分子晶體結構之理論研究
116
0
0
全文
(2) 目錄 目錄 ....................................................................................................................I 圖目錄 ............................................................................................................. IV 表目錄 ............................................................................................................. VI 附錄圖 A 目錄 .............................................................................................. VII 附錄圖 B 目錄 ............................................................................................. VIII 附錄圖 C 目錄 ................................................................................................ IX 附錄圖 D 目錄 ................................................................................................. X 附錄表 E 目錄 ................................................................................................ XI 中文摘要 ........................................................................................................... 1 英文摘要 ........................................................................................................... 2 第一章 分子間作用力 ..................................................................................... 3 1.1 超分子化學 ......................................................................................... 3 1.2 分子間相互作用力 ............................................................................. 4 2.2.1 氫鍵 .......................................................................................... 5 2.2.2 π-π 相互作用力 ....................................................................... 6 2.2.3 凡德瓦力 ................................................................................. 9 第二章 能量分解 ........................................................................................... 11 2.1 交互作用能(Interaction Energy) ..................................................... 11 2.2 雙突變循環(Double-Mutant Cycles,DMC) .................................... 12 2.3 對稱性匹配微擾理論(Symmetry Adapted Perturbation Theory).... 14 I.
(3) 第三章 拓樸分析(Topology Analysis) .......................................................... 19 3.1Hirshfeld Suface ................................................................................. 19 3.1.1 3D-Graph: Mapping on the Surface ....................................... 21 3.1.2 2D-Graph:Fingerprint Plot ..................................................... 21 3.2 Atom in Molecules (AIM) ................................................................. 23 3.3 Electron Localization Function (ELF) .............................................. 24 3.4 Localized Orbital Locator (LOL) ...................................................... 26 3.5 Non-Covalent Interactions Plot (NCIPLOT) .................................... 26 3.5.1 區分非共價相互作用 ........................................................... 26 3.5.2 非共價相互作用類型 ........................................................... 27 第四章 結果討論 ........................................................................................... 29 4.1 不同取代基 Benzoylleucine Diethyl Amides 探討 ......................... 31 4.1.1 由 X-Ray 晶體資訊探討 ...................................................... 31 4.1.2 Hirshfeld Surface 與指紋圖之探討 ...................................... 33 4.2 不同位置含氟取代效應 ................................................................... 37 4.2.1 由 X-Ray 晶體資訊探討 ...................................................... 37 4.2.2 Hirshfeld surface 與指紋圖之探討 ....................................... 37 4.3 Reduced Model ................................................................................. 40 4.4 Ring Moldel....................................................................................... 43 4.4.1 ππ 相互作用力 ....................................................................... 43 4.4.2 ππ 相互作用力之能量分解................................................... 49 4.7 拓撲分析 .......................................................................................... 52 II.
(4) 4.7.1 單取代基探討 ....................................................................... 53 4.7.2 雙取代基探討 ....................................................................... 54 4.7.3 三取代基探討 ....................................................................... 55 第五章 結論 ................................................................................................... 57 第六章 參考文獻 ........................................................................................... 59 附錄 A ............................................................................................................. 64 附錄 B ............................................................................................................. 84 附錄 C ............................................................................................................. 88 附錄 D ............................................................................................................. 92 附錄 E.............................................................................................................. 98. III.
(5) 圖目錄 圖 1 描述氫鍵分子間的作用示意圖,雙箭頭表示次級的相互作用。D 表示氫鍵供給者,A 表示氫鍵提供者 ................................................... 6 圖 2 Double-Mutant Cycle 示意圖 ................................................................ 12 圖 3 Double-Mutant Cycle 示意圖 ................................................................ 13 圖 4 Benzoylleucine Diethyl Amides 的分子結構......................................... 29 圖 5 分子晶體以一個氫鍵形成二聚體之結構 (a)u-4Br (b)u-4Cl,(氫鍵以 虛線表示)................................................................................................ 32 圖 6 二聚體之結構(a)為 u-3OMe 為交叉雙氫鍵 (b)為 u-4CN 為平行雙氫 鍵(氫鍵以虛線表示) .............................................................................. 33 圖 7 u-4Me 分子晶體為 heterochiral,卻具有 π-stack(氫鍵以虛線表示) 35 圖 8 將八個取代 benzoylleucine diethyl amides 所有分子與分子接觸的作 用力用 2D Fingerpint Plot 表示 ............................................................. 39 圖 9 reduced model 結構圖 ........................................................................... 40 圖 10 MP2 及 SAPT0 之 π 結合能與 Σσp 關係圖,(a)為 jun-cc-pV(D+d)Z (b)為 6-31G*(0.25) ................................................................................. 45 圖 11 MP2 及 SAPT0 之相對 π 結合能與 Σσp 關係圖,(a)為 jun-ccpV(D+d)Z (b)為 6-31G*(0.25) ............................................................... 48 圖 12 SAPT0 之 π 結合能分解成各個能量,分別為色散能、交換能及靜 電能 ......................................................................................................... 51 圖 13 (a)3Me AIM 計算之臨界點 (b) 3OMe AIM 計算之臨界點 (c). IV.
(6) 4NO2 AIM 計算之臨界點 ...................................................................... 53 圖 14 (a) 3,5Me AIM 計算之臨界點(b) 3,5OMe AIM 計算之臨界點(c) 3,5NO2 AIM 計算之臨界點 ................................................................... 55 圖 15 (a)4-F AIM 計算之臨界點 (b) 3,5-F AIM 計算之臨界點 (c) 3,4,5-F AIM 計算之臨界點 ................................................................................ 56. V.
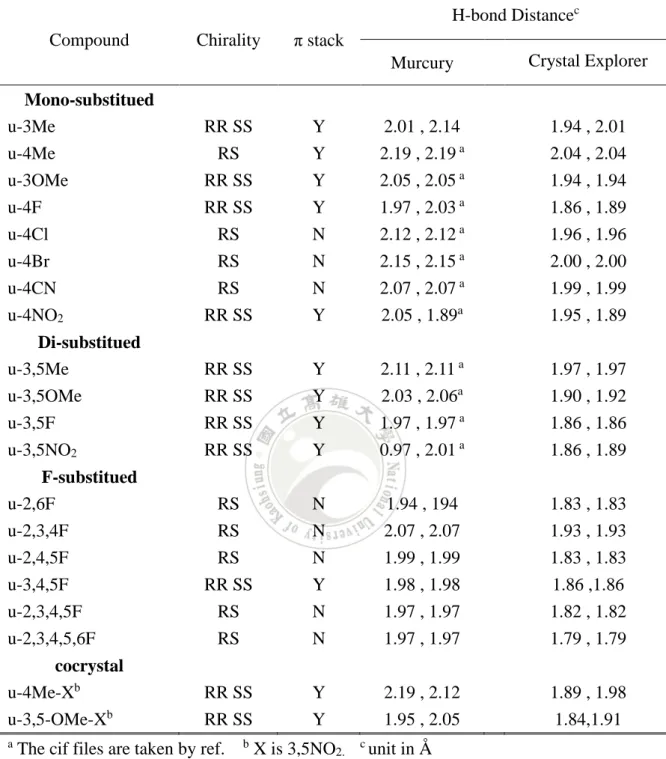
(7) 表目錄 表 1 Benzoylleucine Diethyl Amides 之 x-ray 晶體分析 .............................. 30 表 2 不同取代基在 Hirshfeld Surface 的相對貢獻度 .................................. 34 表 3 氟取代分子之分子間作用力比例 ......................................................... 37 表 4 r-3OMe 分子間作用力、氫鍵作用力、ππ 相互作用力 a.................... 41 表 5 不同模型 π 結合能計算 a 與晶體結構資訊 .......................................... 42 表 6 ring model 分子間結合能 a .................................................................... 44 表 7 以 SAPT 計算不同取代基的 ring model 結合能 ................................ 50. VI.
(8) 附錄圖 A 目錄 圖 A- 1 u-4NO2 Fingerprint plots ................................................................... 64 圖 A- 2 u-4CN Fingerprint plots ..................................................................... 65 圖 A- 3 u-4Br Fingerprint plots ...................................................................... 66 圖 A- 4 u-4Cl Fingerprint plots ....................................................................... 67 圖 A- 5 u-3OMe Fingerprint plots .................................................................. 68 圖 A- 6 u-4F Fingerprint plots ........................................................................ 69 圖 A- 7 u-3Me Fingerprint plots ..................................................................... 70 圖 A- 8 u-4Me Fingerprint plots ..................................................................... 71 圖 A- 9 u-3,5NO2 Fingerprint plots ................................................................. 72 圖 A- 10 u-3,5F Fingerprint plots ................................................................... 73 圖 A- 11 u-3,5OMe Fingerprint plots ............................................................. 74 圖 A- 12 u-3,5Me Fingerprint plots ................................................................ 75 圖 A- 13 u-2,3,4,5,6F Fingerprint plots .......................................................... 76 圖 A- 14 u-2,3,4,5F Fingerprint plots ............................................................. 77 圖 A- 15 u-2,3,4F Fingerprint plots ................................................................ 78 圖 A- 16 u-2,4,5F Fingerprint plots ................................................................ 79 圖 A- 17 u-3,4,5F Fingerprint plots ................................................................ 80 圖 A- 18 u-2,6F Fingerprint plots ................................................................... 81 圖 A- 19 u-3,5OMe-X Fingerprint plots ......................................................... 82 圖 A- 20 u-4Me-X Fingerprint plots ............................................................... 83. VII.
