國
立
交
通
大
學
生物資訊及系統生物研究所
博
士
論
文
同源蛋白質交互作用與複合體剖析蛋白質交互作用體行為
Homologous protein-protein interactions and protein
complexes reveal interactome behavior
研 究 生:羅宇書
指導教授:楊進木 教授
同源蛋白質交互作用與複合體剖析蛋白質交互作用體行為
Homologous protein-protein interactions and protein
complexes reveal interactome behavior
研 究 生:羅宇書 Student:Yu-Shu Lo
指導教授:楊進木 Advisor:Jinn-Moon Yang
國 立 交 通 大 學
生物資訊及系統生物研究所
博 士 論 文
A Thesis Submitted to Institute of Bioinformatics and Systems Biology National Chiao Tung University in partial Fulfillment of the Requirements
for the Degree of Ph.D. in Bioinformatics and Systems Biology
August 2012
Hsinchu, Taiwan, Republic of China
Homologous protein-protein interactions and protein complexes
reveal interactome behavior
Student : Yu Shu Lo Adviser : Dr. Jinn-Moon Yang
Institute of Bioinformatics and Systems Biology National Chiao Tung University
Abstract
Protein-protein interaction (PPI) networks provide key insights into complex biological systems, from how different processes communicate to the function of individual residues on a single protein. Therefore, several large network databases (e.g. IntAct, DIP, and BioGRID) record hundreds of thousands of physical and genetic interactions from a wide variety of organisms have been purposed. However, these PPI databases are dominated by few species and usually could not provide the binding mechanisms. Therefore, constructing the structure resolved PPI networks across multiple organisms should provide a great value for investigating the behavior of PPI network.
To address the issues, we proposed the concepts of protein interaction family (i.e. protein-protein interaction family and protein complex family) to construct a structure resolved PPI networks and study the behaviors of a specific PPI network. The protein interaction family is a group of protein interactions (PPI or protein complex) which share the consensus interacting domain, binding environment, and have similar biological processes. According to the concept "3D-domain interolog mapping" with a scoring system, we are able to explore all homologous protein-protein interaction pairs (protein-protein interaction family) between two homolog families, derived from a known 3D-structure dimmer (template), across multiple species. Then, we also identify the homologous protein complexes with the binding models (e.g. hydrogen bonds and conserved amino acids in the interfaces), functional modules, and the conserved interacting domains and Gene Ontology annotations in multiple organisms.
Based on the PPIs derived from "3D-domain interolog mapping" and "protein complex family", we are able to construct structure resolved PPI networks in multiple organisms (e.g.
Homo sapiens, Mus musculus, and Danio rerio). In each network, the PPIs with residue-based
binding models have a highly agreement in Gene Ontology similarities. Furthermore, the architecture (i.e. scale-free network properties) of these networks is consistent with some cellular networks of previous studies. In addition, the consensus proteins and PPIs derived form on our method are highly related to the essential genes and disease related proteins recorded in OMIM. We also indicate that the disease related mutations are more enrichment on
network, we also provided a new characterization (named MS-matrix) to describe the modularity and relative importance of proteins. We believe that structure resolved PPI networks derived from the PPI family would provide the insight for understanding the mechanism of biological processes within a given PPI network.
同源蛋白質交互作用與複合體剖析蛋白質交互作用體行為
研究生:羅宇書 指導教授:楊進木博士
國立交通大學 生物資訊及系統生物研究所 博士班
中文摘要
透過蛋白質交互作用網路(protein interaction network)可以對於複雜的生物系統有 更進一步的了解,例如探討不同生化途經之間的協同作用、蛋白質上特定殘基對功能的 影響。因此,大量的交互作用資料庫(如:IntAct、DIP 和 BioGRID 等)被建立來探討 蛋白質交互作用網路。然而,這些資料庫中的蛋白質交互作用資料往往集中在少數的物 種,而且也缺乏對於交互作用介面機制的解釋。
針對此議題,我們提出了蛋白質交互作用家族的觀念(包含蛋白質-蛋白質交互作 用家族(protein-protein interaction family)和蛋白質複合體家族(protein complex family))以 協助多個物種中建立具有結構解析的蛋白質交互作用網路,並探討單一物種的蛋白質交 互作用網路行為。蛋白質交互作用家族為一群有擁有保留性交互作用區塊、結合環境和 相似生物途徑的蛋白質交互作用所組成。而透過“3D-domain interolog mapping"與一個 新的能量函式,我們將透過已知結構的模板(template),來探索多個物種間所有的同源 蛋白質相互作用。此外,我們也在多個物種中找尋同源蛋白複合體,並描述了結合模型
(例如在交互作用介面上的氫鍵和保留性氨基酸配對)、功能模塊、保留性相互作用區塊
(interacting domain)和 Gene Ontology。
透過由“3D-domain interolog mapping"與蛋白質複合體家族,我們在人類、老鼠與 斑馬魚中建立了具有結構解析的蛋白質交互作用網路。在每一個網路中,這些具有結構 解析的蛋白質交互作用與 Gene Ontology 相似度有很高的一致性。此外,這些網路也都具 有之前對於生化網路研究中所指出的拓譜特性(scale-free network)。而透過蛋白質交互作 用家族我們也可指出網路中在跨物種間具有高度保留的蛋白質與交互作用,而這些蛋白 質往往是生存必須基因(essential gene)或是跟疾病相關。更進一步的,這些跟疾病相關的 基因突變往往位於蛋白質交互作用介面,並擔任重要的交互作用(例如氫鍵)。此外,對 於單一蛋白質交互作用網路,我們提出了一個新的概念“MS-matrix"來描述網路上重要 的蛋白質以及模組化特性。基於上述這些研究,我們認為透過交互作用家族所構建的結 構解析交互作用網路對於了解生化途徑的機制是很有幫助的。
誌謝
在順利撰寫完這份博士論文並取得博士學位的過程中,我獲得非常多的貴人相助。 首先,我必須感謝我的指導教授楊進木老師,老師是一位非常有研究熱誠的研究者與教 育家,在我遭遇到瓶頸或是對事情產生消極態度的時候,總是可以透過跟老師的開導, 而讓我重新燃起希望或是突破瓶頸。除了在研究專業上的教導與訓練之外,在生活與人 生態度上,更是教導了我許多,指出了我所欠缺的積極思考、積極爭取的態度與其他那 些該糾正的缺點。更感謝老師在我博士班研讀期間所提供的學習環境、研究資源以及出 國參與研討會的機會,不僅使我在求學時能夠更為順利亦增廣了我的見聞。 接著我要感謝我的口試委員,包含我的指導教授楊進木教授、口試召集人熊昭教授、 江安世教授、黃鎮剛教授、莊永仁教授、陳豐奇教授。感謝每位教授在百忙之中抽空擔 任我的口試委員並且評鑑我的論文,更感謝各位教授在口試期間對我的研究所提出的寶 貴建議,有了各位教授的建議與指導才使得這份研究論文能夠更臻完美。 我也要感謝我的實驗室其他一起努力的夥伴們,特別感謝系統生物組的峻宇、怡馨、 星翰、尚文、采凌、俊辰、怡瑋。我們系統生物組都是靠全組成員一起努力才能有不錯 的研究成果。特別感謝峻宇,他是一位非常優秀的學弟,在研究上跟我可以互相討論、 互相激勵,也可以容忍我這個組長沒有耐心的脾氣,讓很多研究可以順利進行。也感謝 怡馨,在研究上她是個非常有耐心跟細心的研究者,可以注意到我沒有想到的一些地方, 彼此相輔相成,也在行政事務上給了我很大的幫助,讓我能輕鬆很多。並感謝星翰跟采 凌兩位學弟妹,他們認真的態度讓我們的研究可以順利的進行。並感謝實驗室其他同仁, 特別是凱程、章維、志達與一原在討論時往往給予一些很有助益的建議與看法,而且一 原也在程式方面給予了我很大的協助。因為實驗室的同仁讓辛苦的博士生活更顯得豐富 與多采多姿。 最後我想特別感謝我父母跟怡馨,我的父母能夠體諒我在博士研讀期間的苦悶,對 我十分的體貼與關心,常常原諒我的無理取鬧並傾聽我很多日常瑣碎的抱怨與訴苦,支 持我讓我能夠面對一切困難。怡馨是一位非常非常好的伴侶,能在我不開心的時候安慰 我、鼓勵我。開心的時候則一起分享喜悅。研究上也可以互相扶持,忍耐與糾正我的很 多缺點。有她陪伴在身邊讓很多很困難的事情,都變成兩個人可以克服的小事。希望我 們兩個可以一直這樣走下去。 最後,僅將此論文獻給我這些敬愛的人以及幫助過我的人。Contents
ABSTRACT ... I
中文摘要 ... III 誌謝 ... IV
CONTENTS ... V LIST OF FIGURES ... VII LIST OF TABLES ... IX
CHAPTER 1. INTRODUCTION ...1
1-1. BACKGROUND ...1
1-2. CURRENT STATE OF CONSTRUCTING PROTEIN-PROTEIN INTERACTION NETWORKS ...3
1-3. THESIS OVERVIEW ...6
CHAPTER 2. 3D-INTEROLOGS: AN EVOLUTION DATABASE OF PHYSICAL PROTEIN-PROTEIN INTERACTIONS ACROSS MULTIPLE GENOMES ... 10
2-1. INTRODUCTION ... 12
2-2. METHODS AND MATERIALS ... 14
2-3. SCORING FUNCTION AND MATRICES ... 17
2-4. INPUTS AND OUTPUTS ... 21
2-5. EXAMPLE ANALYSIS ... 21
2-6. RESULTS ... 24
2-7. CONCLUSIONS ... 31
CHAPTER 3. PCFAMILY: A WEB SERVER FOR SEARCHING HOMOLOGOUS PROTEIN COMPLEXES 32 3-1. INTRODUCTION ... 32
