行政院國家科學委員會專題研究計畫 成果報告
腺核甘三磷酸感受性鉀離子通道膜內區域之表現,純化,及結
晶
計畫類別: 個別型計畫 計畫編號: NSC91-2311-B-002-046- 執行期間: 91 年 08 月 01 日至 92 年 07 月 31 日 執行單位: 國立臺灣大學醫學院口腔生物科學研究所 計畫主持人: 樓國隆 報告類型: 精簡報告 處理方式: 本計畫可公開查詢中 華 民 國 92 年 10 月 17 日
Structural Basis of Binding and Inhibition of
Novel Tarantula Toxins in Mammalian
Voltage-Dependent Potassium Channels
Yu-Shuan Shiau1,2, Po-Tsang Huang2,3, Horng-Huei Liou4, Yen-Chywan Liaw5, Yuh-Yuan Shiau2 and Kuo-Long Lou2,3*
1
Institute of Entomology, National Taiwan University; 2Graduate Institute of Oral
Biology, Medical College, National Taiwan University; 3Institute of Biochemistry and
Molecular Biology, College of Medicine, National Taiwan University; 4Department
and Graduate Institute of Pharmacology, Medical College, National Taiwan
University; 5Institute of Molecular Biology, Academia Sinica, Taipei, Republic of
China.
*CORRESPONDING AUTHOR FOOTNOTE:
Mailing Address: Dr. Kuo-Long Lou, GIOB, Chang-Teh Street #1, Taipei 10042,
Republic of China. Tel:+886-2-23123456 ext. 6611/6616/7691.
Fax:+886-2-23820785. E-mail address: [email protected].
ABSTRACT:
Voltage-dependent potassium channel Kv2.1 is widely expressed in mammalian
neurons and was suggested responsible for mediating the delayed rectifier (IK)
currents. Further investigation of the central role of this channel requires the
development of specific pharmacology, for instance, the utilization of spider venom
toxins. Most of these toxins belong to the same structural family with a short peptide
reticulated by disulfide bridges and share a similar mode of action. Hanatoxin 1
(HaTx1) from a Chilean tarantula was one of the earliest discussed tools regarding
this and has been intensively applied to characterize the channel blocking not through
the pore domain. Recently, more related novel toxins from African tarantulas like
heteroscordratoxins (HmTx) and stromatoxin 1 (ScTx1) were isolated and shown to
act as gating modifiers like HaTx on Kv2.1 channels with electrophysiological
recordings. However, further interaction details are unavailable due to the lack of
channels. Therefore, in the present study, we explored structural observation via
molecular docking simulation between toxins and Kv2.1 channels based upon the
solution structures of HaTx1 and a theoretical basis of an individual S3C helical
channel fragment in combination with homology modeling for other novel toxins. Our
results provide precise chemical details for the interactions between these tarantula
toxins and channel, reasonably correlating the previously reported pharmacological
properties to the 3-D structural interpretation. In addition, it is suggested that certain
subtle structural variations on the interaction surface of toxins may discriminate
between the related toxins with different affinities for Kv channels. Evolutionary links
between spider peptide toxins and a “voltage sensor paddles” mechanism most
recently found in the crystal structure of an archaebacterial K+-channel, KvAP, are
also delineated in this paper.
INTRODUCTION
More than 70 mammalian genes encoding potassium channels have been cloned,
in which four different subfamilies (Kv1, Kv2, Kv3 and Kv4) are responsible for
functional voltage-dependent K+ channels (Kv) (1). Kv channels are homotetramers
comprising six putative transmembrane segments termed S1 through S6 in each
K+-selective ion conduction pathway(2-4). The first four transmembrane segments
(S1-S4) of voltage-gated K+ channels do not contribute to the simple pore, and appear
to underlie their unique voltage-sensing capability (5). S4 is an unusual
transmembrane segment that contains a large number of basic residues, which has
been suggested by many studies to be strongly involved in sensing changes in
membrane voltage(6-8).
The physiological role of the Kv channels is now better understood thanks to
the discovery of animal toxins that bind to Kv channels with high affinity and
specificity. The Kv2 subfamily, including Kv2.1, encodes delayed rectifier (IK)
currents (9). Kv2.1 is widely expressed in mammalian neurons and is present in the
soma and proximal dendrites, particularly in of the hippocampus (10). Indirect
evidence suggests a major role for Kv2.1 in the regulation of electrical transmission to
and from the soma (10). However, the specific contribution of Kv2.1 to delayed
rectifier currents in neurons has not been properly investigated until recent years
because of a lack of suitable pharmacological effectors. Regarding this, more
emphasis has been put on the value of spider venoms as unique sources of toxins for
Kv2.1 channels (11). Hanatoxins were first discovered to show specific inhibition on
Kv2.1 through a region other than the pore domain (12,13). Such toxins act on the
therefore change the channel gating towards its depolarization state (11-13). With the
availability of a solution structure for hanatoxin 1 (HaTx1) (16), detailed approaches
have revealed the possibility of conformational change for S3C helix in Kv2.1
interfering with the spatial freedom of S4 translocation during gating upon HaTx1
binding (17,18). All this confirms and emphasizes the importance of an individual S3C
helix in voltage sensing and in gating (19), as well as toxin binding and regulation.
