國 立 交 通 大 學
生
物 科 技 學 院
生
物 科 技 學 系
碩
士 論 文
血管收縮素
II 與血管收縮素 1-7 於人類心臟纖維母細胞中對
心臟血管收縮素轉化酶
II 的表現調節
Interplay of Angiotensin II and Angiotensin 1-7 in the
Modulation of Cardiac Angiotensin-Converting Enzyme II of
Human Cardiofibroblasts
研
究 生:温証皓
指導教授:林志生
博士
血管收縮素
II 與血管收縮素 1-7 於人類心臟纖維母細胞中對心臟
血管收縮素轉化酶
II 的表現調節
Interplay of Angiotensin II and Angiotensin 1-7 in the Modulation
of Cardiac Angiotensin-Converting Enzyme II of Human
Cardiofibroblasts
研 究 生:温証皓 Student:Cheng-Hao Wen
指導教授:林志生 Advisor:Chih-Sheng Lin Ph.D.
國 立 交 通 大 學
生 物 科 技 學 系
碩 士 論 文
A ThesisSubmitted to Department of Biological Science and Technology College of Biological Science and Technology
National Chiao Tung University in partial Fulfillment of the Requirements
for the Degree of Master in
Biological Science and Technology July 2009
Hsinchu, Taiwan, Republic of China
Acknowledgement
當碩士論文寫到此頁,代表我的碩士生涯要正式落幕了,二年的研究所時光看似漫 長,實則如過眼雲煙轉瞬即逝。人生的美好不在於延續生命的永恆不滅,而在於瞬間璀 璨所散發出來的繽紛光芒,雖然二年時間匆匆而逝,但已在我的人生之中留下許多不可 磨滅的奪目光采。很慶幸我可以有這個機會進到這間實驗室,在進到研究所生活之前, 本來一直對研究所抱持著會很艱辛、很歹命的心理準備。不過事實卻不然,因為我遇到 了一群很親切的實驗室夥伴,好多新朋友,還有身兼系主任又對我無私付出的指導教 授,讓我在研究所生活充滿美好回憶。 首先要感謝的人是心臟組的超級leader 俊旭學長!! 學長專業又不失風趣的實驗討 論,以及有問必答的人體百科全書,讓我對於實驗設計有更多的sense,才能順利的完 成這份論文。當然還要感謝已經畢業且幸福甜蜜的建龍學長,學長真的很搞笑,我也不 會忘記那段水深火熱、昏天黑地照confocal,甚至必須靠笑話來醒腦的日子。還要感謝 紹全學長,除了訓練我穩健抓老鼠的技術,對實驗追求完美、近乎苛求的標準,也讓我 實驗的誤差降到最低。宜貞學姊,在妳稚嫩的臉頰上永遠看不出歲月的痕跡,祝妳工作 順利,遇到好的上司!! 再來還要謝謝思豪學長,實驗室永遠的大學長,有著極高的專業及實驗室管理能 力,也是實驗室裡時速160 的紀錄保持者。阿關學長,是實驗室的溫柔漢,凡事都以朋 友為重,有你實驗室多了一個熱心的傾聽者,還有記得把小圓教會,叫他不要畏懼我對 她的寵愛!! 筱晶學姊,人稱馬來妹,是本實驗室的外交官,也是博愛校區外籍生的宿舍 長,很盡責的妳每天都要處理眾多投訴信,也祝妳如期畢業、有個好婚姻。豆豆學長, 這是學妹們對你細膩的稱呼,我還是叫你斗宅(豆仔)好了,是一個才高八斗的宅科新貴, 舉凡電腦、電器、電動,只要是電字輩的通通一把罩,也是我的良師益友!! 騙鬼學長, 為人熱心公益,對學長姊彬彬有禮,對學弟妹則疼愛有加,在球場上熱情揮灑的魅力無 人能敵。 還有我的同學千雅,妳真的是實驗室永遠的助理!!有妳我們才能準時吃飯,有妳挺 胸站出來我們才不會被外人占便宜,少了妳我們說話都大聲不起來了。榕均,我們的實 驗室一哥,也是跑zymo 膠達人,帥氣又正點的外表,簡直就是男女通吃啦,預祝妳之 後工作順利。達達,跟我相依為命的唯二男生,為人隨性,人小志氣高,未來可是生質柴油界的扛壩子。 其實在我碩二這年,特別感謝的人就是子慧,妳是個細心又乖巧的學妹,雖然妳已 經死會,而且我也不敢活標,還是很謝謝妳於實驗上的幫忙與陪伴,妳是很棒的實驗小 助手,期待收到妳的紅色炸彈!! 還有小媽,人如其名,善良體貼也很照顧人,也是陪了 我五年的學妹更是我的好朋友,很感謝妳在生活上和大大小小事情的幫忙,也希望妳實 驗能鴻圖大展,keep in touch。小妤,也是從大學就認識的學妹,但是因為一些特殊原 因,已經變成小白了,我們的系女籃也要靠妳了。小韓,曾經也是我的小組員之一,很 有自己想法,你跟阿大湊在一起就一整個好笑,也祝福妳實驗順利進行。 還要感謝大學部的學弟妹,竣瑋、怡萱和祥兒,你們都是心臟組的優秀小組員,謝 謝你們在實驗上的幫忙,你們的前途不可限量,期待聽到你們在學術上的好成就,或許 將來還有機會可以再一起共事。修兆跟唯婷,你們是實驗室的搞笑二人組也是出團的固 定班底,有你們在實驗室總是充滿歡樂的氣氛。唯婷妳跟宜貞喊「學長~~~」的經典語 錄,我也一定不會忘記的。欣儒和政庭,看你們目前實驗也都有一定的成果,相信你們 會申請上很好的研究所,加油啦。最後還要感謝dg,雖然碩二才認識你,不過我們的 交情你也知道,謝謝你平時提供我免費的受話筒和出氣筒,讓我得以紓解苦悶,專心致 志於研究。還有建宇、阿大、靜敏、Kerry、Linda,謝謝你們帶來這麼多的歡笑。要感 謝的人真的很多,族繁不及備載,如有被遺忘的朋友在此亦一併感謝。 當然,最感謝的還是我的指導教授─ 林志生教授。很榮幸可以跟隨老師您在心血 管疾病的專長直接臨摹受教,從老師身上學習到的不只是實驗的想法與設計,有更多寶 貴的社會歷練以及處世態度是我們平常所學不到的,尤其是老師在接下系主任後所散發 的責任感與對系上的抱負,都是值得我學習的表率。也謝謝老師百忙中在研究上對我的 耐心指導與包容,更由衷感謝老師對於我的極力提拔,因為有您的指導與幫助,才造就 了現今稍有成就的我。在老師的訓練與帶領之下,真的教了我很多,相信未來就算遭遇 任何困境,都能化險為夷、迎刃而解。 我也非常感謝 吳介信院長、毛仁淡講座教授能擔任我的口試委員。吳教授於大學 時期就很照顧我,也很栽培我,這份情我不會忘記。毛教授的課程更精進我的英語報告 能力,也訓練我許多思考問題的能力。再次謝謝各位口委對於我碩士論文的建議與指 教,讓我順利通過口試。
最後,將此論文獻給我最親愛的家人,謝謝你們在背後默默支持我,供給我讀書與 生活所需,以我為榮,使我能無後顧之憂地順利完成學業。還要感謝我的超級好朋友─ 拍狼,認識你之後,除了笑容變多,研究所生活也更多采多姿。再次感謝所有曾經幫助 過我的人,謝謝你們!! 因為有你們,一切都值得了!! 僅以此篇論文表達內心最誠摯的 感激。 温証皓 謹誌 交通大學生物科技學系碩士班 中華民國九十八年七月
血管收縮素
II 與血管收縮素 1-7 於人類心臟纖維母細胞中
對心臟血管收縮素轉化酶
II 的表現調節
研究生:温証皓 指導教授:林志生 博士
國 立 交 通 大 學
生 物 科 技 學 院
生 物 科 技 學 系 碩 士 班
中
文 摘 要
腎素-血管收縮素系統 (renin-angiotensin system, RAS) 中的組成要素已經被廣泛使 用在作為治療多種不同疾病的藥物標靶。這些治療方式常藉由抑制特定的受器 (receptor) 或其合成酵素來減低血管收縮素II (angiotensin II, Ang II) 的胜肽含量並達到抑制高血 壓的效應。在西元兩千年,一個嶄新的酵素被發現,並被命名為血管收縮素轉化酶II (angiotensin-converting enzyme II, ACE2)。ACE2 與其耳熟能詳的類似物血管收縮素轉化 酶I (ACE) 同樣成為眾所矚目的焦點。與 ACE 相同的是,ACE2 同樣是一種第一型跨膜 金屬肽酶 (type I transmembrane metallopeptidase),並可作為一羧基胜肽水解酶
(carboxypeptidase),切除特定受質 (substrate) C 端的殘基 (residue),但兩者所切除的 residue 數目不同。ACE2 之所以可以作為一個調節心血管疾病的潛力標靶,是由於其可 扮演將Ang II 代謝成具有保護血管效用的另一胜肽血管收縮素 1-7 (angiotensin 1-7, Ang 1-7)。Ang II 和 Ang 1-7 同為 RAS 中的重要調控胜肽。
Ang II 已經被證實在心臟重塑 (remodeling) 過程中扮演要角,在許多疾病例如心肌 梗塞 (myocardial infarction, MI)、心臟衰竭 (heart failure, HF) 或是心房顫動 (atrial fibrillation, AF) 的病理狀態下,都可以同時測得 Ang II 以及 ACE2 的高量表現。有研究 顯示,心臟內高量表現的ACE2 可能參與防止 Ang II 異常表現所引起的高血壓或心臟纖 維化,這些現象指出ACE2 直接參與心臟保護的角色,並提供在醫療上可能的新契機。 更進一步的證據指出,在心臟衰竭病患的心臟組織中可同時測得高量表現的ACE2 以及 Ang 1-7,這也顯示了 ACE2 在心血管疾病中可能是藉由調節 Ang II 的含量來維持體內 的自我平衡。因此,我們提出一個假設,ACE2 表現量的提升可能是心臟為抵抗異常高
量表現的Ang II 所產生的自我保護機制。
在目前的研究中,我們使用人類心臟纖維母細胞 (human cardiofibroblast, HCF) 作 為探討Ang II 以及 Ang 1-7 對於 ACE2 在轉錄及轉譯調節上的重要模型。而當前的實驗 結果也證實Ang II 可以提高 ACE2 在人類心臟纖維母細胞的表現量,而此活化機制是經 由血管收縮素II 第一型受器 (angiotensin II type 1 receptor, AT1R) 進行調控。Ang II 所引 起的ACE2 高量表現更可以被 AT1R 及其下游諸如菸醯胺腺嘌呤二核酸磷酸氧化酵素 (Nicotinamide adenine dinucleotide phosphate oxidase, NADPH oxidase)、Extracellular signal-regulated kinase - Mitogen-activated protein kinase, ERK−MAPK 的拮抗劑所阻斷, 這樣的結果更確立Ang II 對於 ACE2 調控可能經由的訊號傳遞路徑。此外,promoter assay 的結果顯示在Ang II 的刺激下,ACE2 promoter 活性顯著提升,而此提升的效應也可在 加入AT1R 的阻斷劑纈沙坦 (Valsartan) 後被阻斷,顯示出 Ang II 參與調控 ACE2 啟動 子 (promoter) 的活性。
