行政院國家科學委員會專題研究計畫 成果報告
以蛋白質序列物化特性為特徵的蛋白質激 kinase-specific
磷酸化位置預測方法與分析
研究成果報告(精簡版)
計 畫 類 別 : 個別型
計 畫 編 號 : NSC 99-2221-E-009-137-
執 行 期 間 : 99 年 08 月 01 日至 100 年 07 月 31 日
執 行 單 位 : 國立交通大學生物科技學系(所)
計 畫 主 持 人 : 黃慧玲
共 同 主 持 人 : 范洪春
計畫參與人員: 碩士班研究生-兼任助理人員:許馨云
碩士班研究生-兼任助理人員:李銘哲
博士班研究生-兼任助理人員:劉一帆
報 告 附 件 : 出席國際會議研究心得報告及發表論文
公 開 資 訊 : 本計畫涉及專利或其他智慧財產權,2 年後可公開查詢
中 華 民 國 100 年 10 月 31 日
中文摘要: 本計畫提出「以蛋白質序列物化特性為特徵的蛋白質激酶磷酸
化位置預測方法與分析」。研究進行的方式為改善蛋白質激酶
磷酸化位置預測為基礎,並以下列四個方向來進行研究,以達
成目標: (1)目前蛋白質激酶磷酸化位置預測工具中,對穿膜蛋
白磷酸化位置預測非常不準確,首先設計針對穿膜蛋白磷酸化
位置預測、(2)研究穿膜蛋白的特性並正確蒐集穿膜蛋白序列,
建立穿膜蛋白序列資料集、(3) 預測蛋白質相對溶劑可接觸性
(RSA,代表蛋白質上某一氨基酸和溶劑接觸程度)可以改善磷酸
化位置預測、(4)探討蛋白質序列的物化特性來了解蛋白質可接
觸性,有助於發展蛋白磷酸化位置預測。本計畫研究進度順
利,已達預期目標,並有相關期刊論文及研討會論文發表。
英文摘要: The project proposes prediction and analysis of identifying protein
kinase-specific phosphorylation sites based on the features of
physicochemical properties of sequences. This study bases on
improving the existing methods and achieves the project goal by
way of the following fours aspects. 1) The existing prediction tools
to identify protein kinase-specific phosphorylation sites have very
low accuracy for the transmembrane proteins. Design an identifying
transmembrane protein kinase-specific phosphorylation sites
system. 2) Study the special properties of transmembrane proteins
and create an up to date transmembrane protein dataset. 3) The RSA
value plays an important role in developing explicit models for
aiding prediction of the phosphorylation sites. 4) Investigate
informative physicochemical and biochemical properties of protein
sequence to understand the RAS of proteins that is helpful in
developing protein kinase-specific phosphorylation sites predicting
method. The goal of this project is achievement conference and
journal papers.
1
以蛋白質序列物化特性為特徵的蛋白質激酶磷酸化位置預測方法與分析
Prediction and Analysis of identifying protein kinase-specific phosphorylation sites
based on the features of physicochemical properties of sequences
計畫編號:99-2221-E-009-137-
執行期限:99 年08 月01 日至100 年07 月31 日
主持人:黃慧玲 國立交通大學
[email protected]
摘要
本計畫提出「以蛋白質序列物化特性為特
徵的蛋白質激酶磷酸化位置預測方法與分
析」。研究進行的方式為改善蛋白質激酶
磷酸化位置預測為基礎,並以下列四個方
向來進行研究,以達成目標: (1)目前蛋白質
激酶磷酸化位置預測工具中,對穿膜蛋白
磷酸化位置預測非常不準確,首先設計針
對穿膜蛋白磷酸化位置預測、(2)研究穿膜
蛋白的特性並正確蒐集穿膜蛋白序列,建
立穿膜蛋白序列資料集、(3) 預測蛋白質相
對溶劑可接觸性(RSA,代表蛋白質上某一
氨基酸和溶劑接觸程度)可以改善磷酸化
位置預測、(4)探討蛋白質序列的物化特性
來了解蛋白質可接觸性,有助於發展蛋白
磷酸化位置預測。本計畫研究進度順利,
已達預期目標,並有相關期刊論文及研討
會論文發表。
關鍵字:物化特性, 蛋白質激酶磷酸化,
基因演算法, 溶劑可接觸性, 蛋白質序列,
預測方法
Abstract
The project proposes prediction and analysis
of identifying protein kinase-specific
phosphorylation sites based on the features of
physicochemical properties of sequences.
This study bases on improving the existing
methods and achieves the project goal by
way of the following fours aspects. 1) The
existing prediction tools to identify protein
kinase-specific phosphorylation sites have
very low accuracy for the transmembrane
proteins. Design an identifying
transmembrane protein kinase-specific
phosphorylation sites system. 2) Study the
special properties of transmembrane proteins
and create an up to date transmembrane
protein dataset. 3) The RSA value plays an
important role in developing explicit models
for aiding prediction of the phosphorylation
sites. 4) Investigate informative
physicochemical and biochemical properties
of protein sequence to understand the RAS of
proteins that is helpful in developing protein
kinase-specific phosphorylation sites
predicting method. The goal of this project is
achievement conference and journal papers.
Keywords: Physicochemical properties,
kinase-specific phosphorylation, genetic
algorithms, relative surface area of solvent
accessibility, protein sequence, prediction
method.
一. 前言
蛋白質的磷酸化是在蛋白質轉譯修
飾中很重要機制,在調控基本進行過程例
如新陳代謝、訊號傳遞、細胞分化和細胞
膜穿透性等扮演重要角色。因此預測蛋白
質的磷酸化作用位置是非常重要議題。能
知道蛋白質磷酸化位置,就可以測出蛋白
質功能。雖然最近有許多的磷酸化位點的
預測工具已經發展出來了,但卻還是沒有
一 個 預 測 工 具 是 針 對 膜 蛋 白 (membrane
protein)而設計,膜蛋白對於一些生理功能
是很重要的,所以針對膜蛋白的磷酸化位
點預測是一項重要且迫切的課題。
二. 研究目的
膜蛋白同於具有兩性(同時有書水及
親水性區段),因此要調整出適合膜蛋白結
晶的環境非常困難。近年來由於蛋白質結
晶技術的進步,讓研究者可以順利取得一
些膜蛋白的結晶資訊。然而針對膜蛋白的
2
後轉譯修飾的工具卻還是不足,因此讓研
究膜蛋白的實驗需要耗費大量的人力和金
錢來進行實驗。
本計畫目標系以「以蛋白質序列物化
特性為特徵的蛋白質激酶磷酸化位置預測
方法與分析」為主題進行研究。研究進行
的方式為改善蛋白質激酶磷酸化位置預測
為基礎,並以下列四個方向來進行研究,
以達成目標: (1)目前蛋白質激酶磷酸化位
置預測工具中,對穿膜蛋白磷酸化位置預
測非常不準確,首先設計針對穿膜蛋白磷
酸化位置預測、(2)研究穿膜蛋白的特性並
正確蒐集穿膜蛋白序列,建立穿膜蛋白序
列資料集、(3) 預測蛋白質相對溶劑可接觸
性(RSA,代表蛋白質上某一氨基酸和溶劑
接觸程度)可以改善磷酸化位置預測、(4)
探討蛋白質序列的物化特性來了解蛋白質
可接觸性,有助於發展蛋白磷酸化位置預
測。
三. 文獻探討
3.1 膜蛋白磷酸化位點分析
在很多的細胞上都會有許多的穿膜
蛋白,例如說離子通道或接收器等等。這
些蛋白質中,磷酸化扮演了很重要的角
色,這些磷酸化的過程不外乎透過自身磷
酸化或是一些磷酸化激酶達到目的。在球
蛋白中,包埋在內部的通常都是疏水性胺
基酸,而暴露在外的都是親水性胺基酸。
然而在穿膜蛋白就不是這樣的情況,有時
疏水的區位會暴露在外,而親水部分反而
包埋在內部。在這兩種迥異的情況下,我
們假設在膜蛋白和球蛋白的磷酸化位點會
十分的不同。目前有存在一些效能不錯的
預測工具和資料庫列表於表1。在當中,每
一個資料庫都含有我們感興趣的膜蛋白,
所以在研究中需要把這些膜蛋白從中擷取
出來。
而現今的預測方法包括兩大項(圖
1),有直接以磷酸化位點為預測對象,
或是先以特定的磷酸化激酶為標的,再預
測有哪些位點會被磷酸化。
圖1. 目前磷酸化位點的預測方法
3.2 溶劑暴露面積預測
預測蛋白質的折疊無論在生物資訊或
者實驗領域中都是很重要的問題。ASA 是了
解蛋白質摺疊的一項重要工具。透過了解
蛋白質上每一個殘基的 ASA,可以對蛋白質
的構型有約略的了解,並且猜測蛋白質中
殘基跟殘基間的交互作用或者是生理功
能,催化位點與催化機制甚至是蛋白質-蛋
白質間的交互作用。例如說 Hikijata et.al.