Recently, more novel spider toxins have been identified from the venoms of
African tarantulas Stromatopelma calceata (for stromatoxin, ScTx1) and
Heteroscordra maculata (for heteroscordratoxins, HmTx1; HmTx2) (20). The first
two of these three toxins (ScTx1 and HmTx1) showed strong inhibition on Kv2.1
currents with affinity (20) as high as hanatoxins (11), whereas the last (HmTx2) only
showed minor inhibition (20), despite the high sequence homology and a similar
pattern of disulfide linkage for all three toxins. How do these tarantula toxins interact
with Kv2.1? Do they exert themselves exactly in the same way as hanatoxin does to
bind on S3C helix? Are they using similar structural components for their modes of
action? Due to the lack of channel structures in high resolution for Kv2.1, such
questions can not be answered very easily if one only relies on electrophysiological
analyses. Therefore, in order to obtain structural details with which to elucidate
toxins with Kv2.1 channels. This was based upon the hypothesis that they may all use
the highly conserved individual S3C helix as a binding target for the inhibition of
Kv2.1 currents. Other possible mechanisms were considered as well. Our data
successfully suggest a coincidental structural-functional correlation to explain the
pharmacological properties previously reported (20). Meanwhile, such structural
information may also account for the discrimination in the inhibitory behaviors
between HmTx1 and HmTx2. Our data are very useful in exploring future
experimental approaches to elucidate further mechanism in molecular and atomic
levels. Finally, a structure model derived from KvAP crystal structure (31) for Kv2.1
was generated and simulated with hanatoxin as ligand.
EXPERIMENTAL METHODS
Structure of drk1 channel /S3C fragments.
The human Kv2.1 (drk1) S3C molecule (Val-271 to Gln-284, amino acid
sequence: VTIFLTESNKSVLQ) was constructed via modification from fragment
dictionary with geometry optimized using the consistent valence force field (CVFF)
with Biopolymer module of Insight II software package (Accelrys, USA). Atomic
based on existence of an α-helix were individually regularized by energy
minimization to give reasonable geometry.
Homology models of drk1 channel were built up based upon coordinates from
crystal structure of KvAP channel (PDB ID: 1ORQ and 1ORS) (30-32) (see below).
Essentially, potassium voltage-gated channel subfamily B member 1 (Kv2.1, h-DRK1;
NCB accession number: AAB88808) from human as search sequence was applied for
homology modeling. Voltage-sensor paddle in model structure: drk1, Leu-175 to
Gln-427, compared to Val-275 to Gly-316 in KvAP.
Toxin structures.
The coordinates for HaTx1 structure were obtained from Brookhaven Protein
Databank in pdb file (PDB ID number 1D1H; solution structure by NMR method)
(16). Structures for ScTx1 and HmTx1 were created via homology modeling based on
the HaTx1 structure as template. HmTx2 was created by following the same
procedure with ω-grammotoxin SIA (GsTxSIA, PDB ID: 1KOZ_A) (33) as template
according to their BLAST search results (see following section).
Search for templates. BLAST algorithm was employed to search in PDB the protein
segments whose sequences are similar to those of tarantula toxins and whose
structures can serve as viable structural templates. The crystal or solution structures of
such template that showed the highest scores in the sequence alignments were chosen
for the determination of structural conserved regions (SCRs). The residues of toxins
used for model building are according to their paired sequence compared to the
template sequence.
Paired sequence alignment. GCG program was used to determine the equivalent
residues. The residue regions of template toxins represented as continuous lines
dominantly observed from GCG results were employed as appropriate regions, and
the corresponding fragments in the target toxins were chosen for alignment
individually. The amino acid sequences of these novel toxins were then included in
the multiple sequence alignment (21) of the appropriate template regions to specify
the residue numbers for model building.
Model building and residue side-chain simulation. Modeling by homology was
performed essentially following the procedures previously described (21-25). Briefly,
the residue fragments of novel toxins were chosen according to the results from GCG
paired sequence alignment. They were then superimposed onto the structure
This generated the secondary structure and relative position of the definite structural
elements in the chosen residue fragments of individual target toxin models. Junctions
between the secondary structural elements were individually regularized by energy
minimization to give reasonable geometry. Further hydrophobic/-philic interactions
between residue side-chains were performed and obtained with molecular dynamics
and simulated annealing. All the calculations and structure manipulations were
performed with the Discover/Insight II molecular simulation and modeling programs
(from Accelrys Inc., San Diego, CA, USA; 950 release) on Silicon Graphics
Octane/SSE and O2/R12000 workstations as well as O-300 server.
Docking Simulation.