除此之外,我們的研究結果更發現Ang 1-7 也可以提升 ACE2 在心臟纖維母細胞的 表現量,而ACE2 的向上調控則可在加入 Mas receptor 的抑制劑 A779 後被阻斷。我們 的結果推論,Ang 1-7 對於 ACE2 的調控是經由 Mas receptor 並可以透過其下游的 NADPH oxidase 以及 ERK−MAPK 的訊息路徑調控 ACE2 的表現。共軛焦螢光顯微鏡的影像結果 也更進一步提供Ang II 和 Ang 1-7 對 ACE2 調控的證據,並呈現出 ACE2 及 AT1R 在心 臟纖維母細胞的實際分布,而此影像的結果也與先前的發現一致。
簡而言之,據我們的實驗結果證實了Ang II 所誘導產生的 ACE2 可以增加 Ang II 代謝成Ang 1-7 的量,而增量的 Ang 1-7 又更進一步增強 ACE2 的表現。根據這樣的結 果,我們提出ACE2 在心臟調控中具備一正回饋機制 (positive feedback loop),以維持人 體內RAS 的穩定平衡。我們的結果也提供產學界一個可發展治療因 RAS 失常所引起心 血管疾病的潛力新標靶。
【關鍵詞】血管收縮素II、血管收縮素 1-7、血管收縮素轉化酶 II、人類心臟纖維母細 胞、腎素-血管收縮素系統
Interplay of Angiotensin II and Angiotensin 1-7 in the
Modulation of Cardiac Angiotensin-Converting Enzyme II
of Human Cardiofibroblasts
Graduate student: Cheng-Hao Wen Advisor: Chih-Sheng Lin Ph.D.
Department of Biological Science and Technology
College of Biological Science and Technology
National Chiao Tung University
Abstract
Components of renin–angiotensin system (RAS) are well established targets for pharmacological intervention in a variety of disorders. Many such therapies abrogate the effects of the hypertensive and mitogenic peptide, angiotensin II (Ang II), by antagonising its interaction with its receptor, or by inhibiting its formative enzyme, angiotensin-converting enzyme (ACE). At the turn of the millennium, a novel homologous enzyme, termed ACE2, was identified which increasingly shares the limelight with its better-known homologue. In common with ACE, ACE2 is a type I transmembrane metallopeptidase; however, unlike ACE, ACE2 functions as a carboxypeptidase, cleaving a single C-terminal residue from a distinct range of substrates. ACE2 is a potential therapeutic target for the control of cardiovascular disease owing to its key role in the formation of vasoprotective peptides angiotensin 1-7 (Ang 1-7) from Ang II [cleavage from angiotensin I by angiotensin-converting enzyme (ACE)]. Ang II and Ang 1-7 are both critical regulatory peptides in RAS.
Ang II has been documented to play important role in the progression of cardiac
remodeling. Elevated Ang II paralleled to cardiac ACE2 upregulation was reported in some pathophysiological conditions, such as myocardial infarction, heart failure and atrial
fibrillation. Intracardiac overexpression of ACE2 prevents Ang II induced hypertension and cardiac fibrosis, implicating a direct in vivo cardioprotective role for ACE2, in addition to suggesting possible therapeutic utility. Further evidence for a role of ACE2 in maintaining cardiovascular homeostasis is via Ang II regulation which detected increased ACE2 and Ang
1-7 forming activity in failing human hearts. Hence, we tested the hypothesis that
upregulation of ACE2 may provide cardio-protection effects to counteract the elevated Ang II. In the present study, human cardiofibroblast (HCF) cells were used to test the
regulatory effects of Ang II and Ang 1-7 on the ACE2 expression at transcriptional and translational level. The results show that Ang II could upregulate ACE2 expression and this action may modulate through the activation of Ang II type I receptor (AT1R). Ang
II-mediated ACE2 upregulation could be blocked by the antagonists of downstream targets of AT1R, NADPH oxidase and ERK−MAPK cascades. To test the Ang II mediated ACE2 promoter activity, our result showed that human cardiac ACE2 promoter activity was significantly upregulation with Ang II stimulation. Additionally, Ang II-induced ACE2 promoter activity could be abolished when the HCF cells pretreated with Valsartan.
Furthermore, Ang 1-7 also could up-regulate ACE2 expression in the HCF cells and this upregulation could be inhibited by Mas receptor blocker, A779. Our result shows that the Ang 1-7−depedent ACE2 upregulation is via Mas receptor signaling pathway and even go through the NADPH oxidase and ERK-MAPK cascades. The confocal fluorescence
imaging results provide further validation for Ang II− and Ang 1-7−mediated ACE2 expression and an actual presentation of AT1R and ACE2 localization in HCF cells. Additionally, the image data also show the consistent results with our previous data.
In conclusion, our observation implicate that Ang II-induced ACE2 may increase Ang 1-7 formation from Ang II and then the ACE2 expression is further enhanced by the Ang 1-7. According to the results, we proposed a positive feedback-like loop on the cardiac ACE2 regulation for heart to maintain a steady state of RAS. Our results may point out new targets and possibilities for developing novel therapeutic strategies in cardiovascular diseases
induced by the dysfunction of RAS.