在預測蛋白質 3D 結構時,除了使用序列比
對的結果之外,同時也使用到溶劑暴露面
積輔助預測結果。在預測蛋白質網絡領域
中,Martin et. al.希望預測會進行交互作用
的殘基(residues),會同時參考蛋白質的二
級結構、實驗結果跟二級結構資訊的參
數,由此可知,在研究蛋白質的功能、結
構等等溶劑暴露面積都扮演很重要的角
色。
目前預測 ASA 的工具可以大多為 two
states 的包埋暴露預測,這一類的預測器是
表 1. 之前所提出預測非穿膜蛋白預測
器
Tools name Category
PHOSIDA For specific a.a
PPSP For kinase
NetPhosK For kinase
KinasePhos For kinase
Musite For specific a.a
GPS For kinase
DISPHOS For specific a.a
3
以預測器的效能而決定出 threshold,在某
些情況下無法滿足想了解特定殘基的特定
功能,因此發展出預測 RSA 實際值的預測
方法,透過對 RSA 直接預測就能解決上述
問題。
除此之外,雖然預測工具很多,但卻沒有
一個研究有將不同胺基酸個別探討其物化
特性在蛋白質摺疊上所扮演的角色。本篇
研究也會針對所挑選來的 featrure 做探
討,了解各個殘基的包埋或暴露的物化特
性,進而發現一些原本在預測殘跡暴露與
否的問題中沒有考慮到的因素。這次我們
的方法不但提供了一個可以預測相對暴露
面積的方法,同時我們的方法也提供了一
些資訊,來了解蛋白質折疊的問題。
目前為止,預測實際相對溶劑暴露面積
的研究很多如表一所示,其中的方法包括
使用 NN,SVR,multi layer regression,
two-stage SVR。
3.3 磷酸結合相關蛋白分析
蛋白質的磷酸化過程中,除了將磷酸運輸
至目標蛋白進行化學變化外,同時也包含
有蛋白質間作用力。為了要了解蛋白質和
磷酸根之間的作用力,研究需要取得一些
蛋白質和磷酸根作用的資料幫助了解。然
而在蛋白質中沒有一個胺基酸是帶有磷原
子甚至是磷酸根為官能基的胺基酸。DNA
在骨幹(backbone)的部分是磷酸以
phospho-diesterbone 連結磷酸所構成,含
有大量的磷酸根,同時在目前的資料庫裡
也有足夠的資料。Kumar et al.就曾經蒐
集 DNA 結合蛋白作為研究對象,使用支援
向量機做出模型。研究裡我們會使用它們
所使用的 dataset 作為研究對象。
四.研究方法
4.1膜蛋白磷酸化位點整理及預測
4.1.1dataset 的取得
資料從 uniprot 中擷取,為了要得到更精確
的目標,因此我們只採用有真實實驗數
表 2. 溶劑暴露面積相關研究和其效能比較表
Work Regression tool Description of features MAE
(%)
CC
Ahmad et al.,2003 NN Amino acid composition 18.8 0.48
Yuan and Huang,2004 SVR Amino acid composition 18.5 0.52
Adamczak et al.,2004 NN PSSM 15.3 --
Wang et al.,2005 MLR Amino acid composition, PSSM and
sequence length
16.2 0.64
Garg et al.,2005 NN PSSM and secondary structure
information
15.9 0.65
Nguyen and Rajapakse,2006
Two-stage SVR PSSM 15.7 0.66
Chang et al.,2008 Two-stage SVR enhance PSSM and sequence length 14.8 0.68
4
據驗證的資料作為磷酸化位點的參考點,
如
果
該
資
料
中
紀
錄
是”potential”、”similarity”或是”probable”的
資 料 將 不 會 被 採 用 。 接 著 , 我 們 再 從
PhosphoSite plus資料庫中取得各種穿膜蛋
白 。 其 中 包 括 anion exchange 、 calcium
transport 、 chloride channel 、 copper
transport 、 hydrogen ion transport 、 ionic
channel 、 iron transport 、 ligand-gated ion
channel、porin potassium transport、sodium
transport、viral ionic channel、voltage-gated
channel、zinc transport、cobalt transport。
最後得到222條蛋白質和其上總共約三萬
個磷酸化位點。如表3所示。從中挑出100
條作為test,剩下的122條作為training。
4.1.2
磷酸化位點預測方法和效能評估
模型的建構分成兩的大部分做預測,其
流程如圖1所示。首先會先分析統計該資料
中顯而易見的特性,同時找尋文獻中可以
使用的知識作為feature之一。而另外一部
份則是使用IBCGA從531個物化特性中挑
選出最具代表性的結果。最後做出恰當的
模型用來進行預測。模型分為兩個預測,
其一為先以磷酸激酶做分類後進行預測,
首先探討以PKA作為磷酸化激酶的目標,
其結果如表4所示。
結果顯示我們如果先用磷酸激酶分類
後進行預測,最後最高的準確率大約是八
成,而在專一性更可以達到八成五。
最後如果以磷酸化位點作為預測目標
做預測,當預測serine和threonine時,結果
如表5所示。結果顯示直接使用位點的結果
會比以磷酸激酶分類後做出的預測好,準
確率約九成,專一性更達到九成六。
4.2 蛋白質與磷酸根交互作用分析
為了想了解蛋白質和磷酸根之間的作
用,所以最直接的辦法就是從帶有磷酸根
的分子和蛋白質間如何作用做分析。在生
物體內帶有最多林酸根的分子為DNA,同
時在蛋白質和DNA的共同結晶和文獻的量
都足夠做為分析之用。故挑選DNA結合蛋
白作為分析的目標。我們dataset使用之前
Kumar et al所使用的dataset。首先我們先從
531個物化特性中挑選出重要的23個,然後
對這23個物化特性做fuzzy rule的分析。其
流程如圖2所示。
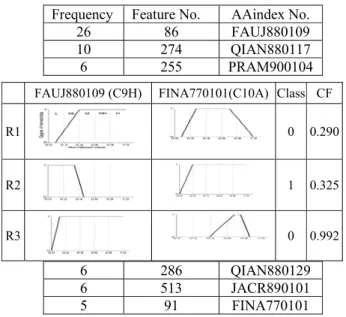
最後fuzzy rule的判讀結果列在表6,最
重要的三項因子分別為電性、凡德瓦力和
溶劑暴露面積。
這項結果說明了,如果可以在與磷酸作
用的位點,特別加入溶劑暴露面積,對於
判斷該位點是否作用有很大的助益。
表 3.222 個 從 phosphosite plus 和
uniprot 中所挑選出來的磷酸化位點
S T Y Total 100 proteins_positive 169 56 101 326 100 proteins_negative 6934 5199 2862 14995 122 proteins_positive 198 68 115 381 122 proteins_negative 8466 6244 3495 18205表 4. 以 PKA 為磷酸化激酶所預測的磷酸
化位點
Spec Sen OAcc MAcc MCC
GPS 0.859 0.423 0.848 0.607 0.121 PPSP 0.667 0.299 0.645 0.232 -0.017 KinasePhos 0.006 0.959 0.024 0.503 -0.063 NetPhosK 0.155 0.280 0.157 0.226 -0.183 Spec = specificity、 Sen = sensitivity、OAcc = Overall accuracy 、MAcc = Mean accuracy 、MCC = Matthews correlation coefficient.