Determination of starting orientations. In principle, three criteria were used to
determine the starting positions: stereochemistry, side-chain charge distribution, and
previous structural information (16-20). Inappropriate possibilities have been
immediately excluded when definitely unreasonable combinations of alignment for
docking were observed. Uncertain orientations were reserved and submitted for
docking calculation to allow the computational results to perform the screening.
Calculation for the energies. Upon docking, the total energies of electrostatic
toxins and S3C (binding models) were compared. Each run was composed of 500
cycles of simulated annealing and 500,000 steps of accepted/rejected configurations.
The values of all other default parameters were used. The alignment between
docked and undocked molecules was performed by manually fitting the atomic
coordinates of groups of residues that may be involved in the conserved interaction
(17,19). Briefly, three-dimensional (3D) surfaces of the binding site enclose the most
active members (after appropriate alignment) of the starting set of molecules. Note
that errors in alignment can lead to incorrect, poorly predictive receptor surface
models. This problem was overcome by using information obtained from previously
related functional data (19,20). The surface was generated from a "shape field", in
which the atomic coordinates of the contributing models were used to compute field
values on each point of a 3D grid using a van der Waals function (26-28). A solvation
energy correction term and the electrostatic charge complementarity’s method were
used for energy evaluation (26,27). And with that, the solvation energy correction
term is a penalty function that attempts to account for the loss of solvation energy
when polar atoms are forced into hydrophobic regions of the receptor surface. All the
calculations and structure manipulations described above were performed with the
(Accelrys, San Diego, CA, USA; 950 release) displayed on a Silicon Graphics
O2/R12000 and Octane/SSE workstations.
THEORETICAL BASIS
Molecular and docking simulation has since more than a decade been
contributing to precise interaction details in atomic levels, provided an appropriate
force field is applied. However, the accurate structural information will be ensured
and reliable only when the starting orientations and positions are correctly determined.
For such reason, distinguished strategies to perform simulations are undoubtedly
crucial and absolutely required to be established before the docking procedure
commences. Sufficient constraints based on the previous functional properties will
assist this to a fairly large extent. In our case, as also described in the methods section,
the hydrophobic patch and the charged belt of these toxins are sufficient as criteria in
both stereochemistry and previous functional data. Thus all the rest is to observe and
make decisions for the best combination possibilities within a restricted area by
rotation and/or translocation of the receptor/ligand molecules, especially for residue
important starting point, the other pivotal concept for how to take free body of the
interaction system is described in the next paragraph.
Many tarantula toxins as hanatoxins bind and inhibit mammalian Kv channels
not through the pore domain. The carboxyl terminal residues of the transmembrane
segment S3 are highly conserved in voltage-dependent potassium channels and have
been suggested for such target of binding sites. However, due to the lack of
high-resolution structure for voltage-sensing domains before in previous stage,
docking simulation for this kind of toxin-channel interaction was thought to be either
impossible or with less significance. Recently, mutagenesis scanning and helical
analyses (19,29) as well as other previous progresses, which led to the observation
that S3C should be regarded as an individual helical fragment, have provided us the
opportunity to consider this interaction system as a free body, and therefore the
molecular simulation could be performed within this space. Furthermore, simulation
results would provide compensation through comparison of the energy states even if
this helix has distortion in three dimensions. We have tested this idea with a system
containing hanatoxin and drk1/shaker S3C (17,18). Chemical details of interaction
correlated successfully in both structural and functional aspects (17,19). Spatial
orientation of S3C helix was appropriately discussed (18) and even structural change
analyzed through our simulation results (17,18). Moreover, existence of S3C helix as
individual fragment seemed to have been strongly confirmed as a result of low energy
for binding. Thus, the expansion of such a concept into the interactions between
various members in tarantula toxin family and mammalian channels in this study is
supposed to be quite rational. Meanwhile, the related algorithms and functions applied
in the simulation procedures are described in detail in the Experimentals section.
The crystal structures of a voltage-gated potassium channel from thermophilic
archaebacteria (30) have been quite recently determined to atomic resolutions (31). A
mechanism for gating based upon “voltage-sensor paddles” was proposed through the
structural observation (31,32). This mechanism gave new directions and possibilities
for further investigations by considering the toxins and entire “paddle” together as an
interaction free body.
RESULTS AND DISCUSSIONS
Structure of novel tarantula toxins.
In order to perform the docking simulation, the atomic coordinates of toxin
structures in three dimensions are required to be assigned as receptor or ligand
homology modeling for HmTx1 and ScTx1. The structures of template toxin and
targets are compared in Fig. 1.
