i
National Chiao Tung university
交通大學
Department of Applied Chemistry,
應用化學
PhD Thesis
博士論文
Studies toward the Synthesis of Five, Six, Seven Member
Heterocyclic Small Molecules as Possible Inhibitors of Vascular
Endothelial Growth Factor Receptor-3 on Soluble Support
Student 研究生:Kaushik Chanda 曾古旭
Advisor 指導教授:Prof. Chung-Ming Sun, 孫仲銘 教授
July 2010
ii
Studies toward the Synthesis of Five, Six, Seven Member Heterocyclic
Small Molecules as Possible Inhibitors of Vascular Endothelial Growth
Factor Receptor-3 on Soluble Support
研究生:
曾古旭 Student
:
Kaushik Chanda
指導教授:孫仲銘 教授
Advisor:Prof. Chung-Ming Sun
國 立 交 通 大 學
應用化學系
PhD DISSERTATION
Presented in Partial Fulfillment of the Requirements for the Degree of
Doctor of Philosophy to the Department of Applied chemistry of
National Chiao Tung University
July 2010
Hsinchu, Taiwan, Republic of China
iii
ACKNOWLEDGEMENTS
First of all, I would like to thank my supervisor, Professor Chung Ming Sun, for giving me this great opportunity of carrying out a PhD in his research group and believing in me throughout these four years. His concern, enthusiasm and encouragement were indispensable for the successful completion of this work. I would like to thank all members of the Sun group (both past and present) who made this time so special and enjoyable. Many thanks to Chih hau, for all the help during this period, and Gorakh yellol “Postdoc” for accepting to check this thesis. Thanks to all the members of the chemistry department for their daily good humour and their dedication in the lab organisation. Special thanks to Barnali, for her tremendous encouragement and help to carry out the research. I would like to wish Barnali, all the best for the end of her PhD; thanks for your friendship and your help.
Finally, I would like to thank my parents and my sister and brother–in law for their never-ending love and support throughout my study. Specially my father who passed away during the middle of my dissertation and who gave me the chance to prove my worth to the world. Thanks for the incredible help you gave me, especially during hard times. I could not have written this thesis without you.
iv
DECLARATION
I, Kaushik chanda, declare that the thesis entitled Studies toward the Synthesis of Five, Six, Seven Member Heterocyclic Small Molecules as Possible Inhibitors of Vascular Endothelial Growth Factor Receptor-3 on Soluble Support and the work presented in it are my own.
I confirm that:
his work was done wholly or mainly while in candidature for a research degree at this University;
Where I have consulted the published work of others, this is always clearly attributed;
Where I have quoted from the work of others, the source is always given. With the exception of such quotations, this thesis is entirely my own work;
I have acknowledged all main sources of help;
where the thesis is based on work done by myself jointly with others, I have made clear exactly what was done by others and what I have contributed myself;
v
TABLE OF CONTENTS
Acknowledgements...iii Declaration...…..iv Table of contents ...v Abstract……….. …..xiChapter One: Soluble Polymer Supported Synthesis of Bis
Heterocyclic Compounds
1.0 Introduction………...11.1. Classification of cancer………3
1.2. Metastatic cancer……….3
1.3. Metastatic cascade………...4
1.3.1. Growth of primary tumor (Angiogenesis)………...5
1.3.2. Detachment………..5 1.3.3. Invasion………...6 1.3.3.1. Attachment………....7 1.3.3.2. Proteolysis………...7 1.3.3.3. Movement (locomotion)………8 1.3.3.4. Intravasion………...8 1.3.3.5. Transport ………..9 1.3.3.6. Lodgement………9
vi
1.4. Factors influencing malignancy in Cancer………10
1.5. Currents treatments of cancer………11
1.6. Cancer and VEGF……….11
1.6.1. VEGF……….12
1.6.2. VEGF receptors………..13
1.6.3. The strategies for inhibiting the VEGF pathway………...14
1.6.4. Factors leading to the production and expression of VEGF…...15
1.6.4.1. The role of VEGFR-1………18
1.6.4.2. The role of VEGFR-2………18
1.6.4.3. The role of VEGFR-3………19
1.7. Solid-Phase methods in organic synthesis………21
1.8. Soluble polymer supported technology in organic synthesis………...25
1.8.1. Application & recent progress of PEG as support………...27
1.8.2. Application of PEG in biological studies………...28
1.8.2.1. PEG-proteins for pharmaceutical use………...29
1.8.2.2. PEG surfaces………...30
1.8.2.3. PEG-liposomes………..30
1.8.2.4. Biological purification………...31
1.8.2.5. Biopolymer synthesis………...31
1.8.2.6. Solubilization of insoluble molecules………...31
1.9. Small molecule synthesis………..32
1.10. Benzoxazols-importance &synthesis………...37
vii
1.12. Results & Discussions………...50
1.2. Quinoxalinones-importance & synthesis………...62
1.21. Chemical methods for Quinoxalinones synthesis………63
1.22. Results & Discussions………..68
1.3. Benzazepine-importance & synthesis………...76
1.31. Chemical methods for Benzazepine synthesis………...77
1.32. Results & Discussions……….79
1.4. Experimental section……….90
1.4.1. General direction………...90
1.5. General procedure for synthesis of Intermediates……….91
1.6. Biological Assay……….121
1.7. References………...125
Chapter Two: Convergent Solution Phase Synthesis of Chimeric
Oligonucleotides by 2+2 Phosphoramidite Strategy.
2.1 Introduction……….1362.2. Introduction and Mechanism of RNA interference……….139
2.2.1. Commonly used RNA Interference Drugs………..140
2.3. Chemical Methods for Synthesizing Oligomeric Nucleotide……….141
2.3.1. Solution Phase Synthetic Methods………141
2.3.2 Polymer Supported Approach to Oligomeric Nucleotides………..148
viii
2.5. Results and Discussion………...159
2.6. Experimental Section……….182
2.7. References………..187
Chapter Three: Ionic Liquid supported Synthesis of Novel Small
Molecules
3.1. Introduction………1893.2. What is an Ionic Liquid?...191
3.2.1. History of Ionic Liquid……….192
3.2.2. Characteristics of ionic liquids………..192
3.2.3. Application of Ionic Liquids………...194
3.2.3.1. Chemical industry………...194 3.2.3.2. Cellulose processing………..195 3.2.3.3. Dispersants………....196 3.2.3.4. Gas handling………...196 3.2.3.5. Gas treatment………....196 3.2.3.6. Nuclear industry………....197 3.2.3.7. Solar energy………..197
3.2.3.8. Food and bio products………..198
3.2.3.9. Waste recycling………...198
3.2.3.10. Batteries………...198
ix
3.2.3.12. Safety……….199
3.3. Structural Manifestation of Ionic Liquids………200
3.4. IL Functionalization………...201
3.4.1. IL Functionalization via Imidazolium Cation Modification…...201
3.4.2. ILs containing alkene and alkyne functionality………...202
3.4.3. ILs containing alcohol, ether and carboxyl functionalities…………..203
3.4.4. ILs containing silicon, nitrogen, phosphous and transition metal substituents……….203
3.4.5. N,N-bis(functionalized)-imidazolium ILs (double armed)…………...205
3.4.6. Alternative methods………..206
3.4.7. Non-imidazolium based functionalized ILs………..208
3.5. Application of ionic liquid in organic synthesis………..210
3.5.1. Friedel-Crafts Acylation in Ionic Liquid………211
3.5.2. Hydrogenation………...212
3.5.3. Cross coupling Reaction………...213
3.5.4. Diol/Carbonyl Protection………...214
3.5.5. Friedlander Synthesis………215
3.5.6. Knoevenagel Condensation/Robinson Annulation………...216
3.5.7. Diels-Alder Reaction………216
3.5.8. Olefin Epoxidation………...217
3.5.9. Swern Oxidation………..217
3.5.10. Ring Closing Metathesis………218
x
3.6.1. The use of Ionic-Liquid as Supported Catalysis………...219
3.6.2. The use of Ionic-Liquid as Supported Reagents………..223
3. 7. Ionic-Liquid-Supported Synthesis of Small Molecules and Combinatorial Synthesis………...224
3.8. Benzimidazoles and its amino derivatives, Importance& Synthesis……….234
3.9. Chemical methods for synthesizing benzimidazole derivatives…………...236
3.10. Results and Discussions………...241
3.11. Hydantoin fused tetrahydroazepino[4,5-b]indoles…….. …………...259
3.12. Chemical synthesis of azepino[4,5-b]indoles and its derivatives…...260
3.13. Results & Discussions………...261
3.14. Experimental Section………269
3.15. References……….308
xi O O PEG NH 2 N H R1 2 HO O OH NO2 N C S R2 MW N N R1 O O O N HN R2
ABSTRACT
This dissertation comprises of three chapters:
The work described in this dissertation involves the design, synthesis, and biological evaluation of novel bi-heterocyclic molecules on soluble support using focused microwave irradiation. In Chapter 1, section A, we report the design of an efficient liquid-phase method for the parallel synthesis of substituted benzimidazolylbenzoxazols using focused microwave irradiation and subsequent bioactivity. A key step in this approach involves the attachment of 4-hydroxy-3-nitrobenzoic acid to polymer immobilized o-phenylenediamine. Mild acidic conditions then promote a ring closure and subsequent reduction to form substituted benzimidazole derivative. The so formed benzimidazole derivatives underwent efficient ring closure with various alkyl, aralkyl, aryl, heteroaryl and unsaturated isothiocyanates to generate the benzimidazolylbenzoxazols.