(9) 附錄圖 B 目錄 圖 B- 1 苯環二聚體之環距離為 3 Å (a) sign(λ2)對 RDG 之散點圖(b) RDG=0.5 之填色等值面圖 .................................................................... 84 圖 B- 2 苯環二聚體之環距離為 3.5 Å (a) sign(λ2)對 RDG 之散點圖(b) RDG=0.5 之填色等值面圖 .................................................................... 84 圖 B- 3 苯環二聚體之環距離為 3.9 Å (a) sign(λ2)對 RDG 之散點圖(b) RDG=0.5 之填色等值面圖 .................................................................... 85 圖 B- 4 苯環二聚體之環距離為 4.5 Å (a) sign(λ2)對 RDG 之散點圖(b) RDG=0.5 之填色等值面圖 .................................................................... 85 圖 B- 5 苯環二聚體之環距離為 5.0 Å (a) sign(λ2)對 RDG 之散點圖(b) RDG=0.5 之填色等值面圖 .................................................................... 86 圖 B- 6 苯環二聚體之環距離為 5.5 Å (a) sign(λ2)對 RDG 之散點圖(b) RDG=0.5 之填色等值面圖 .................................................................... 86 圖 B- 7 苯環二聚體之環距離為 6.0 Å (a) sign(λ2)對 RDG 之散點圖(b) RDG=0.5 之填色等值面圖 .................................................................... 87 圖 B- 8 苯環二聚體之環距離為 6.5 Å 之 sign(λ2)對 RDG 之散點圖 ......... 87. VIII.
(10) 附錄圖 C 目錄 圖 C- 1 水二聚體之環距離為 1.95 Å (a) sign(λ2)對 RDG 之散點圖(b) RDG=0.5 之填色等值面圖 .................................................................... 88 圖 C- 2 水二聚體之環距離為 2.5 Å (a) sign(λ2)對 RDG 之散點圖(b) RDG=0.5 之填色等值面圖 .................................................................... 88 圖 C- 3 水二聚體之環距離為 3.0 Å (a) sign(λ2)對 RDG 之散點圖(b) RDG=0.5 之填色等值面圖 .................................................................... 89 圖 C- 4 水二聚體之環距離為 3.5 Å (a) sign(λ2)對 RDG 之散點圖(b) RDG=0.5 之填色等值面圖 .................................................................... 89 圖 C- 5 水二聚體之環距離為 4.0 Å (a) sign(λ2)對 RDG 之散點圖(b) RDG=0.5 之填色等值面圖 .................................................................... 90 圖 C- 6 水二聚體之環距離為 4.5 Å (a) sign(λ2)對 RDG 之散點圖(b) RDG=0.5 之填色等值面圖 .................................................................... 90 圖 C- 7 水二聚體之環距離為 5.0 Å (a) sign(λ2)對 RDG 之散點圖(b) RDG=0.5 之填色等值面圖 .................................................................... 91. IX.
(11) 附錄圖 D 目錄 圖 D- 1 3Me (a) sign(λ2)對 RDG 之散點圖(b) RDG=0.5 之填色等值面 .... 92 圖 D- 2 3OMe (a) sign(λ2)對 RDG 之散點圖(b) RDG=0.5 之填色等值面 . 92 圖 D- 3 4F (a) sign(λ2)對 RDG 之散點圖(b) RDG=0.5 之填色等值面圖 ... 93 圖 D- 4 4NO2 (a) sign(λ2)對 RDG 之散點圖(b) RDG=0.5 之填色等值面圖93 圖 D- 5 3,5Me(a) sign(λ2)對 RDG 之散點圖(b) RDG=0.5 之填色等值面圖94 圖 D- 6 3,5OMe (a) sign(λ2)對 RDG 之散點圖(b) RDG=0.5 之填色等值面 圖 ............................................................................................................. 94 圖 D- 7 3,5F (a) sign(λ2)對 RDG 之散點圖(b) RDG=0.5 之填色等值面圖 95 圖 D- 8 3,5NO2 (a) sign(λ2)對 RDG 之散點圖(b) RDG=0.5 之填色等值面 圖 ............................................................................................................. 95 圖 D- 9 3,4,5F (a) sign(λ2)對 RDG 之散點圖(b) RDG=0.5 之填色等值面圖96 圖 D- 10 4Me-X (a) sign(λ2)對 RDG 之散點圖(b) RDG=0.5 之填色等值面 圖 ............................................................................................................. 96 圖 D- 11 3,5OMe-X (a) sign(λ2)對 RDG 之散點圖(b) RDG=0.5 之填色等 值面圖 ..................................................................................................... 97. X.
(12) 附錄表 E 目錄 表 E- 1 3Me AIM 拓撲分析 (unit in a.u.) ..................................................... 98 表 E- 2 4F AIM 拓撲分析 (unit in a.u.) ........................................................ 98 表 E- 3 3OMe AIM 拓撲分析 (unit in a.u.) ................................................ 99 表 E- 4 4NO2 AIM 拓撲分析 (unit in a.u.).................................................. 99 表 E- 5 3,5Me AIM 拓撲分析 (unit in a.u.) ................................................ 100 表 E- 6 3,5OMe AIM 拓撲分析 (unit in a.u.) ............................................. 101 表 E- 7 3,5F AIM 拓撲分析 (unit in a.u.) ................................................... 102 表 E- 8 35NO2 AIM 拓撲分析 (unit in a.u.)................................................ 102 表 E- 9 345F AIM 拓撲分析 (unit in a.u.) .................................................. 103 表 E- 10 4Me-X AIM 拓撲分析 (unit in a.u.) ............................................. 103 表 E- 11 35OMe-X AIM 拓撲分析 (unit in a.u.) ........................................ 104. XI.
(13) 分子晶體結構之理論研究 指導教授:莊曜遠博士 國立高雄大學應用化學系 學生:羅珮溱 國立高雄大學應用化學系 摘要 本研究進行晶體分析與量子化學計算,一系列具不同取代基的 Benzoylleucine Diethyl Amides(BDA)的晶體,利用 Hirshfeld Surface 分析,由% C…C 值篩選出具 有 π-π 相互作用的分子晶體,大部分具有 π-π 相互作用 BDA 二聚體為相同掌性, 此與固有文獻所得結果相同;然相較於以往之研究,本文利用指紋圖定量分析多種 類型作用力於分子堆疊的貢獻度。 為量化不同取代基對 π-π 相互作用的影響,利用 MP2 計算二聚體,發現結合 能與 Hammett constat(Σσp)具有相關性,也以 SAPT 進行分子間相互作用能量分解分 析,發現色散力在分子間交互作用是重要的。經由 AIM、NCI 電子密度拓撲法來圖 解相互作用力,可視化這些不同類型的作用力。從計算結果發現分子間的鍵臨界點 的 potential energy 與二聚體的結合能的關聯。 藉由分析 ring model 與 reduced model 的相互作用力,進一步了解分子晶體堆 疊的方式與不同分子間作用力的協同作用,以達到利用理論計算從事分子識別研究 的目的。. 關鍵字:Hirshfeld表面、π-π相互作用力、取代基效應、拓撲分析、能量分解、分子 晶體. 1.
(14) Theoretical Study of Molecular Crystal Structure Advisor: Dr. Yao-Yuan Chuang Department of Applied Chemistry National University of Kaohsiung Student: Pei-Chen Lo Department of Applied Chemistry National University of Kaohsiung ABSTRACT. We carried out both quantum calculations and crystal engineering analysis to a series of substituted Benzoylleucine Diethyl Amides (BDA) molecules which were synthesized for the Chiral Solid Phase usage. Compare to previous crystal structure analysis, we apply the Hirshfeld Surface analysis to the BDA and found the BDA dimers with π-π interaction are homochiral which is in consistent with literature. We further quantify the contributions of distinct atoms to different types of the intermolecular forces between BDA dimmers by means of fingerprint plots. We performed Sencond-Order Moller-Plesset Perturbation Theory (MP2) calculations to the ring model with various substituents.Good correalation is observed between the Hammett constant (Σσp) and the binding energy. We also analyzed the energy components of binding energy by Symmetry Adapted Perturbation Theory (SAPT) which indicated that dispersion forces is important. In order to visualize the noncovalent interactions, topological analysis such as Atom In Molecule and Non-Covalent Interaction were applied. We found the electron density at the Bond Critical Point (BCP) can be used to estimate the binding energy. We hope by investigating the cooperation between the intermolecular forces, we are able to explain the formation of BDA dimers and hence to assist the design of new system for Chiral Solid Phase separation and at the same time gain more insight on molecular recognition using theoretical methods. Keywords: Hirshfeld Surface, π-πinteraction, substituent effect, topological analysis, energy decompose, molecular crystal. 2.