3-2. METHOD AND IMPLEMENTATION ... 33
3-3. INPUT,OUTPUT AND OPTIONS ... 37
3-4. EXAMPLE ANALYSIS ... 39
3-5. RESULTS ... 42
3-6. CONCLUSIONS ... 45
CHAPTER 4. STRUCTURAL INTERACTOME OF MULTIPLE VERTEBRATE GENOMES THOUGH HOMOLOGOUS PROTEIN-PROTEIN INTERACTIONS ... 46
4-1. INTRODUCTION ... 47
4-4. CONCLUSIONS ... 81
CHAPTER 5. MODULARITY STRUCTURE MATRIX FOR INVESTIGATING PROTEIN INTERACTION NETWORK ... 82 5-1. INTRODUCTION ... 82 5-2. METHODS ... 84 5-3. RESULTS ... 88 5-4. CONCLUSIONS ... 96 CHAPTER 6. CONCLUSION ... 98 6-1. SUMMARY ... 98
6-2. DISCUSSION AND FUTURE WORK ... 100
LIST OF PUBLICATIONS ... 102
List of Figures
Figure 1-1. The overview of constructing the structure resolved PPI networks and studying the interactome
behavior ...2
Figure 2-1. Two frameworks of template-based methods for protein-protein interactions (PPI). ... 14
Figure 2-2. Overview of the 3D-interologs database for protein-protein interacting evolution, protein functions annotations and binding models across multiple species. ... 16
Figure 2-3. Knowledge-based protein-protein interacting scoring matrices: (A) sidechain-sidechain van-der Waals scoring matrix; (B) sidechain-backbone van-der Waals scoring matrix; (C) sidechain-sidechain special-bond scoring matrix; (D) sidechain-backbone special-bond matrix scoring. ... 19
Figure 2-4. The 3D-interologs database search results of using human NXT1 as query. ... 23
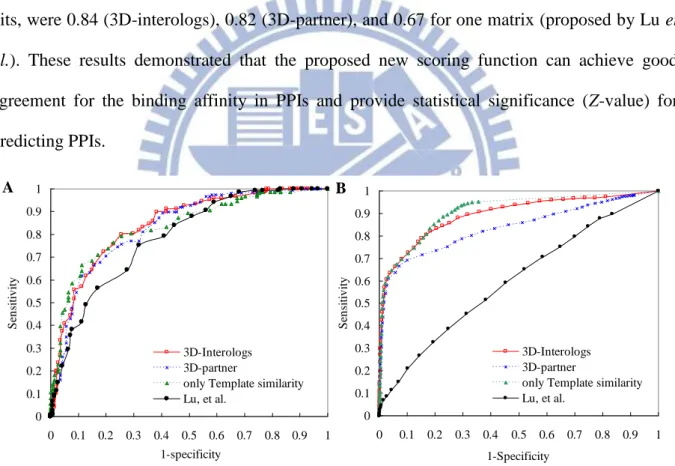
Figure 2-5. Evaluation of the 3D-interologs in binding affinities. ... 26
Figure 2-6. The ROC curves of the 3D-interologs for protein-protein interactions. ... 27
Figure 2-7. Precisions and recalls of 3D-interologs the on Integr8. ... 28
Figure 3-1. Overview of the PCfamily server for homologous complexes search using proteins Skp1, Skp2, and Cks1 of Rattus norvegicus as the query. ... 34
Figure 3-2. Binding models and multiple sequence alignments of PPI family in Skp1-Skp2-Cks1 complex (PDB code 2ast). ... 35
Figure 3-3. The PCfamily server search results using proteins Epor, Epo, and Epor of Mus musculus as the query. ... 38
Figure 3-4. Binding models and multiple sequence alignments of PPI family in Skp1-Skp2-Cks1 complex (PDB code 2ast). ... 40
Figure 3-5. Multiple sequence alignments of the (Epo-Epor) A-C interface of template cytokine/receptor complex (PDB code 1eer). ... 41
Figure 3-6. Evaluations of the PCFamily server on 941 protein complex families. ... 42
Figure 3-7. The distributions of the biological process (BP) and cellular component (CC) RSS scores on 84,082 protein-protein interactions selected from the IntAct database. ... 44
Figure 4-1. The overview of constructing structure resolved PPI networks in three vertebrates though "3D-domain interolog mapping" ... 49
Figure 4-2. Conceptual overview of alignment procedure. ... 52
Figure 4-3. The distributions of relative specificity similarity (RSS) of BP, CC, and MF of the interacting protein pairs in the derived structural PPI networks ... 55
Figure 4-4. The distributions of BP, CC, and MF RSS scores on interacting protein pairs and all protein pairs within the mouse and zebrafish networks ... 56
Figure 4-5. The node degree distributions of three structure resolved PPI networks: (A) H. sapiens, (B) M. musculus, and (C) D. rerio ... 56
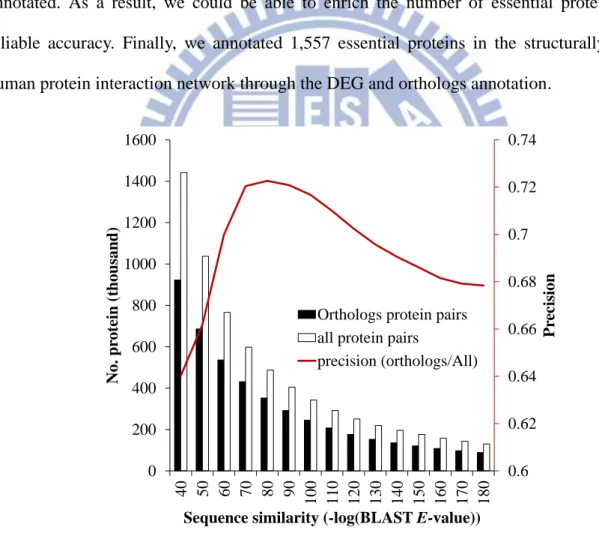
Figure 4-8. The distribution of orthologs protein pairs under different sequence similarities ... 60 Figure 4-9. Six major cellular processes in consensus proteins and PPIs. ... 63 Figure 4-10. The distributions of protein dynamic ability (A), degree (B), closeness centrality (C), and
betweenness centrality (D) in network of 1,887 consensus proteins ... 64 Figure 4-11. The odds ratios of in-frame and truncating mutations on the binding interface ... 66 Figure 4-12. The pathways and proteins involved in a great amount of diseases, especially the cancers ... 68 Figure 4-13. The pathways and proteins involved in a great amount of diseases, especially the cardiovascular-
related diseases ... 69 Figure 4-14. The mapping pathways and proteins which are related to the cancers of M. musculus. ... 71 Figure 4-15. The mapping pathways and proteins which are related to the cancers of D. rerio. ... 72 Figure 4-16. The mapping pathways and proteins which are involved in the cardiovascular-related diseases of M.
musculus. ... 73 Figure 4-17. The mapping pathways and proteins which are involved in the cardiovascular-related diseases of D.
rerio. ... 74
Figure 4-18. Binding models and multiple sequence alignments of PPI family derived from FGF2-FGFR2 heterodimer (PDB code: 1ev2) ... 75 Figure 4-19. Binding models and multiple sequence alignments of PPI family derived from TNNT2-TNNI3
heterodimer (PDB code: 1j1d) ... 77 Figure 4-20. The specific proteins among the complement and coagulation pathway ... 78 Figure 4-21. Binding models and multiple sequence alignments of PPI family derived from F2-SERPINA5
heterodimer (PDB code: 3b9f) ... 79 Figure 4-22. The mapping pathways and proteins involved in the complement and coagulation pathway of M.
musculus. ... 80
Figure 4-23. The mapping pathways and proteins involved in the complement and coagulation pathway of D. rerio. ... 80 Figure 5-1. The overview of the evaluating the importance of each node in a simple network through the
"MS-matrix" ... 84 Figure 5-2. The relationship between importance of protein and essential proteins ... 89 Figure 5-3. Evaluation importance of protein by (A) Degree centrality (B) Closeness centrality (C) Between
centrality ... 90 Figure 5-4. The distribution of gene ontology similarities (i.e. RSS of BP, CC, and MF) and the shortest path
between protein pairs under different modular similarity ... 91 Figure 5-5. The distribution of the number of gene ontology annotations (i.e. (A)BP, (B)CC, and C(MF) within a
given module derived from MS-matrix and MIPS ... 94 Figure 5-6. The modules derived from the MS-matrix ... 95
List of Tables
Table 1-1. The list of the members of proteins and protein-protein interactions in 11 common used organisms ...4 Table 2-1. Statistics of 3D-interologs database on 19 species commonly used in research projects ... 24 Table 2-2. 3D-interologs search results using human calcineurin heterodimer as the query ... 30 Table 4-1. Statistics of proteins and PPIs derived from our result, public databases, and Wang, X. J. et al. on H.