The derived polymer-bound compounds were finally cleaved from the support with KCN in methanol at room temperature resulted in the generation of molecular library with two points of structural diversity (Section A). Interestingly, all the members of the molecular library exhibited moderate to high inhibition against VEGFR-3. This novel synthetic methodology offers an easy access to benzimidazolylbenzoxazols on soluble polymeric support, from which a new class of anti-cancer drugs may be developed.
xii N N R1 O O NH 2 F R2 O R2 1. PTSA, Toluene 2. KCN/MeOH N N R1 O O N R2 R2 N N R1 NH HN O O O R2 6 examples 80-99% ee O O NH2 N H R1 PEG 2 O HO NO2 F HCl.H2N R2 O O +
Section B involves the focused microwave irradiation to a multistep synthetic sequence of reactions designed to generate benzimidazolyl quinoxalinones using a soluble polymer support. They were obtained by the ipso-fluoro (SNAr) displacement of the immobilized ortho-nitro
fluoro benzimidazoles with chiral alpha amino esters under microwave irradiation. When subjected to neutral reduction
,
intermediate chiral organic-polymer conjugates underwent a spontaneous intramolecular ring closure. Cleavage of the polymer support, at room temperature did not cause any significant racemization resulting in the generation of a chiral molecular library with two points of structural diversity (Section B).
Section C involves the synthesis of Benzazepines, the subject of our interest having seven-membered aza-heterocyclic ring fusing aromatic unit, are of considerable interest owing to their varied biological activities. Polymer anchored 3-amino-4-fluorophenyl benzimidazole derivatives underwent intramolecular cyclisation with 1,3-disubstituted 2-butenone to generate the polymer anchored benzazepine derivatives.
Subsequent cleavage from polymer support results the formation of architecturally diverse seven member heterocyclic molecules (Section C).
xiii O ABz HO OTBDMS O P O O O T O O O O NC + O ABz DMTO OTBDMS O P O O O T O P O NC O N NC
5'-OHABzo=pTo=pABzo=pT-3'-O-Lev
In chapter 2, we have developed a general procedure for the solution-phase synthesis of chimeric oligonucleotides (oNA) analogues using readily available phosphoramidite reagents with 2+2 and 3+3 protocol. The key feature of this method is using the solution-phase phosphoramidite procedure to assemble linear oligonucleotide sequences and sequential removal of 3΄ levulinyl group as well as 5΄ dimethoxytrityl group without the contamination of n−1 or shorter failures. This methodology offers an easy access to the scale up synthesis of oligonucleotides for clinical as well as commercial purposes.
In Chapter 3, of section A, approaches towards the synthesis of 2-(substituted amino) benzimidazoles derivatives under focused microwave irradiation are described. Ionic liquid were employed as soluble support. Ionic liquid bound 4-fluoro-3-nitro benzoic acid underwent nucleophillic substitution reaction with various primary amines which after reduction and subsequent cyclisation with various isothiocyanates generates the desired compounds in high purity and yields. In the same way an alternative strategy on ionic liquid support for the parallel synthesis of thioanlogues of benzoxazol derivatives under focused microwave irradiation. Herein, we have replaced the NH group of R2 in chapter 1 of section A with sulphur analogues. A series
of 16 member library were successfully synthesized with two structural variability present on benzoxazol moiety.
xiv MW N H NHBOC OH O N + N OH BF4 -N H N N O O O O R1 R2 N N NHR2 R1 O O R2NCS N N R1 O O O N S R2
Section B develops the novel ionic-liquid supported synthetic protocol for hydantoin analogs tethered with azepino[4,5-b]indoles by the use of focused microwave irradiation. Ionic liquid bound tryptophan underwent Pictet-Spengler reaction and subsequent basification with various keto esters to generate the ionic liquid immobilized azepino[4,5-b]indoles. The so formed azepino[4,5-b]indole derivatives underwent cyclisation with various isocyanates to generate the three dimensional molecular architecture in traceless fashion (Section B).
1
Chapter One
Soluble Polymer Supported Synthesis of Bis Heterocyclic
Compounds
1. Introduction
Cancer is a universal disease which has been known and written about for thousands of years. The word cancer is a generic term used to describe as many as 200 malignant diseases which arise in different tissues and cause different illnesses.1 The disease cancer is characterized by abnormal cell growth and division that produces an expanding mass of disorganized tissues know as a tumor. As such, it is a disease of the cell and can develop in any tissue of the body. Cancerous cells multiply rapidly due to uncontrolled cellular growth and division. By the very nature of the disease, cancerous cells can infiltrate into other tissue types and leading to the spread of cancers.2
Sixty percent of patients with newly diagonised solid tumors have clinically evident or microscopic metastasis on diagnosis of primary tumors.3 It is this statistic, and the fact that the metastases are the major cause of treatment failure and death in cancer patients, this makes the cancers is one of the deadly diseases that exists today. Once tumor have metastasized and spread to different tissues, treatment is very difficult, as local therapy can no longer be directed at the disease, making metastasis the most life threatening aspects of cancer.
A way of identifying the various causes of cancers is by studying populations and behaviours.This approach compares peoples of various groups of people exposed to
2
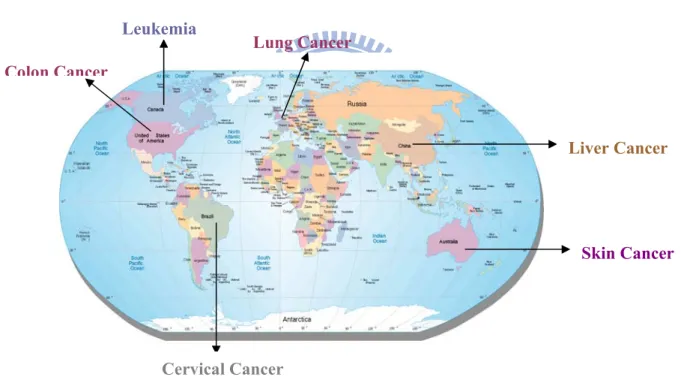
Colon Cancer
different factors or exhibiting different behaviors. A striking finding to emerge from population studies is that cancers arise with different frequencies in different areas of the world. For example, stomach cancer is prominent in Japan, colon cancer is prominent in USA, and skin cancer is common in Australia.4 The reason for the high rates of the 2specific kinds of cancer in certain countries is explained by the several factors specific for each country. An example of this is Australia, where the rate of skin cancer is highest in the world, due to the amount of sunlight to which people are exposed.
Regions of Highest Incidence
Figure 1.1. Regions of higher incidence of different forms of Cancers
As metastasis often proves fatal, there is an important need for improved understanding of this complicated process. The mechanism of invasion and metastasis, and patterns of spread of cancers need to be better understood in order to develop new therapies. Potential treatment interfering with this process could lead to better efficacy and
Leukemia
Skin Cancer Liver Cancer
Cervical Cancer
3
selectivity against cancers. The ultimate outcome would be to develop the treatment strategies that inhibit the metastasis and prevent the spread of cancers allowing the eradication of primary tumor and along with improved techniques capable of irradiating the secondary tumor metastasis.