Keywords: angiotensin II, angiotensin 1-7, angiotensin-converting enzyme II, human
Abbreviation
ACE Angiotensin-converting enzyme
ACE2 Angiotensin-converting enzyme II
ACEIs Angiotensin-converting enzyme inhibitors AF Atrial fibrillation
Ang 1-7 Angiotensin 1-7 (Asp-Arg-Val-Tyr-Ile-His-Pro)
Ang 1-9 Angiotensin 1-9 (Asp-Arg-Val-Tyr-Ile-His-Pro-Phe-His) Ang II Angiotensin II (Asp-Arg-Val-Tyr-Ile-His-Pro-Phe-His-Leu) ARBs Angiotensin receptor blockers
AT1R Angiotensin II type I receptor AT2R Angiotensin II type II receptor CR Cardiac remodeling ECM Extracellular matrix ERK Extracellular signal-regulated kinase HCF Human cardiac fibroblasts
HCM Human cardiac myocytes HF Heart failure MAPK Mitogen-activated protein kinase
MEK Mitogen-activated/ERK kinase MI Myocardial infarction
NADPH Nicotinamide adenine dinucleotide phosphate RAS Renin-angiotensin system
ROS Reactive oxygen species
IDC Idiopathic dilated cardiomyopathy ICM Ischemic cardiomyopathy
Content
Acknowledgement ... i Chinese Abstract ... iv English Abstract ... vi Abbreviation ... viii Content ... ixList of Figures ... xii
List of Tables ... xiv
I. Literature review 1-1. Cardiac remodeling ... 1
1-2. Renin-angiotensin system ... 6
1-3. Angiotensin peptides and core enzyme ... 9
1-4. Angiotensin associated receptors ... 16
1-5. Oxidative stress in cardiovascular disease ... 18
1-6. ACE2−Ang 1-7 axis in regulation of cardiovascular disease ... 19
II. Research Purpose and Strategy 2-1. Develop the specific targets in reversing the abnormalities in cardiac regulation.. 33
2-2. ACE2 as a novel target in the regulation of Ang II-induced negative effect ... 33
2-3. The schematic representation of research strategy and experimental flowchart .. 33
III. Materials and Methods 3-1. Chemicals and reagents ... 36
3-2. Cell culture ... 36
3-3. Total RNA extraction ... 37
3-4. Reverse transcription-polymerase chain reaction ... 37
3-6. Protein extraction and electrophoresis ... 42
3-7. Western blotting ... 42
3-8. Human ACE2 promoter constructs ... 42
3-9. Transient transfection and luciferase reporter assay ... 45
3-10. Immunocytochemistry ... 45
3-11. Statistics ... 46
IV. Results 4-1. Ang II-mediated cardiac ACE2 upregulation in human cardiofibroblast ... 47
4-2. Human AT1R and AT2R could be markedly increased after Ang II stimulation .. 49
4-3. ERK−MAPK cascade is involved in Ang II-mediated upregulation of cardiac ACE2 ... 51
4-4. NADPH oxidase signaling pathway is concerned with Ang II-stimulated ACE2 upregulation ... 53
4-5. Mas receptor is involved in the effect of Ang 1-7−mediated upregulation of ACE2 ... 54
4-6. ERK−MAPK cascade and NADPH oxidase signaling pathway were involved in the Ang 1-7–ACE2 axis ... 55
4-7. ACE2 upregulation stimulated by Ang 1-7 might be independent to Ang II−AT1R pathway ... 57
4-8. The interference of each specific inhibitor or blocker was ruled out ... 59
4-9. ACE2 was major represented at the peripheral of cell membrane with both Ang II and Ang 1-7 treatment ... 60
4-10. AT1R representing at the boundary of cell membrane was increased by Ang II treatment but not Ang 1-7 ... 60
4-11. Deletion mutation of ACE2 promoter and figure out the intense promoter activity with Ang II and Ang 1-7 treatment ... 64
V. Discussion 5-1. The role of ACE2 in the cardiovascular system may be more complex ... 68
5-2. Ang II−AT1R modulated ACE2 upregulation was affirmed ... 68
5-3. Cardiac ACE2 upregulation is associated with the modulation of Ang II to antagonize the effects of increased Ang II ... 68
5-4. The upregulated ACE2 might play a compensatory role in maintaining a steady state of RAS ... 69
5-5. Ang 1-7 provides counter-regulatory effects to Ang II-induced deleterious effects on the cardiac functions ... 69 5-6. Ang 1-7−enhanced ACE2 expression might be independent to the Ang II−AT1R
signaling transduction pathway ... 70 5-7. ERK-MAPK cascade could be the main pathway to stimulate ACE2 expression . 70 5-8. Distinguishing the signaling pathway in the ACE2 regulation between Ang II and
Ang 1-7 ... 71
VI. Conclusions ... 73 VII. References ... 75
List of Figures
Figure 1-1 Schematic represents conversion of angiotensin peptides and balance
between ACE/ACE2 in RAS ... 8 Figure 1-2 Role of Ang II in the inflammatory response in vascular injury ... 10 Figure 1-3 Abnormal Ang II generation results in cardiac and renal damage ... 11 Figure 1-4 Stellar plot illustrating the mRNA copy number in logarithmic form in 72
human tissues ... 13 Figure 1-5 Cascade of the processing of angiotensin peptides and their interaction with
AT1R and Ang 1-7 receptor systems ... 15 Figure 2-1 The schematic representation of research strategy and experimental
flowchart ... 35 Figure 3-1 The vector map of the pGL3-ACE2 constructs ... 43 Figure 4-1 The mRNA expression of human ACE2 in the HCF cells treated with Ang II
... 48 Figure 4-2 The mRNA expression of human AT1R and AT2R in the HCF cells treated
with Ang II ... 50 Figure 4-3 Role of ERK−MAPK signaling of AT1R in the ACE2 regulation by Ang II .. 52 Figure 4-4 Role of NADPH oxidase in the regulation of ACE2 by Ang II ... 53 Figure 4-5 The regulation of ACE2 in HCF cells after Ang 1-7 treatment ... 54 Figure 4-6 Role of ERK-MAPK signaling of Mas receptor and NADPH oxidase in the
ACE2 regulation by Ang 1-7 ... 56 Figure 4-7 The regulation of angiotensin II type I receptors in the HCF cells treated
with Ang 1-7 ... 57 Figure 4-8 Role of the possibility of AT1R-dependent effect in the ACE2 regulation by
Ang 1-7 ... 58 Figure 4-9 The influence of each signaling specific inhibitor on ACE2 regulation ... 59 Figure 4-10 Localization and regulation of ACE2 and AT1R in HCF cells treated with
Ang II ... 61 Figure 4-11 Localization and regulation of ACE2 and AT1R in HCF cells treated with
Ang 1-7 ... 62 Figure 4-12 Quantification of the fluorescence expression of ACE2 and AT1R in HCF
cells treated with Ang II and Ang 1-7 ... 63 Figure 4-13 Deletion mutation analysis of the ace2 promoter region in the HCF cells ... 65 Figure 4-14 The regulation of ACE2 promoter activity in the HCF cells treated with Ang
Figure 4-15 The upstream region of the ACE2 gene ... 67 Figure 4-16 Schematic representation of interplay of Ang II and Ang 1-7 on the cardiac
List of Tables
Table 1-1 Processes occurring in ventricular remodeling ...1 Table 1-2 Studies of ACE2 in regulation of cardiovascular disease in human studies and
animal models ...21 Table 1-3 Studies of Ang 1-7 in regulation of cardiovascular disease in human studies
and animal models ...28 Table 3-1 The nucleotide sequences of the PCR primers used to assay gene expression
by RT-PCR are shown ...39 Table 3-2 The nucleotide sequences of the PCR primers used to assay gene expression
by Real-time PCR are shown ...41 Table 3-3 Sequences of the primers used for construction of human ACE2 promoter
I. Literature review
1-1. Cardiac remodeling
1-1-1. Concepts of cardiac remodeling
Cardiovascular disease will be the greatest health care burden of the twenty-first century [Crackower et al., 2002]. The term “remodeling” implies changes that result in the
rearrangement of normally existing structures [Swynghedauw, 1999]. Cardiac remodeling (CR) is defined as genome expression resulting in molecular, cellular and interstitial changes and manifested clinically as changes in size, shape and function of the heart resulting from cardiac load or injury, cardiac remodeling is influenced by hemodynamic load,
neurohormonal activation and other factors still under investigation [Cohn et al., 2000]. The concept of myocardial remodeling excludes concomitant changes in the cardiac atria, valves, blood vessels, and pericardium [Swynghedauw, 1999].
Cardiac remodeling is generally accepted as a determinant of the clinical course of heart failure (HF). Heart failure is an all-too-frequent outcome of hypertension and arterial vascular disease, making it a major concern in public heath and preventive medicine. It is a common cause of morbidity and mortality, and the incidence is increasing [Tyagi et al., 1995; Kannel, 2000; Rodeheffer, 2003; Izzo and Gradman, 2004; Mathew et al., 2004; Franklin and Aurigemma, 2005; Hunt et al., 2005; Weir et al., 2006]. Following a specific cardiovascular stress, a cascade of compensatory structural events occurs within the myocardium and
contributes to eventual left ventricular (LV) dysfunction and the manifestation of the heart failure syndrome.
The time course of events is influenced, however, by the severity of the underlying disease, secondary events (such as recurrent MI), other factors (such as ischemia or
neuroendocrine activation), genotype and treatment [Hutchins and Bulkley, 1978; Weisman et
al., 1985]. Animal studies also show that infarct expansion, regional dilation and thinning of
the infarct zone can occur within one day of an MI [Weisman et al., 1985]. Severe
impairment of global ventricular function, a functional and clinical phenomenon that can be differentiated clearly from LV remodeling, can be observed within two days of an insult [Anversa et al., 1991]. The changes that occur after an insult are summarized in Table 1-1.
multi-mechanistic and complex. Few clinical trials have specifically addressed the role of remodeling in disease progression. The key next steps will be the determination of how the information generated from cellular and molecular models can be used, together with data from clinical trials, to ensure that patients receive optimal therapy at an appropriate time to slow disease progression [Cohn et al., 2000].
Table 1-1. Processes occurring in ventricular remodeling
Processes occurring Description References
Cardiomyocyte lengthening Cardiomyocyte lengthening due to series addition of new sarcomeres and consequent fall in the short/long axis ratio.
Weisman et al., 1985; Anversa
et al., 1991
Ventricular wall thins Ventricular compliance depends upon the thickness of the ventricular wall and
on factors, such as fibrosis, that alter the stiffness of the ventricle. Weisman et al., 1985; McKay et al., 1986; Anversa et al.,
1991 Infarct expansion rather than
extension occurs
Infarct expansion and infarct extension are events early in the course of myocardial infarction with serious short- and long-term consequences. Expansion has an adverse effect on infarct structure and functional infarct size is increased because of infarct segment lengthening, and expansion results in over-all ventricular dilatation. Infarct extension is defined clinically as early in-hospital reinfarction after a myocardial infarction.
Hutchins and Bulkley, 1978; Weisman et al., 1985
Inflammation and resorption of necrotic tissue
If early thinning and dilatation did not occur after myocardial infarction, the process of remodeling with resorption of necrotic tissue, laying down of granulation tissue and scar formation would probably result in a healed area that was somewhat thinned but generally preserved normal LV contour.
Hochman and Bulkley, 1982; Weisman et al., 1985
Scar formation Scar formation is a natural part of the healing process. A scar forms from excess amounts of collagen in the wound as the body attempts a repair.
Zdrojewski et al., 2002
Continued expansion of infarct zone
Infarct expansion is a progressive thinning and dilation of the infarcted zone. A progressive increase in infarct expansion is associated with increased left ventricular volume and predisposes to remodeling of the non infarcted segment.
Table 1-1. Continued
Processes occurring Description References
Dilation and reshaping of the left ventricle
Surgically reshaping the adversely remodeled dilated left ventricle is a concept that holds promise in the management of patients with dilated cardiomyopathy.
Weisman et al., 1985; McKay
et al., 1986; Olivetti et al.,
1990; Gaudron et al., 1993 Myocyte hypertrophy Hypertrophy of the surviving myocytes is an important adaptive response to
loss of contractile fuction. A decrease in cardiac fuction leads to increased levels of norepinephrine and activation of the renin-angiotensin system, leading to release of angiotensn II. Angiotensin II and mechanical stress induce a number of cellular signaling pathways important in the development of cellular hypertrophy.