(15) 第一章 分子間作用力 有機分子晶體近年來發展快速,其潛在的應用前景亦有著明顯的優 勢,它的構型與組成方式是一極為新興的議題。結晶相關的研究在進行 的過程中,預知晶體的構型對相關結晶的生成及性質具有極為重要的影 響;結晶經常被應用在各種產品上如壓克力顏料 1、醫藥 2、半導體材 料 3 等等。研究晶體幾何排列不僅有助於我們了解對晶體生長的過程, 對晶體性質的認識更有極為重要的意義。 分子間作用力經常被利用於討論分子識別 4 現象,例如酶催化過程 中,酵素與其基體分子之間可透過不同種類的分子作用力結合,或者可 利用具掌性化合物的管柱以高效能液相層析法分離 5-6。分子間的作用 力依強弱可分為:凡德瓦力(含:分散力、取向力與誘導力)及次級鍵如氫 鍵(XH…Y)。我們利用能量分解方法(如 Symmetry Adapted Perturbation Theory)及電子密度的拓撲分析(如 Atom in Molecule、Electron Localize Function),試圖分析此些作用力,以探討分子間作用力在分子晶體中的 堆疊方式。以下,本章將針對分子間作用力與拓樸分析方法分別為其定 義及說明。. 1.1 超分子化學 超分子化學(Supramolecular Chemistry)是一研究分子間相互作用而 形成複雜有序且具有特定功能的分子聚集體的科學,它是由 Lehn7 所定 義出來的,Lehn 也因而被稱為「超分子化學之父」 。因分子間作用力極 3.
(16) 微弱,如 DNA 雙股螺旋結構內鹼基對堆積作用形成有序的組合 8(在 DNA 中,A 與 T 配對 C 與 G 配對,其配對都以氫鍵聯繫)、酶的催化 作用、神經系統的訊息傳遞等等,故些許能量的輸入便可改變,造成實 驗量測的困難。超分子化學主要係研究超分子體系中分子間相互作用, 並從這些研究衍生出一些新的概念,如分子識別(Molecular Recognition); 分子識別是指兩個或兩個以上的分子透過例如氫鍵、金屬鍵、親水性、 凡德瓦力、π-π 相互作用及靜電力等的非共價鍵結特殊反應。結晶過程 是一種高度精確的分子識別實例,可以想像分子識別如同是”鎖與鑰匙” 的分子間專一性的結合,是形成超分子結構的基礎,且有關超分子化學 這方面的研究已被應用在許多科學領域中,可謂化學分支中目前最矚目 的科學領域。9. 1.2 分子間相互作用力 晶體工程係指"通過分子堆積了解分子間相互作用(Intermolecular Interaction),用以設計具有特定的物理性質和化學性質的新晶體。"晶 體工程一詞是於 1971 年由 Schmidt 在感光二聚作用(photodimerization) 的文章中所提出 10,晶體工程和分子識別的概念極為相似,皆是以分子 間的相互作用操縱超分子組裝的技藝。在已知晶體結構下合理化這些分 子間相互作用,此方法必須滿足 Desiraju 在 1997 發表的文章 11 中所提 到的,我們所需要的是能將整個晶體可視化,而非只選擇觀察被認定為 重要的相互作用而已。從而,「晶體工程」的研究可以幫助我們去了解. 4.
(17) 分子間作用力與分子堆疊的關係,並藉此來設計新的固態超分子結構與 材料。分子間相互作用相當複雜,控制分子在晶體中位向之排列,除了 最熟悉的氫鍵以外,π-π 相互作用亦為另一種會影響分子晶體的堆疊。 因此,氫鍵與 π-π 作用力為本研究中主要探討的作用力,此外,尚有一 些分子間作用力如靜電作用力、偶極-偶極相互作用力(Dipole-Dipole Interactions)、鹵素之間等等的作用力,在分子晶體中這些大大小小的 作用力都會影響它們在固態空間排列與堆疊的方式。. 2.2.1 氫鍵 一般分子設計均利用氫鍵以控制及設計 Synthon,因其作用力較強 且為我們目前最了解的部分,氫鍵係定義為氫原子接在電負度較高的原 子與電負度較高的原子所形成,如 N-H…O/N 或 O-H…N/H 等等。氫鍵 供 給 者 (Hydrogen Bond Donor , D) 和 氫 鍵 接 受 者 (Hydrogen Bond Acceptor,A),來描述氫鍵分子間的作用表示,以圖 1 為例 12,氫鍵以 虛線表示,次級的相互作用以雙箭頭表示,圖 1 可用 DA-AD 表示。本 研究中晶體氫鍵形成 Homochiral Dimer 類型為 DD-AA,而 Heterochiral 類型為 DA-AD。. 5.
(18) 圖 1 描述氫鍵分子間的作用示意圖,雙箭頭表示次級的相互作用。D 表示氫鍵供給者,A 表示氫鍵提供者. 氫鍵主要是靜電作用所造成,當多個氫鍵存在時,除了要考慮主要 吸引的相互作用,次級相互作用也必須加入探討,因這些次級相互作用 有可能是吸引力或排斥力。當系統含有三個平行氫鍵時,計算次級相互 作用能量約為 7kJmol-113,藉由這些次級相互作用的計算可預測育有兩 個氫鍵的 DD-AA 排序會比 DA-AD 排序更有利,三個氫鍵的排序以此 類推。本文所欲探討的分子晶體都是具有掌性且產生的兩個氫鍵呈現交 叉,而非如上所述氫鍵都是平行鍵結的,但依然可用上述方法來來描述 氫鍵分子間的作用。. 2.2.2 π-π 相互作用力 經過文獻搜尋,我們發現大部分研究 π-π 相互作用力,均討論三種 苯環二聚體的排列方式,Sandwich configuration(S,或稱 face-to face)是 兩個苯環彼此平行重疊,T shaped configuration (T,或稱 edge-to-face) 是一個苯環指向另一苯環的中心,Parallel-displaced configuration (PD, 6.
(19) 或稱 offset stacking)是一個苯環平行位移後的結構。14 Hunter 和 Sanders 提出了 π 系統三明治構型靜電模型,用以說明為 何芳香環會面對面堆疊(stacking)15,兩個芳香環間是互相排斥的,因為 兩端皆為帶負電的 π 電子雲,而非我們一直所認知的兩個芳香環是互 相吸引進而堆疊。在 Hunter 和 Sanders 確認這些模型的有效性後,他們 統整成一個定則以廣泛解釋這三種構型形成的原因,y 軸代表的是上方 帶正電的 π 原子逆時針旋轉的角度,而 x 軸代表 π 原子往右手邊平移 的距離,我們可清楚的看到 face-to-face(角度=0,偏移=0)落在排斥區, 主要屬於 π-π 排斥所致,而 T 型(角度=90,偏移=0)和 Paralleldisplaced(角度=0,有偏移)落在相互吸引區,則屬於 π-σ 吸引所致。 取代基的效應對於芳香環堆疊方式的影響也是很重要的,普遍對苯 環二聚體取代基影響的看法是 Hunter 等人所提出的 polar/π 模型 15。他 們認為在 polar/π 模型中,若取代基是拉電子基(如 CN、NO2 等)會減少 π 電子雲的電子密度,而降低與另一 π 系統中相互作用的相互斥力,因 此與無取代基的二聚體比較,其 π-π 結合能會增強;反之,若為推電子 基(如 CH3、OCH3 等),則 π-π 結合能會減弱。 但 Sherrill 等人利用理論計算探討取代基效應,得出的結果卻與 Hunter 和 Sander 所提出的理論相矛。利用 CCSD(T)/aug-cc-pVTZ 計算 三明治構型的苯環-單取代苯環二聚體系統,發現取代基為 OH 和 CH3(推電子基)時,其相互作用的能量竟比不具有取代基的苯環二聚體 時穩定, Sherrill 等人認為大部分的取代基,不管是推電子基亦或是拉. 7.
(20) 電子基都會增加 π-π 相互作用的強度 14。 Sherrill 接著以 Symmetry Adapted perturbationtheory (SAPT)將 π-π 相互作用分成四個部分,靜電力(Electrostatic)、分散力(Dispersion)、誘 導力(Induction)、交換排斥力(Exchange-epulsion),發現分子間作用力主 要為分散力,將再後面章節詳細說明 SAPT。 Houk 和 Wheeler 在 2008 年利用 M05-2X/6-31+G(d)計算三明治構 型之單取代苯環-苯環二聚體及單取代苯環-六氟苯二聚體,發現 Hammett Constant(σm)與結合能有良好線性相關性(R2=0.94),可能是因 取 代 基 和 未 取 代 的 芳 香 環 直 接 靜 電 交 互 作 用 (direct electrostatic interactions)與其分散力相互作用,才是堆疊成三明治構型的原因。16 Hammett Constant 是各種取代基常數 σm、σp 是通過量測間位、對位 取代苯甲酸上的氫,在 25 度水溶液中的 pKa 值所得到的。σ 為負代表 取代的苯甲酸之酸性比苯甲酸小;σ 為正則表示取代的苯甲酸之酸性比 苯甲酸大。一般取代基常數絕對值 σp 會比 σm 大,其原因為在對位的取 代基除了具有 inductive effect,還具有 resonance effect,然而間位取代 只有 inductive effect。以甲氧基(-OCH3)為例,若在間位,取代基屬於拉 電子基(inductive effect);若在對位,取代基則屬於推電子基(resonance effect)。17 Houk 和 Wheeler 在 2008 年提出取代基效應是由於三明治構型中, 取代基與另一環上的相互作用而使二聚體更穩定。16Wheeler 又單獨提 出了另一種模型來解釋取代基效應稱之為 Local,Direct Interaction Model,. 8.