sapiens, M. musculus, and D. rerio ... 54
Table 4-2. The ratios of essential proteins and disease related proteins in consensus and non-consensus proteins . 62 Table 4-3. The distribution of in-frame and truncating mutations in human protein interaction network ... 66 Table 4-4. The diseases recorded in OMIM of each protein in FGF-FGFR and upstream proteins of MAPK1 and
MAPK3 ... 69 Table 5-1. The degree, clustering coefficient, closeness centrality, betweenness centrality, and dynamic property of
each node in the simple network (Fig. 5-1) ... 85 Table 5-2. Connectivity of module and proteins which include the module and the proteins connecting to the
Chapter 1. Introduction
1-1. Background
Protein-protein interaction (PPI) networks provide key insights into complex biological systems, from how different processes communicate to the function of individual residues on a
single protein. For instance, the systematic identification of protein-protein interactions1-3 or
protein complexes4-7 has been a widely used strategy for understanding the physical
architecture of the cell. Therefore, several large network databases such as IntAct8, DIP9, and
BioGRID10 record hundreds of thousands of physical and genetic interactions from a wide
variety of organisms have been purposed.
A wealth of investigations have been undertaken to deepen our understanding of hereditary diseases. As a result of that, databases such as the Online Mendelian Inheritance in
Man (OMIM)11 and UniProt12 together contain almost 30,000 experimentally verified
mutations. Nevertheless, the exact mechanisms by which mutations alter a protein's function are in many cases poorly understood. Therefore, researchers have recently begun to use PPI
networks to explore the genotype-to-phenotype relationships13-16, on the basis that many
proteins function by interacting with other proteins. However, this idea has only been applied in Human based on the requirement of high-quality PPI with the binding mechanism.
In addition, the concept "homologs" is useful for identifying consensus proteins across multiple organisms and could provide the key residues related to the functions within a given protein. Previous studies have been compared PPI network across multiple organisms to
identify the essential pathways and the mechanisms of evolution17-19. For example, Peterson, G.
J. et al. have shown that interaction change through binding site evolution is faster than
However, these studies only focused on a small sub-network or on few organisms which have an enrichment PPI data (e.g. Homo sapiens and Saccharomyces cerevisiae).
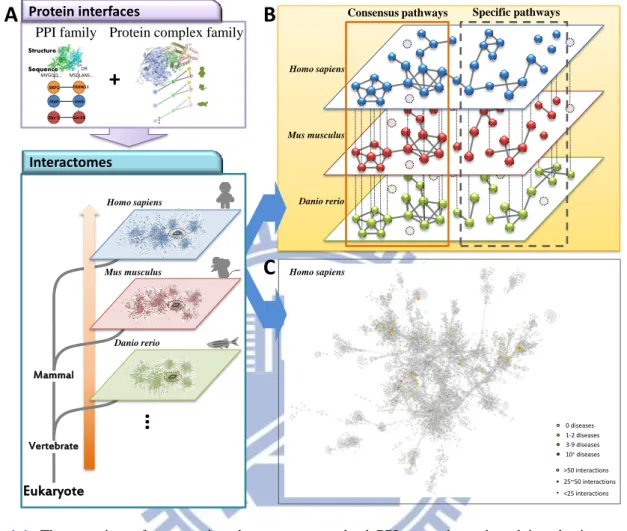
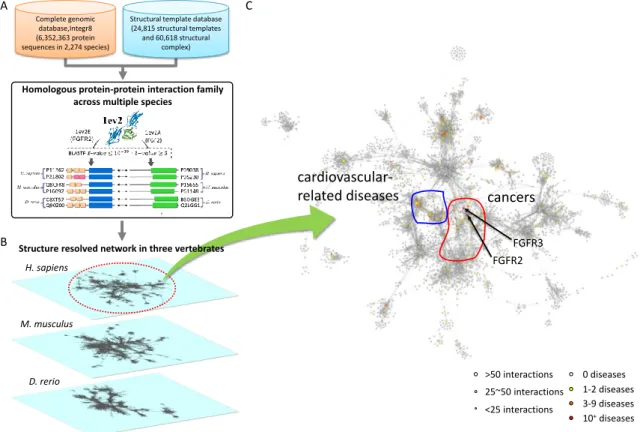
Figure 1-1. The overview of constructing the structure resolved PPI networks and studying the interactome behavior
(A) Using protein-protein interaction family and protein complex family to construct the structure resolved PPI networks in multiple oragnsims. (B) The "interactome behavior" through the consensus component. (C) The structure resolved PPI networks would provide the insight for understanding the mechanism of biological processes.
To address these issues, the structure resolved interaction family (i.e. protein-protein interaction family and protein complex family) are the basic elements and the core idea of our research to construct structure resolved PPI networks and study the behaviors of a specific PPI
network. The PPI family is a group of molecular interactions which share the consensus
interacting domain, binding environment, and have similar biological processes. The concepts
Vertebrate
Protein interfaces
…
PPI family Protein complex family
… D C B A D C B A D C B A Structure Sequence Mammal Eukaryote Interactomes Homo sapiens
+
Mus musculus Skr-1 1in-23 skpA slmb SKP1 FBXW11 MVGQQ... MSDLANS... OR Danio rerio Homo sapiens Mus musculus Danio rerioConsensus pathways Specific pathways
Homo sapiens >50 interactions 25~50 interactions <25 interactions 0 diseases 1-2 diseases 3-9 diseases 10+diseases
A
B
C
of PPI families not only help us to construct the highly reliable PPI network in a specific organisms (e.g. Homo sapiens, Mus musculus, and Danio rerio) but also provide the consensus
and the diversity behavior of interactome through comparing with multiple species (Fig. 1-1).
The methods of inferring interface families and interactomes are briefly summarized as follows.
In protein-protein interaction family, the concept of PPI families is similar to that of
protein sequence family20,21 and protein structure family22. Here, the members of a PPI family
are conserved on specific functions and in interacting domain(s). Using these conservations of homologous PPIs, it can be used to annotate the protein functions and provide high quality PPIs.
Protein complexes are fundamental units of macromolecular organization and their
composition is also known to vary according to cellular requirements7. According to these
homologous complexes across multiple species, protein complex family provides the binding models (e.g. hydrogen bonds and conserved amino acids in the interfaces), functional modules, and the conserved interacting domains and Gene Ontology annotations of the members.
Based on the members (protein-protein or protein complexes) of protein-protein interface
family23 and protein complex family24 that are consensus of functional annotation across
multiple species, we are able to identify the conserved components in the PPI networks across multiple species and indicate the changes of the conserved components at the interspecific level. Therefore, we would use the strategies to reveal "interactome behavior".
1-2. Current state of constructing protein-protein interaction networks
Many high throughput experimental and computational approaches, such as
proposed to construct the PPI network within an organism. These large-scale methods are often unable to respond how a protein interacts with another one and describe the relationship between the mutation of proteins and disease syndrome. Previous studies have combined protein structure information with protein interaction data to investigate how mutations affect
protein interactions in disease14-16. For instance, Wang, X. J. et al. generated a structurally
resolved human protein interaction network to systematically examine relationship genes,
mutations and associated disorders16.
Table 1-1. The list of the members of proteins and protein-protein interactions in 11 common used organisms NCBI
Taxonomy ID Organisms
No. Proteins in Integr8 database
No. PPIs in five annotated database 9606 Homo sapiens 56,006 67,596 10090 Mus musculus 36,379 7,535 3702 Arabidopsis thaliana 35,825 6,985 6239 Caenorhabditis elegans 23,154 10,095 7227 Drosophila melanogaster 15,155 37,674 7955 Danio rerio 21,601 221 10116 Rattus norvegicus 13,807 2,199 9913 Bos taurus 12,235 281 9031 Gallus gallus 6,279 70 36329 Plasmodium falciparum 5,353 2,956 4932 Saccharomyces cerevisiae 5,727 237,193 Total 231,521 372,805
However, the experimental PPI data is necessary for these methods. The experimental PPI
databases (e.g. IntAct8, DIP9, MIPS28, BioGRID10, and MINT29) are dominated by few species,
especially Saccharomyces cerevisiae. Table 1-1 presents the number of PPIs and proteins in
organisms that are commonly used in molecular researches. For example, there are 56,006 proteins (24.19% of 11 common organisms) and 67,596 PPIs (18.1% of 11 common organisms)
of Homo sapiens are recorded in Integr8 database 30 (which are collected the complete
sequencing genomes) and the five public interaction databases, respectively. On the contrary, the Saccharomyces cerevisiae only has 5,727 proteins (2.4%), but it has the dominant experimental PPI recorded in the databases (i.e. 237,193; 63.6% of 11 common organisms). This statistical data indicate that current interaction databases are overestimated and have many false-positive recorded PPIs in some organisms (e.g. Saccharomyces cerevisiae). Moreover,
these databases are underestimated and incomplete in most organisms (e.g. Homo sapiens and
Mus musculus). Both of the overestimated and underestimated protein interaction data could
influence the low reliable construction of protein interactome in a specific organism.