表 5. 以磷酸化位點直接進行預測之結果
Spec Sen OAcc MAcc MCC
DISPHOS 0.958 0.141 0.941 0.455 0.069
PHOSIDIA 0.318 0.735 0.326 0.548 0.016
MuSite 0.837 0.165 0.787 0.170 0.001
5
圖2. FRKAS流程圖。輸入的物化特性可以經過FRKAS得到一個了解該特性值對於分類影
響的規則。
表6. FRKAS挑選出來的物化特性。結果說明在磷酸化位點的結合部份,其帶有電荷的物
化特性十分鐘要,其中也需要溶劑舖暴露面積做為判斷的物化特性之一,以提高分類
器的效能。
6
圖3. 模型建構方法
4.3 溶劑暴露面積預測器
4.3.1製作流程
先前的研究很多都使用Barton502作為
預測和建構模型的資料。該資料使用的人
多,因此適合用來比較預測器的效能。
同時因為資料量也夠大,因此使用該資
料所得到的結果可信度也高。所有的蛋白
質會按照圖3所列出來的方式,照上面的胺
基酸分別建構模型和預測模型。實驗流程
如圖4所示。
其中的三分之二作為 traininig,剩下
的三分之一作為 test。各種不同的預測器效
能的比較都是以 test 所做出來的結果。經
過 IBCGA 挑選 features 後,所選出來的如
表 7 所列之 features。
4.3.2
溶劑暴露面積預測器效能評比
以MAE的平均值來看,如果將序列長
度加上PSSM或是序列長度加上AAindex,
表現都沒有比PSSM加上aaindex和序列長
度 的 資 訊 ( 表 8) , 顯 示 如 果 PSSM 搭 配
AAindex和序列長度在預測蛋白質溶劑暴
露面積上有非常好的效果。相較於Chang et.
al.和Nguyen et.al的方法,我們和他們不一
樣的地方在於他們都只是使用PSSM,即使
是Chang et.al.的enhance PSSM,其實也是
從PSSM中擷取資訊,嚴格講起來也都是單
一feature進行預測;而Wang et.al.是使用多
組feature一起進行預測,但使用的方法是
Multi-Learning regression的方法,相對於後
來發展的方法都是使用SVR的情況來看,
也許預測溶劑暴露面積的問題,使用SVR
會比MLR更合適。最後比較相關係數和總
MAE(這邊的MAE是把所有的胺基酸實際
值跟預測值一起算),如表8所列,使用
PSSM搭配aaindex和長度資訊也確實得到
不錯的結果。
4.3.3
溶劑暴露面積預測器物化特性分
析
按照先前的研究將531個aaindex分成20個
clusters,觀察其分布狀況(圖5)。
圖4. 溶劑暴露面積模型製作流程
7
表 7. 用來建立 model 的 feature set
amino acid feature number Best feature set
A 31 12,18,39,89,102,114,141,164,169,199,206,209,210,251,266,267,294,305,309,333,338,352,356,364,369,373,403,415,447,481,523, R 21 10,41,93,105,113,152,194,259,287,317,327,334,352,358,381,387,398,404,408,409,451, N 34 11,31,41,60,99,100,102,124,126,172,175,176,231,253,260,262,264,265,274,310,321,322,323,336,351,385,410,412,414,419,483,512,521,523, D 25 23,94,95,113,124,146,178,179,196,210,221,257,274,275,285,295,300,318,340,352,404,442,481,490,531, C 30 12,18,29,41,54,61,95,102,106,151,169,221,242,245,249,278,296,306,379,391,405,420,447,473,477,491,492,497,503,522, Q 33 78,86,101,102,106,161,166,178,198,201,219,238,294,295,300,315,328,342,343,352,362,376,381,389,398,403,419,423,434,450,452,461,481, E 28 13,44,66,71,79,108,199,200,209,211,221,247,289,302,318,332,355,371,381,387,399,404,410,414,446,501,521,522, G 32 21,31,42,66,82,110,114,165,196,200,233,262,267,268,295,304,306,320,333,364,371,372,416,420,423,432,442,454,455,461,469,530, H 28 3,6,18,30,69,75,83,108,113,126,200,230,252,286,300,373,374,376,402,419,420,423,447,462,467,497,515,530, I 34 14,26,28,29,30,45,74,79,88,92,107,165,187,218,235,237,252,255,266,267,268,289,290,322,323,334,341,386,415,418,446,452,453,526, L 26 3,10,61,69,113,168,176,194,200,236,239,305,315,318,322,323,339,349,356,370,390,419,480,508,512,518, K 23 27,32,64,114,140,163,179,186,219,223,235,258,267,273,284,322,330,414,443,480,488,495,515, M 29 71,90,144,147,149,151,185,221,222,272,279,284,285,294,319,348,349,352,357,386,410,422,477,481,483,484,488,516,529, F 30 10,39,46,54,93,149,169,174,193,215,223,238,275,277,327,331,336,369,371,384,419,432,443,446,450,488,503,508,527,528, P 21 22,40,53,61,63,80,83,110,209,228,259,260,299,300,351,361,404,418,424,443,521, S 26 12,25,71,96,103,114,168,175,189,200,221,345,351,376,389,405,409,440,450,452,474,477,514,516,522,531, T 27 10,11,57,58,60,98,105,106,115,159,194,224,229,231,296,309,318,339,352,362,380,383,392,400,409,422,433, W 26 3,4,37,80,114,124,178,186,190,206,228,229,274,276,277,285,304,308,333,336,352,381,387,483,510,525, Y 31 18,28,40,53,140,173,206,209,211,218,231,238,324,328,333,352,364,387,391,414,416,422,436,477,481,497,506,510,516,522,531, V 12 26,117,153,235,303,316,405,447,448,450,452,509,
表8. 溶劑暴露面積預測器和相關研究之效能比較
pssm + + + -aaindex + - - + sequence length + - + + 1 A 12.2199 13.4740 13.4723 18.8132 13.3 14.4 15.6 2 R 16.8119 16.8845 17.318 24.3333 17 17 17.1 3 N 18.4971 20.3228 20.446 25.0654 19.6 20.2 21 4 D 18.0767 20.2810 20.2245 24.5231 19.2 19.5 20.8 5 C 8.87168 10.0537 10.0537 9.61839 8.9 9.9 14.2 6 Q 16.2366 17.7884 22.1026 21.6074 17.2 17.6 18 7 E 15.9341 18.1813 18.2301 22.2619 17.8 18.3 19.3 8 G 18.0293 19.7634 19.7632 24.0462 19.5 19.6 21.1 9 H 15.8715 15.8185 20.5737 19.8794 15.1 15.4 15.7 10 I 8.09382 8.9793 8.9742 10.2656 8.7 9.7 10.6 11 L 9.79063 12.5323 10.2074 11.8409 9.8 10.8 11.6 12 K 15.7747 15.7908 15.8488 18.4192 15.8 16.4 16.3 13 M 11.3245 11.3839 11.3408 13.7533 11.3 12.1 12.9 14 F 10.0539 9.9997 9.99919 11.0815 10.2 11.2 11.9 15 P 16.6900 17.9133 17.9515 21.2484 17.4 17.7 18.2 16 S 16.0847 18.2228 18.8323 23.5514 18.3 18.8 19.8 17 T 15.8687 16.5704 16.5863 21.049 16 16.7 17.1 18 W 12.1712 12.4307 12.3633 13.4755 11.8 12.4 13.2 19 Y 11.5132 12.1179 12.1725 13.8647 13 12.9 13.3 20 V 9.89251 10.187 10.1642 12.1627 9.6 10.7 11.2 average 13.742981 15.7183133 15.3312295 18.0430245 14.475 15.065 15.945 MAEChang(2008) Nguyen(2006 Wang(2005) no. amino acid