Fig. 1a
Fig. 1b In addition to the crucial residues that may be involved in the toxin-channel binding
illustrated in Fig. 1a, the charge distributions of HaTx1, HmTx1 and ScTx1 are
represented with the electrostatic surface in Fig. 1b. Compared with HaTx1, the
hydrophobic patch is apparently well conserved in HmTx1 and ScTx1. It is also
fascinating to note that residues tending to form a charged belt, as seen in HaTx1, are
also flanking the corresponding hydrophobic patch exactly in the same way and as
HmTx2 was not suggested for the similar modeling procedure via using HaTx1
as template. Instead, BLAST results revealed a preference for GsTxSIA (33) as
template. Comparing Fig. 1a with Fig. 2a, a major difference can be easily observed
between the two sets of models.
Fig. 2a
Fig. 2b
In the HmTx2 structure, a longer protruding loop containing residues 25-31 was
observed (Figs. 2a-b). This fact leads to two significant consequences: (i) HmTx2 was
suggested to be an inappropriate target for template HaTx1 in homology modeling; (ii)
Apparent disturbance in binding for inhibition on Kv2.1 gating currents may occur.
We will come back to this point in more details later in this section.
In Fig. 3, the most reasonable docking results between novel toxins (ScTx1,
HmTx1, and HmTx2) and channels are illustrated in comparison with the
HaTx1-Kv2.1 S3C interaction. Total binding energies are listed in Table 1. All the
first three complexes (Figs.3a-3c: HaTx1-, HmTx1-, and ScTx1-Kv2.1 S3C,
respectively) demonstrate a very similar binding mode: the residues on the
hydrophobic patch of the toxin (e.g., Tyr-27 in Fig. 3a) form hydrophobic interactions
with non-polar residues from Kv2.1 S3C, whereas polar residues on S3C form
hydrophilic interactions with residues from the charged belt flanking the patch on
toxin surface. In comparison with the interaction details in Table 1, it is interesting to
note that the magnitudes of inhibition (some are represented as IC50) by various toxins
are in line with the binding energies, which are further explained by the strength of
interaction categorized as number of bonds/interactions formed by residues from both
sides of participating molecules (Table 1). HaTx1 and ScTx1 both show very strong
inhibition on Kv2.1 currents, therefore a reasonable docking for them should be
implied from lower total binding energies, which have been successfully and clearly
Fig. 3a
Fig. 3b
Fig. 3d Activity of spider toxin against drk1 channel as IC50 (nM) or percent-age of current inhibition (20)
Van der Waals / Electro-static / Total energies (Kcal/mol) after
docking Number of Salt-bridges and H-bonds Number of resi-dues forming hy-drophobic inter-actions ScTx1- drk1 S3C 12.7 322.05 / -219.183 / 102.87 6 15 HaTx1- drk1 S3C 42 268.85 / -35.71 / 233.14 7 14 HmTx1-drk1 S3C 23% (100nM) 402.27 / -65.20 / 337.07 5 10 HmTx2-drk1 S3C 18% (300nM) 1343.16 / -419.74 / 923.421 4 8 Table 1 Interaction of HmTx2.
The extraordinarily weaker interactions observed for HmTx2 can be
existence of a longer protruding loop 25-31, compared to HaTx1, ScTx1 and HmTx1,
makes HmTx2 itself very distinct in shape from others (Figs. 1 & 2). When the toxins
are bound onto the channel by interacting with S3C fragment, this part may become an
obstacle for HmTx2 to approach towards S3C. In other words, this loop will form
steric hindrance to prevent a tight binding while approaching the channel. From the
complex structure of HmTx2 and Kv2.1 S3C depicted in Fig. 3d, one can easily find
that the longer 25-31 loop interferes with the binding of the two molecules not only
through steric hindrance regarding the main bodies, but also through perturbation of
residue types for detailed interaction. And therefore, the docking energies will
certainly not favor HmTx2 with binding to Kv2.1 (Table 1). This concept can be
further emphasized by the reduced number of residues involved in forming
hydrophobic interactions compared to those for HmTx1 and ScTx1. However, the
electrostatic energy is very low for HmTx2 regarding binding with Kv2.1 S3C, despite
that it can be compensated by the extremely high van der Waals’ energy and thus
produced much higher total energies. This seems to contradict the concept described
previously. Therefore, we examined the complex structure of HmTx2-S3c in more
details.
If one looks into the complex structure in Fig. 3d even more carefully, it is not
relationship for residue side chains around the hydrophobic patch (Fig. 3d). This
brings the van der Waals’ energy to the very high value listed in Table 1. Another
disturbing factor affecting hydrophobic interactions can be observed in the Loop
25-31 for residues adjacent to Lys-25. At this position, S3C provides several polar or
charged residues to form H-bonds or salt-bridges with polar residues from toxins for
HaTx1, HmTx1 and ScTx1. However, in HmTx2, a few aromatic residues are located
nearby, and as a consequence, van der Waals’ energy could be brought to an even
higher value (Fig.3d). The second feature is with respect to the “high-positional” salt
bridge (Lys-25 from HmTx2 and Gln-286 from drk1 S3C). This unusual interaction,
due to the longer loop protruding from the Lys-25 side chain on tip of loop to a height
about to form interaction with Gln-286 from almost the C-terminal end of S3C has not
been seen in all other toxin-channel complexes (Figs. 3a-c). It might provide a
rational explanation for such an unexpected low electrostatic energy listed in Table 1.