Protein Data Bank (PDB)31 stores three-dimensional (3D) structure complexes, from
which physical interacting domains can be identified to study DDIs and PPIs using
comparative modeling32,33. As the number of protein structures increases rapidly, some
domain-domain interaction databases, such as 3did34, and iPfam35, have recently been derived
from PDB. Additionally, some methods have utilized template-based methods (i.e. comparative
modeling32 and fold recognition33), which search a 3D-complex library to identify homologous
templates of a pair of query protein sequences, in order to predict the protein-protein interactions by accessing interface preference, and score query pair protein sequences
according to how they fit the known template structures. However, these methods32,33 are
time-consuming to search all possible protein-protein pairs in a large genome-scale database.
For example, the possible protein-protein pairs on the UniProt12 database (4,826,134 sequences)
are about 2.33×1013. In addition, these methods are unable to form homologous PPIs to explore
the protein-protein evolution for a specific structure template.
In this thesis, we presented the "3D-domain interologs mapping" and "protein complex family" to construct the structure resolved PPI networks across multiple organisms. "3d-domain interolos mapping" is a concept for efficiently enlarging protein interactions annotated through the homologous PPIs with residue-based binding models. We verified the
structure resolved PPI networks on Gene Ontology annotations36 and the architecture of
topology (i.e. scale-free network properties). In addition, we also provide the consensus proteins across three networks based on "3D-domain interologs mapping". These consensus proteins are highly related to the essential genes and disease related proteins. We believe that structure resolved PPI networks would provide the insight for understanding the mechanism of
biological processes within a given PPI network.
1-3. Thesis overview
The thesis is organized as follows. In Chapter 2, for efficiently enlarging protein interactions annotated with residue-based binding models, we proposed a new concept "3D-domain interolog mapping" with a scoring system to explore all homologous protein-protein interaction pairs between the two homolog families, derived from a known 3D-structure dimmer (template), across multiple species. Each family consists of homologous proteins which have interacting domains of the template for studying domain interface evolution of two interacting homolog families. The 3D-interologs database records the evolution of protein-protein interactions database across multiple species. Based on “3D-domain interolog mapping” and a template-based scoring function, we infer 173,294 homologous protein-protein interactions by using 1,895 three-dimensional (3D) structure heterodimers to search the UniProt database (4,826,134 protein sequences). The 3D-interologs database comprises 15,124 species and 283,980 protein-protein interactions, including 173,294 interactions (61%) and 110,686 interactions (39%) summarized from the IntAct database. For a protein-protein interaction, the 3D-interologs database shows functional annotations (e.g. Gene Ontology), interacting domains and binding models (e.g. hydrogen-bond interactions and conserved residues). Additionally, this database provides couple-conserved residues and the interacting evolution by exploring the interologs across multiple species. Experimental results reveal that the proposed scoring function obtains good agreement for the binding affinity of 275 mutated residues from the ASEdb. The precision and recall of our method are 0.52 and 0.34, respectively, by using 563 non-redundant heterodimers to search on the Integr8
protein-protein interaction evolution across multiple species. In addition, the top-ranked strategy and template interface score are able to significantly improve the accuracies of identifying protein-protein interactions in a complete genome.
In Chapter 3, we presented the PCFamily server to identify template-based homologous protein complexes (called protein complex family) and infer functional modules of the query proteins. This server first finds homologous structure complexes of the query using BLASTP to search the structural template database (11,263 complexes). PCFamily then searches the homologous complexes of the templates (query) from a complete genomic database (Integr8 with 6,352,363 protein sequences in 2,274 species). According to these homologous complexes across multiple species, this sever infers binding models (e.g. hydrogen bonds and conserved amino acids in the interfaces), functional modules, and the conserved interacting domains and Gene Ontology annotations of the protein complex family. Experimental results demonstrate that the PCFamily server can be useful for binding model visualizations and annotating the query proteins. We believe that the server is able to provide valuable insights for determining functional modules of biological networks across multiple species.
In chapter 4, we provide the structure resolved PPI networks across multiple species, including H. sapiens, M. musculus, and D. rerio. According to structure-based homologous PPIs in multiple species, the PPIs with atomic residue-based binding models in the derived structure resolved network achieved highly agreement with Gene Ontology (BP, CC, and MF terms) similarities. Furthermore, the architecture of these networks is a scale-free network which is consistent with most of the cellular networks. In addition, our derived networks can be used to observe the consensus proteins and modules (a fundamental unit forming with highly connected proteins) which are high conserved appearing in multiple organisms. These consensus proteins are often the essential genes and related to diseases recorded in OMIM. Experimental results also indicate that the mutations of interacting residues on the PPIs often
related to diseases are often on. Our results demonstrate that the structure resolved PPI networks can provide valuable insights for understanding the mechanisms of biological processes.
In chapter 5, we provide a method to characterize a given PPI network. Although, many graphic features have been purposed to measure the role of proteins and identify local modularity structures of high connectivity in a PPI network, the pseudoinverse of the Laplacian matrix plays a key role, has a nice interpretation in terms of random walk on a network, and defines the kernels on a given network. Therefore, we proposed the modularity structure matrix (MS-matrix), which is the pseudoinverse of the Laplacian matrix for a given network, to evaluate the modularity structure properties of a PPI network. According to our knowledge, the
MS-matrix is the first property to identify both global important proteins and local density
regions within a network. For a given PPI network of S. cerevisiae, our results demonstrate that the important proteins identified by the MS-matrix are related to the essential biological processes (i.e. essential genes) and highly consistence with the topology features (i.e. degree, closeness centrality, and betweenness centrality). Then, the relationship between proteins derived from the MS-matrix could reflect the similarity of Gene Ontology and could be useful for the module identification. Furthermore, biological characterization (e.g. Gene Onotology) of the modules derived from the MS-matrix is similar to the modules collected from the experiment database (e.g. MIPS). Our results demonstrate that the MS-matrix would provide the insight for investigating a PPI network through important proteins and local modularity structures.
In the final chapter, we summarized the results of this thesis, and then discuss the future works. To further investigate the behavior of PPI network within a given cell, gene expression data would provide an aspect of in-depth understanding of the dynamic organization of the PPI network and its role in the regulation of cellular processes. For example, the Connectivity Map
(also known as cmap) provided by Lamb, J. et al. is a collection of genome-wide transcriptional expression data from cultured human cells treated with bioactive small molecules and simple pattern-matching algorithms that together enable the discovery of functional connections between drugs, genes and diseases through the transitory feature of
common gene-expression changes 37. Therefore, we will combine the gene expression data into
the PPI network. We will try to illustrate the behavior of PPI networks under different cell types and different conditions. For example, because the Connectivity Map could provide the up-regulated and down-regulated proteins of given drugs and diseases, combining these data with our structure resolved PPI networks should be able to explain the mechanism of relationship between the drugs, genes and diseases.
Chapter 2. 3D-interologs: An evolution database of
physical protein-protein interactions across multiple
genomes
Interactions between proteins are critical to most biological processes. To identify and characterize protein-protein interactions (PPIs) and their networks, many high-throughput experimental approaches, such as yeast two-hybrid screening, mass spectroscopy, and tandem
affinity purification, and computational methods (phylogenetic profiles38, known 3D
complexes39, and interologs40) have been proposed41. Some PPI databases, such as IntAct8,
BioGRID10, DIP9, MIPS28, and MINT29, have accumulated PPIs submitted by biologists, and
those from mining literature, high-throughput experiments, and other data sources. As these interaction databases continue growing in size, they become increasingly useful for analysis of newly identified interactions.
The discovery of sequence homologs to a known protein often provides clues for understanding the function of a newly sequenced gene. As an increasing number of reliable PPIs become available, identifying homologous PPIs should be useful to understand a newly determined PPI. Recently, several PPI databases (e.g., IntAct and BioGRID) allow users to input one or a pair of proteins or gene names to acquire the PPIs associated with the query
protein(s). Few computational methods42,43 applied homologous interactions to assess the
reliability of PPIs.
To address this issue, we proposed the concept called "homologous protein-protein
interaction"23. We define a homologous PPI as follows: (1) homologs of A and B are proteins
protein pair (A and B) and their respective homologs (A1' and B1') recorded in annotated PPI
databases. In addition, we constructed the PPISearch server for searching homologous PPIs across multiple species and annotating the query protein pair. According to our knowledge, PPISearch is the first public server that identifies homologous PPIs from annotated PPI databases and infers transferability of interacting domains and functions between homologous PPIs and the query. Our results demonstrate that this server achieves high agreements on interacting domain-domain pairs and function pairs between query protein pairs and their respective homologous PPIs.