8
圖 5.cluster 分析
分析過後,可以發現不管是 hydrophilic
或 hydrophobic 的胺基酸,分數最高的前三
名都是 cluster 3,12,17。我們分別查出餓三
個 cluster 裡的成員列在圖 6,這三個 cluster
並不是 aaindex 分類中元素最多。有趣的
是,這三個 cluster 當中的元素全部都是屬
於 hydrophobic index。
圖 6. 在第 3、12、17 群中,成員都是疏水
作用力相關的物化特性
這樣證據顯示不管是在親水性和疏水
性的胺基酸,hydrophobic properties 都
很重要。也應證了第一張圖所提出的不管
親水性胺基酸跟疏水性胺基酸,對於判斷
其親疏水性是非常重要的。
四. 論文發表
這一年計劃皆有完成預定的目標。也
如期投出期刊論文並且接受刊出論文。感
謝國科會給予資源才能順利完成並且豐碩
收穫。執行論文這一年期間,發表相關的
論文如下
1 H.-L. Huang, F.-L. Chang, S.-J. Ho, L.-S.
Shu, W.-L. Huang, and S.-Y. Ho*,
“
FRKAS:
Knowledge Acquisition Using
a Fuzzy Rule Base Approach to Insight of
DNA-Binding Domains/Proteins,”
accepted by Protein and Peptide Letters,
2011. (SCI)
2 H.-L. Huang, I-C. Lin, Y.-F. Liou, C.-T.
Tsai, K-T. Hsu, W.-L. Huang, S.-J. Ho,
and S.-Y. Ho*, “Predicting and analyzing
DNA-binding domains using a systematic
approach to identifying a set of
informative physicochemical and
biochemical properties”, BMC
Bioinformatics, 12(Suppl 1):S47,
2011.(SCI)
雖 然 我 們 致 力 於 國 科 會 的 研 究 題
目,但另外在其他的非直接相關的論文和
研討會成果也同樣豐碩,期刊論文共一篇:
1. S.-Y. Ho,C.-Y. Chao, H.-L. Huang, T.-W.
Chiu, P.Charoenkwan, and E. Hwang*,
“NeurphologyJ: an automatic
neuronalmorphology quantification
method and its application in
pharmacological discovery,”BMC
Bioinformatics, 12:230, 2011. (SCI)
另外有研討會論文共八篇
1.
H.-L.Huang, T.-F.Kao
.,P.Charoenkwan
.,
W.-L. Huang, S.-J. Ho and S.-Y. Ho*,
2012, “Estimating solubility scores of
dipeptides and residues for predicting
proteins solubility,”The Tenth Asia
Pacific Bioinformatics Conference,
Melbourne, Australia, 17-19 January
2012.
2.
C.-T. Tsai, W.-L. Huang, C.Liaw, C.-W.
Tung, H.-L. Huang and S.-Y. Ho*,
2012, “Virulence-iGO: Predicting
virulence factors in pathogenic bacteria
using informative Gene Ontology
9
terms,”The Tenth Asia Pacific
Bioinformatics Conference, Melbourne,
Australia, 17-19 January 2012.
3.
H.-C. Lee, S.-J. Ho, L.-S.Shu, F.-L.
Chang, S.-Y. Ho and H.-L.
Huang*,2012, “Optimization method of
predicting enzyme mutant activity from
sequences by identifying a set of
informative physicochemical
properties,”The Tenth Asia Pacific
Bioinformatics Conference, Melbourne,
Australia, 17-19 January 2012.
4.
H.-L. Huang,Y.-H. Lin, W.-L. Huang
and S.-Y. Ho*, 2011, “Intelligent
triple-objective genetic algorithm for
selecting informative Tag SNPs,” The
22
ndInternational Conference on
Genome Informatics, Korea, Dec. 5-7,
2011.
5.
H.-L. Huang, S.-B. C., Y.-H.Chen, and
S.-Y. Ho*, 2011, “Optimization
approach to estimation of kinetic
parameters for modelling metabolic
pathways of muscle
glycogenolysis,”The 22
ndInternational
Conference on Genome Informatics,
Korea, Dec. 5-7, 2011.
6.
L.-S. Shu, H.-L. Huang, S.-J. Ho, and
S.-Y. Ho*, 2011, “Establishing
large-scalegene regulatorynetworks
using a gene-knowledge-embedded
evolutionary computation method,”
IEEE International Conference on
Computer Science and Automation
Engineering, June 10-12, 2011,
Shanghai, China.
7.
H.-L. Huang, F.-L. Chang, S.-J. Ho,
L.-S. Shu, and S.-Y. Ho*, 2011,
“Interpretable knowledge acquisition for
predicting DNA-binding domains using
an evolutionary fuzzy classifier
method,” IEEE International
Conference on Computer Science and
Automation Engineering, June 10-12,
2011, Shanghai, China.
8.
H.-L. Huang, I-C. Lin, Y.-F. Liou, C.-T.
Tsai, K.-T. Hsu, W.-L. Huang, S.-J. Ho,
and S.-Y. Ho*, 2011, “Predicting and
analyzing DNA-binding domains using
a systematic approach to identifying a
set of informative physicochemical and
biochemical properties, APBC 2011,
Korea, Jan. 11-14.
參考資料
[1] Wong YH, Lee TY, Liang HK, Huang
CM, Wang TY, Yang YH, Chu CH, Huang
HD, Ko MT, Hwang JK, "KinasePhos 2.0: a
web server for identifying protein
kinase-specific phosphorylation sites based
on sequences and coupling patterns," Nucleic
Acids Res,vol 35,(Web Server issue),
pp.W588-594, 2007.
[2] Blom N, Gammeltoft S, Brunak S,
"Sequence and structure-based prediction of
eukaryotic protein phosphorylation sites," J
Mol Biol,vol 294,(5), pp.1351-1362, 1999.
[3] Iakoucheva LM, Radivojac P, Brown CJ,
O'Connor TR, Sikes JG, Obradovic Z,
Dunker AK, "The importance of intrinsic
disorder for protein phosphorylation,"
Nucleic Acids Res,vol 32,(3), pp.1037-1049,
2004.
10
[4] Linding R, Jensen LJ, Ostheimer GJ, van
Vugt MA, Jorgensen C, Miron IM, Diella F,
Colwill K, Taylor L, Elder K et al,
"Systematic discovery of in vivo
phosphorylation networks," Cell,vol 129,(7),
[5] Dor O, Zhou Y, "Real-SPINE: an
integrated system of neural networks for
real-value prediction of protein structural
properties," Proteins,vol 68,(1), pp.76-81,
2007.
[6] Chen K, Kurgan M, Kurgan L, "Sequence
based prediction of relative solvent
accessibility using two-stage support vector
regression with confidence values," J
Biomedical Science and Engineering,vol
1,pp.9, 2008.
[7] H.-L. Huang, F.-L. Chang, S.-J. Ho, L.-S.
Shu, W.-L. Huang, and S.-Y. Ho*, “
FRKAS:
Knowledge Acquisition Using a Fuzzy Rule
Base Approach to Insight of DNA-Binding
Domains/Proteins,” accepted by Protein and
Peptide Letters, 2011. (SCI)
[8] H.-L. Huang, I-C. Lin, Y.-F. Liou, C.-T.