Considering as a whole, the docking interaction between HmTx2 and Kv2.1 should be
inappropriate or unnatural, although such orientation was the best possibility we could
find to carry out simulation for this combination. The sequence of HmTx2 seems not
to be designed by nature to inhibit Kv2.1 through binding on S3C.
This discussion provides an explanation for the highest energy result in Table
also imply the possibility for a totally different orientation for binding? Conservation
of the hydrophobic patch and the charged belt in HmTx2 seems to suggest a similar
binding mode as observed in all the other tarantula toxins acting on S3C. On the other
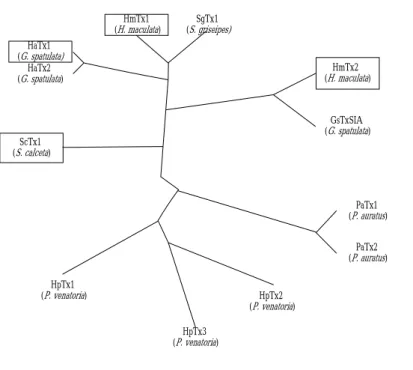
hand, evolutionary links delineated in Fig. 4 might support the point that a different
kind of channel serves as binding target for HmTx2, with only subtle structural
changes on S3C. And this may reflect the adaptation of toxins in evolution for various
environments, based on the relationship in the phylogenic tree (more close to
GsTxSIA, Fig. 4b). This assumption remains to be verified through more experiments
in electrophysiology. Nevertheless, the close relation between HaTx, ScTx, and
HmTx1 illustrated in Fig. 4, rather than with HmTx2, indeed accounts for their
pharmacological properties (20) and the structural observation described in this study.
HaTx2 (G. spatulata) HaTx1 (G. spatulata) HmTx1 (H. maculata) SgTx1 (S. griseipes) ScTx1 (S. calceta) GsTxSIA (G. spatulata) HmTx2 (H. maculata) PaTx1 (P. auratus) PaTx2 (P. auratus) HpTx2 (P. venatoria) HpTx3 (P. venatoria) HpTx1 (P. venatoria) Fig. 4b Evolutionary links.
Fig. 4a represents the sequence comparison for related tarantula toxins.
Apparently the typical structural arrangement for such small peptide toxins is strictly
and extensively conserved throughout widely spread species as toxin sources based on
the disulphide linkage. This has been known for the Cysteine-rich proteins
superfamily (34-38). In addition, the charged/polar residues required for the charge
belt and aromatic/hydrophobic ones for the hydrophobic patch are also highly
conserved in all the related spider toxins (Fig. 4a). Using ClustalW (39) by manual
and Bioedit (40), a phylogenic tree, based on comparison of amino acid sequences for
such related tarantula toxins delineated in Fig. 4a, can be generated (Fig. 4b). HaTx1,
neighboring branching between ScTx1 and the others. Meanwhile, HmTx2 deposits
itself on the other side of the tree, even though it comes from the same venom source
as HmTx1 does. This may probably conduct the structural role of a molecule into its
evolutionary necessity: even a subtle change in structure (here, a small part on the
interaction surface) could presumably suffice to satisfy the demands of selection.
Speculations for putative mechanism.
Previously, we have proposed a hypothesis describing a possible molecular
mechanism for how hanatoxin may affect the gating behavior of Kv2.1 channels via
binding on S3C fragment (17,18). In this hypothesis, we derived a C-terminal helical
movement of drk1 S3C (17,18) through observed conformational change (17) and
binding pocket (crevice) analysis (24) from simulation data, reducing the spatial
freedom for S4 translocation (18), and therefore a more depolarized potential for the
open gate may arise as anticipated (11,20). However, most recently, Jiang and
MacKinnon disclosed a crystal structure and therefore the principle of voltage gating
of an archaebacterial K-channel, KvAP was proposed (30-32). This has a significant
impact on our hypothesis. The major challenge was based on the observation that S3b
(i.e., S3C) and S4 (or S4N) should “translocate” as an unity upon voltage-sensing and
From such a point of view, a short S3-S4 linker will be absolutely required,
considering the free energy for an en bloc movement. This is true for the situation in
KvAP (30,31), but not necessarily for mammalian channels, as suggested by their
much more complicated kinetics and regulatory behaviors (18,41). To compare these
two different ideas, we have carried out homology structural modeling and
re-performed the docking simulation of drk1 complexed with hanatoxin (Fig. 5).
Although it is still difficult to have successful interpretation of how flexible this linker
is during activation, we found a very interesting structural essential in the S3C helix
(Fig. 5c): the helical arrangement becomes a random coil after the mid-point of S3C.
In addition, this feature seems not to interfere with the toxin binding (Fig. 5a, b). This
new finding is based on energy minimization results from both modeling and docking
procedures, and the reasons could be possibly comprehended in the following ways.