(21) 其認為取代基只會與靠近苯環的位置作用而使 π-π 交互作用穩定,而非 與整個苯環作用。所以苯環-具取代苯環二聚體可視為取代基與丙烯的 作用,在進行多種類型芳香環堆疊相互作用計算後結果也與其一致,此 模型是以一個簡單且直觀的觀點來預測芳香環堆疊的相互作用。18. 2.2.3 凡德瓦力 凡德瓦力(Van der Waals Force)是一種分子間相互作用力,其來源主 要有三種方式,第一種是極性分子跟極性分子間相互作用產生的取向力 (Keesom Force 或 Permanent Dipole Force),第二種是非極性分子與極性 分子間的產生的誘導力(Debye Force 或 Induced dipole Force),第三種是 非極性與非極性分子間產生的分散力(Dispersion Force),亦是凡德瓦力 最主要的來源,因其在所有分子中皆會發生,故又將分散力俗稱為凡德 瓦力 19。 分散力的產生是因為原子的電子雲會瞬間分布不均,產生短暫的偶 極,此瞬間偶極矩會使附近也出現類似的偶極矩,而偶極-偶極相互作 用就是 van der Waals Force。 如何準確地定量 π-π 相互作用力是本文所主要探討與計算的部分; 然而,由於實驗數據的缺乏,要精確地描述色散力具有一定的難度,因 目前廣泛使用 Kohn-Sham Density Functional Theory (DFT)理論計算,因 其區域性電子密度泛涵(Local Density Functional Approximation)無法描 述色散力,如何準確地估算 Dispersion Force 亦為本文所欲討論之議題。 DFT 方法中,利用近似電子密度泛涵簡化其計算方式,因此可以計算. 9.
(22) 中大型系統,這些密度泛涵稱為 Exchange-Correlation(XC) Functional, 標準的 XC functional 包括局部密度近似法(LDA)、廣義梯度近似法 (GGA)或 hybrid XC functional(如 B3LYP),但基於區域性的假設因素, 中遠程相互作用無法利用目前已知的 XC Functional 來加以計算。. 10.
(23) 第二章 能量分解 2.1 交互作用能(Interaction Energy) 我們要計算單體 A 與 B 的分子間作用力,其計算統一稱為 π 結合 𝐴𝐵 能(π Binding Energy),其公式如(1),𝐸𝐴𝐵 (𝐴𝐵)表示用 AB 雙分子能量是. 用 A 與 B 基底函數計算出的能量,𝐸𝐴𝐴 (𝐴)代表用單體 A 的基底函數計 算單體 A 能量,𝐸𝐵𝐵 (𝐵) 代表用單體 B 的基底函數計算單體 B 能量。 𝐴𝐵 ∆Ebind(𝐴𝐵) = 𝐸𝐴𝐵 (𝐴𝐵) − 𝐸𝐴𝐴 (𝐴) − 𝐸𝐵𝐵 (𝐵). (1). 但(1)所算出來的能量會有 Basis Set Superposition Error (BSSE),此 誤差來源是由於作用的二聚體與其所構成的單分子所用基底函數數目不 同,而高估了分子之間真正的交互作用能量。而移除這個誤差的其中一 個方法為 Counterpoise (CP)20,單體 A 及單體 B 的 BSSE 可用(2)及(3)表 示,𝐸𝐴𝐴𝐵 表示以 A 的基底函數加上 B 的基底函數計算單體 A 的能量, 單體 B 則依此類推。 E𝐵𝑆𝑆𝐸(𝐴) = 𝐸𝐴𝐴𝐵 (𝐴) − 𝐸𝐴𝐴 (𝐴). (2). E𝐵𝑆𝑆𝐸(𝐴) = 𝐸𝐵𝐴𝐵 (𝐴) − 𝐸𝐵𝐵 (𝐵). (3). 𝐶𝑃 (𝐴𝐵)如(4),在計算單體 A、單體 B、雙聚 修正後的能量為∆E𝑏𝑖𝑛𝑑. 體 AB 都使用相同的基底函數,計算的能量才會較為準確,因此在本研 究中計算皆使用 counterpoise 校正。 𝐶𝑃 𝐴𝐵 (𝐴𝐵) = 𝐸𝐴𝐵 ∆E𝑏𝑖𝑛𝑑 − 𝐸𝐴𝐴𝐵 − 𝐸𝐵𝐴𝐵. 相對於位取代的 π 結合能則可以用下式表示. 11. (4).
(24) 𝐶𝑃 𝐶𝑃 𝐶𝑃 ∆∆E𝑟𝑒𝑙𝑎𝑡𝑖𝑣𝑒 = ∆E𝑏𝑖𝑛𝑑 (𝐴𝐵𝑠𝑢𝑏 ) − ∆𝐸𝑏𝑖𝑛𝑑 (𝐴𝐵𝑛𝑜𝑠𝑢𝑏 ). (5). 2.2 雙突變循環(Double-Mutant Cycles,DMC) 雙突變循環(Double-Mutant Cycle,DMC)方法被使用於在定量蛋白質 中的非共價鍵相互作用和協同效應(Cooperativity),在 1984 年 Fersht 提 用 DMC 來評估酶的協同相互作用 21 藉此了解其結合特性如下圖 2,此 方法是利用將單一的 X-mutation 用 X'-mutation 取代所改變的自由能(即 ∆G𝑋𝑌→𝑋′𝑌 )會與 Y-mutation 用 Y'- mutation 取代改變的自由能(∆G𝑋𝑌→𝑋𝑌′ ) 不同,因此兩個自由能的差值(即∆G𝑋𝑌→𝑋′𝑌 − ∆G𝑋𝑌→𝑋𝑌′ )直接或間接提供 測量 X-mutation 與 Y-mutation 之間的相互作用力大小。使用此方法來 定量蛋白質之間非共價相互作用現在已成為標準作法 22。. 圖 2 Double-Mutant Cycle 示意圖. DMC 也應用在超分子複合物(supramolecular complexes),如圖 323, 利用 DMC 對複合物 A 的 x 和 y 官能基相互作用力進行定量,粗線代表 12.
(25) 主要的非共價相互作用,而細線代表次級效應(secondary effect)會在循 環中被抵銷掉,此方法一個最主要的優點是小的熱力學貢獻(即大約 1kJmol-1)也可測量。. 圖 3 Double-Mutant Cycle 示意圖. Hunter 等人利用 DMC 測量 Edge-To-Face 構型苯環雙聚體的取代 基效應 24,他們合成出具氫鍵的 zipper complexes,以 1H NMR 及 CDCl3 中,溫度為 295K 下的條件,定量不同取代基取代於苯環所造成 相互作用力的自由能,其結果顯示與 Hammett substituent constants 有良 好的相關性。後來,也用來測量 Face-to-Face 芳香環相互作用力的大小 ,在條件為 CDCl3 中,溫度為 293K 下,量測合成具氫鍵 zipper. 25. complexes 的自由能,其結果也顯示了苯環的堆疊交互作用與 Hammett substituent constants 有良好的相關性,在本研究中也建構 DMC 來定量 13.
(26) π-π 相互作用探討取代基效應,將芳香環的取代基以氫原子進行取代進 行量子計算。. 2.3 對稱性匹配微擾理論(Symmetry Adapted Perturbation Theory) 對稱性匹配微擾理論(Symmetry Adapted Perturbation Theory,SAPT) 是計算分子間相互作用的一種方法。SAPT 將相互作用能量分解為靜電 能(electrostatic)、誘導能(induction)、色散能(dispersion)和交換能 (Exchange)。SAPT 直接計算分子間相互作用,二聚體的 Hamiltonian 算 符可以下式表示 26 H = 𝐹𝐴 + 𝐹𝐵 + 𝑊𝐴 + 𝑊𝐵 + 𝑉. (6). 其中𝐹𝐴 、𝐹𝐵 分別是單體 A、B 的 Fock 算符,𝑊𝐴 、𝑊𝐵 是分子內單體 微擾算符,V 是分子 A、B 間相互作用的算符。SAPT 有不同的計算公 式,常用的有 SAPT0、SAPT2 及 SAPT,SAPT0 適用於較大體系,綜 合考慮計算資源及計算精度的需求,於本文中所計算的皆採用 SAPT0 完成,其公式可表達為 (10). (10). (20). (20). (20). (20). 𝐸𝑆𝐴𝑃𝑇0 = 𝐸𝑒𝑙𝑠𝑡 + 𝐸𝑒𝑥𝑐ℎ + 𝐸𝑖𝑛𝑑 + 𝐸𝑒𝑥𝑐ℎ−𝑖𝑛𝑑,𝑟𝑒𝑠𝑝 + 𝐸𝑑𝑖𝑠𝑝 + 𝐸𝑒𝑥𝑐ℎ−𝑑𝑖𝑠𝑝 式中括號第一個數字為 V 的微擾階數,第二個為 W 為擾階數,根 據上述公式其物理意義可分為下列四種能量. 14. (7).