Tsai, K-T. Hsu, W.-L. Huang, S.-J. Ho, and
S.-Y. Ho*, “Predicting and analyzing
DNA-binding domains using a systematic
approach to identifying a set of informative
physicochemical and biochemical properties”,
BMC Bioinformatics, 12(Suppl 1):S47,
2011.(SCI)
國科會補助教師出席國際會議結案心得報告
報告人姓名
黃慧玲
所屬學校
學系(所)
交通大學
生物科技學系
會議期間
及地點
2011/01/14 至
2011/01/14
南韓
補助項目
及金額
■ 機票費
□ 註冊費
■ 生活費
會議名稱
(中文)2011 第九屆亞太生物資訊會議
(英文)2011 The Ninth Asia-Pacific Bioinformatics
Conference (APBC2011)
發表論
文題目
用一套系統化方法找一組相關的物化生化學特性集來預
測與分析DNA-binding domains
Predicting and analyzing DNA-binding domains
using a systematic approach to identifying a set
of informative physicochemical and biochemical
properties
報告內容:(1、參加會議經過;2、與會心得3、建議4、攜回資料)
吾人發表的論文是在01/12 日下午2 點50分的SONGDO CONVENSIA會議廳1F
1. 參加會議經過
本次會議APBC2011 為第九屆亞太生物資訊會議此次主辦單位是KSBSB (Korean Society for Bioinformatics and Systems Biology)協辦單位是KRIBB (Korean Research Institute of Bioscience and Biotechonology)、KOBIC (Korea Bioinformation Center) 、 和 Chungbuk BIT Research-Oriented University Consortium 。 贊 助 機 構 有 IBC Journal(Interdisciplinary Bio Central)以及BioMed Central。創辦人Professor Phoebe Chen (Professor and Chair & Head of Department, Department of Computer Science and Computer Engineering, La Trobe University, Melbourne, Victoria 3086, Australia)。亞太生物資訊會議每年都在不同國家舉辦,2011年在韓國舉辦。今年大會共有5個Room,包含口頭報告 區、演講區、與海報展示走廊。本次共收55篇口頭報告論文,很榮幸我們的論文被接受口頭報告。至於海報共接受 127篇。其中52個program committee來自各個國家、4個chair分為6 group負責大會工程。6個Steering committee統籌大 會運作。在本次會議共有8個tutorials及六位keynote speaker及17個Session,及8場演講。
會議最後共有6場IBC
Journal的教授演講。
我們由首爾機場搭飯店公車抵達飯店,首爾溫度大約-5。 C~-17。 C,看著飄雪的景象,優美的雪景令疲勞的身體 舒解些許。首爾的消費額比台北多些,這點個人頗有點感慨。會場的地點是在仁川所以搭地鐵大約一小時多便可以 到達。仁川堆積的雪比首爾高,所以沿路的雪景比首爾更有一番風味。到會場報到後因為我們的報告是第三天,因 此我們有許多時間可以聽演講。比較令人深刻映像是Keynote1 speaker Steven Jones,與chair Phoebe Chen談論生物資 訊與演癌症基因體,癌症基因病變與突變,如何由運用生物資訊能力,以及資訊發現探討。覺得對於自己投入生物資訊研究,有獲得支持與可持續發展的力量。演講後有一場知性的音樂演奏做為中場結束。
演講分為Biological
Data Evaluation與Translational Bioinformatics二大主軸,其中交通大學生科系系主任黃憲達教授任第
一場首位演講。我們在會場亦與系主任討論問題,同時也分享他們演講前必須先開會討論,再進行
演講流程相當嚴謹。
第三天我們報告時間是下午2:50,這次大會將每場演講與報告都錄影起來,由網站亦可觀賞精彩
演講。在我們這場報告以辨認圖樣識別為主,我們主要是提出由蛋白質序列轉物化特存在問題,對
此提出解決方法並應用DNA-binding domains序列資料去分析及發現來探討問題。我對這session的一
個主題發現Phosphorylation Motifs感到有興趣,因為我目前正在研究的是Phosphorylation sites研究。
會議結束後今天大會的晚餐相當特別的是,搭遊覽車到仁川海港邊搭遊輪出海,並且在郵輪上享用
美味海鮮大餐,欣賞聲樂家演唱,夜色在美食美聲下越來越美,帶著滿滿的陶醉結束這一天會議行
程。以下照片由右到左分別為交大生科副院長何信瑩教授、系主任黃憲達教授、APBC創辦人
Professor Phoebe Chen、我、台大研究員、以及台大博士班學生,在一樓會場留影。
2、與會心得
感謝國科會補助參加國際會議之出國補助,使本人得以出席跨領域生物資訊國際會議,開拓眼
界及促進國際觀。每次參加國計會議除了努力讓世界知道臺灣人在研究方面非常認真與相當有能力
為心則。此次交大生科副院長何信瑩教授、系主任黃憲達教授深為APBC創辦人
Professor Phoebe Chen的愛戴,除暢談她的每年經驗以及她個人行程的滿檔,亦對此次舉辦的不充足之處作分享。並希望2015年由交大舉辦 亞太生物資訊會議。 個人覺得生物資訊這領域,由此次舉辦國韓國,這國家對生物資訊投入組織相當龐大,也可見他們對這領域的 企圖心與團結。反觀台灣生物資訊投入與組織結構發展還需更努力。而我們與系主任、副院長的討論彷彿將系院擴 大到國際空間進行情境探討與未來計畫,相當難得的收穫。
3、建議
近年來國科會、教育部和學校積極鼓勵年輕研究人員,除鼓勵教師參與會議外,特別是博士班
學生,參與大型國際會議,及早進入研究領域的核心,吸取國際研究經驗,以提高國人的研究水
準。參加生物資訊國際會議對老師及學生是非常重要的,會議中不但可以得到相關研究的最新發展
資訊,認識結交許多相關領域的學者,彼此交換研究心得,更可找到跨領域的學者國際合作,在跨
領域的生物資訊研究更是重要。目前研究生已有多管道獲(部份)補助出席國際會議,建議繼續擴大
進行。而國際化的學術交流是往後的趨勢,也能有所激勵國人學界能力與國際觀。
4、攜回資料
1. 期刊一本
2. Tutorial 一本
3.記事本一本。
日 期: Sat, 16 Oct 2010 12:15:55 +0100 [2010/10/16 19 時 15 分 55 秒 CST] 寄 件 人: APBC2011 <[email protected]> 收 Hui-Ling Huang <[email protected]>件 人:
主
旨: APBC2011 notification for paper 120 - Accept
Dear Hui-Ling Huang Paper: 120
Title: Predicting and analyzing DNA-binding domains using a systematic approach to identifying a set of informative physicochemical and biochemical properties
We are very pleased to inform you that your submission has been accepted for an oral presentation at APBC 2011. The reviews are appended below. Further, the revised version of your submission, taking the comments of the reviewers into account, is invited to be published in BMC Bioinformatics. Congratulations!
Please follow these instructions carefully to avoid any possible delays or problems: 1. As per the tradition of APBC, at least one author must register by 25 Oct 2010 for the paper to be accepted. Registiation information can be found at
http://sysbio.kribb.re.kr/APBC2011/regist.php
Please note that it is APBC's policy that at least one of the authors must be able to present the paper for it appear in the proceedings.
2. Please take all the comments of the reviewers into account while preparing the revised version. If unsatisfactory, the submission may be returned to you, delaying the publication of your paper. (If substantial, include a bulleted list of changes in the body of the email.)
3. The final version in the BMC Bioinformatics format is due from you by 25 Oct 2010. This is a STRICT DEADLINE to enable timely publication. The final version should be directly emailed to [email protected] with Subject: "APBC2011 Final Version - Paper ID". Format details are in the submission guidelines of the conference webpage
(http://sysbio.kribb.re.kr/APBC2011/CFP.php ).
4. To enable prompt handling of your paper publication in BMC Bioinformatics, please also include the following in the body your email:
a) Paper id (the submission id)
b) Name, address and phone number of CONTACT author c) VAT number, if the address is in EU
5. Finally, charges - BMC Bioinformatics
The online publication fee payable to BioMed Central shall be £540 (Five Hundred and Forty Pounds Sterling) per Article. Such fees are payable regardless of any BioMed Central membership arrangements and it
shall be the responsibility of the author to pay this to BioMed Central. BMC Bioinformatics will contact you for this charge very soon.
We look forward to seeing you in Incheon, Korea, 11-14 January 2011! Best Regards,
Phoebe Chen, La Trobe University, Melbourne, Australia Kwang-Hyun Cho, KAIST, Korea
Program Co-Chairs of APBC2011
The 9th Asia Pacific Bioinformatics Conference http://sysbio.kribb.re.kr/APBC2011/index.php
P
REDICTING AND ANALYZING
DNA-
BINDING DOMAINS USING A SYSTEMATIC APPROACH
TO IDENTIFYING A SET OF INFORMATIVE PHYSICOCHEMICAL AND BIOCHEMICAL
PROPERTIES
Hui-Lin Huang1,2, I-Che Lin1, Yi-Fan Liou2, Chia-Ta Tsai2, Kai-Ti Hsu2, Wen-Lin Huang3, Shinn-Jang Ho4, and Shinn-Ying Ho1,2§
1Department of Biological Science and Technology, National Chiao Tung University, Hsinchu, Taiwan 2Institute of Bioinformatics and Systems Biology, National Chiao Tung University, Hsinchu, Taiwan
3Department of Management Information System, Chin Min Institute of Technology, Miaoli, Taiwan 4Department of Automation Engineering, National Formosa University, Yunlin 632, Taiwan
§Corresponding author Email addresses: HLH: [email protected] ICL: [email protected] YFL: [email protected] CTT: [email protected] KTH: [email protected] WLH: [email protected] SJH: [email protected] SYH: [email protected]
I. ABSTRACT A. Background
Existing methods of predicting DNA-binding proteins used valuable features of physicochemical properties to design support vector machine (SVM) based classifiers. Selection of physicochemical properties and determination of their corresponding feature vectors rely mainly on known properties and experience of designers. However, there exists a troublesome problem for designers that some different physicochemical properties have similar vectors of representing 20 amino acids and closely related physicochemical properties in the same group have dissimilar vectors.