Furthermore, a known 3D structure of interacting proteins provides interacting domains and atomic details for thousands of direct physical interactions. It is usually possible to build the binding model of a protein-protein interaction by comparative modeling if a known
complex structure comprising homologs of these two sequences is available32,45. Therefore, we
developed a new scoring function39, which includes the contact residue interacting score (e.g.
the steric, hydrogen bonds, and electrostatic interactions) and the template consensus score (e.g. couple-conserved residue and the template similarity scores), to evaluate how well the interfaces between the query and interacting candidates.
For efficiently enlarging protein interactions annotated with residue-based binding models,
we proposed a new concept "3D-domain interolog mapping" with a scoring system39 to explore
all possible homologous protein-protein interaction pairs between the two homolog families, derived from a known 3D-structure dimmer (template), across multiple species. Each family consists of homologous proteins which have interacting domains of the template for studying domain interface evolution of two interacting homolog families.
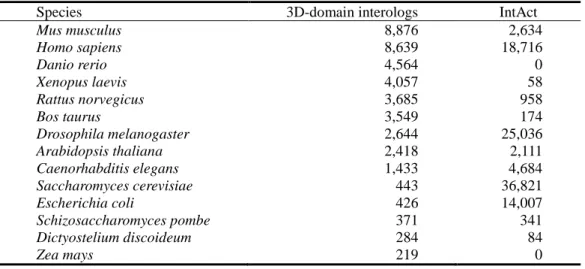
The 3D-interologs database records the evolution of protein-protein interactions database across multiple species. Based on “3D-domain interolog mapping” and a new scoring function, we infer 173,294 homologous protein-protein interactions by using 1,895 three-dimensional
(3D) structure heterodimers to search the UniProt database (4,826,134 protein sequences). The 3D-interologs database comprises 15,124 species and 283,980 protein-protein interactions, including 173,294 interactions (61%) and 110,686 interactions (39%) summarized from the IntAct database. For a protein-protein interaction, the 3D-interologs database shows functional annotations (e.g. Gene Ontology), interacting domains and binding models (e.g. hydrogen-bond interactions and conserved residues). Additionally, this database provides couple-conserved residues and the interacting evolution by exploring the interologs across multiple species. Experimental results reveal that the proposed scoring function obtains good agreement for the binding affinity of 275 mutated residues from the ASEdb. The precision and recall of our method are 0.52 and 0.34, respectively, by using 563 non-redundant heterodimers to search on the Integr8 database (549 complete genomes).
Experimental results demonstrate that the proposed method can infer reliable physical protein-protein interactions and be useful for studying the protein-protein interaction evolution across multiple species. In addition, the top-ranked strategy and template interface score are able to significantly improve the accuracies of identifying protein-protein interactions in a
complete genome. The 3D-interologs database is available at
http://3D-interologs.life.nctu.edu.tw.
2-1. Introduction
A major challenge of post genomic biology is to understand the networks of interacting genes, proteins and small molecules that produce biological functions. The large number of
protein interactions 8,9,28, generated by large-scale experimental methods 26,46,47, computational
methods 32,38,39,44,48-50, and integrated approaches 51,52, provides opportunities and challenges in
(DDI), and in modeling the cellular signaling and regulatory networks. An approach based on
evolutionary cross-species comparisons, such as PathBLAST 53,54 and interologs (i.e.
interactions are conserved across species 40,44), is a valuable framework for addressing these
issues. However, these methods often cannot respond how a protein interacts with another one across multiple species.
Protein Data Bank (PDB) 31 stores three-dimensional (3D) structure complexes, from
which physical interacting domains can be identified to study DDIs and PPIs using
comparative modeling 32,33. Some DDI databases, such as 3did 34, and iPfam 35, have recently
been derived from PDB. Additionally, some methods have utilized template-based methods (i.e.
comparative modeling 32 and fold recognition 33), which search a 3D-complex library to
identify homologous templates of a pair of query protein sequences, in order to predict the protein-protein interactions by accessing interface preference, and score query pair protein sequences according to how they fit the known template structures. However, these methods
32,33
are time-consuming to search all possible protein-protein pairs in a large genome-scale
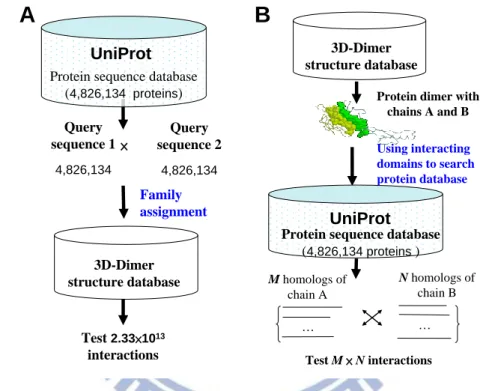
database (Fig. 2-1A). For example, the possible protein-protein pairs on the UniProt database
(4,826,134 sequences) are about 2.33×1013 12. In addition, these methods are unable to form
homologous PPIs to explore the protein-protein evolution for a specific structure template.
To address these issues, we proposed a new concept "3D-domain interolog mapping" (Fig.
2-1B): for a known 3D-structure complex (template T with chains A and B), domain a (in chain
A) interacts with domain b (in chain B) in one species. Homolog families A' and B' of A and B
are proteins, which are significant sequence similarity BLASTP E-values ≤10-10 and contain
domains a and b, respectively. All possible protein pairs between these two homolog families are considered as protein-protein interaction candidates using the template T. Based on this concept, protein sequence databases can be searched to predict protein-protein interactions across multiple species efficiently. When the genome was deciphered completely for a species,
we considered the rank of protein-protein interaction candidates in each species into our
previous scoring system 39 to reduce a large number of false positives. The 3D-interologs
database which can indicate interacting domains and contact residues in order to visualize molecular details of a protein-protein interaction. Additionally, this database can provide couple-conserved residues and evolutionary clues of a query sequence and its partners by examining the interologs across multiple species.
Figure 2-1. Two frameworks of template-based methods for protein-protein interactions (PPI).
(A) For each query protein sequence pair, the method searches 3D-dimer template library to identify homologous templates for exploring the query protein pair, such as MULTIPROSPECTOR 33. (B) For each structure in 3D-dimer template library, the method searches protein sequence database to identify homologous PPIs of the query structure, such as 3D-interologs.
2-2. Methods and Materials
Figure 2-2 illustrates the overview of the 3D-interologs database. The 3D-interologs
allows users to input the UniProt accession number (UniProt AC 12) or the sequence with
FASTA format of the query protein (Fig. 2-2A). When the input is a sequence, 3D-interologs
uses BLAST to identify the hit interacting proteins. We identified protein-protein interactions
Query sequence 2 4,826,134 Query sequence 1 4,826,134 Family assignment 3D-Dimer structure database Test 2.33×1013 interactions Using interacting domains to search protein database
Protein dimer with chains A and B M homologs of chain A N homologs of chain B Test M × N interactions 3D-Dimer structure database × … …
A
B
Protein sequence database
(4,826,134 proteins ) UniProt
UniProt Protein sequence database
in 3D-interologs database through structure complexes and a new scoring function using the
following steps (Fig. 2-2B). First, a 3D-dimer template library comprising 1,895 heterodimers
(3,790 sequences, called NR1895) was selected from the PDB released in Feb 24, 2006. Duplicate complexes, defined by sequence identity of above 98%, were removed from the
library. Dimers containing chains shorter than 30 residues were also excluded 33,55. Interacting
domains and contact residues of two chains were identified for each complex in the 3D-dimer library. Contact residues, in which any heavy atoms should be within a threshold distance of 4.5 Å to any heavy atoms of another chain, were regarded as the core parts of the 3D-interacting domains in a complex. Each domain was required to have at least 5 contact residues and more than 25 interacting contacted-residue pairs to ensure that the interface between two domains was reasonably extensive. After the interacting domains were determined,
its SCOP domains 22 were identified, and its template profiles were constructed by PSI-BLAST.
PSI-BLAST was adopted to search the domain sequences against the UniRef90 database 12, in
which the sequence identity < 90% of each other and the number of iteration was set to 3.
After 3D-dimer template library and template profiles were built, we inferred candidates of interacting proteins by 3D-domain interolog mapping. To identify the interacting-protein candidates against protein sequences in the UniProt version 11.3 (containing 4,826,134 protein sequences), the chain profile was used as the initial position-specific score matrix (PSSM) of
PSI-BLAST in each template consisting of two chains (e.g. CA and CB, Fig. 2-2C). The number
of iterations was set to 1. Therefore, this search procedure can be considered as a profile-to-sequence alignment. A pairing-protein sequence (e.g. S1 and S2) was considered as a protein-protein interaction candidate if the sequence identity exceeded 30% and the aligned
contact residue ratio (CR) was greater than 0.5 for both alignments (i.e. S1 aligning to CA and
S2 aligning to CB). For each interacting candidate, the scoring function was applied to calculate
interacting score. An interacting candidate was regarded as a protein-protein interaction if its
Z-value was above 3.0 and it ranked in the Top 25 in one species. The candidate rank was
considered in one species to reduce the ill-effect of the out-paralogs that arose from a
duplication event before the speciation 56. These inferred interacting protein pairs were
collected in the database.