1.1. Classification of Cancer
Cancer cells are classified into two main categories depending on whether or not they can spread by invasion and metastasis, as being either benign or malignant. Benign tumors are tumors that cannot spread by invasion or metastasis; hence, they only grow locally. They are not life threatening and are often treated by surgery. In some instances, particularly in benign brain tumors, the size of the tumors can lead to severe problems and ultimately death if untreated.5
Figure 1.2. Malignant Vs Benign tumors 1.2. Metastatic Cancer
Joseph Claude Recamier, a French physician, introduced the term “metastasis in 1898.6 Before that, physicians believed that secondary tumors were separate from the primary
4
tumor. Recamier showed that the secondary tumors (metastases) are caused by the spread of cancer cells from a primary tumor, and he described the mechanism of the invasion into the blood circulation and the growth of the secondary tumor (metastasis). The metastatic mechanism has been explained by two theories. The first is the “seed-and-soil” theory by Stephan Paget, first described in 1889 (for a review of this theory see ref 3). James Ewing proposed a second explanation in 1929, which described the process of metastasis on purely anatomical grounds. This theory suggests that most secondary growths occur at the first organ that tumor cells encounter. Researchers world wide have found that both theories are partially correct and both are used these days to explain the spread of cancer. Nowadays it is recognized that metastasis is a complicated process that involves interaction and response between both cancerous and normal cells. Recently researchers have produced very important results towards developing an understanding of the mechanisms and individual steps of metastasis, which is referred to as metastatic cascade.
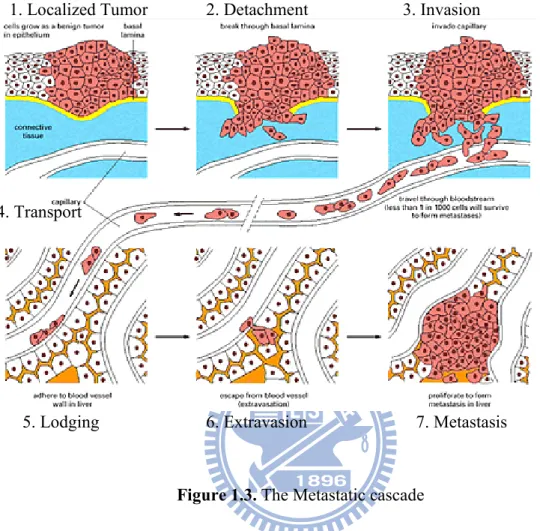
1.3. The Metastatic Cascade
Metastasis is a complex process, which consists of several sequential steps beginning from the growth of the primary tumor at a localized site, invading the local host tissue, invading blood or lymphatic vessels travel in the circulatory system, settling at a different site in the body, and finally, growing a new colony.7 Each step in the metastatic cascade is rate-limiting, and failure to complete any of these-steps prevents the tumor cell from producing a metastasis. Each of the processes leading to metastasis, as shown in Figure 1.3 will be described in turn.
5
1.3.1. Growth of primary tumor (Angiogenesis)
The first step shown in Figure 1.3 represents the growth of the primary tumor ata a localized site.7 The primary tumor canot grow beyond the size of 1-2 mm without acquiring a blood supply. The acquisition of a new blood supply via the growth of new blood vessels is called angiogenesis (neovascularization), and allows tumor growth, invasion and metastasis to occur.8 Tumor angiogenesis is the proliferation of a network of
blood vessels that penetrates into the cancerous cells growth, supplying nutrients and oxygen and removing waste products.9-13 Angiogenesis starts with cancer cells sending signals to surrounding normal cells. These signals activate some genes in the host tissue that, in turn, makes proteins to encourage the growth of new blood vessels. As a tumor becomes vascularized, the number of cells released into circulation correlates with the density of blood vessels in the primary tumor. The rate of proliferation of vascular endothelial cells is 20-2000 times faster in host-induced tumor endothelium than in normal endothelial cells.14
1.3.2. Detachment
The primary growth of cancerous cells is accompanied by several changes, both on the surface of the cells and with aberrant secretion of some materials. The underlying processes of invasion and metastasis are dependent on the modified behavior of tumor cells with regard to cell-cell and cell-matrix interaction.5 Malignant cells have a reduced ability to adhere to each other, which helps them detach from the primary tumor and invade local tissue. These changes cause drastic alteration in aspects of the biological behaviors of the cells.
6
Figure 1.3. The Metastatic cascade
The most important change is the decrease of E-cadherin (a cell surface glycoprotein) that mediates the adhesion between cells. Loss of E-cadherin expression relates to more invasive and metastatic phenotype.8,15 This occurs due to the over branching of the cell surface oligosaccharide, results in the change of negative charge of the cell surface. With the increase of negative charge on the cell surface, there is repulsion between cells which results to detachment of metastasizing tumor cells.
1.3.3. Invasion
1. Localized Tumor 2. Detachment 3. Invasion
4. Transport
7
Historically, invasion was thought of as a passive force, where tumor cells were believed to be pushed by the pressure of growth into the circulation. Much research and experiments tested this growth-pressure theory, and showed that blocking the growth of metastatic tumor cells had no effect on their invasive and metastatic potential.6 Since the pressure of a tumors growth does not effect the invasion of local tissue, tumor invasion is clearly an active process. Once malignant cells have detached from the primary tumor they will penetrate the extra cellular matrix (ECM) and enter the blood or lymph system. The invasion involves three steps: attachment, proteolysis and movement.
1.2.3.1. Attachment
The first step in invasion is the attachment of the tumor cells to the ECM or basement membrane. The ECM provides cell attachment ligands for extracellular receptors such as fibronectins, laminas and integrins. Some of these adhesion molecules including all ECM molecules and their cell surface receptors have been characterized.16-17 All of these are
important surface receptors that mediate the adhesion of cells to the ECM or basement membrane, and over expression of these receptors during tumor metastasis causes an increase in the progression of the disease. Fibronectins and lamins are cell-surface glycoprotein receptors that have the responsibility for activation of the host cells to produce proteolytic enzymes.6 Inhibition of fibronectin or laminin has experimentally inhibited the metastasis.
8
The second step in invasion is the degradation of the basement membrane. The breakdown of proteins including collagen, fibronectin, proteoglycans and other components in the ECM is mediated by degradative enzymes that are produced by the stimulative action of laminin or fibronectin to the host cells. There are three types of proteases produced from the tumor cells: serine proteases, collagenases (metalloproteinase), and cysteine proteases. The most important serine proteases are the highly regulated plasminogen activators, which catalyze the conversion of the extracellular plasminogen to plasmin, leading to the degradation of the ECM.18 The metalloproteinase are the most important proteinases associated with the malignant process. Liotta and colleagues found that augmented levels of metalloproteinase correlated with the development of invasion and metastasis in human breast, colon, stomach, thyroid, lung and liver cancers.19
1.3.3.3. Movement (locomotion)
After completion of the degradation process the cells begin moving and this movement is the defining characteristic of the invasion stage. The cells are then free to circulate via the blood stream and invade other tissues in the body. Direction of locomotion is influenced by tumor cell derived motility factors, host derived chemotactic factors, components of ECM and their proteolytic digestion products and growth factors. The stimulation of the tumor cells movement is known as chemotactic activity.
9
Intravasion is the process by which tumor cells enter the blood or lymph system. The tumor cells attack the membrane of the venules or capillaries in the same way that occurred during the invasion process by degradation of the ECM. The tumor vessels formed in angiogenesis are often defective and leaky, allowing malignant cells to cross their walls fairly easily and enter the circulation.6
1.3.5. Transport
Most of the tumor cells that reach the blood circulation die, due to the presence of several resistance forces, such as mechanical trauma, attack by natural killer cells and anoikis(loss of th anchorage dependence). Only the most aggressive of tumor cells can successfully survive all the stages of the metastatic cascade, and it is estimated that less than0.01% of the tumor cells that reach the circulatory system survive. This vulnerability in the blood mean that anchorage independence of malignant cells is not complete, and many may die through apoptosis.8
1.3.6. Lodgement
The process of lodgement is not a random process. Different tumors exhibit different attachment factors and adhesion mediating molecules, specific cell recognition and affinity between adhesion molecules.15 The patterns of spread of metastasis are explained to some extent on the basis of circulatory anatomy and the organ of first encounter. Since gastrointestinal tract tumors penetrate the portal venous system they lead to liver
10
metastasis, where as other tumors penetrate the systemic veins, eventually draining into the Vena Cava, leading to lung metastasis.20 However, some tumor cells do not lodge in
the first capillary bed they reach, but are transported to more distant organs and have a more selective pattern of metastatic colony formation.