B. Methods
This study proposes a systematic approach (named Auto-IDPCPs) to automatically identify a set of physicochemical and biochemical properties in the AAindex database to design SVM-based classifiers for predicting and analyzing DNA-binding domains/proteins. Auto-IDPCPs consists of 1) clustering 531 vectors in AAindex into 20 classes using a fuzzy c-means algorithm, 2) utilizing an efficient genetic algorithm based optimization method IBCGA to select an informative set of feature vectors of representing sequences from the viewpoint of machine learning, and 3) analyzing the selected feature vectors to identify the related physicochemical properties which may affect the binding mechanism of DNA-binding domains/proteins.
C. Results
The proposed Auto-IDPCPs identified m=22 features of properties belonging to five classes for predicting DNA-binding domains with a five-fold cross-validation accuracy of 87.12%. If m=5 that one representative property is selected from each class, the accuracy of 83.59% is also promising compared with the accuracy of 82.07% of the existing method PSSM-400 using 400 features. For predicting DNA-binding sequences, three additional classes (totally eight classes) were needed, and the accuracies of 75.50% and 73.24% were obtained using m=28 and 8 features, respectively, where PSSM-400 has the accuracy of 74.22%. When applied on an independent test data set of DNA-binding domains, Auto-IDPCPs and PSSM-400 have accuracies of 80.73% and 82.81%, respectively. Some typical physicochemical properties discovered are hydrophobicity, secondary structure, charge, solvent accessibility, polarity, flexibility, normalized Van Der Waals volume, pK (pK-C, pK-N, pK-COOH and pK-a(RCOOH)), etc.
D. Conclusions
The proposed approach Auto-IDPCPs would help designers to investigate informative physicochemical and biochemical properties by considering both prediction accuracy and analysis of binding mechanism simultaneously. The approach Auto-IDPCPs can be also applicable to predict and analyze other protein functions from sequences.
II. BACKGROUND
DNA-binding domains/proteins are functional proteins in a cell, which plays a vital role in various essential biological activities, such as DNA transcription, replication, packaging, repair and rearrangement [1]. The computational methods using support vector machine (SVM) in conjunction with evolutionary information of amino acid sequence in terms of their position-specific scoring matrices (PSSMs) for predicting DNA-binding sites were successfully developed [2]. Several methods of using machine learning approaches were developed to predict DNA-binding domains/proteins from given sequences of variable lengths [3-7], shown Table 1. Due to different design aims and data sets used, it is difficult to assess which feature type is the most informative cooperated with SVM by comparing with prediction accuracies only. The PSSM is an effective feature type of representing DNA-binding sequences, but its ability of interpretability is not satisfactory enough in analyzing the binding mechanism [5]. Besides PSSMs, the physicochemical properties with the characteristics of high interpretability were commonly used [3-4, 6-7]. Some issues are concerned in designing prediction methods, described below. 1) Selection of physicochemical properties: Generally, effective physicochemical properties of amino acids are selected as
prediction features by using known properties of DNA-binding mechanism and knowledge of related binding mechanism [3-4, 6-7]. However, it is desirable to explore undiscovered properties by machine learning approaches to further advance the prediction accuracy and understand the binding mechanism.
2) Representation of sequences: How to effectively represent sequences of variable lengths as a feature vector using physicochemical properties play an important role in advancing prediction accuracy. The pseudo-amino acid
composition (PAAC) is an efficient representation method of coupling physicochemical properties, which was used to represent a sequence as a 40-dimensional feature vector for discriminating DNA-binding proteins from non-binding proteins [3]. The combined descriptor was proposed using amino acid composition and a series of associated physicochemical properties to form a 132-dimensional feature vectors [7]. The conjoint triad descriptor of 343-dimensional feature vector was proposed that 20 amino acids were clustered into seven classes according to their dipoles and volumes of side chains [6].
3) Values of amino acids for specific physicochemical properties: The AAindex database [8-9] collected 531 physicochemical properties (ignoring 13 properties without available values) with corresponding values of amino acids. Recently, some computational methods of predicting protein functions were successfully developed by mining informative physicochemical properties from AAindex [10-11].
Besides pursuit of high prediction accuracy, discovering potential properties to further understand the binding mechanism are also taken into account in this study. We present a troublesome problem in using the AAindex database and propose an effective method to solve. We found that some different physicochemical properties have similar vectors of representing the 20 amino acids and the closely related physicochemical properties in the same group have dissimilar vectors. For example, the determination of values of the 20 amino acids to represent a sequence by coupling the hydrophobicity property is highly related to prediction performance. Similarly, if a different property with a similar vector replaces the known one without significantly degrading prediction performance, it means that the replaced property may be also important to the binding mechanism from the viewpoint of machine learning. The detailed explanation by using a real quantization example is described below.
Figure 1 shows an illustration example. The 402 properties in AAindex were classified into six groups according to their biological meanings classified by Tomii et al. [9], as shown in Fig. S1 [see additional file]. According to the vectors of amino acids for 531 properties, we clustered them into 20 clusters by a fuzzy c-means algorithm [12] based on normalized Euclidean distances. The properties H88 and A392 are two different properties but their distance 0.0178 is small belonging to the same cluster 7. On the other hand, H88 and H178 belonging to the same group Hydrophobicity in AAindex have a large distance 0.0877 belonging to clusters 7 and 18, respectively. Although H88 and H151 (used in [3]) are in the same group Hydrophobicity, their distance 0.0299 is larger than that between H88 and A392.
For the aim of designing accurate prediction methods, the major concern is to identify feature vectors with high discrimination abilities for classifying positive and negative samples. This task can be done well for computational methods by an optimization approach to feature selection. If the feature vectors were identified by predetermined properties based on prior knowledge, the selected vectors of representing amino acids may be not the best. Considering the other aim of discovering potential properties to further look insight the binding mechanism, we proposed a systematic, optimization approach (named Auto-IDPCPs) to automatically identifying a set of feature vectors and analyzed the feature vectors to find properties of affecting the DNA-binding mechanism.
The proposed approach Auto-IDPCPs can identify a small number m of feature vectors and discover the related hydrophobicity properties with comparable performance, compared with the PSSM feature. Auto-IDPCPs would help designers to investigate informative physicochemical and biochemical properties by considering both prediction accuracy and analysis of binding mechanism simultaneously. Auto-IDPCPs can be also applicable to predict and analyze other protein functions from sequences.
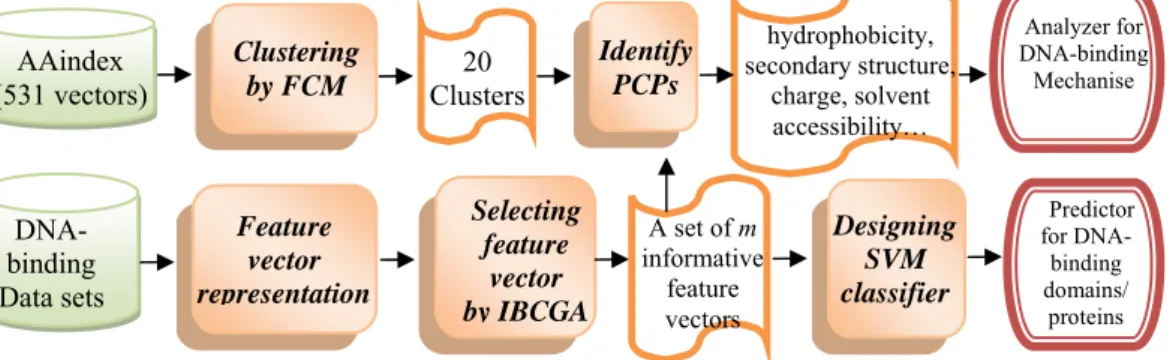
III. METHODS
The system flowchart of the proposed approach Auto-IDPCPs is shown in Fig. 2. The input of the method comprises the AAindex database and three data sets, including DNA-binding domains and sequences, and one independent test data set. The output has two parts: 1) a predictor of DNA-binding domains/proteins with a set of m informative feature vectors and the
parameter setting of SVM by an efficient feature selection algorithm IBCGA, and 2) a set of physicochemical and biochemical properties in the AAindex database for analyzing the DNA-binding mechanism.