Figure 2-2. Overview of the 3D-interologs database for protein-protein interacting evolution, protein functions annotations and binding models across multiple species.
Finally, for the hit interacting partner derived from 3D-domain interolog mapping, this database provides functional annotations (e.g. UniProt AC, organism, descriptions, and Gene
Ontology (GO) annotations 36, Fig. 2-2D), and the visualization of the binding models and
interaction evolutions (Fig. 2-2C) between the query protein and its partners. We then
constructed two multiple sequence alignments of the query protein and its interacting partner
Users input a query sequence or UniProt accession number
3D-interologs Database
Create a two-chain complex library from PDB and create profiles of two chains (e.g. chains A and B) of each complex (e.g. 1jkg) in this library
by PSI-BLAST
Identify all interacting partners of each complex by using our scoring system and PSI-BLAST
to find homologous proteins of two chains
Create interacting evolutions, binding models and functional
annotations for all derived protein-protein interactions for
each complex in the library CA
CB
Output interactive partners of
the query sequence with
interacting evolution, binding models and functional
annotations
A
B
C
D
Interacting domain Interacting domain
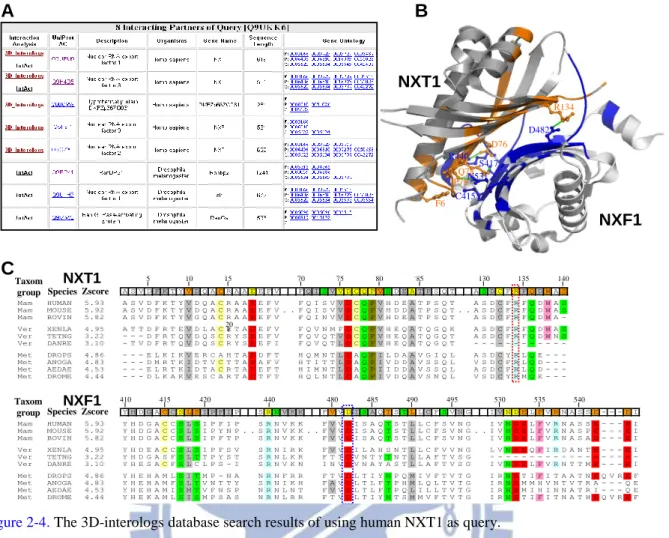
> Q9UKK6 | NXT1_HUMAN NTF2-related export protein 1 - Homo sapiens (Human). MASVDFKTYVDQACRAAEEFVNVYYTTMDKRRRLLSRLYMGTATLVWNGNAVSGQESLSE FFEMLPSSEFQISVVDCQPVHDEATPSQTTVLVVICGSVKFEGNKQRDFNQNFILTAQAS PSNTVWKIASDCFRFQDWAS 1jkg Chain A(1jkg-A) Chain B (1jkg-B)
(Fig. 2-2C) across multiple species. Here, the interacting-protein pair with the highest Z-score in a species was chosen as interologs for constructing multiple sequence alignments using a
star alignment. The chains (e.g. Chains A and B, Fig. 2-2C) of the hit structure template were
considered as the centers, and all selected interacting-protein pairs across species were aligned to respective chains of the template by PSI-BLAST. The 3D-interologs database annotates the important contact residues in the interface according to the following formats: hydrogen-bond residues (green); conserved residues (orange), conserved residues with hydrogen bonds (yellow) and other (gray).
Data Sets
Two data sets were used to assess 3D-domain interolog mapping and the scoring functions. To determine the contribution of a residue to the binding affinity, the alanine-scanning mutagenesis is frequently used as an experimental probe. We selected 275 mutated (called
BA-275) residues from the ASEdb 57 with 16 heterodimers whose 3D structures were known.
Those mutated residues are contact residues and positioned at protein–protein interfaces. ASEdb gives the corresponding delta G value representing the change in free energy of binding upon mutation to alanine for each experimentally mutated residue. Residues that contribute a large amount of binding energy are often labeled as hot spots.
In addition, we selected a non-redundant set (NR-563), comprising 563 dimer protein structures from the set NR1895 to evaluate the performance of our scoring functions for predicting PPIs in S. cerevisiae and in 549 species collected in Integr8 database (2,102,196 proteins 30).
2-3. Scoring Function and Matrices
protein-protein interaction 39. This study enhances this scoring by dividing the template consensus score into the template similar score and the couple-conserved residue score. Based on this scoring function, the 3D-interologs database can provide the interacting evolution across multiple species and the statistical significance (Z-value), the binding models and functional annotations between the query protein and its interacting partners. The scoring function is defined as cons sim SF vdw tot E E E wE E (1)
where Evdw and ESF are the interacting van der Waals energy and the special interacting bond
energy (i.e. hydrogen-bond energy, electrostatic energy and disulfide-bond energy),
respectively; and Esim is the template interface similar score; and the Econs is couple-conserved
residue score. The optimal w value was yielded by testing various values ranging from 0.1 to 5.0; w is set to 3 for the best performance and efficiency on predicting binding affinity (BA-275) and predicting PPIs in S. cerevisiae and in 549 species (Integr8) using the data set
NR-563. The Evdw and ESF are given as
CP j i ji ij ij vdw Vss Vsb Vsb E , ) (
CP j i ji ij ij SF Tss Tsb Tsb E , ) (where CP denotes the number of the aligned-contact residues of proteins A and B aligned to a
hit template; Vssij and Vsbij (Vsbji) are the sidechain-sidechain and sidechain-backbone van der
Waals energies between residues i (in protein A) and j (in protein B), respectively. Tssij and Tsbij
(Tsbji) are the sidechain-sidechain and sidechain-backbone special interacting energies between
i and j, respectively, if the pair residues i and j form the special bonds (i.e. hydrogen bond, salt
bridge, or disulfide bond) in the template structure. The van der Waals energies (Vssij, Vsbij,
and Vsbji) and special interacting energies (Tssij, Tsbij, and Tsbji) were calculated from the four
knowledge-based scoring matrices (Fig. 2-3), namely sidechain-sidechain (Fig. 2-3A) and
2-3C) and sidechain-backbone special-bond scoring matrices (Fig. 2-3D).
Figure 2-3. Knowledge-based protein-protein interacting scoring matrices: (A) sidechain-sidechain van-der Waals scoring matrix; (B) sidechain-backbone van-der Waals scoring matrix; (C) sidechain-sidechain special-bond scoring matrix; (D) sidechain-backbone special-bond matrix scoring.
The sidechain-sidechain scoring matrices are symmetric and sidechain-backbone scoring matrices are non-symmetric. For sidechain-sidechain van-der Waals scoring matrix, the scores are high (yellow blocks) if large-aliphatic residues (i.e. Val, Leu, Ile, and Met) interact to large-aliphatic residues or aromatic residues (i.e. Phe, Tyr, and Trp) interact to aromatic residue. In contrast, the scores are low (orange blocks) when nonpolar residues interact to polar residues. For sidechain-sidechain special-bond scoring matrix, the scores are high when an interacting resides (i.e. Cys to Cys) form a disulfide bond or basic residues (i.e. Arg, Lys, and His) interact to acidic residues (Asp and Glu). The scoring values are zero if nonpolar residues interact to other residues.