First, it may be only due to the energy minimization similar as in a flexible loop. We
would not be satisfied by such simple explanation. Second, it can reflect a
compromised transitional structure preceding the S3-S4 linker in a more dynamic way,
which was thought to be extremely crucial in the regulation of activation kinetics (41).
Such a point of view may partially support our previously observed conformational
change (17,18) in a more preserved manner, because a longer S3-S4 linker either
regulation and thus allow a certain range of motions. We would not speculate
further the role of this looser structural arrangement with such limited data.
Nevertheless, all the issues discussed above may provide hints that there is still quite a
large space for researchers to work on regarding the regulatory roles of the area
around the vicinity of S3C and S4N. In other words, it is not an ending, but on the
contrary, just a fresh beginning for further investigations after the unraveling of this
first crystal structure of an archaebacterial Kv channel containing the whole
voltage-sensing domains. And for tarantula toxins, they will be even more useful in
the future study of the related gating phenomena they have shown to us in the past
decade.
Fig. 5b
Fig. 5c
Conclusion. Based upon an in silico study, we have provided a structural basis for
the inhibition and binding of mammalian Kv channels by novel tarantula toxins,
successfully correlating their previously reported pharmacological properties to a 3-D
structural-functional interpretation. By combining this structural model with
evolutionary considerations, discrimination between subtypes of toxins due to subtle
structure changes in the interaction surface can be proposed. Finally, a
“voltage-sensor paddles” mechanism deduced from the crystal structure of
archaebacterial channel has been used as reference for further consideration on
ACKNOWLEDGEMENT. The authors thank Dr. Wang Pao-Hsiang at Dept. of
Foreign Languages and Literature in NTU with deepest sincerity for his careful
reading through the manuscript and his useful suggestions in further correction of the
sentences. This work was supported in part by the Taiwanese NSC fundings
92-2914-I-002-077-A1/91-2914-I-002-191-A1 and the Grants 912C012/902A006 and
92G017 from Teaching Improvements Projects Package by AOE, R.O.C. for LKL.
FIGURE CAPTIONS
Figure 1. Comparison for overall structures of novel tarantula toxins: HaTx1,
HmTx1, and ScTx1. (a) Ribbon diagrams for template structure of HaTx1 and
homology structure models of HmTx1 and ScTx1. Crucial residue side chains that
may involve in the interaction with channels are emphasized with residue number
indicated. Residues in HmTx1 and ScTx1 structures not appearing as conservative
substitutions are labeled in yellow (otherwise in white). (b) Electrostatic surface of
circles in black. On the surface of molecules, red corresponds to an electrostatic
potential of < -4.0 kBT/e, white to 0 kBT/e, and blue for >3.0 kBT/e.
Figure 2. Comparison of overall structures of novel tarantula toxins: HmTx2
and GsTxSIA. As shown in Fig.1, the schematic diagrams for both template
GsTxSIA and target HmTx2 are illustrated in (a) with crucial residue side chains
opened and residue types, number emphasized; (b) Electrostatic surface potentials
represented: red for < -4.0 kBT/e, white for 0 kBT/e, and blue for > 13.0 kBT/e.
Figure 3. Stereo views for complex structures of various tarantula toxins and
drk1 S3C from docking simulation results. (a) HaTx1-S3C (cyan), (b) HmTx1-S3C
(dark blue), (c) ScTx1-S3C (magenta), and (d) HmTx2-S3C (violet). Kv2.1 S3C is
colored in orange. Crucial residue side chains involved in the interactions are opened
and drawn in colors according to their atom types with residue number indicated. It is
important to note that in Fig. 3d, a distinct longer Loop 25-31 in HmTx2 displays an
unusual structural characteristics regarding steric hindrance for binding (see text for
more details).
Figure 4. Evolutionary links between related tarantula toxins. (a) Sequence
alignments. (b) Phylogenic tree. Software used to produce these figures has been
Figure 5. Further study of HaTx1-Kv2.1 interaction based on crystal structural
information from archaebacterial KvAP channel. (a) Overview of Kv2.1 (drk1)
structure in complex with HaTx1. Homology structures for drk1 were generated as
described in Experimental section. Red cylinders represent the α-helices. Connection
loops are built up with yellow ribbons with energy minimization to give reasonable
geometry. HaTx1 molecule is shown in blue. (b) Enlarged view for drk1
voltage-sensor paddle in ligand with HaTx1. (c) Comparison of voltage sensors
between KvAP and Kv2.1 (drk1). Close attention is suggested for the different
structural arrangement in the C-terminal part of S3C (or S3b) (see more details in text).
Residues applied: Pro-99 to Ala-140 in KvAP, whereas in Kv2.1, Val-275 to Gly-316.
Besides, gap in modeling was suggested between Gly-114 and Leu-115 in KvAP for
S3-S4 linker in drk1 replaced by residues between Leu-287 and Val-295. All the
structural manipulations were performed with Insight II software package as
described in Experimental section.