1.3.7. Growth of secondary tumor (Matastasis)
After progressing through all the stages outlined above and shown in Figure 1.3 most tumors thereafter remain dominant and fail to complete the final stage of metastasis in the metastatic cascade. The failure may be due to lack of appropriate stimulatory growth factors. As with primary tumor cells, in order for the secondary tumor to grow larger than 0.5 mm in diameter a new blood supply is needed.17 This process of angiogenesis is therefore crucial for the growth of the secondary tumor cells, and for this reason there is intensive research aimed at designing anti-angiogenic drugs to prevent the growth of both primary and secondary tumor cells.
1.4. Factors Influencing Malignancy in Cancer
There are two important factors that effect the development of a malignancy. These two factors are cell-to-cell adhesion and cell to extracellular matrix interaction. The interaction between malignant cells is particularly important, since they have a low ability to adhere to each other. This facilitates their detachment from the primary tumor site and subsequent entry into the circulatory system.
11
1.5. Current treatments of Cancer
The current treatment regimes for cancer are dependent upon the type of cancer being treated. Surgery is useful for large tumors that have not begun to metastasize. For small tumors or metastatic tumors, radiotherapy and/or chemotherapy are usually required. Both of these treatments cause serious side effects, primarily because they are targeting all cells that are rapidly dividing. Whilst cancerous cells are targeted, cells such as hair follicles or those cells lining gastrointestinal tract and bone marrow are also rapidly dividing cells and are therefore also affected during radiotherapy or chemotherapy regimes. The need for effective chemotherapeutic agent that can destroy the cancer cells without affecting the normal cells is paramount.
1.6. Cancer and VEGF
It is a well established fact in cancer biology that the tumor growth depends on the advancement of neovasculature. While there are more than 100 distinct types of cancer (and considerable heterogeneity within each tumor), the mechanisms that fuel tumor growth and survival are relatively similar. Across all cancers, sustained angiogenesis is reported to be the hall mark of cancer. Angiogenesis, a physiological process involves the formation of new blood vessel from the pre-existing ones is important for the supply of nutrients, oxygen, hormones and growth factors. For a tumor to grow beyond 1 to 2 mm diameter needs continuous supply of blood, which is acquired by expressing growth factor that recruit new vasculature from existing vessels as show in figure 1.4.21-24
12
Figure 1.4. The Angiogeneic Process
Among the many factors implicated in angiogenesis, VEGF has been identified as the most important one. The scope of scientific research involving VEGF continues to grow dramatically.
1.6.1. VEGF
VEGF (also known as VEGF-A, but commonly referred to simply as VEGF) stands for “vascular endothelial growth factor.” Vascular endothelial growth factor (VEGF) is a chemical signal produced by cells that stimulates the growth of new blood vessels. It is part of the system that restores the oxygen supply to tissues when blood circulation is inadequate. This protein plays an important role in angiogenesis. As the name suggests, VEGF stimulates vascular endothelial cell growth, survival, and proliferation. The VEGF is a member of a family of 6 structurally related proteins that regulate the growth and differentiation of multiple components of the vascular system, especially blood and
13
lymph vessels. The six members are A, placenta growth factor, B, VEGF-C, VEGF-D and VEGF-E. 25
VEGF Family Members Receptors Functions
VEGF (VEGF-A) VEGFR-1, VEGFR-2, neuropilin-1
Angiogenesis, vascular maintenance
VEGF-B VEGFR-1 Not established
VEGF-C VEGFR-2, VEGFR-3 Lymphagiogenesis
VEGF-D VEGFR-2, VEGFR-3 Lymphagiogenesis
VEGF-E (viral factor) VEGFR-2 Angiogenesis Placental growth factor
(PIGF)
VEGFR-1, neuropilin-1 Angiogenesis Inflammation
1.6.2. VEGF receptors
Vascular endothelial growth factor (VEGF) ligands mediate their angiogenic effects by binding to specific VEGF receptors, leading to receptor dimerization and subsequent signal transduction. VEGF ligands bind to 3 primary receptors and 2-co-receptors. These six members can bind and activate the tyrosine kinase receptors, VEGF receptors 1, 2, and 3, which promotes the propagation, endurance, and migration of endothelial cells.26 VEGFR-1 is able to bind VEGF-A, VEGF-B, and PlGF. VEGFR-2 is activated basically by VEGF-A, but of VEGF-C, VEGF-D, and VEGF-E may also activate this receptor. So, basically the angiogenesis is regulated by VEGFR-1 and VEGFR-2 (Figure 1).
14
Endothelial expression of VEGF receptors varies among the 3 primary recetors; VEGFR-2 is expressed on almost all endothelial cells, where as VEGFR-1 and -3 are selectively expressed in distinct vascular beds. The neuropilin-1 (NP-1 or NRP-1) and NP-2 (or NRP-2) receptors are thought to increase the binding affinity of the various VEGF ligands to these primary receptors, although the site specific roles of NP-1 and NP-2 in angiogenesis are not known.
1.6.3. The strategies for inhibiting the VEGF pathway
There are two primary pathways for inhibiting the VEGF signaling pathway which includes inhibiting either the VEGF ligand or the VEGF receptor. These are explained as below.
Extra cellular targeting of the VEGF ligand
Anti-VEGF strategies that directly target the VEGF ligand include ligand-binding antibodies and soluble receptors. These agents work extracellularly to provide specific inhibition of the VEGF pathway without disrupting other non-VEGF related targets. Therefore, they may inhibit angiogenesis without affecting other secondary or “off-target”pathways.27 VEGF also promotes angiogenesis by signaling through neuropilin, a co-receptor to VEGFR-1 and -2 on endothelial cells. The presence of neuropilin is associated with tumor aggressiveness and poor prognosis. Neuropilin receptors lack a targetable intracellular kinase domain. Therefore, anti-VEGF strategies that target the ligand extracellularly may be capable of attenuating VEGF signaling that is mediated through neuropilin in Fig. 1.5.27 Neuropilin receptors lack an intracellular kinase domain
15
but are able to induce signaling by recruiting ligands to the cell membrane. Emerging evidence also now suggests that neuropilin may be able to transduce VEGF signals in the absence of tyrosine kinase VEGF receptors. Accordingly, prevention of neuropilin-mediated signaling may require an extracellular strategy of VEGF inhibition.
Intracellular targeting of the VEGF receptor
Anti-VEGF strategies that target the VEGF receptor include tyrosine kinase inhibitors (TKIs) and receptor antibodies. Agents that target the VEGF receptor intracellularly, such as TKIs, have a wider range of inhibitory effects and may disrupt other secondary pathways that are also mediated through receptor kinases.28
Figure 1.5. Stategies for inhibiting the VEGF pathways
1.6.4. Factors leading to the production and expression of angiogenesis by VEGF
Vascular endothelial growth factor (VEGF) production and subsequent angiogenesis can be triggered by a number of factors, including both genes and gene products, in the cellular microenvironment.