Olivetti et al., 1992; Kajstura et
al., 1994
Ongoing myocyte loss Recent studies in experimental animals have shown that cardiac myocyte loss through apoptosis, or programmed cell death, occurs following myocardial infarction, in the presence of cardiac hypertrophy, in the aging heart, and in the setting of chronic heart failure.
Weisman et al., 1985; McKay
et al., 1986; Olivetti et al.,
1990; Anversa et al., 1991
Excessive accumulation of collagen in the cardiac interstitium
The accumulation of excess collagen is believed tobe an important
pathophysiological process that contributesto diastolic heart failure. Diastolic heart failure accountsfor 30% to 50% of heart failure in clinical practice, and hypertensivedisease is the major cause of this type of heart failure.
Weber and Brilla, 1991; Dostal, 2001
1-1-2. The effect of cardiac remodeling
Cardiac remodeling can be described as a physiologic and pathologic condition that may occur after myocardial infarction (MI), pressure overload (aortic stenosis, hypertension), inflammatory heart muscle disease (myocarditis), idiopathic dilated cardiomyopathy or volume overload (valvular regurgitation) [Fedak et al., 2005].
With an increased workload during hypertension, the heart eventually undergoes hypertrophic (enlargement) and fibrotic responses. Myocyte hypertrophy, when
accompanied by fibrosis can lead to a decrease in cardiac function. This cardiac hypertrophy and inappropriate interstitial collagen formation can contribute to increased wall stiffness and diastolic dysfunction. Thus the remodeling process, which could accompany hypertension, would consist of changes in the architecture of the heart, including myocardial fibrosis, and medial thickening of intramyocardial coronary arteries, in addition to the myocyte
hypertrophy. Therefore, ventricular remodeling after myocardial infarction is a risk factor for development of heart failure and sudden cardiac death [Cohn et al., 2000; Fedak et al., 2005; Grobe et al., 2007]. The prevention of ventricular remodeling after myocardial infarction is an important strategy in reducing mortality from myocardial infarction.
1-1-3. Critical factors involved in cardiac remodeling
The renin-angiotensin system (RAS) has previously been established to play an
important role in the progression of cardiac remodeling, and inhibition of a hyperactive RAS provides a protection from cardiac remodeling and subsequent heart failure [Dzau, 1993; Cockcroft et al., 1995; Parmley, 1998; Bader et al., 2001; Ruiz-Ortega et al., 2001a; Grobe et
1-2. Renin-angiotensin system (RAS)
1-2-1. Physiological and patho-physiological roles of local RAS
RAS is a coordinated hormonal cascade in the control of cardiovascular, renal, and adrenal function that governs body fluid and electrolyte balance, as well as arterial pressure [Peach, 1977]. RAS is well known for its effects on the cardiovascular system and fluid
homeostasis. Classically, these effects were thought to result primarily from the systemic production of angiotensin II (Ang II) [Paul et al., 2006]. Circulating Ang II stimulates Ang II type 1 receptors (AT1R) present in the kidney and the vasculature to produce
vasoconstriction but also water and salt reabsorption [Davisson, 2003; Lavoie and Sigmund, 2003].
It has become clear that a local RAS is present in several tissues, for example, the heart, adipose, vasculature, and bone marrow, with similar effects to the endocrine RAS but also more specific functions depending on the individual system [Paul et al., 2006]. One of these local systems, the brain RAS, has long been considered pivotal in cardiovascular regulation and important in the pathogenesis of hypertension and heart failure [Davisson, 2003]. Yet the brain RAS remains poorly understood, because of the difficulty in experimentally dissecting the brain RAS at the cellular, regional, and whole organism levels.
1-2-2. RAS in regulation of cardiovascular homeostasis
Numerous clinical and laboratory data are now available supporting the hypothesis that the renin-angiotensin system (RAS) is relevant in the pathogenesis of cardiovascular diseases [Ruiz-Ortega et al., 2001a; Boos and Lip, 2004; Levy, 2004; Healey et al., 2005]. RAS plays a major role in regulating the cardiovascular system, and disorders of the RAS contribute largely to the pathophysiology of hypertension, renal diseases, myocardial
infarction [Hanatani et al., 1995; Sutton and Sharpe, 2000], atrial fibrillation [Freestone et al., 2004; Savelieva and John Camm, 2004], and chronic heart failure [Weber et al., 1993; Shi et
al., 2002]. This is to say that the emergence of cardiovascular diseases is largely related to
the regulation of RAS.
1-2-3. Heart failure and RAS
Most cardiovascular diseases are multifactorial quantitative traits controlled by both genetic and environmental factors [Jacob, 1999]. One major factor for cardiovascular disease is the RAS. Due to this continuing morbidity and mortality, significant efforts have
been made to identify new drug targets in the RAS. We note that in human patients, inhibition of angiotensin-converting enzyme (ACE) or Ang II receptors can improve the outcome of heart failure [Garg and Yusuf, 1995; Boos and Lip, 2004; Madrid et al., 2004; Healey et al., 2005]. To solve the severe issue in the increasingly cardiovascular diseases such as heart failure, the endless stage of heart diseases, a novel efficient approach may be considered.
1-2-4. The components of peptide converting involved in RAS
The protease renin is synthesized and released from the kidney and acts on a circulating inactive peptide, angiotensinogen [angiotensinogen (Asp-Arg-Val-Tyr-Ile-His-Pro-Phe-His- Leu-Val-Ile)], produced by the liver, giving rise to angiotensin I [angiotensin I; Ang I (Asp-Arg-Val-Tyr- Ile-His-Pro-Phe-His-Leu)]. Ang I is then transformed into the
biologically active octapeptide, Ang II [Ang II; Asp-Arg-Val-Tyr-Ile-His-Pro-Phe], through enzymatic cleavage by ACE [Skeggs et al., 1980; Carey and Siragy, 2003a; Lambert et al., 2008], which is a critical regulator of the RAS and the target of a number of highly effective therapeutic agents used to treat cardiovascular and renal diseases. ACE is also a
metalloproteinase which converts the inactive decapeptide Ang I into the potent
vasoconstrictor and mitogen Ang II [Skeggs et al., 1980; Lambert et al., 2008], which can contribute to hypertension by promoting vascular smooth muscle vasoconstriction and renal tubule sodium reabsorption [Skeggs et al., 1980].
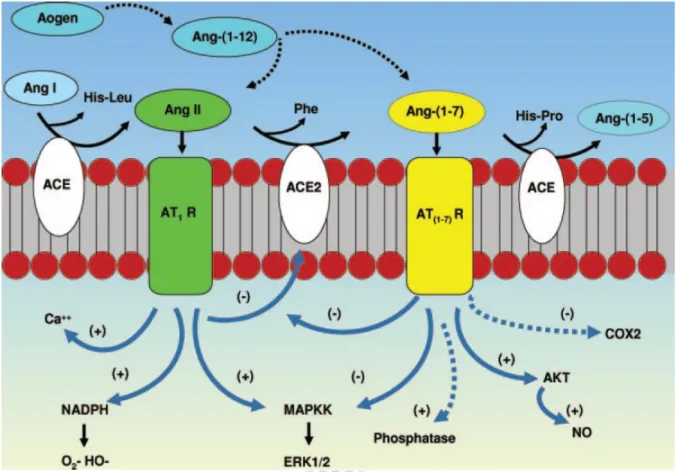
In common with ACE, ACE2 is a type I transmembrane metallopeptidase; however, unlike ACE, ACE2 functions as a carboxypeptidase, cleaving a single C-terminal residue from a distinct range of substrates. One such substrate is angiotensin II, which is hydrolysed by ACE2 to the vasodilatory/anti-hypertrophic peptide angiotensin 1-7 [Ang 1-7;
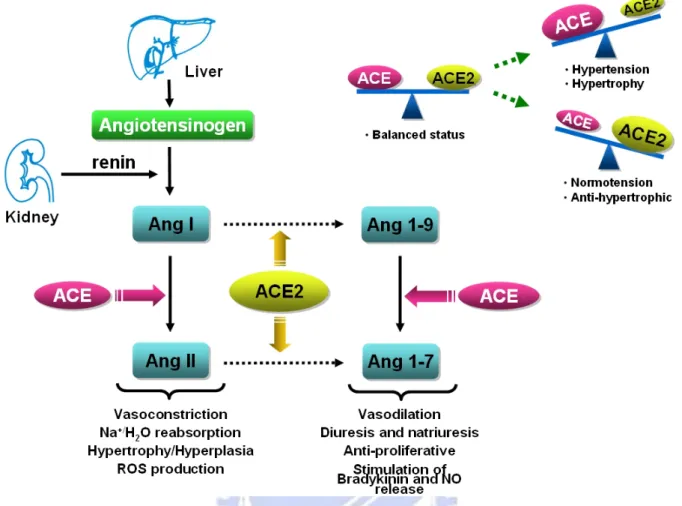
Asp-Arg-Val-Tyr-Ile-His-Pro] [Lambert et al., 2008]. The schematic conversion of angiotensin peptides and balance between ACE/ACE2 was shown in Figure 1-1.