(27) (10). E𝑒𝑙𝑠𝑡 = 𝐸𝑒𝑙𝑠𝑡. (20). (8) (20). E𝑖𝑛𝑑 = 𝐸𝑖𝑛𝑑 + 𝐸𝑒𝑥𝑐ℎ−𝑖𝑛𝑑,𝑟𝑒𝑠𝑝 (20). (9). (20). E𝑑𝑖𝑠𝑝 = 𝐸𝑑𝑖𝑠𝑝 + 𝐸𝑒𝑥𝑐ℎ−𝑑𝑖𝑠𝑝. (10). (10). E𝑒𝑥𝑐ℎ = 𝐸𝑒𝑥𝑐ℎ. (11). 靜電能(E𝑒𝑙𝑠𝑡 )是計算兩個分子上電荷之間的庫倫作用所產生的能量, 在 SAPT 理論架構下屬於 Polarization Energy 的一階校正項,其公式為 3. 1. 3. 𝑡𝑜𝑡 𝑡𝑜𝑡 E(10) 𝑒𝑙𝑠𝑡 = ∫ ∫ 𝑄𝐴 (𝒓𝟏 ) 𝑟 𝑄𝐵 (𝒓𝟐 )𝑑 𝒓𝟏 𝑑 𝒓𝟐. (12). ( ) ∑ ( ) Q𝑡𝑜𝑡 𝐴 𝒓 = 𝛼 𝑍𝛼 𝛿 𝒓 − 𝑹𝛼 − 𝑄𝐴 (𝒓). (13). 12. 𝑡𝑜𝑡 其中𝑄𝑡𝑜𝑡 𝐴 、𝑄𝐵 未考慮微擾之單體 A 與 B 的電荷分布、R 為原子核. 座標、r 為電子座標。 誘導能(E𝑖𝑛𝑑 ) 則是考慮單體本身的電子雲也受其他單體所影響,造 成單體本身的電子雲重新排列產生極化現象產生永久多極矩,在此重新 (20). 排列下與另一單體所產生的庫倫作用力我們稱之誘導能,E𝑖𝑛𝑑 屬於 (2). (2). (2). Polarization Energy 的二階校正項,E𝑖𝑛𝑑 = 𝐸𝑖𝑛𝑑 (𝐴) + 𝐸𝑖𝑛𝑑 (𝐵),針對單體 A 公式如下,其中Ω𝐵 為未微擾之 B 單體產生的靜電位能 ̂. (20). E𝑖𝑛𝑑 (𝐴) = −< 𝛷𝐴 |𝛺𝐵 𝑅𝐴0 𝛺𝐵 |𝛷𝐴 >. (14). Ω𝐵 = ∑𝑖∈𝐴 𝜔𝐵 (𝒓𝒊 ). (15). 1. 3. ω𝐵 (𝒓𝒊 ) = ∫ 𝑟 𝑄𝑡𝑜𝑡 𝐵 (𝒓𝒋 )𝑑 𝒓𝒋. (16). 𝑖𝑗. 色散能(E𝑑𝑖𝑠𝑝 )指分子間電子的 Correlation Energy,微觀上因改變一 個電子位置而其他電子位置也會馬上改變所修正的能量,我們稱之為 15.
(28) Correlation Energy,也可看成是單體間瞬間偶極矩的交互作用,其公式 如下 (20) 𝐴𝐵 E𝑑𝑖𝑠𝑝 = −< 𝛷0 |𝑉𝑅̂ 0 𝑉|𝛷0 >. (17). 交換能(E𝑒𝑥𝑐ℎ )是交換排斥能量(Exchange-Repulsion Energy),但有些 學者會簡稱為交換能,交換能的產生是因為兩個單體靠近時必須遵守包 立不相容原理(Pauli exclusion principle),在兩個閉殼層(colsed-shell)分 子相互作用的電子必為相同自旋,而使得電子必須重新排列此能量即為 交換能。除了上述校正,前述每一項能量如誘導力及色散力也可以加入 (20). (20). 考慮 exchange effect 即𝐸𝑒𝑥𝑐ℎ−𝑖𝑛𝑑,𝑟𝑒𝑠𝑝 及𝐸𝑒𝑥𝑐ℎ−𝑖𝑛𝑑,𝑟𝑒𝑠𝑝 。 Sherrill 利用 SAPT2/aug-cc-pVDZ′計算不同構型的苯環二聚體, 其結果顯示分散力決定二聚體結合能的主要貢獻,而靜電力是其次,因 隨著距離增加三明治構型苯環二聚體靜電力會 destabilizing,因此單憑 靜電效應的模型預測 π-π 作用力是不可靠的。27 Sherill 等人也做了一系列不同單邊單取代苯環三明治構型二聚體的 SAPT 計算,發現屬於取代基為拉電子基的 CN、F 符合 Hunter-Sanders 所提出的模型,但是對於弱推電子基 CH3,相互作用能最大的改變並非 是靜電作用而是色散作用,因此他們認為色散對於了解在氣相計算二聚 體結合很重要。28 此後 Houk 和 Wheeler 在 2008 年利用 M05-2X/6-31+G(d)計算三明 治構型之單取代苯環-苯環二聚體及單取代苯環-六氟苯二聚體,其結果 成 功 複 製 Sherrill 等 人 所 計 算 結 合 能 的 結 果. 16. 14. ,同時他們也發現.
(29) Hammett Constant(σm)與結合能有良好相關性(R2=0.94),可能是因為取 代基和未取代的芳香環直接靜電交互作用(direct electrostatic interactions) 與其分散力相互作用,才是苯環堆疊成三明治構型的原因。16 而 Lewis 等人則作了一系列以多鹵素取代基的苯環,形成苯環三明 治構型研究,發現∑ 𝜎𝑝 與結合能呈現良好的線性相關性,其中較特別的 是,有計算 BSSE 相關係數(R2=0.56)竟比為計算 BSSE(R2=0.82)低。 這些結果被解釋為支持了 Hunter-Sanders 模型,因芳香環之間的作 用力仍以靜電力為主。在 Houk 和 Wheeler 發表後此結果後,Sherrill 等 人作了一系列單取代以及多取代基苯環的研究,並無發現取代基與 Hammett 𝜎𝑚 有線性相關。29 其實他們研究得出的結果不同最主要的原 因是在於 Sherill 等人所計算的取代基拉電子及推電子數目是相同的, 而 Houk 和 Wheeler 所作的計算 24 個取代基裡只有 5 個是推電子基, 其餘則為拉電子基。 此後,Lewis 等人提出了另一個看法,他們不否認 Sherill 等人認為 結合能主要是色散力主導,但他們也認為要預測苯環二聚體之結合能需 要考慮靜電能,因此利用了 MP2(full)/6-311**計算了不同取代基二聚體 結合能,其結果顯示 Hammett Constant(∑|𝜎𝑚 |)與結合能有良好的線性 相關(R2=0.9)。 Lewis 等人也利用 SAPT 去探討取代基效應,利用 CCSD/6-311G** 計算不同取代基的能量分解,從計算結果顯示分子間主要的吸引力為色 散力,但將色散力、交換能、誘導能能量相加可發現其總和大約為一個. 17.
(30) 常數值,而靜電能有大幅度的變化,這些顯示當預測苯環取代的結合能 (𝐸𝑏𝑖𝑛𝑑𝑖𝑛𝑔 )靜電能是重要的。30 但 Sherill 等人利用 SAPT0/aug-cc-pVDZ’ 進行如上結構計算,其結 果與 Lewis 等人結果相似,但在之前並無人使用∑|𝜎𝑚 |當作預測相互作 用能量的例子,因此無法確定在未來是否可證明其可使用。同時,該研 究也顯示 ππ 相互作用中的靜電力與其取代基效應,利用 Distributed Multipole Analysis(DMA)計算,發現關鍵 Charge Penetration Effect,也 就是當兩個分子很靠近時,單體上電子會受另一個單體的原子核吸引, 當這個吸引力大過電子-電子與原子核-原子核的排斥能時則二聚體體可 以穩定,Charge Penetration Effect 隨著單體之間距離變近會更明顯。31. 18.
(31) 第三章 拓樸分析(Topology Analysis) 3.1Hirshfeld Suface 近年來,Hirshfeld Surface32 已成為分析分子晶體結構重要的工具, 對於可視化、探索、合理化分子晶體結構的組成,其提供很大的潛力, 如比較萘(Naphthalene)相似於蔥(Anthracene)的相似度大於萘相似於苯 (Benzene)的程度,這種分析方法賦予抽象的類型比對概念一種定量的 指標,這種定量指標具有一定的重要性,因晶體結構相似也可能代表他 們擁有相似的性質。Hirshfeld Surface33 是在 1997 年由 Spackman 所提 出 為 劃 分 分 子 在 晶 體 的 佔 據 空 間 而 所 產 生 的 一 種 方 法 , Hirshfeld Surface 是以 F.L Hirshfeld 的名字命名的,因 Hirshfeld 提出一種在分子 中劃分原子(Atom in Molecules)的方法(Stockholder Partition Scheme),這 使 Spackman 聯想出可以將其擴大定義在晶體中劃分出分子(Molecule in Crystal)。 Hirshfeld 藉由數值積分分子內原子的電子密度得到原子的電荷和其 它性質,它對分子中每一個原子定義了一個 weight function 𝑎𝑡. w𝑎 (r) = ρ𝑎𝑡 𝑎 (𝑟)/ ∑𝑖∈𝑚𝑜𝑙𝑒𝑐𝑢𝑙𝑒 ρ𝑖 (𝑟). (18). ρ𝑎𝑡 𝑖 (𝑟):原子電子密度對角度方向取平均的徑向電子密度(spherically averaged electron densities)以此方式可將分子片段(molecule fragment)的 電子密度定義為如下 ρ𝑎 (𝑟) = w𝑎 (r)ρ𝑚𝑜𝑙 (𝑟). (19). ρ𝑚𝑜𝑙 (𝑟):為分子電子密度 19.