These four knowledge-based matrices, which were derived using a general mathematical
structure 58 from a nonredundant set of 621 3D-dimer complexes proposed by Glaser et al. 59,
are the key components of the 3D-interologs database for predicting protein-protein interactions. This dataset is composed of 217 heterodimers and 404 homodimers and the
GLY ALA VAL LEU ILE MET PRO PHE TRP TYR CYS SER THR ASN GLN HIS ARG LYS ASP GLU GLY 0.1 -0.7 -0.8 -0.8 -0.5 -0.4 -0.4 -0.3 0.2 0.0 -0.2 -0.8 -0.3 -0.5 -0.2 -0.1 0.1 -0.4 -0.8 -1.1 ALA -0.7 0.3 -0.3 0.1 -0.2 0.3 -0.5 0.6 0.3 0.0 -0.6 -1.1 -0.9 -0.6 -0.5 -0.2 -0.6 -1.2 -1.0 -1.0 VAL -0.8 -0.3 1.5 1.1 0.9 1.1 -0.2 1.2 0.3 0.6 -0.3 -0.9 -0.1 -0.8 0.1 -0.4 -0.1 -0.6 -1.0 -0.5 LEU -0.8 0.1 1.1 2.3 1.4 1.3 0.0 1.3 1.2 0.6 -0.2 -0.6 -0.2 -0.4 0.3 0.6 0.2 -0.9 -1.3 -0.3 ILE -0.5 -0.2 0.9 1.4 2.3 1.2 0.1 1.7 1.5 1.0 -0.9 -1.2 0.1 -0.5 0.1 -0.3 -0.1 -0.8 -0.9 -0.4 MET -0.4 0.3 1.1 1.3 1.2 3.0 0.5 1.8 1.2 1.0 0.4 -0.5 0.0 -0.4 -0.2 0.3 0.1 0.0 -1.3 0.0 PRO -0.4 -0.5 -0.2 0.0 0.1 0.5 0.4 0.5 1.5 0.9 0.0 -0.5 -0.2 0.0 0.5 -0.3 0.0 -0.7 -0.8 -0.1 PHE -0.3 0.6 1.2 1.3 1.7 1.8 0.5 2.7 2.1 1.2 -0.1 -0.4 0.6 0.1 0.5 0.1 0.5 -0.6 -0.9 -0.1 TRP 0.2 0.3 0.3 1.2 1.5 1.2 1.5 2.1 2.9 1.1 0.3 -0.3 0.3 0.9 0.7 0.6 0.9 0.1 0.0 -0.6 TYR 0.0 0.0 0.6 0.6 1.0 1.0 0.9 1.2 1.1 1.4 0.3 0.1 0.1 0.6 0.9 0.8 1.0 0.5 0.5 0.5 CYS -0.2 -0.6 -0.3 -0.2 -0.9 0.4 0.0 -0.1 0.3 0.3 -0.1 -0.3 -1.1 -1.6 -1.0 0.5 -0.9 -1.3 -2.2 -1.1 SER -0.8 -1.1 -0.9 -0.6 -1.2 -0.5 -0.5 -0.4 -0.3 0.1 -0.3 0.4 -0.5 0.0 -0.3 -0.3 -0.1 -0.3 -0.1 -0.1 THR -0.3 -0.9 -0.1 -0.2 0.1 0.0 -0.2 0.6 0.3 0.1 -1.1 -0.5 0.6 0.0 0.0 0.1 0.1 -0.6 -0.2 -0.2 ASN -0.5 -0.6 -0.8 -0.4 -0.5 -0.4 0.0 0.1 0.9 0.6 -1.6 0.0 0.0 1.4 0.4 -0.2 0.3 0.0 0.3 -0.2 GLN -0.2 -0.5 0.1 0.3 0.1 -0.2 0.5 0.5 0.7 0.9 -1.0 -0.3 0.0 0.4 1.6 0.2 0.7 0.2 0.4 0.1 HIS -0.1 -0.2 -0.4 0.6 -0.3 0.3 -0.3 0.1 0.6 0.8 0.5 -0.3 0.1 -0.2 0.2 2.1 0.3 -0.8 0.8 0.8 ARG 0.1 -0.6 -0.1 0.2 -0.1 0.1 0.0 0.5 0.9 1.0 -0.9 -0.1 0.1 0.3 0.7 0.3 1.3 -0.5 1.8 1.9 LYS -0.4 -1.2 -0.6 -0.9 -0.8 0.0 -0.7 -0.6 0.1 0.5 -1.3 -0.3 -0.6 0.0 0.2 -0.8 -0.5 0.0 1.2 1.6 ASP -0.8 -1.0 -1.0 -1.3 -0.9 -1.3 -0.8 -0.9 0.0 0.5 -2.2 -0.1 -0.2 0.3 0.4 0.8 1.8 1.2 0.0 -0.9 GLU -1.1 -1.0 -0.5 -0.3 -0.4 0.0 -0.1 -0.1 -0.6 0.5 -1.1 -0.1 -0.2 -0.2 0.1 0.8 1.9 1.6 -0.9 0.7
GLY ALA VAL LEU ILE MET PRO PHE TRP TYR CYS SER THR ASN GLN HIS ARG LYS ASP GLU
GLY 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 ALA 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 VAL 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 LEU 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 ILE 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 MET 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 PRO 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 PHE 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 TRP 0 0 0 0 0 0 0 0 0.0 2.2 0.0 3.2 3.7 4.4 3.1 0.0 0.0 0.0 5.1 4.5 TYR 0 0 0 0 0 0 0 0 2.2 3.7 4.0 4.4 4.0 4.7 5.0 4.7 4.6 4.2 5.5 5.5 CYS 0 0 0 0 0 0 0 0 0.0 4.0 8.0 2.4 0.0 2.6 2.8 3.9 0.0 0.0 3.2 3.1 SER 0 0 0 0 0 0 0 0 3.2 4.4 2.4 4.9 4.3 4.5 4.1 4.6 4.4 4.1 5.4 5.4 THR 0 0 0 0 0 0 0 0 3.7 4.0 0.0 4.3 4.8 4.7 4.8 3.2 4.4 4.0 5.2 4.8 ASN 0 0 0 0 0 0 0 0 4.4 4.7 2.6 4.5 4.7 5.6 4.7 4.5 4.6 4.4 5.2 4.7 GLN 0 0 0 0 0 0 0 0 3.1 5.0 2.8 4.1 4.8 4.7 5.7 4.6 4.9 4.5 4.8 4.7 HIS 0 0 0 0 0 0 0 0 0.0 4.7 3.9 4.6 3.2 4.5 4.6 5.3 4.3 1.9 5.9 6.2 ARG 0 0 0 0 0 0 0 0 0.0 4.6 0.0 4.4 4.4 4.6 4.9 4.3 0.0 0.0 6.9 7.0 LYS 0 0 0 0 0 0 0 0 0.0 4.2 0.0 4.1 4.0 4.4 4.5 1.9 0.0 0.0 6.5 6.7 ASP 0 0 0 0 0 0 0 0 5.1 5.5 3.2 5.4 5.2 5.2 4.8 5.9 6.9 6.5 0.0 0.0 GLU 0 0 0 0 0 0 0 0 4.5 5.5 3.1 5.4 4.8 4.7 4.7 6.2 7.0 6.7 0.0 0.0
GLY ALA VAL LEU ILE MET PRO PHE TRP TYR CYS SER THR ASN GLN HIS ARG LYS ASP GLU
GLY 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 ALA 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 VAL 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 LEU 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 ILE 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 MET 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 PRO 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 PHE 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 TRP 0.0 4.5 0.0 2.8 3.3 0.0 3.2 3.4 0.0 0.0 3.3 3.1 3.1 0.0 3.5 0.0 0.0 3.2 3.2 0.0 TYR 4.1 3.8 4.4 3.6 4.0 3.6 4.5 3.7 4.1 3.8 3.6 2.8 2.8 4.8 3.1 3.5 4.3 4.0 4.4 3.8 CYS 4.7 4.4 4.5 4.5 4.0 3.8 4.4 4.2 5.2 2.9 3.7 4.5 3.9 4.1 4.5 4.0 4.4 4.9 5.1 3.5 SER 4.6 4.9 3.8 4.7 4.7 4.4 3.9 2.7 4.2 2.8 4.3 5.1 4.7 3.9 2.1 0.0 3.8 4.8 3.9 4.4 THR 4.9 4.1 4.1 3.9 4.0 3.7 3.5 2.0 4.2 4.2 4.5 4.4 4.8 3.3 2.8 3.9 3.8 3.8 4.7 4.0 ASN 4.8 5.3 4.7 5.0 4.2 4.6 4.0 4.8 4.7 5.2 4.9 4.9 5.3 5.4 5.1 4.6 4.9 4.9 4.9 4.3 GLN 5.1 4.8 5.1 5.3 5.3 5.0 4.4 5.6 4.6 5.1 4.4 5.5 5.3 4.8 5.0 4.6 4.6 4.4 4.7 4.7 HIS 4.9 4.8 4.6 4.5 3.8 4.0 4.4 4.5 5.0 4.9 2.7 3.9 4.1 4.1 4.2 5.2 3.5 4.0 4.6 3.8 ARG 5.9 5.3 5.2 5.3 4.8 5.2 5.3 5.3 5.4 4.5 5.2 5.4 5.6 5.0 5.0 5.2 4.8 4.5 5.2 5.4 LYS 4.9 3.8 3.5 4.2 3.6 2.7 4.5 4.4 4.3 4.7 3.6 4.9 4.4 5.0 4.6 4.0 3.7 5.2 4.7 5.0 ASP 4.9 4.5 3.4 3.4 4.5 2.6 0.0 2.7 4.3 3.9 2.7 5.1 4.6 4.5 4.0 4.2 4.3 4.0 3.3 3.9 GLU 4.7 5.0 4.7 4.6 4.4 4.6 0.0 4.8 4.2 3.4 3.5 5.1 4.4 4.5 4.3 2.5 4.4 4.4 4.0 4.1 A D C B
GLY ALA VAL LEU ILE MET PRO PHE TRP TYR CYS SER THR ASN GLN HIS ARG LYS ASP GLU
GLY 0.1 -0.7 -0.8 -0.8 -0.5 -0.4 -0.4 -0.3 0.2 0.0 -0.2 -0.8 -0.3 -0.5 -0.2 -0.1 0.1 -0.4 -0.8 -1.1 ALA -0.7 1.1 -0.6 -1.3 -1.1 -0.8 -0.7 -1.4 -1.4 -1.9 -0.5 -0.3 -0.7 -0.9 -0.9 -0.7 -1.8 -1.4 -1.1 -0.8 VAL -0.8 0.6 0.7 -0.4 0.1 0.2 -0.2 -0.7 -1.0 -1.6 0.3 0.1 0.1 -0.8 -0.8 -0.8 -0.8 -0.8 -0.8 -0.6 LEU -0.8 0.7 0.1 1.1 -0.1 0.5 0.0 -0.4 -0.4 -1.5 0.2 0.4 -0.1 -0.7 0.0 0.2 -0.6 -0.4 -0.5 -0.5 ILE -0.5 0.6 0.3 0.2 0.6 0.1 -0.3 -0.7 -1.3 -0.9 -1.1 0.0 0.2 -0.4 -0.4 -0.4 -1.3 -0.7 -0.6 -0.4 MET -0.4 1.5 1.0 0.5 0.5 1.3 0.5 0.2 0.4 0.2 0.6 0.4 0.2 -1.2 -0.3 0.3 -0.4 0.3 -0.2 0.1 PRO -0.4 0.4 -0.8 -0.7 -1.1 0.2 0.6 -0.2 -0.6 -1.0 0.3 0.0 0.0 0.2 -0.3 -0.5 -0.4 -0.1 -0.7 -0.6 PHE -0.3 1.5 0.3 0.7 0.6 0.9 0.9 1.0 0.8 0.1 0.2 0.5 0.6 0.2 0.3 -0.2 -0.5 0.2 -0.2 0.0 TRP 0.2 1.3 0.6 0.8 1.4 1.5 2.2 0.4 1.1 0.9 2.3 0.5 0.9 1.6 0.4 1.2 1.1 0.7 1.1 -0.5 TYR 0.0 1.2 0.3 0.0 0.2 0.7 1.1 0.7 -0.8 0.4 1.3 0.9 0.5 0.9 0.8 0.6 0.5 1.0 0.6 0.3 CYS -0.2 0.8 -1.3 -1.0 -0.5 -1.3 -0.1 -0.4 -3.1 -1.3 1.0 1.2 -0.9 -0.6 -0.9 -1.2 -0.4 -1.4 -1.2 -1.2 SER -0.8 0.6 -0.8 -0.6 -0.9 -0.5 0.0 -1.4 -1.8 -1.2 -0.2 0.8 0.0 -0.5 -1.8 -1.5 -1.3 -1.0 -0.1 -0.7 THR -0.3 0.4 -0.3 -0.4 -0.4 -0.5 -0.1 -1.0 -0.8 -0.7 0.2 0.5 0.5 -0.1 -1.2 -0.9 -0.8 -0.5 0.3 -0.9 ASN -0.5 0.9 -0.1 -0.1 -0.4 -1.0 0.2 0.1 -0.4 -0.3 0.5 0.8 0.7 1.4 -0.3 0.3 -0.3 -0.1 0.4 -0.6 GLN -0.2 0.9 0.4 0.5 0.2 0.4 0.8 0.7 -0.1 -0.2 0.7 1.3 0.4 0.7 1.1 0.0 -0.4 -0.2 0.4 0.4 HIS -0.1 1.2 -0.4 0.2 -0.8 -0.8 -0.3 -0.2 0.1 -0.7 0.8 0.9 0.0 0.0 -0.2 1.4 -0.4 -0.5 0.4 -0.3 ARG 0.1 1.3 0.4 0.5 0.2 0.3 1.0 0.4 0.1 -0.2 0.5 1.1 1.0 0.7 0.3 0.5 0.5 0.0 1.6 0.9 LYS -0.4 -0.1 -0.7 -0.7 -1.3 -0.6 0.0 -0.7 -0.5 -1.1 -0.1 0.8 -0.5 0.0 0.0 -0.3 -1.7 0.3 0.3 0.6 ASP -0.8 0.3 -1.4 -1.6 -1.0 -2.4 -0.3 -2.3 -1.5 -1.3 -0.7 0.6 0.1 -0.2 -0.4 -0.1 -0.6 -1.2 -0.1 -1.9 GLU -1.1 0.4 -0.2 -0.5 -0.7 0.1 0.0 -0.3 -1.2 -1.2 0.2 0.7 0.1 -0.2 -0.6 -0.8 -0.3 -0.1 -0.6 -0.2