Figure 1-1. Schematic represents conversion of angiotensin peptides and balance between
1-3. Angiotensin peptides and core enzyme
1-3-1. Angiotensin II
Ang II is the main effector peptide of the RAS, acting in an endocrine,
autocrine/paracrine, and intracrine hormone involved in the regulation of blood pressure, vascular tone, waster as well as electrolyte balance [Skeggs et al., 1980; Parfrey, 2008]. Historically, Ang II was only seen as a regulatory hormone that regulates blood pressure, aldosterone release, and sodium reabsorption. Now it is generally accepted that locally formed Ang II could activate the cells regulating the expression of many substances,
including growth factors, cytokines, chemokines, and adhesion molecules, which are involved in cell growth/apoptosis, procoagulation, fibrosis, and inflammation (Figure 1-2)[Matsubara, 1998; Sadoshima, 2000; Bader et al., 2001; Ruiz-Ortega et al., 2001b].
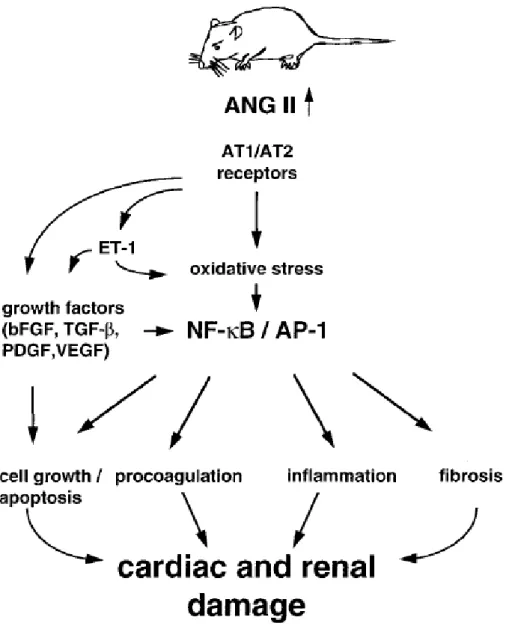
Figure 1-2. Abnormal Ang II generation results in cardiac and renal damage. Ang II
activates the transcription factors nuclear factor κB (NF-κB) and activator protein 1 (AP-1) as well as proinflammatory and profibrotic genes. Ang II signaling also leads to impaired balance of cell growth and apoptosis and pro- and anti- coagulative systems. bFGF Basic fibroblast growth factor; TGF-β transforming growth factor β; PDGF platelet-derived growth factor; VEGF vascular endothelial growth factor [Bader et al., 2001].
Ang II binds and activates G protein–coupled receptors, the AT1R and angiotensin II type 2 receptor (AT2R), to mediate its actions [Carey et al., 2000; Shi et al., 2002]. Activation of AT1R mediates most of the cardiovascular responses attributed to Ang II (ie, vasoconstriction, mitogenic and hypertrophic effects, fibrosis, inflammation, and fluid retention) [Booz and Baker, 1995; Booz and Baker, 1996; Unger, 2002]. In contrast, AT2R activation may cause opposing physiological responses that are increased in several disease processes [Ohkubo et al., 1997; van Kesteren et al., 1997]. Multiple lines of evidence indicate that stimulation of Ang II plays a key role in the development of pathological ventricular remodeling [Pfeffer and Braunwald, 1990]. Role of Ang II in the inflammatory response and cardiac damage was shown in Figure 1-3.
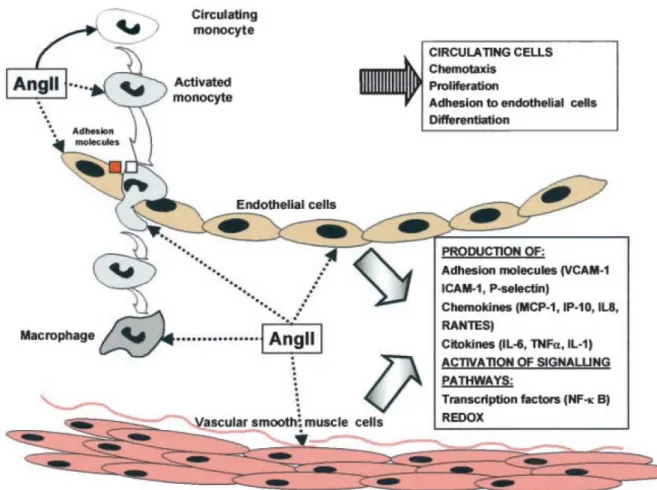
Figure 1-3. Role of Ang II in the inflammatory response in vascular injury. Ang II activates
mononuclear cells, causing direct chemotaxis and proliferation. In both resident and infiltrating cells, Ang II, by NF-κB pathway and redox mechanisms, upregulates proinflammatory mediators, such as adhesion molecules, chemokines, and cytokines.
1-3-2. Angiotensin 1-7
Ang 1-7 is a biologically active peptide of the RAS that is known to potentiate the vasodilatory effects of bradykinin [Greco et al., 2006], stimulate NO and prostaglandin release [Rajendran et al., 2005], and antagonize the actions of Ang II [Grobe et al., 2007]. Ang 1-7 has been reported to act as an antagonist to the AT1R and may also work by
antagonizing ACE, which is involved in both the production of Ang II and the degradation of Ang 1-7 [Ferrario, 1998; Castro et al., 2005; Igase et al., 2005].
In the current study, Ang 1-7 infusion prevented cardiac hypertrophy and fibrosis
without having any effect on the elevated blood pressure induced by chronic Ang II treatment. It has been well documented that Ang 1-7 levels are elevated during pharmacological ACE inhibition and blockade of AT1R [Ferrario et al., 2005a; Ferrario et al., 2005c; Igase et al., 2005], and it has been proposed that these cardioprotective inhibitors may actually work through the actions of increased Ang 1-7 [Ferrario, 1998]. Correlative studies have shown that ACE2 and Ang 1-7 levels are increased by cardiac myocytes in hearts following
myocardial infarction in both rats and human [Averill et al., 2003; Burrell et al., 2005]. Iwata et al. [2005] recently demonstrated that Ang 1-7 attenuates profibrotic signaling within the myocardium, through direct actions on cardiac fibroblasts. In the current study, chronic in vivo administration of Ang 1-7 also appears to have effects on hypertrophic actions on the cardiomyocytes that are induced by Ang II. This observation has also been observed
in vitro, as Tallant et al. [2005a] showed that Ang 1-7 acts on cultured cardiac myocytes to
inhibit hypertrophic responses through the Mas receptor.
Collectively, these findings suggest that elevated Ang 1-7 may also protect against cardiac hypertrophy in some forms of hypertension. Ang 1-7 delivery has been shown to delay development of cardiac hypertrophy [Santos et al., 2004], inhibit vascular growth [Tallant and Clark, 2003], attenuate development of heart failure [Loot et al., 2002], reduce cardiac Ang II levels [Mendes et al., 2005], and reduce Ang II receptor populations [Clark et
al., 2003]. Evidence presented here would support the hypothesis that Ang 1-7 is a
cardioprotective peptide.
1-3-3. Angiotensin-converting enzyme II (ACE2)
At the turn of the millennium, a homologous enzyme, termed ACE2, was identified which increasingly shares the limelight with its better-known homologue, ACE [Donoghue et
al., 2000b; Tipnis et al., 2000; Lazartigues et al., 2007]. In vivo, ACE2 is predominantly
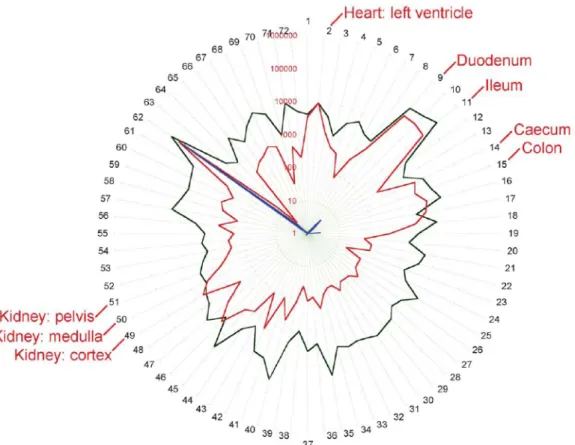
expressed in the heart, kidneys and testes. In human study, confirming that ACE2 is expressed in human heart, kidney and testis, consistent with a possible role in cardio-renal function [Harmer et al., 2002]. The quantitative expression map for ACE 2 across 72 human tissues is shown in Figure 1-4.
Figure 1-4. Stellar plot illustrating the mRNA copy number in logarithmic form in 72 human
tissues. For ACE (black), ACE 2 (red) and ACE testicular (blue). Each point represents the geometric mean copy number from determinations in three donors. Gene copy number increases logarithmically moving from the centre to the periphery of the circle. The tissues used are: 1. heart: left atrium; 2. heart: left ventricle; 3. blood vessel: coronary artery; 9. duodenum; 11. ileum; 14. caecum; 15. colon; 49. kidney: cortex; 50. kidney: medulla; 51. kidney: pelvis [Harmer et al., 2002].
In the heart, ACE2 is essentially confined to the endothelia [Donoghue et al., 2000b; Tipnis et al., 2000; Guy et al., 2008; Pan et al., 2008].
ACE2 is able to cleave both Ang I and Ang II, to Ang 1-9 and Ang 1-7, respectively. The high level of expression of ACE2 in the heart together with its ability to hydrolyse angiotensin peptides have suggested a role for ACE2 in maintaining cardiovascular physiology [Lambert et al., 2008]. The potential role of Ang 1-7 as a cardioprotective peptide having vasodilator, anti-growth and antiproliferative actions has been recognized [Ferrario, 1992b; Ferrario, 1992a; Clark et al., 2001; Carey and Siragy, 2003b; Burrell et al., 2004; Lambert et al., 2008]. It was shown that ACE2 provides a counter-regulatory system to Ang II [Carey and Siragy, 2003b; Burrell et al., 2004]. Crackower et al. [2002] showed that deletion of ACE2 in mice resulted in elevated cardiac and plasma Ang II together with impaired cardiac contractility which increased with age. These changes were associated with an upregulation of hypoxia-induced genes, consistent with a role for Ang II in the ACE2 null phenotype.