TABLES.
Table 1. Comparison of the magnitudes of inhibition by novel tarantula toxins on
drk1 currents with the structural features (energies, bonds, etc.) after docking
REFERENCES
(1) Jan, L. Y., and Jan. Y. N. (1997) Cloned potassium channels from eukaryotes and
prokaryotes. Annu. Rev. Neurosci. 20, 91-123.
(2) Liman, E. R., Tytgat, J., and Hess, P. (1992) Subunit stoichiometry of a
mammalian K+ channel determined by construction of multimeric cDNAs. Neuron
9, 861-871.
(3) Heginbotham, L., Lu, Z., Abramson, T., and MacKinnon, R. (1994) Mutations in
the K+ channel signature sequence. Biophys. J. 66, 1061-1067.
(4) Ranganathan, R., Lewis, J. H., and MacKinnon, R. (1996) Spatial localization of
the K+ channel selectivity filter by mutant cycle-based structural analysis. Neuron
16, 131-139.
(5) Armstrong, C. M. and Hille, B. (1998) Voltage-gated ion channels and electrical
(6) Bezanilla, F., Perozo, E., Papazian, D. M., and Stefani, E. (1996) Molecular basis
of gating charge immobilization in shaker potassium channels. Science 254,
679-83.
(7) Cha, A., Snyder, G. E., Selvin, P. R., and Bezanilla, F. (1999) Atomic scale
movement of the voltage-sensing region in a potassium channel measured via
spectroscopy. Nature 402, 809-813.
(8) Bezanilla, F. (2000) The voltage sensor in voltage-dependent ion channels.
Physiol. Rev. 80, 555-592.
(9) Frech, G. C., van Dongen, A. M., Schuster, G., Brown, A. M., and Joho, R. H.
(1989) A novel potassium channel with delayed rectifier properties isolated from
rat brain by expression cloning. Nature (Lond) 340, 642-645.
(10) Murakoshi, H. and Trimmer, J. S. (1999) Identification of the Kv2.1 K+ channels
as a major component of the delayed rectifier K+ currents in rat hippocampal
neurons. J. Neuronsci. 19, 1728-1735.
(11) Swartz, K. J., and Mackinnon, R. (1995) An inhibitor of the Kv2.1 potassium
(12) Swartz, K. J., and Mackinnon, R. (1997) Hanatoxin modifies the gating of a
voltage-dependent K+ channel through multiple binding sites. Neuron 18,
665-673.
(13) Swartz, K. J., and Mackinnon, R. (1997) Mapping the receptor site for hanatoxin,
a gating modifier of voltage-dependent K+ channels. Neuron 18, 675-682.
(14) Rogers, J. C., Qu, Y., Tanada, T. N., Scheuer, T., and Catterall, W. A. (1996)
Molecular determinants of high affinity binding of alpha-scorpion toxin and sea
anemone toxin in the S3-S4 extracellular loop in domain IV of the Na+ channel
alpha subunit. J. Biol. Chem. 271, 15950-15962.
(15) Winterfield, J. R. and Swartz, K. J. (2000) A hot spot for the interaction of
gating modifier toxins with voltage-dependent ion channels. J. Gen. Physiol. 116,
637-644.
(16) Takahashi, H., Kim, J. I., Min, H. J., Sato, K., Swartz, K. J., and Shimada, I.
(2000) Solution structure of Hanatoxin1, a gating modifier of voltage-dependent
K+ channels: common surface features of gating modifier toxins. J. Mol. Biol. 297,
771-780.
(17) Huang, P.-T., Liou, H.-H., Lin, T.-B., Shiau, Y.-S., Spatz, H.-Ch., Chen, T.-Y.,
hanatoxin binding on the carboxyl terminus of S3 segment in voltage-gated
potassium channel Kv2.1. Recept. Channels 8, 79-85.
(18) Lou, K.-L., Huang, P.-T., Shiau, Y.-S., French, R. J., Liaw, Y.-C., Shiau, Y.-Y.,
and Liou, H.-H. (2003) A possible molecular mechanism of hanatoxin
binding-modified gating in voltage-gated potassium channels. J. Mol. Recognit.
(in press).
(19) Li-Smerin, Y., and Swartz, K. J. (2001) Helical structure of the COOH terminus
of S3 and its contribution to the gating modifier toxin receptor in voltage-gated
ion channels. J. Gen. Physiol. 117, 205-218.
(20) Escoubas, P., Diochot, S., Célérier, M.-L., Nakajima, T., and Lazdunski, M.
(2002) Novel tarantula toxins for subtypes of voltage-dependent potassium
channels in the Kv2 and Kv4 subfamilies. Mol. Pharmacol. 62, 48-57.