16
Entry VEGF triggers Description/subsequent
effect
Factors involved
1 Hypoxia Shortage of oxygen in the tumor environment
HIF-1α, HIF-1β
2 Oncogenes/Tumor suppressor genes
Genes that stimulate or suppress tumor formation
c-Src oncogene Bcr-Abl oncogene
Ras oncogene p53 tumor suppressor gene 3 Cellular receptors Proteins on the cell
surface that are part of signaling pathways
EGFR HER-2 IGF-IR 4 Other growth factors
and cytokines
Proteins secreted by cells that stimulate cellular
signaling
COX-2 PDGF
Hypoxia triggers VEGF expression
Without an independent blood supply, tumors must rely on diffusion to obtain oxygen and other nutrients, and typically cannot grow more than 2 mm3 in size. Thus, a growing tumor without sufficient vasculature will have hypoxic or lacking in oxygen areas. In
17
response to hypoxic conditions, tumors secrete vascular endothelial growth factor (VEGF) in order to recruit new vasculature, which then provides a supply of oxygen.29 Hypoxia
remains an important trigger of VEGF expression even once a tumor becomes vascularized. As the tumor grows, it continually outgrows its existing blood supply, leaving a rim of necrotic and hypoxic tissue. The tumor responds by up regulating VEGF gene expression, primarily through the activity of hypoxia inducible factor-1 (HIF-1), a protein consisting of 2 subunits (HIF-1α and HIF-1β).30
Oncogenes and tumor suppressor genes trigger VEGF expression
Oncogenes (genes that contribute to the production of a cancer) and tumor suppressor genes (genes encoding a protein that normally suppress tumor formation) are associated with increased vascular endothelial growth factor (VEGF) production. Oncogenes are generally mutated forms of proto-oncogenes (normal cellular genes capable of transforming a cell when activated). Some examples of oncogenes and tumor suppressor genes include
• c-Src is a proto-oncogene that appears to directly stimulate VEGF expression. • Bcr-Abl is an oncogene formed from fusion of two proto-oncogenes31 A
preclinical study in tumor cell lines showed that transfection of Bcr-Abl caused an increase in VEGF expression, whereas blocking the function of Bcr-Abl reduced VEGF expression
• Ras oncogene: Ras proteins are part of the signaling cascade of growth factor— induced angiogenesis. The genes that encode for Ras proteins have been
18
associated with induction of VEGF expression in many solid tumors, including pancreatic, colorectal, and non-small cell lung cancers32
• p53 tumor suppressor gene: Dysregulation of p53, normally a regulator of the cell cycle and trigger of apoptosis in damaged cells, has been implicated in the pathology of solid malignancies, including colorectal, breast, and endometrial carcinomas. Genetic alteration of tumor suppressor genes, including p53, has been shown to induce VEGF production33
1.6.4.1. The Role of VEGFR-1
Vascular endothelial growth factor (VEGF) receptor-1 (VEGFR-1) is a receptor for VEGF-A and can also bind VEGF-B and placental growth factor (PlGF). VEGFR-1 is a key receptor in developmental angiogenesis, but does not appear to be critical to pathogenic angiogenesis. Its role appears to vary with stages of development, physiologic and pathophysiologic conditions, and cell type.34
1.6.4.2. The Role of VEGFR-2
Vascular endothelial growth factor (VEGF) receptor-2 (VEGFR-2) mediates the majority of the downstream angiogenic effects of VEGF, including:
• Permeability
• Endothelial cell proliferation • Invasion
• Migration • Survival
19
Recent work suggests that VEGFR-2 can stimulate angiogenesis on its own. The activation and signaling of VEGFR-2 may be positively or negatively influenced by co- expression and activation of VEGFR-1
1.6.4.3. The Role of VEGFR-3
Vascular endothelial growth factor (VEGF) receptor-3 (VEGFR-3) promotes lymphangiogenesis. The term lymphangiogenesis involves the formation of new lymphatics, includes the spreading and invasion from tumors into surrounding stromal tissues, incursion into lymphatic walls and followed by lymph nodes implantation, and proliferation in the parenchyma of target organs. Recent studies have identified that because of single-layered endothelial cells having incomplete basal membranes, the lymphatic vessel composed, it lacks the tight junctions between endothelial cells. That’s why, the tumor cells can easily enter the lymphatic vessels and metastasize to lymph nodes.35 Unlike blood vascular system, the lymphatic system does not receive much attention owing to the lack of knowledge about the molecular mechanism of its development and function. Expression of VEGFR-3 is mostly restricted to lymphatic endothelium in normal tissues. However, recent studies have suggested that increased expression of VEGF-C and VEGF-D promotes tumor lymphangiogenesis and one can only curb this by inhibiting the VEGFR-3 signaling pathways. VEGFR-3 activation has been observed in several solid tumor types, including melanoma and breast cancer. In these tumors, elevated levels of VEGFR-3 ligands VEGF-C and VEGF-D are associated with lymph node metastases.36 In recent years a promising approach to the therapeutic intervention of cancer has focused on antiangiogenesis therapies. This approach to
20
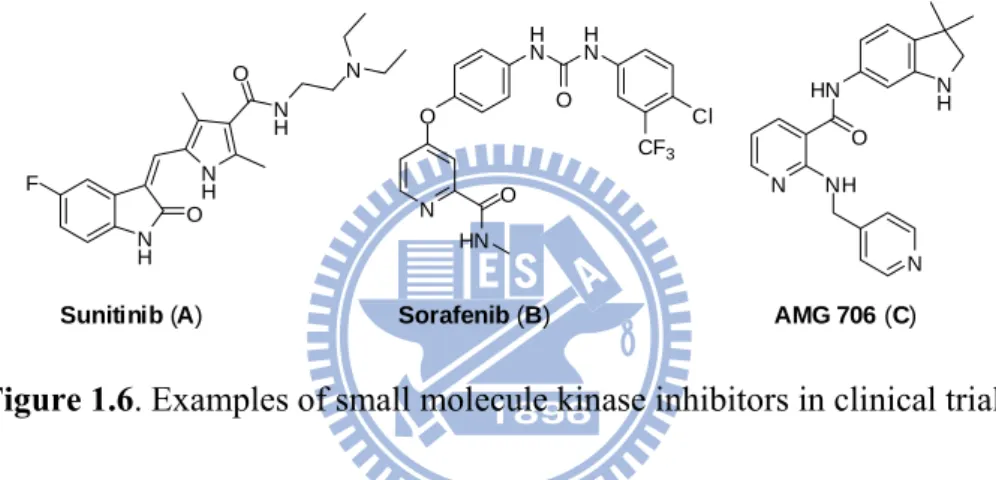
intervening in cancer progression takes advantage of the idea that inhibiting the blood supply to tumors will deplete the oxygen and nutrients. Resultantly it arrest tumor cell growth and proliferation. This approach has been found to be effective and there are presently twenty antiangiogenic drugs undergoing various stages of evaluation in clinical trials and numerous others in preclinical development. Recently two small molecule inhibitors sunitinib (A) and sorafenib (B) have been approved, in addition to AMG 706 (C), as antiangiogenic drugs (Figure 1).37
N H O N H O N H N F N O O HN H N O H N Cl CF3 N HN O N H NH N
Sunitinib (A) Sorafenib (B) AMG 706 (C)
Figure 1.6. Examples of small molecule kinase inhibitors in clinical trials
In spite of these achievements, it remains an important challenge to develop new drugs in order to overcome the drug resistance and maintain the steady progress in the cancer research. Hence the synthesis of large number of molecules for screening and ‘lead’ generation plays an important role in cancer drug research. Concomitantly, coupled with high throughput screening, development of effective methodologies for rapid synthesis of diversified libraries of small heterocyclic molecules is of great importance.
21
1.7. Solid-Phase Methods in Organic Synthesis
Since the genesis of modern organic chemistry, classical organic reactions have been carried out in solution phase, implying the separation of the desired product from reagents and by-products after the reaction. This purification step can however turn out to be incredibly time consuming and often rigorous. In 1963 Merrifield first introduced the solid-phase synthesis of peptide and oligosaccharide.38 This methodology, which was
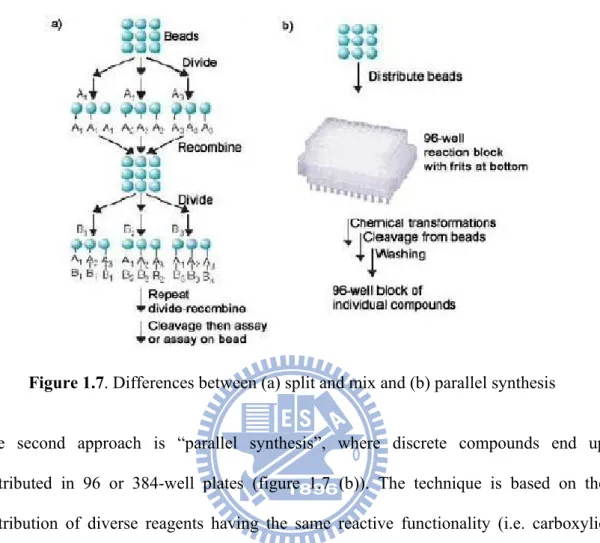
limited to the synthesis of peptides39 and oligosaccharides, however remained predominantly limited to this field until the introduction of combinatorial techniques. There are two different ways of combining reagents to achieve chemical diversity. One is “Split-and-mix” synthesis (Figure 1.7 (a)), which was first developed by Furka, Lam and Houghten has now been used extensively in the pharmaceutical industry for the generation of thousands of compounds.40-42 At every step, all beads are split equally into the number of reactions carried out (3 in the example). After reaction and washing, the beads are mixed together and then separated again into the next number of vessels required (3 again). The process continues until the last step with each bead containing a different compound (one bead / one compound). The advantage of this technique is the ability to generate exponentially growing numbers of compounds (3n after n steps) while keeping the number of reactions quite low (3 × n in the case of n steps). With split and mix, however, all compounds end up in the same mixture.