sequence identity is less than 30% to each other. The entry (Sij), which is the interacting score
for a contact residue i, j pair (1≤i, j≤20), of a scoring matrix is defined as
ij ij ij e q S ln , where qij
and eij are the observed probability and the expected probability, respectively, of the occurrence
of each i, j pair. For sidechain-sidechain van-der Waals scoring matrix, the scores are high (yellow blocks) if large-aliphatic residues (i.e. Val, Leu, Ile, and Met) interact to large-aliphatic residues or aromatic residues (i.e. Phe, Tyr, and Trp) interact to aromatic residue. In contrast, the scores are low (orange blocks) when nonpolar residues interact to polar residues. The top two highest scores are 3.0 (Met. interacting to Met) and 2.9 (Trp interacting to Trp).
The value of Esim was calculated from the BLOSUM62 matrix 58 based on two alignments
between two chains (A and B) of the template and their homologous proteins (A' and B'),
respectively. The Esim is defined as
CP j i ii jj jj ii sim K K K K E , ' ' (2)where CP is the number of contact residue pairs in the template; i and j are the contact residue
in chains A and B, respectively. Kii' is the score of aligning residue i (in chain A) to i' (in protein
A') and Kjj' is the score of aligning residue j (in chain B) to j' (in protein B') according to
BLOSUM62 matrix. Kii and Kjj are the diagonal scores of BLOSUM62 matrix for residues i
and j, respectively. The couple-conserved residue score (Econs) was determined from two
profiles of the template and is given by
) ) ( ) ( , 0 (max( , '
CP j i jj jp ii ip cons M K M K E (3)where CP is the number of contact residue pairs; Mip is the score in the PSSM for residue type i
at position p in Protein A; Mjp′ is the score in the PSSM for residue type j at position p′ in
Protein B, and Kii and Kjj are the diagonal scores of BLOSUM62 matrix for residue types i and
To evaluate statistical significance (Z-value) of the interacting score of a protein-protein interaction candidate, we randomly generated 10,000 interfaces by mutating 60% contact residues for each heterodimer in 3D-dimer template library. The selected residue was substituted with another amino acid residue according to the probability derived from these 621
complexes 59. The mean and standard deviation for each 3D-dimer were determined from these
10,000 random interfaces which are assuming to form a normal distribution. Based on the mean and standard deviation, the Z-value of a protein-protein candidate predicted by this template can be calculated.
2-4. Inputs and Outputs
The 3D-interologs database server is easy-to-use. Users input the UniProt AC or the
FASTA format of the query protein (Fig. 2-2A). The server generally returns a list of
interacting partners with functional annotations (e.g. the gene name, the protein description and
GO annotations) (Fig. 2-2D) and provides the visualization of the binding model and contact
residues between the query protein and its partner by aligning them to respective template sequences and structures. Additionally, the 3D-interologs system indicates the interacting evolution analysis by using multiple sequence alignments of the interologs across multiple
species (Fig. 2-2C). The significant contact residues in the interface are indicated. If Java is
installed in the user’s browser, then the output shows the structures, and users can dynamically view the binding model, interacting domains and important residues in the browser.
2-5. Example Analysis
Figure 2-4 show the search results using the human protein NXT1 (UniProt AC Q9UKK6)
between the nucleus and cytoplasm, accumulates at the nuclear pore complexes60. For this
query, 3D-interologs database yielded 8 hit interacting partners (Fig. 2-4A), comprising 5
partners derived from 3D-interologs database and 5 partners from the IntACT database. Thus, two partners were present in both databases. Among these 8 hits, 3 partners (i.e. Uniprot AC Q68CW9, Q5H9I1 and Q9GZY0) were not recorded in IntAct database, but they very likely interact with NXT1. The Q68CW9, which is part of the protein NXF1 (UniProt AC Q9UBU9), consists of the UBA-like domain and the NTF-like domain, which is responsible for
association with the protein NXT1 61. The sequence of the protein Q5H9I1 is the same as that
of the protein Q9H4D5 (i.e. nuclear RNA export factor 3), which binds to NXT1 62. The
protein Q9GZY0 (nuclear RNA export factor 2) binds protein NXT1 to export mRNA cargoes
from nucleus into cytosol 63.
The protein NXT1 interacts with the protein NXF1 to form a compact heterodimers (PDB
code 1jkg 63)and an interacting β surface, which is lined with hydrophobic and hydrophilic
residues (Fig. 2-4B). Twenty hydrogen bonds or electrostatic interactions are formed in this
compact interface. The salt bridge formed by NXT1 Arg134 and NXF1 Asp482 is especially
important in the interface 57. The interacting evolution analysis built by 10 interologs reveals
that two residues (Arg134 and Asp482) are conserved in all species (Fig. 2-4C). Additionally,
some interacting residues forming the hydrogen bonds are also couple-conserved, for example NXT1 Asp76 and NXF1 Arg440; NXT1 Gln78 and NXF1 Ser417; NXT1 Pro79 and NXF1
Asn531 57. The evolution of interaction is valuable to reflect both couple-conserved and critical