(21) Voorhorst, W. G., Warner, A., de Vos, W. M., and Siezen, R. J. (1997)
Homology modelling of two subtilisin-like proteases from the hyperthermophilic
archaea Pyrococcus furiosus and Thermococcus stetteri. Prot. Engineer. 10,
(22) Tsai, Y.-W., Chia, J.-S., Shiau, Y.-Y., Chou, H.-C., Liaw, Y.-C., and Lou, K.-L.
(2000) Three dimensional modeling of the catalytic domain of S. mutans
glucosyltransferase B. FEMS Microbiol. Lett. 188, 75-79.
(23) Lou, K.-L., Chou, H.-C., Tsai, Y.-W., Shiau, Y.-S., Huang, P.-T., Chen, T.-Y.,
Shiau, Y.-Y., and French, R. J. (2001) Involvement of a novel C-terminal kinase
domain of Kir6.2 subunit in K-ATP channel rundown reactivation. J. Mol. Model.
7, 20-25.
(24) Lou, K.-L., Huang, P.-T., Shiau, Y.-S. and Shiau, Y.-Y. (2002) Molecular
determinants of hanatoxin binding in voltage-gated potassium channel drk1. J.
Mol. Recognit. 15, 175-179.
(
(2255)) Chia, J.-S., Shiau, Y.-S., Huang, P.-T., Shiau, Y.-Y., Lou, K.-L. (2003)
Structural analysis of the influence of enzyme activity by surface peptide Gtf-P1
in S. mutans glucosyltransferase GtfC. J. Mol. Model. DOI
10.1007/s00894-003-0121-5.
(26) Hahn, M. (1995) Receptor surface models. 1. Definition and constructions. J.
Med. Chem. 38, 2080-2090.
(27) Hahn, M. and Rogers, D. (1995) Receptor surface models. 2. Application to
(28) Costantino, G., Macchiarulo, A., Camaioni, E., and Pellicciari, R. (2001)
Modeling of poly-(ADP-ribose)-polymerase (PARP) inhibitors: docking of
ligands and quantitative structure-activity relationship analysis. J. Med. Chem. 44,
3786-3794.
(29) Li-Smerin, Y. and Swartz, K. J. (1998) Gating modifier toxins reveal a
conserved structural motif in voltage-gated Ca2+ and K+ channels. Proc. Natl.
Acad. Sci. USA 95, 8585-8589.
(30) Ruta, V., Jiang, Y., Lee, A., Chen, J., and MacKinnon, R. (2003) Functional
analysis of an archaebacterial voltage-dependent K+ channel. Nature 422,
180-185.
(31) Jiang, Y., Lee, A., Chen, J., Ruta, V., Cadene, M., Chait, B. T., and MacKinnon,
R. (2003) X-ray structure of a voltage-dependent K+ channel. Nature 423, 33-41.
(32) Jiang, Y., Ruta, V., Chen, J., Lee, A., and MacKinnon, R. (2003) The principle
of gating charge movement in a voltage-dependent K+ channel. Nature 423,
42-48.
(33) Takeuchi, K., Park, E. J., Lee, C. W., Kim, J. I., Takahashi, H., Swartz, K. J.,
and Shimada, I. (2002) Solution structure of ω-Grammotoxin SIA, a gating
(34) Terras, F. R., Eggermont, K., Kovaleva, V., Raikhel, N. V., Osborn, R. W.,
Kester, A., Rees, S. B., Torrekens, S., van Leuven, F., and Vanderleyden, J. (1995)
Small cysteine-rich antifungal proteins from radish: their role in host defense.
Plant Cell 7, 573-588.
(35) Froy, O. and Gurevitz, M. (1998) Membrane potential modulators: a thread of
scarlet from plants to humans. FASEB J. 12, 1793-1796.
(36) Hwa, V., Oh, Y., and Rosenfeld, R. G. (1999) Insulin-like growth factor binding
proteins: a proposed superfamily. Acta Paediatrica Suppl. 88, 37-45.
(37) Boisbouvier, J., Blackledge, M., Sollier, A., and Marion, D. (2000)
Simultaneous determination of disulphide bridge topology and three-dimensional
structure using ambiguous intersulphur distance restraints: possibilities and
limitations. J. Biomol. NMR 16, 197-208.
(38) Fariselli, P. and Casadio, R. (2001) Prediction of disulfide connectivity in
proteins. Bioinformatics 17, 957-964.
(39) Higgins, D., Thompson, J., Gibson, T., Thompson, J. D., Higgins, D. G., and
Gibson, T. J. (1994) CLUSTAL W: improving the sensitivity of progressive
multiple sequence alignment through sequence weighting, position-specific gap
(40) Hall, T. A. (1999) BioEdiit: auser-friendly biological sequence alignment editor
and analysis program for Window 95/98/NT. Nucleic Acids Symposium Series 41,
95-98.
(41) Gonzalez, C., Roseman, E., Bezanilla, F., Alvarez, O., and Latorre, R. (2000)
Modulation of the shaker K+ channel gating kinetics by the S3-S4 linker. J. Gen.