22
Figure 1.7. Differences between (a) split and mix and (b) parallel synthesis
The second approach is “parallel synthesis”, where discrete compounds end up distributed in 96 or 384-well plates (figure 1.7 (b)). The technique is based on the distribution of diverse reagents having the same reactive functionality (i.e. carboxylic acids, amines, etc.) at each step of the synthesis. At the end of the sequence, each well will have followed a specific order, different from all other wells.
Since then, worldwide organic chemists have been widely adopted solid-phase organic synthesis (SPOS) to achieve combinatorial synthesis of structurally diverse heterocyclic molecular libraries. The key perception for SPOS methodology is to tether a simple molecule onto an insoluble polymer-support and then build-up density by planned chemical modifications and then finally cleave the final product off the resin (Merrifield’s protocol). There are several advantages of SPOS over existing solution phase approaches. One major advantage is the facile separation and purification of
23 OH O N H O N H O H N NH Dde Fmoc O N H O N H O H N HN O O X Y H N O N O I Pd(0) H2N N H O H N HN O O X Y H N O N O 15 Examples 20-24 membered rings
intermediates by simple filtration of the reaction mixture. Moreover, due to this easiness of purification, all transformations applied during SPOS can be achieved using a large excess of reagents to drive reactions to completion. With the introduction of ′pseudo-dilution effect′43 facilitates macrocyclisation in SPOS relative to in solution phase synthesis. For example in 1995, Hauske introduced an acid-labile carbamate linker to deliver, high purity products with high purity following macrocyclisation of ′drug-like′ libraries mediated by Pd(0) under mild conditions.44
Figure 1.8. Hauske’s macrocyclisation on solid-support resins.
All of the macrocycles (20- to 24-membered rings) were liberated from the resins under traceless cleavage conditions and obtained in good overall yields (Figure 1.8).
Although Merrifield’s protocol for SPOS has many advantages over conventional solution phase techniques, it has some drawbacks: functionalized resins are difficult to
24
characterize and reactions are difficult to monitor using normal spectroscopic techniques in comparison with solution phase chemistry; reaction rates are slow; extra steps are required to attach and to release substrates from the resin; convergent synthetic strategies are not applicable; and certain reactions can only be carried out on particular types of resin. However, some of these disadvantages can be overcome by introducing solid-supported reagents. These are particularly reactive chemicals immobilized on a resin for carrying out desired chemical transformations in solution (Ley’s protocol).45 This concept also encompasses the development of solid-supported scavengers to remove excess organic or metallic reagents from solution phase reactions. Solid-supported reagents and scavengers facilitate in situ purification of reaction mixtures, leaving the desired products in solution for convenient identification. Conventional multi-step transformations in solution can be performed using sequential solid-supported reagents and scavengers to telescope workup and purification procedures. In addition, this approach can also be applied to convergent syntheses to achieve higher yields than in the corresponding linear counterparts. Prof Steven Ley demonstrated the former facet by employing solid-supported reagents to effect the clean and efficient total synthesis of several natural products (e.g. oxomartidine and epimartidine) (Figure 1.9).46
25 OH OMe MeO NMe3RuO4 CH2Cl2 100% O OMe MeO + HO H2N NMe3BH4 MeOH 90% HO N H OMe MeO N N (CF3CO)2O 99% HO N OMe MeO O CF3 (OCOCF3)2 CF3CH2OH N MeO MeO COCF3 O NMe3(CO3-2)0.5 MeOH 100% N MeO O MeO H (+)-Oxomaritidine -NMe3BH4 CuSO4 or NiCl2 MeOH 80% N MeO OH MeO H (+)-Epimaritidine
-Figure 1.9. Ley’s total synthesis of (±)-oxomaritidine and (±)-epimaritidine.46
Both the Merrifield and the Ley approaches to SPOS have been widely applied in automated combinatorial and parallel synthesis in industry for rapidly preparing drug-like small molecules for property screening (e.g. as new pharmaceuticals). However, slower conversion rates than for the corresponding transformations in solution remains a weakness due to the biphasic interactions between the polymer and solution.
1.8. Soluble Polymer Supported Technology in Organic Synthesis
To circumvent the drawbacks inherent to solid-supported technologies such as, nonlinear kinetics, unequal distribution to the chemical reaction, solvation problems, alternative
26 On n n OH N H n n OH O n NH2 O O n H N O Cl H N O O O n m O HO OH OH O n Polyethylene glycol (PEG) Polypropylene
Oxide Polyvinyl alcohol Polyethylene imine Polyacrylic acid
Polyacrylamide Polystyrene PEG with 3,5-diisocyanatobenzyl chloride Cellulose
approaches using homogeneous ′beadless′ phase-tagged chemistry have been introduced to facilitate separation whilst retaining solution phase kinetics. Among all of these beadless approaches, soluble polymer phase anchoring of substrates to enable easy separation, monitoring, analysis and characterisation has become the method of choice.47 Normally the polymers employed as soluble supports in liquid phase organic synthesis should possess some basic qualities such as easy availability, good mechanical and chemical stabilities, having appropriate functional groups for easy anchoring to organic moiety and more importantly showing high solubility to dissolve molecular entities etc. Moreover, it has been observed that polymer supports normally used in organic synthesis should have macromolecules of varying sizes. These supports should withstand the reaction condition used in solution phase chemistry and consequently most polymer supports used in liquid phase synthesis possess alkyl ether backbone structures. By variation of functional groups of backbone structures, polymer properties are determined and may provide sites for attachment of organic moieties. There are number of polymers which are normally used for small molecule organic synthesis. These includes polyethylene glycol (PEG), polystyrene, poly(propylene oxide), poly(vinyl alcohol),
polyethylene imine, polyacrylic acid, polyacryl amide, PEG with 3,5-diisocyanatobenzyl
27
Figure 1.10. Different Soluble Supports used in Small Molecule Organic Synthesis 1.8.1. Application and recent development of polyethylene glycol as soluble support in organic synthesis
A number of polymeric reagents have been used for the simplification of organic synthesis. The liquid phase method of peptide synthesis on polyethylene glycol (PEG) was first introduced in 1971 and the PEG method has emerged as one of the most effective supports for the synthesis of oligopeptide, oligonucleotide, oligosaccharide as well as small molecules for the construction of combinatorial libraries.48 Polyethylene
glycol is cheap as compared to polyethylene oxide and polyoxyethylene. Normally for the supported synthesis PEG5000 and PEG4000, PEG6000 are used as soluble supports based
on the loading capacity and hydroxyl functionalities contained. PEG5000 contains one
hydroxyl group and its loading capacity is 0.2 mmol/g, whereas PEG4000 and PEG6000
consist of two hydroxyl groups and their loading capacities are 0.5 mmol/g and 0.33 mmol/g, respectively. Employed as a protecting group, this linear homopolymer exhibits solubility in a wide range of organic solvents and water. PEG is insoluble in hexane,
diethyl ether and tert-butyl methyl ether, and these solvents have been used to induce
PEG precipitation. Careful precipitation conditions or cooling of polymer solutions in ethanol or methanol yields crystalline PEG due to the helical structure of the polymer
that produces a strong propensity to crystallize. Thus, as long as the polymer backbone remains unaltered during liquid-phase synthesis, then purification by crystallization can be utilized at each reaction step. Furthermore, the solubilizing power of PEG not only allows homogeneous
reactions
under numerous reaction28
conditions, but these solubility properties permit individual reaction steps to be monitored without requiring cleavage of product from the polymer support. The characterization of PEG-bound organic moieties is often straight forward as the polymer does not interfere with spectroscopic or chemical methods of analysis. In
addition, MeOPEG (PEGME: polyetylene glycol monomethyl ether) contains a single methoxy group (δ = 3.38 ppm) and ethyl protons of PEG backbone (δ = 3.64 ppm) that provide internal standards for easy monitoring of reactions by 'H NMR spectroscopy. Depending on polymerization conditions, PEG terminal may consist of a hydroxyl group or may be selectively functionalized. Commercially available PEG is produced through anionic polymerization of ethylene oxide to yield a polyether structure possessing either hydroxyl groups at both ends or a methoxy group at one end and a hydroxyl group at the other (MeO-PEG). The polymer MeO-PEG is considered mono functional, as typically the methoxy group of MeO-PEG remains unchanged throughout chemical manipulations. For identical chain lengths, the loading capacity of PEG is twice that of MeO-PEG as two hydroxyl groups serve as anchoring sites on PEG. Recently, varieties of PEG derivatives have been developed and are commercially available.