A. Data sets
To evaluate effectiveness of the identified physicochemical properties by comparing with the famous PSSM features, we used the benchmark data sets used in the PSSM-400 method [5], as shown in Table 2. The data set DNAset has 146 DNA-binding domains (or protein chains) and 250 non-DNA-DNA-binding domains. No two domains have the similarity more than 25%. The data set DNAaset consists of 1153 DNA-binding proteins and 1153non-binding proteins. 3) An independent data set DNAiset is additionally used, having 92 DNA-binding domains and 100 non-DNA-binding domains [5].
B. Feature vector representation
All the domains/sequences have a variable length l. A sequence forms an l-dimensional profile where
the value of each amino acid is obtained from the specified property in the AAindex database. The
l-dimensional profiles are transformed into vectors with the same constant length L for utilizing SVM.
The transformation can be any known effective representation [3-4, 6-7] provided that the L features can
effectively classify the l-dimensional profiles of positive and negative sequences. The simplest feature is
the mean of the profile that L=1 [10-11]. Therefore, the sequences with m properties are represented as
an m-dimensional feature vectors. Finally, all values of the feature vectors are normalized into [-1, 1] for
applying SVM.
C. Feature selection by IBCGA
Selecting a minimal number of informative features while maximizing prediction accuracy is a
bi-objective 0/1 combinatorial optimization problem. An efficient inheritable bi-bi-objective combinatorial
genetic algorithm IBCGA [13] is utilized to solve this optimization problem. IBCGA bases on an
intelligent genetic algorithm IGA [14] with an inheritable mechanism. The IGA algorithm uses a
divide-and-conquer strategy and an orthogonal array crossover to efficiently solve large parameter optimization
problems. In this study, the IGA algorithm can efficiently explore and exploit the search space of C(n, r),
where n=531 in this study. IBCGA can efficiently search the space of C(n, r
1) by inheriting a good
solution in the space of C(n, r) [13]. Therefore, IBCGA can economically obtain a complete set of
high-quality solutions in a single run where r is specified in an interesting range such as [10, 30]. The
chromosome encoding scheme of IGA consists of both binary genes for feature selection and parametric
genes for tuning SVM parameters and C [10]. The performance of selected properties associated with
the parameter values of SVM is measured by five-fold cross-validation (5-CV) for comparing with the
method PSSM-400 [5].
IBCGA with the fitness function f(X) can simultaneously obtain a set of solutions, X
r, where r=r
start,
r
start+1, …, r
endin a single run. In this study, the parameter settings r
start=10, r
end=30, N
pop=50, pc =0.8
and pm =0.05. The output contains a set of m selected properties from AAindex and an SVM classifier
with associated parameter settings. The IBCGA algorithm is given in Fig. 3. The best one of R solutions
can be determined by considering the accurate one S
awith the highest accuracy or the robust one with
the highest score S
rfor identifying informative properties.
D. Designing SVM classifiers
For evaluating the effectiveness by comparing with the commonly used feature sets, we implemented
the predictor using the same single-classifier SVM with the feature types, amino acid composition (AAC)
and PSSM [5]. Additionally, the selected physicochemical properties (PCPs) combined with AAC and
PSSM were also evaluated.
E. Clustering properties by the FCM method
The application of cluster approaches is to partition 531 vectors of physicochemical properties into
clusters, where similar vectors are assigned to the same cluster. An index vector of amino acids is a set
of 20 numerical values representing some physicochemical property of amino acids. All data were
normalized in such a way that every physicochemical property had an average profile value of zero and
a standard deviation equal to 1.
The fuzzy derivative of k-means, known as fuzzy c-means (FCM) [12], has an objective functional of
the form, J(X;U,v)=
21 1
( , )
K n s ij i j i ju d v x
, where n=531 is the number of data vectors, K is the number of
cluster represented by the cluster prototype vi, s[1,∞) is a weighting exponent called the fuzzifier and
d(v
i, x
j) is the distance of x
jfrom the cluster prototype v
i. Dembélé and Kastner [15] suggested the
parameters setting s=1.12 and K=20 clusters, adopted in this study.
F. Identifying physicochemical properties
It is not easy to discover related physicochemical properties for analyzing DNA-binding mechanism by
computational methods with a relatively small size of data sets. Therefore, we present a hybrid method
by combining evidences from the viewpoints of both machine learning and biological meanings.
Auto-IDPCPs identifies m properties belong to c of 20 clusters. We examine all properties P1 by considering
the identified m properties P2 if they satisfying the criteria, P1 is a promising property to be further
investigated: 1) P1 and P2 have a small distance and 2) if P2 is replaced with P1 one at a time, the
prediction accuracy is not significantly decreased.
Only 402 of 531 properties were classified into six groups, (A): Alpha and turn propensities, (B): Beta
propensity, (C): Composition, (H): Hydrophobicity, (P): Physicochemical properties, and (O): Other
properties. We classified the other 129 properties into the six groups according their distance of vectors
using a nearest-neighbor rule. The mapping of 531 feature numbers and AAindex identity and their
classified result into six groups are given in Tables S1 and S2, respectively, and their statistic result is
given in Table S3 [see additional file]. The statistical results of property distribution in the six groups for
531 and 402 amino acid indices are given in Fig. S2 [see additional file].
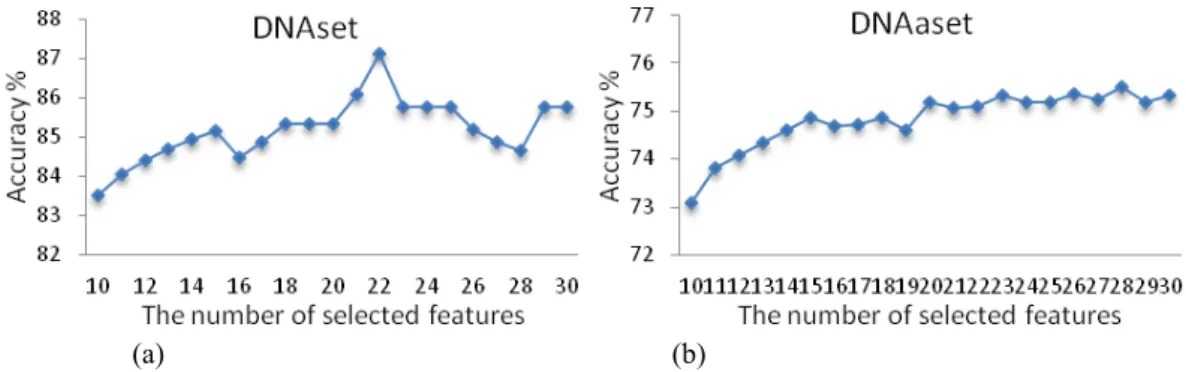
IV. RESULTS A. Identified properties by IBCGA
The statistical result of Sr in selecting property sets from R =30 independent runs on DNAset and DNAaset are given in Fig. 4. The 18th and 6th runs for DNAset and DNAaset, respectively, are selected, and their prediction accuracies for various numbers of selected properties are given in Fig. 5. The m=22 and 28 properties selected for DNAset and DNAaset, respectively, are given in Tables S4 and S5 [see additional file] in which the AAindex identity numbers and their property description are provided. An efficient way to study the effects of several factors simultaneously is to use the main effect difference (MED) that the most effective property has the largest value of MED. The m properties are ranked by using MED is shown in Fig. 6. The properties of rank 1 are feature numbers 86 (FAUJ880109, Localized electrical effect) and 39 (CHOP780202, Normalized frequency of beta-sheet) for DNAset and DNAaset, belonging to groups Hydrophobicity and Beta propensity in the six groups, respectively.