In humans, single nucleotide polymorphisms associated with increased risk of
cardiovascular disease have been identified within the ACE2 gene locus [Yang et al., 2006]. Disturbance of the balance of expression of ACE2 and its homologue ACE could alter the levels of Ang II and contribute to the development of a range of pathologies.
1-3-4. The counterbalance between Ang II and Ang 1-7
Further evidence for a role of ACE2 in maintaining cardiovascular homeostasis via Ang II regulation is provided by studies conducted by Zisman et al. [2003] which detected
increased ACE2 and Ang 1-7 forming activity in failing human hearts. Hence, the ability of ACE2 to degrade Ang II and simultaneously increase Ang 1-7 would effectively oppose the actions of ACE, suggesting the balance of the levels of the two enzymes would be critical in pathologies in the aetiologies of which Ang II is implicated. It is likely that ACE2 may play a protective role in the early stages of heart failure by elevating Ang 1-7 levels. In another
study, Grobe et al. [2007] suggested that infusion of Ang II into adult Sprague-Dawley rats resulted in significantly increased blood pressure, myocyte hypertrophy, and midmyocardial interstitial fibrosis. Coinfusion of Ang 1-7 resulted in significant attenuations of myocyte hypertrophy and interstitial fibrosis, without significant effects on blood pressure. Another findings demonstrate that, in human endothelial cells, Ang 1-7 negatively modulates Ang II/AT1R–activated c-Src and its downstream targets ERK1/2 and NADPH oxidase. These
phenomena may represent a protective mechanism in the endothelium whereby potentially deleterious effects of Ang II are counterregulated by Ang1-7 [Sampaio et al., 2007]. The cascade of the processing of angiotensin peptides and their interaction with AT1R and Ang 1-7 receptor systems was shown in Figure 1-5.
Figure 1-5. Cascade of the processing of angiotensin peptides and their interaction with AT1R
and Ang 1-7 receptor systems. ACE cleaves Ang I, releasing the dipeptide His-Leu to form Ang II, and ACE2 subsequently hydrolyzes Ang II to Ang1-7. ACE also metabolizes Ang 1-7 to Ang 1-5 and the dipeptide His-Pro. Ang 1-12 may be cleaved from angiotensinogen (Aogen) and potentially processed (Æ) directly to Ang II or Ang 1-7. Ang1-7 may attenuate the inflammatory and fibrotic actions of the Ang II-AT1R pathway through inhibition (-) of the MAP kinase kinase (MAPKK) pathway, the potential stimulation (+) of cellular
phosphatases, the inhibition of cyclooxygenase-2 (COX2) and other proinflammatory agents, as well as the stimulation of NO. Although not shown, the AT2R and bradykinin receptor systems may interact with these pathways as well [Chappell, 2007].
1-4. Angiotensin associated receptors
1-4-1. Angiotensin receptors and blockades
Angiotensin II has two major receptor subtypes, the angiotensin II type 1 receptor (AT1R) and angiotensin II type 2 receptor (AT2R). Two subtypes of angiotensin II (Ang II) receptors have been defined on the basis of their differential pharmacological and biochemical properties. AT1R, which are involved in most of the well-known physiological effects of Ang II, and AT2R, which have a less well-defined role but appear capable of
counterbalancing some of the effects of AT1R stimulation [Levy, 2004]. Importantly, AT1R antagonists are associated with a rise in plasma Ang II concentration due to the inhibition of the AT1R–mediated negative feedback on renin release [Campbell, 1996]. Drugs that block Ang II actions, such as ACE inhibitors or angiotensin receptor antagonists, are currently employed in the treatment of hypertension, heart failure, atherosclerosis, and other cardiovascular diseases [Matsubara, 1998; Sadoshima, 2000; Ruiz-Ortega et al., 2001b].
1-4-2. Angiotensin II type 1 receptor (AT1R)
AT1R are widely distributed throughout the body, including vascular smooth muscle, kidney, heart, and brain. It is to say that AT1R are responsible for mediating most of the known actions of Ang II, including vasoconstriction and aldosterone release [Griendling et al., 1996]. AT1R gene has been mapped to chromosome 3, and is highly expressed in smooth muscle cells, fibrolasts, as well as in atrial and ventricular myocytes [Allen et al., 1999]. The amino terminal portion and the first and third loops of the transmembrane domain of this glycoproteic receptor are responsible for the interaction with Ang II [Hjorth et al., 1994]. AT1R transactivates growth pathways and mediates major Ang II effects such as
vasoconstriction, increased cardiac contractility, renal tubular sodium reabsorption, cell proliferation, vascular and cardiac hypertrophy, inflammatory responses, and oxidative stress [Levy, 2004].
Ang II, through its interactions with the AT1R, has been demonstrated to increase fibroblast gene expression (including collagen), fibroblast density and proliferation, and myocyte hypertrophy, all of which are hallmarks of myocardial fibrosis and remodeling [Sun
et al., 1997; Kawano et al., 2000; Gonzalez et al., 2002]. AT1R activation triggers a variety
of intracellular systems, including tyrosine kinase-induced protein phosphorylation,
and fluxes in intracellular Ca2+ concentrations [Berry et al., 2001]. Interestingly, Ang II/AT1R activates the Jak-STAT pathway, which is also part of the signaling pathway of cytokine receptors, leading to activation of growth response genes which could contribute to cardiac tissue remodeling [Berk, 1999].
1-4-3. Angiotensin II type 2 receptor (AT2R)
Using in situ hybridization techniques, Shanmugam et al. [1995] reported that AT2R mRNA was detectable in the large arteries, in the mesenchymal tissues, such as the kidney and the urogenital tract, and variably in the cardiomyocytes of fetal rats. Vascular AT2R mRNA was most abundant in late gestation and in the early postnatal period, becoming undetectable in the cardiovascular system of adult rats.
The AT2R is thought to counteract the signals transmitted by the AT1R, eliciting vasodilatation [Brede et al., 2001], inhibition of proliferation [Nakajima et al., 1995; Stoll et
al., 1995; Mukawa et al., 2003], NO production [Kurisu et al., 2003] and apoptosis [Yamada et al., 1996; Horiuchi et al., 1997; Lehtonen et al., 1999; Wang et al., 2001; Suzuki et al.,
2002]. The AT2R may play a homeostatic role in the regulation of blood pressure in animal models of hypertension [Barber et al., 1999]. In studies in rats with heart failure induced by coronary artery ligation, treatment with an AT1R antagonist was associated with
improvements in left ventricular (LV) systolic function, LV end-diastolic diameter, and LV end-systolic volume. The beneficial effects on cardiac dimensions, but not function, were prevented by cotreatment with an AT2R antagonist [Liu et al., 1997].
Recently, a number of studies have implicated the AT2R as having an opposing role to the AT1R in certain experimental settings, including endothelial cell proliferation and neointimal formation. In both situations, the AT1R causes stimulation, while the AT2R mediates inhibition of the response [Stoll et al., 1995; Matsubara, 1998]. Therefore, it has been suggested that, at therapeutic doses of AT1R antagonists, endogenous Ang II may stimulate unopposed AT2R and thereby contribute to the decrease in blood pressure [de Gasparo and Levens, 1998].
1-4-4. Mas receptor
An anti-remodeling role for Ang 1-7 in cardiac tissue, which is not mediated through modulation of blood pressure or altered cardiac angiotensin receptor populations and may be at least partially mediated through an Ang 1-7 receptor [Grobe et al., 2007]. Santos et al.
receptor Mas. The genetic deletion of Mas abolished the binding of Ang 1-7 to mouse kidneys and abrogated the antidiuretic effect of Ang 1-7 in mice after acute water load. Santos et al. [2006] report impaired cardiac function in the Mas-deficient mouse that was associated with increased forms of collagen and fibronectin. Ang 1-7 also induced an increase of 3H-arachidonic acid release, endothelial NO synthase activation and NO release from Mas-transfected cells that were blocked by the specific Ang 1-7 receptor Mas. These findings further substantiate the role of the Mas receptor mediating the Ang 1-7 dependent activation of endothelial NO synthase.
Although, these studies clearly elevate the relevance of an Ang 1-7-Mas pathway by demonstrating a functional pathway in human cells, the acute vasodilatory effects of the peptide have not been demonstrated in vivo [Wilsdorf et al., 2001]. Tallant et al. [2005a] have shown that Ang 1-7 reduces the growth of cardiomyocytes through activation of the Mas receptor. Ang 1-7-Mas axis may contribute to their beneficial effects on cardiac dysfunction and ventricular remodeling after myocardial infarction.
1-5. Oxidative stress in cardiovascular disease
1-5-1. Effect of oxidative stress in RAS
Several lines of evidence have been indicated that oxidative stress and reactive oxygen species (ROS) participate in the pathogenesis of cardiovascular diseases, including
hypertension and atherosclerosis [Alexander, 1995; Griendling et al., 2000b]. ROS is currently recognized as a modulator of intracellular redox state, which plays an important role as a second messenger in regulating signal transduction pathways and subsequent gene
expression [Sundaresan et al., 1995; Kunsch and Medford, 1999; Griendling et al., 2000a]. Recent work has shown that NAD(P)H oxidases are major sources of superoxide in vascular cells and myocytes. It has recently been shown that many of these effects of Ang II are mediated by generation of ROS through the activation of vascular reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase. For example, Ang II-induced VSMC hypertrophy is inhibited by blocking NADPH oxidase or scavenging intracellular ROS [Ushio-Fukai et al., 1996], and Ang II-induced ERK1/2 activation and subsequent gene expression in VSMCs is also inhibited by pretreatment with antioxidants [Chen et al., 1998; Frank et al., 2001], suggesting its redox-sensitive signaling in VSMCs.