1.8.2. Applications of PEG in biological studies
A key property of PEG is that attachment to other molecules and surfaces provide a
biocompatible, protective coating. This protective coating slows rejection of materials in biological systems (such as the human body), greatly reduces protein, cell and bacterial adsorption, and reduces the rate of kidney clearance (because of larger size). PEG also is
29
nontoxic and has been approved by the FDA for topical and interna1 use in humans. PEG is soluble in water and many organic solvents, and it forms aqueous two-phase systems when paired with certain other polymers (such as dextran). It is insoluble in ethyl ether and hydrocarbons such as hexane. The water solubility, lack of toxicity, high flexibility and well-defined chemistry of bifùnctional PEG makes it ideally suited for many cross linking or tethering applications. Seven technologies that have resulted from use of these properties are: (1) PEG-proteins for pharmaceutical use; (2) PEG-surfaces for electricdy-controlled, nonfouling materials; (3) PEG-liposomes for drug delivery; (4) molecule-molecule or molecule-molecule-surface coupling for drug and materials applications; (5) PEG-molecules for biological purifications; (6) biopolymer synthesis on PEG supports; and (7) PEG attachment for control of solubility (e.g., enzymes into organic solvents or water solubilization of enzyme substrates, dyes, flavors and chemotherapeutic agents).49
1.8.2.1 PEG-proteins for pharmaceutical use
It has been demonstrated that proteins with PEG attached remain active and have a greatIy diminished or negligible immune response. The result is that these proteins have greatly increased serum lifetimes. Examples include SOD, PEG-asparaginase, PEG-IL- 2 and PEG-hemoglobin. In addition, PEG attachment makes proteins much larger and thus reduces their rate of clearance through the kidney. PEG has also been attached to many small molecules (such as vitamin E, cholesterol, fluorouracil, etc.). The goal here is to reduce rate of kidney clearance and impart water solubility.
30
1.8.2.2 PEG-surfaces
In addition to the molecular modifications, PEG can also be attached to surfaces to form protective, bio-compatible coatings. A variety of applications result, including PEG coatings for arterial replacements, diagnostic apparatus and blood contacting devices. Similarly, capillary zone electrophoresis has emerged as an important new analytical technique in biochemistry, and PEG coatings on the capillaries prevent protein adsorption and provide critical control of electro osmosis.
1.8.2.3. PEG-liposomes
There has been intense interest in use of liposomes for controlled-release and selective delivery of drugs. A problem with this application is that liposomes, especially larger ones, are quickly attacked and cleared from the body. Recent research has shown that incorporation of PEG into the outer coating of liposomes can greatly increase serum lifetime, thus solving a critical problem blocking application of this promising drug delivery technique. The hydrophilic, biocompatible nature of PEGs and their mild, well-defined chemistry makes them ideal for coupling or tethering molecules to molecules or molecules to surfaces. This technology is critical for the next generation of drugs and biomaterials. Research has shown that use of PEG as a coupler to bind molecules to other molecules and surfaces provides highly active materials.
31
1.8.2.4 Biological Purification
The genetic engineering revolution has led to methods for production of a variety of physiologically active proteins. There is, however, a critical need in this industry for improved methods for isolation of the proteins produced. An approach to this problem that has recently received much interest is purification by partitioning in aqueous two-phase systems (analogous to oil and water) made by solution of PEG, other polymers and salts in water. In this approach, a PEG-ligand is made (such as a PEG-antibody), which binds specifically with the desired protein and pulls the protein into the PEG-rich phase.
1.8.2.5. Biopolymer syntheses
The three bio-oligomers (peptides, oligonucleotides, and oligosaccharides) can al1 be
grown on PEG as a soluble carrier. The PEG-oligomer is precipitated after each step to isolate the product, which can then be cleaved or taken to the next addition step. Advantages
of
this method are that fewer errors result, chemistry is faster, and large quantities of materials can be handled. A variation on this theory is to build the bio-oligomer on a PEG chain that is bound to a solid polystyrene particle. This approach apparently provides advantages of both solid-phase and liquid-phase synthesis.32 MeO-PEG-OH +O C N S Cl O O cat. Dibutyltinlaurate CH2Cl2 O N H O PEG S Cl O O MeO R-NH2, pyridine CH2Cl2 O N H O PEG S NHR O O MeO 0.5N NaOH H2N S NHR O O
PEG is soluble both in water and in many organic solvents. This property has been utilized to solubilize other molecules by PEG attachment. An interesthg biotechnical application is solubilization of enzymes in organic solvents such as chlorinated
hydrocarbons. Additionally, water-insoluble materials may become water-soluble after PEG attachment. Examples here include dyes, flavors, substrates for enzymes, cofactors, pharmaceuticals etc.
1.9. Small molecule Syntheses
Polyethylene glycol has long been applied as soluble supports for the synthesis of oligopeptide, oligosaccharide as well as small molecules. Till date numerous research efforts has been published using PEG as soluble supports. Janda et al. first reported the synthesis of pentapeptide and sulfonamide libraries using the PEG as a support. This was the first report of the use of PEG in small organic molecule synthesis, and the pentapeptide library preparation was the first application of this polymer in a combinatorial sense. Synthesized libraries were act as ligands for a monoclonal antibody against â-endorphin (Figure 1.11).48
33
MeO-PEG-OH [a-c] MeO PEG O SH [d,e] MeO PEG O S
O O O O O PEG MeO S O O Cl OH OH [h] MeO PEG O S Cl N NH O O [i] O PEG MeO S Cl N NH O O O O [j] Cl N NH O O
Similar methodology was applied for the synthesis of a new class of peptidomimetics called azetides, alkylated malonates and 3,5-pyrazolidinediones which could be used for rheumatoid arthritis.50 (Figure 1.12.)
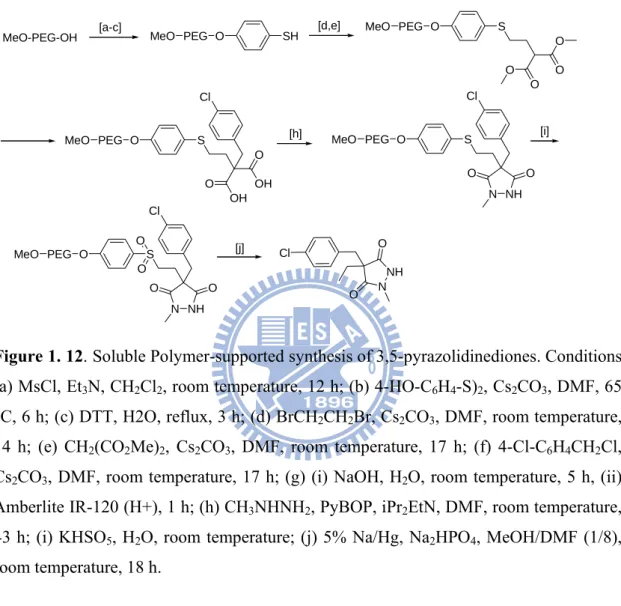
Figure 1. 12. Soluble Polymer-supported synthesis of 3,5-pyrazolidinediones. Conditions:
(a) MsCl, Et3N, CH2Cl2, room temperature, 12 h; (b) 4-HO-C6H4-S)2, Cs2CO3, DMF, 65
°C, 6 h; (c) DTT, H2O, reflux, 3 h; (d) BrCH2CH2Br, Cs2CO3, DMF, room temperature,
14 h; (e) CH2(CO2Me)2, Cs2CO3, DMF, room temperature, 17 h; (f) 4-Cl-C6H4CH2Cl,
Cs2CO3, DMF, room temperature, 17 h; (g) (i) NaOH, H2O, room temperature, 5 h, (ii)
Amberlite IR-120 (H+), 1 h; (h) CH3NHNH2, PyBOP, iPr2EtN, DMF, room temperature,
43 h; (i) KHSO5, H2O, room temperature; (j) 5% Na/Hg, Na2HPO4, MeOH/DMF (1/8),
room temperature, 18 h.