(32) 因原子的電子密度在核附近為尖峰,離核距離增加會成指數衰減, 但(17)的 wa(r)在空間中是一個連續的純量函數(Scalar Function),範圍從 1(原子核)到 0(遠離原子核),式(18)分母是分子中的各個原子密度總和 (原子的 weight function 總和在空間上任一點都必須為 1),因此式 4ρ𝑎 (𝑟)總和會等於ρ𝑚𝑜𝑙 (𝑟),意味著 Hirshfeld 的 Partitioning Scheme 詳 盡討論了所有空間,但被劃分的原子(atomic fragment)會重疊而不像 QT-AIM(Quantum Theory of Atoms in Molecules)中是分開的。 將 Hirshfeld Surface 定義為 WA(r)=0.5 時,這時原子電子密度在這 個表面的變化最小,而且它能確保最接近相鄰分子體積,惟體積不會重 疊。Spackman 等人也做了 Hirsfeld Surface 與其他 surface 比較,他們將 Hirsfeld Surface 與 0.002au(使用 ab initio 計算)及 CPK 比較, Hirsfeld Surface 一般都會包覆住 CPK 和 0.002au 的等電子密度表面,除了與相 鄰分子緊密接觸的部分,這就是 Hirsfeld surface 與其他 surface 不同的 地方,因為其他分子表面只定義分子本身,而 Hirsfeld Surface 是由分 子和鄰近最近的分子來做定義,因此這個表面具有有關分子間作用的資 訊。34 用來 de、di 距離與 Hirshfeld Surface 關係,di 為從表面到表面內部 最近原子的距離,de 為從表面到表面外部最近原子的距離,將這些距離 應用在二維和三維的圖上,使我們便於在紙上閱讀,又三維的圖能對於 在空間中整個分子一目了然。35 以下分別介紹其優缺點及發展。. 20.
(33) 3.1.1 3D-Graph: Mapping on the Surface de 及 di 最早的發展是應用於 3D 表面上,依據不同顏色區分距離長 度,從短距離接觸(紅色)經綠色到長距離接觸(藍色)為其顏色範圍,分 別以 de 及 di 繪圖在 Hirshfeld Surface。de 可以清楚的觀察 hydrogen accepter 的資訊;而 di 給的資訊是 hydrogen donor 的資訊。35 雖然用 de 及 di 可以清楚地看出分子間相互作用較強的地方,但無 法凸顯強調 H,C,O 與較大的原子(如 Br 或 I)作緊密接觸,因此又定義出 dnorm 來對接處距離作歸一化 𝑑𝑛𝑜𝑟𝑚 =. 𝑑𝑖 −𝑟𝑖𝑣𝑑𝑊 𝑟𝑖𝑣𝑑𝑊. +. 𝑑𝑒 −𝑟𝑒𝑣𝑑𝑊. (20). 𝑟𝑒𝑣𝑑𝑊. dnorm 是以 de、di、van der Waals 半徑來作定義,其繪圖在 Hirshfeld Suface 上的顏色範圍與 dnorm 的關係為從紅色(距離小於凡得瓦半徑總合) 經白色到藍色(距離大於凡得瓦半徑總合)。以β-oxalic acid 為例,在 de 圖只能看到一個氫鍵接受者緊密接觸,但從 dnorm 只要是氫鍵接觸的位 置馬上就能看到,因此一般 Hirshfeld Surface 都會以 dnorm 來繪製。36. 3.1.2 2D-Graph:Fingerprint Plot 即使是很簡單的分子晶體結構也可能包含許多不同分子間的作用, 因此二維的 Fingerprint Plot 把 de 、di 資訊置入到一張圖中,它是由 Spackman 和 McKinnon 在 2002 所提出。35 在 Fingerprint Plot 出現之前, 將 分 子 間 的 作 用 力 可 利 用 NIPMAT(Non-Bonded Interaction Pattern MaTrix)圖可視化,它所使用的方法是以不同程度灰階方格來解釋分子 21.
(34) 間接觸,每個方格與接觸的距離和凡德瓦半徑都有關係,NIPMAT 的對 角線為對稱軸,因為兩個原子相互作用力是相同的因此方格顏色必相同, 雖然 NIPMAT 對於整合分子間作用力於一張圖中是可使用的,不過它 仍有一些受限制的因素,NIPMAT 圖對於每一個分子晶體是不是唯一性, Desiraju 在 1997 年被使用此比較 Naphthalene andTerephthalic Acid 堆疊 的相異性。11 NIPMAT 圖對於每一個分子晶體並非是唯一性,因為圖取決於原子 的排列順序,只要把矩陣內的原子順序調換圖就會改變,且 NIPMAT 不適合於大分子,隨著分子越大其矩陣也會成平方倍增大。 Fingerprint Plot 是將 Hirshfeld Surface 上每個點上的 di 和 de,當作 坐標的 x 軸及 y 軸來作圖於範圍從 0 到 3Å 、間隔為 0.01 Å 的二維圖中, 圖中顏色範圍是以比例來區分,從藍色(較少的點)經綠色到紅色(較多的 點);在 3D 的圖上因無法將不同原子的大小考慮進去而無法用 de 及 di 表示,但在 2D 的 Fingerprint Plot 卻是個優點,因其可以將不同性質的 分布分隔開。23 在 2007 年 Parking 等人首先用 Fingerprint Plot 定量比較完全不同晶 體結構相似性,並把這個方法稱為 Structural Genetic Fingerprinting37, 用這個方法不僅可以說明假設分子 A 和 B 相似度為 x,分子 C 和 D 相 似度為 y,如果 x 大於 y,就代表 A 對 B 的相似度比 C 對 D 高,他們 是用較粗略的 Fingerprint Plot(間隔從 0.01Å 變 0.04Å ),計算各個區塊之 間的相關係數(Correlation Coefficient),並使用樹狀圖將相關係數可視化。. 22.
(35) 14. 利用此方法證明了 Naphthalene 與 Anthracene 相似度(值為 0.888)高於. Naphthalene 與 Benzene 相似度(值為 0.869),這個方法所使用的軟體為 dSNAP37,我們所使用的軟體為 CrystlalExplorer36,與實驗合成出的晶 體結構作定量分析。. 3.2 Atom in Molecules (AIM) 分子中的原子理論 38(Atom in Molecules, AIM)是由 Bader 首先提出 的電荷密度拓撲分析,它以電子密度來劃分各個原子所包含的區域,而 每個原子之間皆被零通量面(即電子密度梯度等於 0)隔開,可以描述分 子中的成鍵,也可應用到有機晶體觀察分子間相互作用。在 AIM 中, 臨界點(Critical Point, CP)是電荷密度梯度為零的點,根據 Hessian 矩陣 中的本徵值可分成四類,以用(X,Y)表示,X 代表 Hessian 矩陣的非零本 徵值個數,Y 代表本徵值符號之和。 a) (3, -3)對應電荷密度函數的局部極大值,通常出現在原子核的位 置,因此又稱為核臨界點(Nuclear Critical Point, NCP)。 b) (3, -1)對應電荷密度函數的二階鞍點,介於有相互作用的兩個原 子之間,因此又被稱為鍵臨界點(Bond Critical Point, BCP),根據 電子密度在臨界點的 Laplacian,可以把鍵結分成兩類,閉殼層 相互作用的 Laplacian 為正,而電子共享相互作用為負。鍵徑是 在核臨界點與鍵臨界點之間區域的局部電子密度極大的連線,以 連接核臨界點與鍵臨界點,其中鍵徑不一定為直線。. 23.
(36) c) (3, +1)對應電荷密度函數的一階鞍點,通常出現在環中如苯環中 心,又稱環臨界點(Ring Critical Point, RCP)。 d) (3, +3)對應電荷密度函數的局部極小點,通常出現在籠狀體系中, 又稱為籠臨界點(Cage Critical Point, CCP)。 每個臨界點在空間中內進行電子密度的積分,可得到許多不同性質 的資訊以進行分析,包含電子密度(𝜌(𝑟))、Laplacian(∇𝜌(𝑟)2 )、約化密 度 梯 度 函 數 (Reduced Density Gradient) 、 電 子 定 域 化 函 數 (Electron Localized Function)、sign(λ2)ρ。若利用𝜌(𝑟)sign(λ2 )投影到 RDG 等值 面不僅可以指出哪裡存在弱相互作用,還可以可視化地了解弱用用力的 強度與類型,因此以下將介紹各種利用電子密度拓撲分析的方法。. 3.3 Electron Localization Function (ELF) 電子定域化函數(Electron Localization Function ,ELF)是一個三維空 間函數,ELF 是由六維的費米穴(Fermi hole)函數簡化而來,可表現分 子內不同位置的電子密度局域化程度,ELF 函數因其易於計算已廣泛 應用至各種體系,為研究電子結構特徵的重要工具。Becke 等人將 ELF 定義如下 ELF =. 1. (21). 1+[𝐷(𝑟)/𝐷0 (𝑟)]2 1. 2. 1 |∇𝜌𝛼 (𝑟)|2. D(r) = ∑𝑖 𝜂𝑖 |∇𝜑𝑖 (𝑟)| − [ 2 8 D0 (r) =. 3 10. 𝜌𝛼 (𝑟). 2. +. |∇𝜌𝛽 (𝑟)| 𝜌𝛽 (𝑟). ]. (6𝜋 2 )2⁄3 [𝜌𝛼 (𝑟)5⁄3 + 𝜌𝛽 (𝑟)5⁄3 ]. 在閉殼層系統中(如離子鍵、氫鍵、凡德瓦力)中,因𝜌𝛼 = 𝜌𝛽 =. 24. (22) (23).