It has been well recognized that Ang II exerts its cardiovascular effects mainly via AT1-mediated generation of reactive oxygen species (ROS) [Yoshimoto et al., 2004; Touyz, 2005; Yoshimoto et al., 2005]. There is evidence that Ang II increases NADPH
oxidase–mediated superoxide production through the activation of the AT1R, whereas inhibition of Ang II production ameliorates oxidative stress in the vasculature [Griendling et
al., 2000b; Keaney, 2005]. Ang II could significantly increase activation of NADPH
oxidase in human endothelial cells [Sampaio et al., 2007].
1-5-2. Downstream signaling of oxidative stress in RAS
One potential target for reactive oxygen species may be the mitogen-activated protein kinase (MAPK) family. MAPKs are serine/threonine kinases that transduce signals from the cell membrane to the nucleus in response to classical growth factors and G protein-coupled receptor agonists, as well as cellular stress [Davis, 1994; Cano and Mahadevan, 1995b; Cobb and Goldsmith, 1995b]. Four groups of MAPKs have been identified in mammalian cells: the extracellular signal-regulated kinases 1 and 2 (ERK1/2, also termed p42/44MAPK), the c-Jun NH2- terminal kinases (JNK, also termed stress-activated protein kinase, SAPK), p38MAPK (also termed CSBP) and Big MAPK 1 (BMK1, also termed ERK5) [Cano and Mahadevan, 1995a; Cobb and Goldsmith, 1995a; Abe et al., 1996]. It is to say that the NADPH oxidase–mediated signaling pathway is associated with ERK−MAPK and p38-MAPK [Abe et al., 1996; Ushio-Fukai et al., 1998; Wenzel et al., 2001].
1-6. ACE2-Ang 1-7 axis in regulation of cardiovascular disease
1-6-1. Role of ACE2 in recent studies of cardiovascular disease
Recently, the relationship between ACE2−Ang 1-7 axis and cardiovascular disease has been largely elucidated. It was shown that ACE2−Ang 1-7 axis play a critical role in heart disease development. The high level of expression of ACE2 in the heart together with its ability to hydrolyse angiotensin peptides have suggested a role for ACE2 in maintaining cardiovascular physiology from the outset, a hypothesis subsequently supported by
experimental data. In the ACE2 studies, Huentelman et al. [2005] utilize transduction with lenti-mACE2 resulted in significant attenuation of the increased heart/body weight and myocardial fibrosis induced by Ang II infusion. In addition, ACE2 also plays an important
Disruption of this regulatory function may accelerate cardiac hypertrophy and shorten the transition period from compensated hypertrophy to cardiac failure. Increase of ACE2 mRNA and its protein expression in hypertrophic myocardium can attenuate cardiac hypertrophy due to pressure overload effectively [Yamamoto et al., 2006; Qin et al., 2008]. These observations demonstrate that ACE2 overexpression results in protective effects on Ang II-induced cardiac hypertrophy and fibrosis.
Targeted disruption of ACE2 in mice causes enhanced susceptibility to Ang II–induced hypertension and results in a severe cardiac contractility defect [Crackower et al., 2002; Gurley et al., 2006]. Oudit et al. showed that the age-dependent cardiomyopathy in ACE2 null mice is related to increased Ang II-mediated oxidative stress, and defined a critical role of ACE2 in the suppression of Ang II-mediated heart failure [Oudit et al., 2007]. These researches showed that ACE2 is an essential regulator of heart function in vivo. Sluimer et
al. [2008] suggested that RAS may play a role in the pathogenesis of atherosclerosis.
During the progression of atherosclerosis, overexpression of ACE2 results in stabilized atherosclerotic plaques and the mechanism is probably the conversion of vasoconstrictive Ang II to vessel protective Ang 1-7 [Dong et al., 2008]. The studies of ACE2 in regulation of cardiovascular disease in human studies and animal models were shown in Table 1-2.
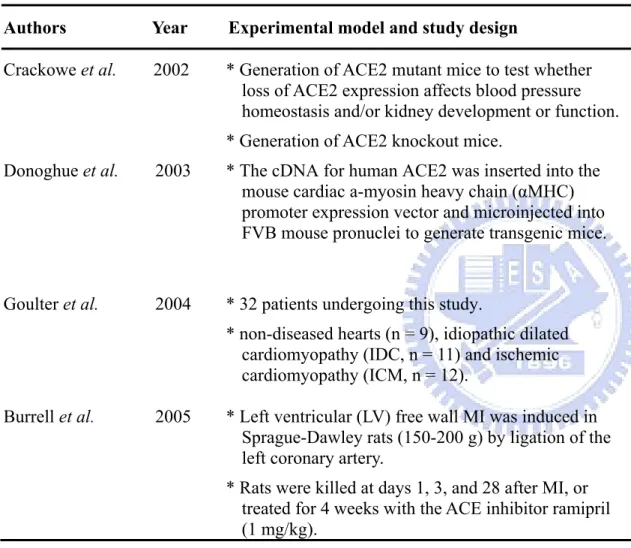
Table 1-2. Studies of ACE2 in regulation of cardiovascular disease in human studies and animal models. Authors Year Experimental model and study design Key findings
Crackowe et al. 2002 * Generation of ACE2 mutant mice to test whether loss of ACE2 expression affects blood pressure homeostasis and/or kidney development or function. * Generation of ACE2 knockout mice.
Targeted disruption of ACE2 in mice results in a severe cardiac contractility defect, increased Ang II levels, and upregulation of hypoxia-induced genes in the heart. ACE2 is an essential regulator of heart function in vivo. Donoghue et al. 2003 * The cDNA for human ACE2 was inserted into the
mouse cardiac a-myosin heavy chain (αMHC) promoter expression vector and microinjected into FVB mouse pronuclei to generate transgenic mice.
Transgenic mice with increased cardiac ACE2 expression had a high incidence of sudden death. Spontaneous downregulation of the ACE2 transgene in surviving older animals correlated with restoration of nearly normal conduction, rhythm, and connexin expression.
Goulter et al. 2004 * 32 patients undergoing this study.
* non-diseased hearts (n = 9), idiopathic dilated cardiomyopathy (IDC, n = 11) and ischemic cardiomyopathy (ICM, n = 12).
ACE2 is upregulated in human IDC and ICM and are consistent with the hypothesis that differential
regulation of this enzyme may have important
functional consequences in heart failure. ACE2 may be a relevant target for the treatment of heart failure. Burrell et al. 2005 * Left ventricular (LV) free wall MI was induced in
Sprague-Dawley rats (150-200 g) by ligation of the left coronary artery.
* Rats were killed at days 1, 3, and 28 after MI, or treated for 4 weeks with the ACE inhibitor ramipril (1 mg/kg).
The increase in ACE2 after MI suggests that it plays an important role in the negative modulation of the renin angiotensin system in the generation and degradation of angiotensin peptides after cardiac injury.
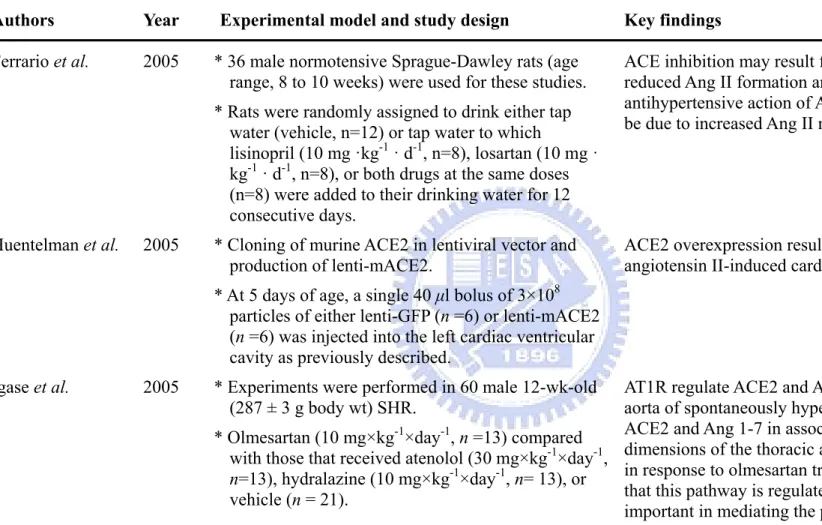
Table 1-2. Continued
Authors Year Experimental model and study design Key findings
Ferrario et al. 2005 * 36 male normotensive Sprague-Dawley rats (age range, 8 to 10 weeks) were used for these studies. * Rats were randomly assigned to drink either tap
water (vehicle, n=12) or tap water to which
lisinopril (10 mg ·kg-1 · d-1, n=8), losartan (10 mg · kg-1 · d-1, n=8), or both drugs at the same doses (n=8) were added to their drinking water for 12 consecutive days.
ACE inhibition may result from the combined effect of reduced Ang II formation and Ang 1-7 metabolism, the antihypertensive action of AT1 antagonists may in part be due to increased Ang II metabolism by ACE2.
Huentelman et al. 2005 * Cloning of murine ACE2 in lentiviral vector and production of lenti-mACE2.
* At 5 days of age, a single 40 μl bolus of 3×108 particles of either lenti-GFP (n =6) or lenti-mACE2 (n =6) was injected into the left cardiac ventricular cavity as previously described.
ACE2 overexpression results in protective effects on angiotensin II-induced cardiac hypertrophy and fibrosis.