B. Prediction performance evaluation
To evaluate the effectiveness of the identified m informative feature vectors (PCPs), three feature types were additionally evaluated, as shown in Table 3. AAC is a 20-dimensional vector of amino acid composition, PSSM is the feature representation [5] of 400 features. PCPs +AAC and PCPs +PSSM are two hybrid feature types by adding individual feature vectors. Considering the DNA-binding domain data set DNAset, the set of m=22 informative properties (PCPs) identified by Auto-IDPCPs performs best where the robust solution Sr with accuracy of 87.12%, compared with ACC, PSSM, PCPs+AAC
and PCPs+PSSM are 80.30%, 82.07%, 81.82% and 86.62%, respectively. For the DNA-binding protein data set DNAaest, the method with PCPs and m=28 informative properties (75.50%) is slightly worse than that with PSSM (76.58%). However, PCPs+PSSM can improve the accuracy to 80.27%. When the predictor trained by DNAset (Sr with m=22 informative
properties) were evaluated by the independent test data set DNAiset, the accuracy is 80.73% (=155/192), slightly worse than 82.81% (=159/192) of PSSM-400. A small, high-performance features set of size c from c clusters is given in Table 4. The properties and their descriptions are given in Tables S6 and S7 [see additional file] where c=5 and 8 for DNAset and DNAaset, respectively.
The experimental results reveal that the identified small set of m physicochemical properties with a simple representation performs equally well, compared with the PSSM feature type. However, the identified physicochemical properties are interpretable for further understanding the DNA-binding mechanism.
C. Analyzing binding mechanism by physicochemical properties
The 30 sets of m properties belonging to the 20 clusters from the results of 30 runs are shown in Fig. S3 [see additional file]. From the statistic result, the clusters 7, 9, 10, 16 and 18 with very high selection frequencies are more important for predicting DNA-binding domains and proteins. The m=22 properties (Table S4) belong to five clusters which are the same as the five clusters 7, 9, 10, 16 and 18. For predicting DNA-binding proteins, the m=28 properties (Table S5) belong to eight clusters with additional three clusters 3, 14 and 17.
An illustration example is given in Fig. 7. The both feature sets S1 (H88, H86, H67, C209, H178) and S2 (A392, A303, A307, C440, H178) are selected for predicting DNA-binding domains in DNAset that one properties selected from one of five clusters 7, 9, 10, 16 and 18. The identified properties H88 and A392 belong to hydrophobicity, and alpha and turn propensities groups but they belong to the same cluster 7 with a relatively small distance 0.0178. The prediction accuracy of S3 by replacing H88 with H151 is 81.05 %. On the other hand, H151 belonging to the cluster 7 and Hydrophobicity group used in [3] can be inferred from feature sets S1 and S2. After carefully analyzing all properties, we identify some properties in the five identified clusters for analyzing DNA-binding domains, shown in Table 5. Some typical physicochemical properties discovered are hydrophobicity, secondary structure, charge, solvent accessibility, polarity, flexibility, normalized Van Der Waals volume, pK (pK-C, pK-N, pK-COOH and pK-a(RCOOH)), etc. Most of identified properties were used in previous works [3-4, 6-7] but a few properties such as the flexibility property H8 BHAR880101 in cluster 7 “Average flexibility indices (Bhaskaran-Ponnuswamy, 1988)” are not utilized yet in existing method of predicting DNA-binding domains. The correlation between protein flexibility and protein function suggests a link between DNA-binding activity and the conformational freedom of the DNA-binding domain [16].
V. DISCUSSION
To avoid from overfitting the small-scale data sets in identifying physicochemical properties using an optimization approach, this study proposes a hybrid method of combining evidences from computational methods of considering robust factors and biological experiments from literature. The future work is to further verify these discovered properties in predicting and analyzing the DNA-binding mechanism.
VI. CONCLUSIONS
This study has proposed a systematic approach Auto-IDPCPs to automatically identify an informative set of physicochemical and biochemical properties in the AAindex database to design SVM-based classifiers for predicting and analyzing DNA-binding domains/proteins.
VII. COMPETING INTERESTS
The authors declare that they have no competing interests. VIII. AUTHORS' CONTRIBUTIONS
HLH designed the system, implemented programs, carried out the analysis, and participated in manuscript preparation. ICL provided biological knowledge and carried out the analysis. YFL developed the web server. CTT, KTH, WLH and SJH
implemented programs and participated in the experimental design. SYH supervised the whole project and participated in manuscript preparation. All authors have read and approved the final manuscript.
IX. ACKNOWLEDGEMENTS
The authors would like to thank the National Science Council of Taiwan for financially supporting this research under the contract numbers NSC 98-2627-B-009-004- and NSC 96-2628-E-009-141-MY3.
REFERENCES
1. G
AOM,
S
KOLNICKJ:
A
THREADING-
BASED METHOD FOR THE PREDICTION OFDNA-
BINDINGPROTEINS WITH APPLICATION TO THE HUMAN GENOME.
PL
OS
C
OMPUTB
IOL2009,
5(11):
E1000567.
2. H
OSY,
Y
UFC,
C
HANGCY,
H
UANGHL:
D
ESIGN OF ACCURATE PREDICTORS FORDNA-BINDING SITES IN PROTEINS USING HYBRID
SVM-PSSM
METHOD.B
IOSYSTEMS2007,
90(1):234-241.
3. C
AIYD,
L
INSL:
S
UPPORT VECTOR MACHINES FOR PREDICTING RRNA-,
RNA-,
AND DNA-BINDING PROTEINS FROM AMINO ACID SEQUENCE.
B
IOCHIMB
IOPHYSA
CTA2003,
1648(1-2):127-133.
4. F
ANGY,
G
UOY,
F
ENGY,
L
IM:
P
REDICTINGDNA-
BINDING PROTEINS:
APPROACHED FROMC
HOU'
S PSEUDO AMINO ACID COMPOSITION AND OTHER SPECIFIC SEQUENCE FEATURES.
A
MINOA
CIDS2008,
34(1):103-109.
5. K
UMARM,
G
ROMIHAMM,
R
AGHAVAGP:
I
DENTIFICATION OFDNA-
BINDING PROTEINS USINGSUPPORT VECTOR MACHINES AND EVOLUTIONARY PROFILES.
BMC
B
IOINFORMATICS2007,
8:463.
6. S
HAOX,
T
IANY,
W
UL,
W
ANGY,
J
INGL,
D
ENGN:
P
REDICTINGDNA-
ANDRNA-
BINDING PROTEINS FROM SEQUENCES WITH KERNEL METHODS.J
T
HEORB
IOL2009,
258(2):289-293.
7. Y
UX,
C
AOJ,
C
AIY,
S
HIT,
L
IY:
P
REDICTING RRNA-,
RNA-,
ANDDNA-
BINDING PROTEINSFROM PRIMARY STRUCTURE WITH SUPPORT VECTOR MACHINES.
J
T
HEORB
IOL2006,
240(2):175-184.
8. K
AWASHIMAS,
P
OKAROWSKIP,
P
OKAROWSKAM,
K
OLINSKIA,
K
ATAYAMAT,
K
ANEHISAM:
AA
INDEX:
AMINO ACID INDEX DATABASE,
PROGRESS REPORT2008.
N
UCLEICA
CIDSR
ES2008,
36(D
ATABASE ISSUE):D202-205.
9. T
OMIIK,
K
ANEHISAM:
A
NALYSIS OF AMINO ACID INDICES AND MUTATION MATRICES FOR SEQUENCE COMPARISON AND STRUCTURE PREDICTION OF PROTEINS.
P
ROTEINE
NG1996,
9(1):27-36.
10. T
UNGCW,
H
OSY:
POPI:
PREDICTING IMMUNOGENICITY OFMHC
CLASSI
BINDING PEPTIDES BYMINING INFORMATIVE PHYSICOCHEMICAL PROPERTIES.