity (arb. units). S2p. disulfide. 167. 166 165. monosulfide. 164. 163. 162 161. 160. Binding Energy (eV). 圖 4-2-10 未經熱處理 Ti/20min (NH4) 2Sx-treated GaN 其界面之 S2p 的 XPS 能譜圖。. 49.
(60) Intensity (arb. units). Ga2p3/2. Ga-N. (NH4)2Sx-treated GaN surface. Ga-S. 1122. 1120. 1118. 1116. 1114. Binding Energy (eV). 圖 4-2-11 20min (NH4) 2Sx-treated GaN 其表面之 Ga2p3/2 的 XPS 能譜圖。. Ti/(NH4)2Sx-treated GaN interface. Intensity (arb. units). Ga2p3/2. Ga-N. Ga-S. 1122. 1120. 1118. 1116. 1114. Binding Energy (eV). 圖 4-2-12 未經熱處理 Ti/20min (NH 4) 2Sx-treated GaN 其界面之 Ga2p3/2 的 XPS 能譜圖。 50.
(61) 150. current (mA). 100 50 0 -50 -100 -150. -6. -4. -2. 0. 2. 4. 6. Voltage (V) Al/Ti/KOH-treated GaN Al/Ti/(NH 4) 2Sx -treated GaN o. 300 C,1 min, in N2. 圖 4-2-13 Al/Ti/KOH-treated GaN 和 2. Al/Ti/(NH 4). Sx –treated GaN 的電特性,其電極間距為. 25 微米。. 51.
(62) (a). (b) 圖 4-2-14 估算經過 20 分鐘硫化以後在氮化鎵近表面處 所 造 成 之 電 阻 的 示 意 圖 。 (a) Al/Ti/KOH-treated GaN (b) Al/Ti/(NH 4) 2. Sx –treated GaN。 52.
(63) 80. as-cleaned GaN (NH4)2Sx-treated GaN. 60 50 40 30. 2. 2. 4. 2. A /C , (cm / µ F ). 70. 20 10 0 -2. -1. 0. 1. Voltage (V). 圖 4-3-1 利用 C-V 法量測未使用硫化處理和經過硫化 處理的蕭特基二極體之 A2/C2 和 V 的關係圖。 1. 10. 0. 10 2. current density (A/cm ). -1. 10. -2. 10. -3. 10. -4. 10. -5. 10. -6. 10. -7. 10. -8. as-cleaned GaN (NH4) 2Sx-treated GaN. 10. -9. 10. -10. 10. 0.0. 0.2. 0.4. 0.6. 0.8. 1.0. voltage (V). 圖 4-3-2 未硫化與硫化之蕭特基二極體於順偏電壓時的 I-V 曲線圖。. 53.
(64) 10. 1. 10. 0. as-cleaned GaN (NH4 )2Sx-treated GaN. 2. current density (A/cm ). -1. 10. -2. 10. -3. 10. -4. 10. -5. 10. -6. 10. -7. 10. -8. 10. -9. 10. -10. 10. 0.0. 0.2. 0.4. 0.6. 0.8. 1.0. voltage (V). 圖 4-3-3 未硫化與硫化之蕭特基二極體於逆偏電壓時的 I-V 曲線圖。. Intensity (arb. units). YL. Band-edge D-A. 1.5. 2.0. 2.5. 3.0. 3.5. 4.0. Photon energy (eV). 圖 4-4-1 於 25K 觀測未硫化處理之氮化鎵試片的 PL。. 54.
(65) 2.4. PL intensity. 2.2 2.0 1.8 1.6 1.4 1.2 1.0 0.8 0.6 as-cleaned. (NH4)2Sx (NH4)2Sx treatment treatment for 10 min for 20 min. (NH4)2Sx treatment for 30 min. 圖 4-4-2 PL 於 25K 下觀測未硫化處理和 10、20 和 30 分鐘硫化處理之氮化鎵試片的能帶邊緣輻射 比例。. PL intensity (arb. units). 25K sulfided for 10min. sulfided for 20min and 30min as-cleaned. 2.9. 3.0. 3.1. 3.2. 3.3. Photon energy (eV). 圖 4-4-3 PL 於 25K 觀測未硫化處理和 10、20 和 30 分鐘硫化處理之氮化鎵試片。. 55.
(66) N 型氮化鎵 Ga/N 原子濃度比 未經硫化處理 20 分鐘硫化處理. 0 1 nm (surface). 2 nm. 3 nm. 0.68. 0.98. 1. 1. 0.38. 0.97. 1. 1. 表 4-1-1 XPS 觀測未硫化處理和 20 分鐘硫化處理之氮 化鎵 Ga/N 原子濃度比與距表面深度之關係。. Schottky barrier height (eV) net donor concentration (cm-3 ). as-cleaned GaN 0.856 6.2×1017. 20 min (NH4 )2 Sx -treated GaN 1.099 7.4×1017. 表 4-3-1 C-V 法測得未硫化處理和 20 分鐘硫化處理之氮化鎵蕭特 基二極體的載子濃度和蕭特基位障。. 56.
(67) surface state density (cm-2 ) 2×1011 1.3×1010. surface band bending (eV) as-cleaned GaN 0.244 20 min (NH4 )2 Sx -treated GaN 0.001. 表 4-3-2 C-V 法測得未硫化處理和 20 分鐘硫化處理之氮化鎵蕭特 基二極體的表面能帶彎曲值與表面態密度估計值。. as-cleaned GaN 20 min (NH4 )2 Sx -treated GaN. ideality factor 1.68 1.08. Schottky barrier height (eV) 0.64 0.92. 表 4-3-3 I-V 法測得未硫化處理和 20 分鐘硫化處理之氮化鎵蕭特基 二極體的理想因子和蕭特基位障。. 57.
(68)
數據


+7


相關文件
現在市面上關於第三煞車燈有相當多的產品,但僅是把傳 統鎢絲燈泡取代成使用 LED
然而,由於美中貿易衝突未完全化解,中國大陸經濟成長 走緩,加上英國脫歐前景未明,影響全球投資信心,仍不利全 球經濟成長,多數經濟預測機構預估 2019
目前數學家所採用的集合論稱為 ZFC 集合論, 這是基於 Zermelo 和 Fraenkel 在 20 世紀初發展出來的 ZF 集合論, 再加上 C 所代表「選擇公設」(axiom of
而此時,對於相對成長率為 k 的族群,其滿足族群成長模型 的解為指數函數 Ce kt ,此時的 k 便是指數中時間 t
而利用 row vectors 的方法, 由於可以化為 reduced echelon form, 而 basis 是由此 reduced echelon form 中的 nonzero vectors 所組成, 所以雖然和來的 spanning
化學風化作用(Chemical Weathering) :係岩石被溶解、氧化及
摘 要 : 在玄奘弟子中,窺基、普光無疑佔據著中心位置,特別是窺
我們認為有幾點值得注意:一是更加社會化,在北朝雕塑渾厚樸拙的基礎上,隋代塑像更加