Igase et al. 2005 * Experiments were performed in 60 male 12-wk-old (287 ± 3 g body wt) SHR.
* Olmesartan (10 mg×kg-1×day-1, n =13) compared with those that received atenolol (30 mg×kg-1×day-1,
n=13), hydralazine (10 mg×kg-1×day-1, n= 13), or
vehicle (n = 21).
AT1R regulate ACE2 and Ang 1-7 expression in the aorta of spontaneously hypertensive rats. Increased ACE2 and Ang 1-7 in association with altered
dimensions of the thoracic aorta but not carotid arteries in response to olmesartan treatment provides evidence that this pathway is regulated by AT1R and may be important in mediating the pressure-independent vascular remodeling effects of angiotensin peptides.
Table 1-2. Continued
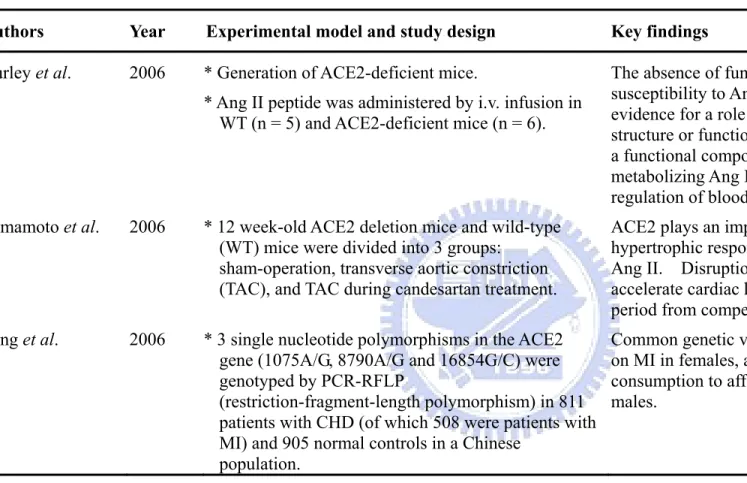
Authors Year Experimental model and study design Key findings
Gurley et al. 2006 * Generation of ACE2-deficient mice.
* Ang II peptide was administered by i.v. infusion in WT (n = 5) and ACE2-deficient mice (n = 6).
The absence of functional ACE2 causes enhanced susceptibility to Ang II–induced hypertension. No evidence for a role of ACE2 in the regulation of cardiac structure or function but the author suggest that ACE2 is a functional component of the renin-angiotensin system, metabolizing Ang II and thereby contributing to
regulation of blood pressure. Yamamoto et al. 2006 * 12 week-old ACE2 deletion mice and wild-type
(WT) mice were divided into 3 groups: sham-operation, transverse aortic constriction (TAC), and TAC during candesartan treatment.
ACE2 plays an important role in dampening the
hypertrophic response to pressure overload mediated by Ang II. Disruption of this regulatory function may accelerate cardiac hypertrophy and shorten the transition period from compensated hypertrophy to cardiac failure. Yang et al. 2006 * 3 single nucleotide polymorphisms in the ACE2
gene (1075A/G, 8790A/G and 16854G/C) were genotyped by PCR-RFLP
(restriction-fragment-length polymorphism) in 811 patients with CHD (of which 508 were patients with MI) and 905 normal controls in a Chinese
population.
Common genetic variants in the ACE2 gene might impact on MI in females, and may possibly interact with alcohol consumption to affect the risk of CHD and MI in Chinese males.
Table 1-2. Continued
Authors Year Experimental model and study design Key findings
Oudit et al. 2007 * ACE2 mutant mice develop a progressive age-dependent dilated cardiomyopathy with increased oxidative stress, neutrophilic infiltration, inflammatory cytokine and collagenase levels, mitogen-activated protein kinase (MAPK) activation and pathological hypertrophy.
* The AT1R blocker, irbesartan, prevented the dilated cardiomyopathy.
The age-dependent cardiomyopathy in ACE2 null mice is related to increased Ang II-mediated oxidative stress and neutrophilic infiltration via AT1R. The combination of genetic and pharmacological approaches defines a critical role of ACE2 in the suppression of Ang II-mediated heart failure.
Takeda et al. 2007 * DS rats and Dahl salt-resistant (DR) rats fed high or low salt diets.
* The rats were treated orally with or without eplerenone (100 mg/kg/d), candesartan (10 mg/kg/d), or both drugs combined for 8 weeks.
In DS rats, blockade of aldosterone or Ang II protects cardiac hypertrophy and fibrosis by inactivation of the local RAAS in the heart.
Dong et al. 2008 * Atherosclerotic plaques were induced in the
abdominal aorta of 114 rabbits by endothelial injury and atherogenic diet.
* Gene therapy was performed in 3 groups, a recombinant ACE2 expressing vector, a control vector AdEGFP and AdACE2+A779.
Overexpression of ACE2 results in stabilized
atherosclerotic plaques and the mechanism is probably the conversion of vasoconstrictive Ang II to vessel protective Ang 1-7.
Table 1-2. Continued
Authors Year Experimental model and study design Key findings
Koka et al. 2008 * 12 patients had been diagnosed with hypertensive nephropathy and 8 with hypertensive
cardiomyopathy.
* All hypertensive patients with unequivocal
hypertension were treated with either ACE inhibitor or AT1R blockers.
The AT1R-mediated ERK/p38 MAP kinase signaling pathway may be a key mechanism by which Ang II down-regulates ACE2 expression, implicating an ACE/ACE2 imbalance in hypertensive cardiovascular and renal damage.
Qin et al. 2008 * Suprarenal abdominal aortic coarctation was performed to create the pressure overload induced left ventricular hypertrophy model in rats.
* Rats were randomly divided into 5 groups: (1) normal control group (2) normal control group treated with atorvastatin (3) sham group (4) atorvastatin given orally by gastric gavage for 4 weeks (5) vehicle group.
ACE2 mRNA and its protein expression increase significantly in hypertrophic myocardium in rats; atorvastatin can attenuate cardiac hypertrophy due to pressure overload in rats effectively, and part of this anti-hypertrophy effect may be attributed to decrease ACE2 mRNA and protein expression.
Sluimer et al. 2008 * A total of 5 human veins, 5 non-diseased mammary arteries and 36 human atherosclerotic carotid arteries were collected from 46 donors undergoing vascular surgery.
Differential regulation of ACE2 activity during the progression of atherosclerosis and suggest that this novel molecule of the RAS may play a role in the pathogenesis of atherosclerosis.
Table 1-2. Continued
Authors Year Experimental model and study design Key findings
Ye et al. 2008 * SHRs, 12 weeks old, were randomly divided into 4 groups: the model control group (A), the Verapamil group (B), and the two puerarin groups (C and D) treated by low dose and high dose of puerarin respectively.
High dose puerarin could increase the mRNA expressions of AT1R and ACE2 in kidney, while low dose puerarin could decrease them in heart; there might be a feed back correlation between AT1R and ACE2.
1-6-2. Role of Ang 1-7 in recent studies of cardiovascular disease
In the studies of Ang 1-7, Ferreira et al. demonstrated that the nonpeptide Ang 1-7 analogue, AVE, attenuates postischemic heart failure and has a cardioprotective effect on ISO-induced cardiac remodeling [Clark et al., 2001; Heitsch et al., 2001; Ferreira et al., 2007a; Ferreira et al., 2007b]. Furthermore, deletion or blockade of Mas receptor markedly induced the changes in contractile function in isolated hearts during ischemia, thus Mas receptor plays an important role in cardiac function and keeps with the cardiac and coronary effects previously described for Ang 1-7 [Castro et al., 2006]. Ang 1-7 can reduce
hypertension-induced cardiac remodeling through a direct effect on the heart [Mercure et al., 2008], and it also has beneficial effects on the failing heart by activating the sodium pump, hyperpolarizing the cell membrane and increasing the conduction velocity.
An optimal generation of Ang 1-7 must be achieved to permit a protective role of Ang 1-7 on cardiac arrhythmias [De Mello et al., 2007]. These findings are in agreement with previous studies demonstrating that Ang 1-7 is a cardioprotective peptide. Moreover, Ang 1-7-forming activity from both Ang I and Ang II was increased in failing human heart ventricles [Zisman et al., 2003]. The studies of Ang 1-7 in regulation of cardiovascular disease in human studies and animal models were shown in Table 1-3.
Table 1-3. Studies of Ang 1-7 in regulation of cardiovascular disease in human studies and animal models Authors Year Experimental model and study design Key findings
Davie and McMurray
1999 * 8 patients with chronic heart failure.
* Patients were undergoing treatment with an ACE inhibitor.
* 5 patients taking enalapril 10 mg BID, 1 patient lisinopril 10 mg QD, 1 patient captopril 25 mg TID, and 1 patient perindopril 4 mg BID.
Ang 1-7 is biologically inactive in the forearm
circulation of patients with heart failure treated with an ACE inhibitor.
Zisman et al. 2003 * 35 patients undergoing open heart surgery. * 14 idiopathic dilated cardiomyopathy (IDC), 8
primary pulmonary hypertension (PPH), and 13 nonfailing human hearts.
Ang 1-7-forming activity from both Ang I and Ang II was increased in failing human heart ventricles.
Oudot et al. 2005 * 14 groups, each composed of six to nine hearts, were subjected to different perfusion protocols at 37°C. * Isolated perfused rat hearts underwent 45 min of
non-ischemic perfusion, or 30 min of global ischemia followed by 30 min of reperfusion.
Ang 1-7 at pharmacological concentration activates NADPH oxidase, an enzyme thought to be involved in several angiotensin II effects.