Furthermore, it has been observed that the intramolecular cyclitive cleavage to generate 3-aminoimidazoline-2,4-diones and intramolecular stille coupling have also occurred on soluble polymer supported synthesis.51 Recently, it has been observed that the problem of low loading capacity was overcome by combining the basic principles of dendrimer chemistry with that of PEG polymers to generate new, soluble PEG supports of expanded functional group capacity as developed by Cozzi.52
34 OMs MsO OH COOMe MeOOC O O R R R R A-E PEG (4600) PEG (4600) A, R = COOMe B, R = COOH C, R = CH2OH D, R = CHO E, R = CH2Cl
Figure 1.13: Synthesis of high molecular weight PEG- polymers.
The bis-mesylate obtained from PEG4600 was reacted (Figure 1.13) with dimethyl 5-hydroxyisophthalate and Cs2CO3 in DMF (50 °C, 15 h) to afford the tetra ester A in 95%
yield. Hydrolysis of A with aqueous KOH at room temperature for 15 h and followed by acidification gave the tetra acid B in 70% yield. Reduction of the ester A with 2 N DIBAL in CH2Cl2 at -78 °C to room temperature for 15 h afforded the tetraol C in 83%
yield. From this compound C, which features four easily removable benzylic hydroxyl groups, the tetra aldehyde D was synthesized by oxidation with MnO2 in CH2Cl2 room
temperature for 72 h in 71% yield, and the tetrachloride E obtained by reaction with SOCl2 in the presence of pyridine in refluxing toluene for 15 h, in 60% yield. Thus five
different functionalities amenable to a variety of synthetic manipulations could be easily attached to the polymer backbone. To further confirm the possibility of performing organic synthesis on this polyfunctionalized PEG, tetraol C was esterified with N-Boc
35 O O PEG (4600) NH2CH2CO2 NH2CH2CO2 O2CCH2NH2 O2CCH2NH2 P N P N P N OR OR RO RO OR RO R = CH2(CH2CH2O)nH n = 14, MW 600 n = 23, MW 1000 n = 45, MW 2000
glycine using DCC as coupling reagent and catalytic amount of DMAP in refluxing CH2Cl2 for 15 h obtained tetra-N-Boc glycinate in 94% yield, which after BOC
Figure 1.14. Reactions anchoring on PEG 4600
deprotection in 2:1 TFA: CH2Cl2 mixture gave tetra amine in 83% yield as shown in
figure 1.14. Here we have observed that loading capacity of polyfunctionalized PEG increases to 0.8 mmol/g as compared to MeO-PEG5000 where loading capacity is 0.2
mol/g. Recently, Janda et. al. have synthesized a new high-loading (1 mmol/g) soluble- polymer based on a cyclotriphosphazene core with PEG arms known as stealth star polymer that exhibit advanced precipitation properties compared with those of linear PEG.53 While it was extremely necessary to increase the loading capacity for soluble supports, Fan et. al. 54 developed the Janus dendrimer via a liquid phase approach which could easily be purified by simple precipitation technique without column chromatography. Their utility as soluble supports was carefully decorated in the Pd catalyzed Suzuki cross coupling reactions giving biaryl products in good yield in figure 1.15.
36
Figure 1. 15. Janus Dendrimers as Soluble Supports for the Pd-Catalyzed Suzuki
37
Section A
1.10. Benzoxazols – Importance and synthesis.
Benzoxazoles are privileged N-O heterocycles known for a wide range of biological activities. Benzoxazoles are categorized as pervasive structural motif, because of their ability to interact with a wide range of different enzymes and receptors. Recently they are found in a number of natural products, agrochemical products, as well as medicinal utility (Figure 1.16). For example, the benzoxazole moiety is established in natural products like antimycobacterial pseudopteroxazole,55 UK-1,56 AJI9561 157 and
salvianen.58 Besides this activity, this moiety has been found as cathepsin S inhibitor 2,59 5-HT3 receptor agonist 3,60 HIV reverse transcriptase inhibitor L-697,661 4,61 anticancer
agent NSC-693638 5,62 estrogen receptor-β agonist ERB-041 6,63 orexin-1 receptor antagonist SB-334867 764, selective peroxisome proliferators-activated receptor γ
antagonist JTP-426467 8,65 Recently, it had been found that the benzoxazol ring 9, is an intriguing heterocycle acts as a novel class of potent KDR inhibitors.66 In spite of this activity it also act as herbicides, such as Fenoxaprop 10 and as fluorescent whitening dyes 11.67 2-Phenylbenzoxazols are known as photo stable, highly efficient UV dyes, and
38 N O O O N O R2 HO R1 R1=CH3, R2=H: UK-1 R1=H, R2=CH3: AJI9561 N O Cl NH Ph HN O CH3 N CH3 H3C F 1 2 N O CH3 Cl N NH 3 N O Cl Cl HN NH O CH3 CH3 L-697,661 N O NH N CH3 N NSC 693638 4 5 N O O N O HN HN O Cl N 9 IC50: 3 nm N N H N HN O N O N O H3C NH O Cl NO2 SB- 334867 JTP-426467 N O O Cl O OH O CH3 Fenoxoprop N O F OH HO ERB-041 N O F3C N O CF3 6 7 8 10 11
Figure 1.16. Biologically active benzoxazol derivatives.
The below section briefly reviews methods to the construction of biologically active benzoxazols rings which involves the condensation of 2-aminophenol with either carboxylic acids or aldehydes by acid or base catalyzed reaction under high temperature conditions or metal induced cyclisation reaction.
39
In 1957, Leavit et. al.68 first synthesized the benzoxazols derivatives using polyphosphoric acid as the condensing agent. The utility of polyphosphoric acid as a remarkably effective condensing agent, particularly for intra- and intermolecular condensations, has been extensively demonstrated in recent years. These condensations generally are carried out by thermal fusion, heating in solvents or in various concentrations of hydrochloric or sulfuric acid, or by heating under pressure in the presence of dilute hydrochloric acid. The catalyst polyphosphoric acid, which also serves as a suitable solvent for the reaction, is equally effective for the condensation of carboxylic acids, amides, esters or nitriles with o-aminophenols to generate the benzoxazol derivatives in good yields in scheme 1.
OH NH2 R2 O N R1 R2 R1 Y + Y = COOH, COOMe, CONH2, CN 250 0C
Scheme 1. Levait’s method of benzoxazol synthesis.
In 1973, Grellmann et. al synthesized the benzoxazols derivatives in photochemical way which involves the reaction of benzylideneanilines substituted in the ortho position of the aniline ring by hydroxyl group in degassed as well as air saturated solution with high quantum yields as shown in scheme 2.69
N
OH O
N light
[O2]
40
In 1997, So et. al. and Kerwin et. al. syntheised the benzoxazol derivatives by the reaction of ortho aminophenols with benzoic acid in presence of phosphorous pentoxide-methane sulfonic acid (P2O5-CH3SO3H), which is a convenient alternative to PPA
reagents under heating conditions and para-toluene sulfonic acid in refluxing benzene conditions in high yields respectively as hown in scheme 3.70
OH NH2 O N R1 R1 COOH + 1. P2O5-CH3SO3H 2. or SO3H
Scheme 3. So et. al. and Kerwin et. al. methods of benzoxazol synthesis.
In 2002, Guzow et.al.have synthesized the same scaffolds using oxidative cyclisation of
azomethines (Schiff’s base) obtained from N-boc-3-nitro-l-tyrosine methyl ester with appropriate aldehyde. They used the three oxidizing agent such as lead tetraacetate, N-bromosuccinimide, and Mitsunobu reagents (triphenylphosphine (TPP) and diisopropylazidodicarboxylate (DIAD) as shown in scheme 4.71