(37) (1/2)𝜌,D 和 D0 項可以簡化成 1. 1 |∇𝜌(𝑟|2 ). 2. 8. D(r) = ∑𝑖 𝜂𝑖 |∇𝜑𝑖 (𝑟)|2 −. D0 (r) =. 3 10. 𝜌(𝑟). (3𝜋 2 )2⁄3 𝜌(𝑟)5/3 ]. (24). (25). ELF 數值範圍在 0 到 1,若具數值較高的 ELF 等值面,代表電子不 易 跑 出 此 區 域 , 分 析 非 共 價 相 互 作 用 時 使 用 的 方 法 為 EDI (ELF Delocalization Index),其方法是觀察在第一階鞍點位置之 ELF 值變化, 但是由於大部分的弱作用力缺乏電子定域化,因此這個方法無法可視化 弱作用力。可利用 ELF 所計算出來的值 f 在三維空間中做等值面分析, 當等值面 f 值開始分裂成較小的區時都會對應一個鍵臨界點(3,-1)稱為 分歧點,此時 f 就等於此鍵臨界點之 ELF 值,若一直增加 f 值的過程中, 不再分歧成更小的區域時稱我們這種區域為不可約域。不可約域只包含 一個核臨界點(3,-3),若兩個以上則稱為可約的。雖然等值面圖直觀清 楚,但其缺點是無法精確做定量分析,無法得到準確的臨界點的位置及 其函數值。直接用拓撲分析雖然準確且快速,但當電子結構複雜時,臨 界點的分布也會較複雜,因此拓撲分析搭配等值面分析是最好的選擇, 前者用於準確的定量分析而後者用於快速定性分析。. 25.
(38) 3.4 Localized Orbital Locator (LOL) Becke 與 Schmider 定義 Localized Orbital Locator39 式(26),D0 是 Thomas-Fermi 電子動能密度,分母則是假設當電子間完全不存在作用 力時的電子動能密度,又稱為 Kohn-Sham 電子動能密度。Becke 發現 τ(r)可以反映出原子的電子殼層結構之位置與大小、鍵結以及孤對電子 對分佈、非共價鍵作用力等有用的化學資訊,但是 τ(r)的大小介於零與 無窮大之間,要做圖分析並不方便,因此 Becke 與 Schmider 便定義了 Localized Orbital Locator(LOL),LOL 的大小介於 0 跟 1 之間,可以充 分地反應出 τ(r)可以反應的化學資訊,與 ELF 類似可以顯示分子內電子 密度具有高度區域性,數值越大代表電子的區域性越大。 τ(r) = LOL =. 𝐷0 (𝑟). (26). (1/2) ∑𝑖|∇𝜑𝑖 (𝑟)|2 τ(r). (27). 1+τ(r). 3.5 Non-Covalent Interactions Plot (NCIPLOT) Johnson 等人利用電子密度及其約化密度梯度函數(Reduced Density Gradient, RDG,s) 40;公式如式(27),其中𝝆(𝑟)代表電子密度,此方法只 需要分子的幾何座標即可繪製與分析非共價作用。可以快速看到分子間 的氫鍵、凡德瓦力及立體排斥作用,也能應用到較大的系統,例如蛋白 質或 DNA。 𝐬=. 𝟏. |𝛁𝝆(𝒓)|. 𝟏 𝟐(𝟑𝛑𝟐 )𝟑. 𝝆(𝒓)𝟒/𝟑. (28). 3.5.1 區分非共價相互作用 在弱作用的區域,其 s 值必須接近於零,但鍵臨界點附近的 s 值也 26.
(39) 極小,以致難以區分出兩部分,但𝜌(𝑟)在此兩區域值存在一定差異,因 此結合 s 和𝜌(𝑟)作圖即可確定分子中哪些區域涉及弱相互作用。以 Methane dimer 為例,它有兩處尖峰(s=0),𝜌(𝑟)接近於零的尖峰代表分 子之間的非共價弱作用力,而𝜌(𝑟)約在 0.27 處的尖峰則是代表 C-H 共 價鍵處(Bond Critical Points)。對於 Benzene dimer 也是相似的。40 類似 AIM 理論,弱相互作用臨界點的電子密度𝜌(𝑟)值可做為相互 作用力強度的指標,卻無法區分出不同類型的相互作用力,例如氫鍵和 空間位阻會出現在相同的區域 s 和𝜌(𝑟),因此需要利用電子密度二次微 分來區分這些相互作用的類型。. 3.5.2 非共價相互作用類型 ρ(r)只能反映出作用力強度,但相互作用力類型必須由 sign(λ2),λ2 為電子密度 Hessian 矩陣之第二大本徵值的符號。在 AIM 理論. 38. 中,. 鍵臨界點的 sign(λ2)= -1 而環的臨界點的 sign(λ2)= +1,利用ρ(r)sign(λ2) 投影到 RDG 等值面不僅可以指出哪裡存在弱相互作用,還可以可視化 地了解弱用用力的強度與類型。投影的色彩刻度為藍到綠到紅,藍色區 域𝜌(𝑟)較大、sign(λ2)= -1 為強吸引的弱相互作用,而符合這個特徵的類 型最常見者為氫鍵。紅色區域為ρ(r)較大、sign(λ2)= +1 是互斥的弱相 互作用,因周圍原子間起互斥作用,也被稱為 Nonbonded Overlap,常 見的類型為苯環中心。綠色區域的𝜌(𝑟)極小代表相互作用強度很弱,由 於此區域電子密度極小,sig(λ2)較不穩定因此可正可負,而凡德瓦作用. 27.
(40) 即符合這個區域特徵。41 sign(λ2)可以很快地看出氫鍵及凡德瓦力作用的區域,再利用投影到 RDG 等值面並擷取自己所要的電子密度範圍,即可把氫鍵作用範圍及 凡德瓦力作用範圍分別顯示。在本文中皆以 RDG = 0.5、密度範圍為0.02 a.u.到 0.02 a.u.。. 28.
(41) 第四章 結果討論 以 Benzoylleucine Diethyl Amides(BDA) 加上不同取代基來做比較, 本文所使用的分子晶體結構如圖 4,各種取代基的 BDA 二聚體前方皆 以 u 來表示分子晶體類別,詳細資訊如表 1,依序為單取代、雙取代、 氟取代與共晶體(cocrystal),並以 Σ|σm|值由小到大排列。. 圖 4 Benzoylleucine Diethyl Amides 的分子結構. 29.
(42) 表 1 Benzoylleucine Diethyl Amides 之 x-ray 晶體分析 H-bond Distancec Compound. Chirality. π stack Murcury. Crystal Explorer. Mono-substitued u-3Me u-4Me u-3OMe. RR SS RS RR SS. Y Y Y. 2.01 , 2.14 2.19 , 2.19 a 2.05 , 2.05 a. 1.94 , 2.01 2.04 , 2.04 1.94 , 1.94. u-4F u-4Cl u-4Br u-4CN u-4NO2. RR SS RS RS RS RR SS. Y N N N Y. 1.97 , 2.03 a 2.12 , 2.12 a 2.15 , 2.15 a 2.07 , 2.07 a 2.05 , 1.89a. 1.86 , 1.89 1.96 , 1.96 2.00 , 2.00 1.99 , 1.99 1.95 , 1.89. Di-substitued u-3,5Me u-3,5OMe u-3,5F. RR SS RR SS RR SS. Y Y Y. 2.11 , 2.11 a 2.03 , 2.06a 1.97 , 1.97 a. 1.97 , 1.97 1.90 , 1.92 1.86 , 1.86. u-3,5NO2. RR SS. Y. 0.97 , 2.01 a. 1.86 , 1.89. F-substitued u-2,6F u-2,3,4F u-2,4,5F u-3,4,5F u-2,3,4,5F u-2,3,4,5,6F. RS RS RS RR SS RS RS. N N N Y N N. 1.94 , 194 2.07 , 2.07 1.99 , 1.99 1.98 , 1.98 1.97 , 1.97 1.97 , 1.97. 1.83 , 1.83 1.93 , 1.93 1.83 , 1.83 1.86 ,1.86 1.82 , 1.82 1.79 , 1.79. cocrystal u-4Me-Xb u-3,5-OMe-Xb. RR SS RR SS. Y Y. 2.19 , 2.12 1.95 , 2.05. 1.89 , 1.98 1.84,1.91. a. The cif files are taken by ref.. b. X is 3,5NO2.. c. unit in Å. 比較分子間相互作用力時,距離就變得很重要,因此晶體結構必須 是 well-characterised,也就是說氫原子必須位在正確的位置上,為了使. 30.
(43) 晶體之間進行有意義的比較,與氫原子鍵結的距離必須標準化成一個實 際值,為此目的 CrystalExplorer 使用中子繞射實驗所得到的平均鍵長, 如 C-H = 1.083 Å 、N-H = 1.009 Å 、O-H = 0.983 Å ,但用 Murcury 所測 量的確有所不同,如 C-H = 0.980 Å 、N-H = 0.834 Å ,這也是為什麼表 1 用兩個不同軟體所測得的氫鍵長度不同的原因,測量的差異值大約為 0.15Å 。. 4.1 不同取代基 Benzoylleucine Diethyl Amides 探討 4.1.1 由 X-Ray 晶體資訊探討 由表 1 我們能推測出一些資訊,如從取代基位置上,只要取代基位 置在 4 號位有取代基(u-4CN、u-4Br、u-4Cl、u-4Me)的分子晶體就非 Homochiral Dimer,也就是由氫鍵所形成的二聚體不具相同掌性,即無 π-stack,但 u-4NO2 和 u-4F 例外。這四個不具 π-stack 的分子晶體當中, 比較特別的是取代基為鹵素的 u4Br 和 u4Cl,僅由一個氫鍵形成二聚體 如圖 5,其中氫鍵以紅色虛線表示。. 31.
(44) (a). (b). 圖 5 分子晶體以一個氫鍵形成二聚體之結構 (a)u-4Br (b)u-4Cl,(氫鍵 以虛線表示). 若兩個分子形成的二聚體具有 π-stack 稱為 Homochiral Dimer,其 中兩個分子具有相同對掌性即 RR 或 SS,且形成的兩個氫鍵互相交叉 (如 u-3OMe,圖 6(a));若兩個分子不具有 π-stack 的二聚體則稱為 Heterochiral Dimer,其中兩個分子具有不同對掌性即 RS,且兩個氫鍵 平行(如 u-4CN,圖 6(b)),從這些資訊我們推測 Homochiral dimer 代表 具有 π-stack,而 Heterochiral dimer 代表分子間不具有 π-stack。. 32.
(45) (a). (b). 圖 6 二聚體之結構(a)為 u-3OMe 為交叉雙氫鍵 (b)為 u-4CN 為平行 雙氫鍵(氫鍵以虛線表示). 4.1.2 Hirshfeld Surface 與指紋圖之探討 我們利用 X-Ray 晶體檔案及 Crystal Explorer 程式將雙聚體的電子 密度以 Hirshfeld Surface 的方式區分為目標分子及其相鄰分子兩部分, 接著 Crystal Explorer 自動計算 Hirshfeld 上每一點距最近的原子核的距 離,因 Hirshfeld surface 已將空間分為兩部分,故每一點有一對(di,de)值, 分原子核種類如 C…C,將每一點的(di,de)值做圖,形成 2D Finger Plot。 表 2 中% (C…C)代表 C…C 在 Hirshfeld Surface 的相對貢獻度,也 可以當作有無π-stack 指標,O…H 代表分子之間的氫鍵作用力,由表 二推測% (C…C)大於零代表此分子晶體具有π-stack,而% (O…H)大於 零代表分子之間存在氫鍵作用力,兩種作用力都會影響晶體的堆疊排列 方式。在此處要注意的是 C…C 指的是一分子中的碳原子和另一分子的 碳原子相互作用,因此並不一定是只有芳香環上的碳原子與另一芳香環 碳原子相互作用,也有可能是芳香環上的碳原子與取代基上的碳原子或 33.
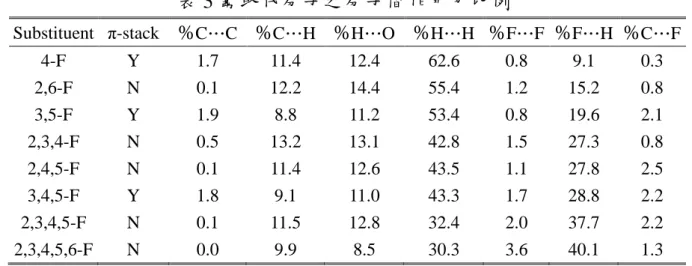
(46) 長鏈上的碳原子作用,最後計算後的% (C…C)是兩者疊加起來。. 表 2 不同取代基在 Hirshfeld Surface 的相對貢獻度 π-stack. % (H…O). % (C…C). % (C…H). % (H…H). Mono-substitued u-3Me u-4Me. Y N. 13.4 14.5. 2.2 1.3. 9.5 12.7. 74.3 71.1. u-4F u-3OMe u-4Cl u-4Br u-4CN u-4NO2. Y Y N Na Y Y. 12.4 17.5 12.5 12.4 12.6 27.2. 1.7 2.3 0.0 0.0 0.0 2.1. 11.4 9.1 16.2 15.7 19.7 8.6. 62.6 70.7 55.9 55.5 54.4 57.6. Di-substitued u-3,5Me u-3,5OMe. Y Y. 11.5 20.2. 2.0 2.2. 8.7 9.2. 76.2 67.8. u-3,5F u-3,5NO2. Y Y. 11.2 31.7. 1.9 2.8. 8.8 4.9. 53.4 47.2. cocrystal u-4Me-Xb u-3,5-OMe-Xb. Y Y. 23.6 29.4. 2.4 2.4. 9.6 6.8. 61.6 58.8. a. RS stack b X is u-3,5-NO2 當% (C…C)為零的分子都不具有 π-stack,但值得注意的是 u-4Me,. 雖然它不是 Homochiral Dimer 但卻具有 π-stack,其%C…C 為 1.3%,圖 7 為三個 4-CH3 分子組成含有氫鍵及 π-stack 的三聚體,由圖中左邊分 子與中間分子形成 Heterochiral Dimer(為平行氫鍵,用虛線表示氫鍵), 但中間分子與右邊分子又形成具有 π-stack 的空間排列,這是在其他晶 體中所沒發現的幾何排列。. 34.
(47) 圖 7 u-4Me 分子晶體為 heterochiral,卻具有 π-stack(氫鍵以虛線表 示). 因此可得到以下結論:具有 π-stack (即% (C…C)非零)且值須大於 1.7 才會形成 Homochiral Dimer Stacking;而% (C…C)小於 1.5 是屬於 Heterochiral Dimer Stacking,但分子間仍具有 π-stack,藉由分子相互作 用力定量分類,能快速且正確的的判斷出這些資訊,而不會因為只由晶 體資訊而誤判含 π-π 相互作用的分子晶體為不具有 π-π 相互作用。 將 Hirshfeld Surface 用 Fingerpint Plot 來 比 較 不 同 取 代 基 的 Benzoylleucine Diethyl Amides,藉由 Fingerpint Plot 快速比較出他們的 相似性及相異性,整理於附錄 A 中。發現在附錄 all 圖中每個分子左下 都具有兩個尖尖的 peak,有些在中間顏色偏綠色,還有的是兩側具有 像翅膀的圖案,這些都有其各自代表的意義。 有 π-π 相互作用表示具有碳原子與碳原子的接觸,且在圖中有 Homochiral 的分子在有顏色的部分中間是藍偏綠色,因為碳的凡德瓦 半徑為 1.8Å ,因此應該在 de 和 di 為 1.8 Å 附近會有格點,因此推測在. 35.
(48) 圖中比較亮的部分是因為有 π-π 相互作用所導致,但亦或者可能是別的 原子的接觸作用力,只是距離與碳碳接觸的相互作用重疊,因此將 Fingerpint Plot 拆解,將不同的相互作用力分開來看更能清楚且快速比 較且能個別探討,圖皆放於附錄 A 中。 以% (C…C)為 2.0 的 u- 3,5Me 為例(即圖 A- 12),將其 Fingerprint Plot 拆解後,發現中間偏綠色不是因為 C…C 而是由 H…H 所造成,即 並非由 π-π 相互作用所導致,H…H 在這些分子晶體中所佔比例都超過 50%因為其氫的數量佔多數因此這現象是合理的,但在 H…H 值得注意 的是只要 4 號位置取代在(de,di)=(1.2,1.2)附近,是屬於一個尖峰但在 3 號位置卻是較平坦的。而 C…C 就如預測具有 π-stack 相互作用顏色範 圍在 1.8Å 附近,對照其他具有 π-stack 的 Fingerprint Plot 也是一樣,而 沒有 π-stack 在圖中 C…C 顯示的就是灰色。 從 Fingerprint Plot 也能大約量測出氫鍵的距離,氫鍵在圖中是 O… H 顏色範圍 34,以 u-3,5Me 較下端尖峰為例(即圖 A- 12),di 大約為 1.16 Å 而 de 大約為 0.8 Å ,相加後推測氫鍵距離應約為 1.96 Å ,再對照表 1, 氫鍵鍵長與 CrystalExplorer 所量測出來的較為接近,接著以較上端的尖 峰計算氫鍵鍵長結果相同,較上端尖峰與較下端尖峰分別代表氫鍵供給 者與氫鍵接受者。. 36.
(49) 4.2 不同位置含氟取代效應 4.2.1 由 X-Ray 晶體資訊探討 本研究以八個含氟取代的分子晶體在不同位置取代進行研究,單從 晶體資訊發現只要氟 取代基接在 2 號位置,其二聚體就不會形成 Homochiral,而氫鍵的鍵長從表 1 看不出與取代數目或取代位置具有 何種關聯。. 4.2.2 Hirshfeld surface 與指紋圖之探討 我們知分子作用力同時具有 π-stack 所形成的 Dimer 不一定代表此 兩個分子間具有氫鍵,而是有另外的堆疊方式(如 u-4Me),根據這些不 同類型的取代基得到這些結果,我們設想也許取代基的位置與取代基數 目和晶體的堆疊方式具有特別的規律性,所以接著探討不同位置含氟作 為取代的 Benzoylleucine Diethyl Amides,計算其 Hirshfeld Surface 得到 各個相互作用力比例如表 3。. 表 3 氟取代分子之分子間作用力比例 Substituent π-stack %C…C %C…H %H…O %H…H %F…F %F…H %C…F 4-F 2,6-F 3,5-F 2,3,4-F 2,4,5-F 3,4,5-F 2,3,4,5-F 2,3,4,5,6-F. Y N Y N N Y N N. 1.7 0.1 1.9 0.5 0.1 1.8 0.1 0.0. 11.4 12.2 8.8 13.2 11.4 9.1 11.5 9.9. 12.4 14.4 11.2 13.1 12.6 11.0 12.8 8.5. 37. 62.6 55.4 53.4 42.8 43.5 43.3 32.4 30.3. 0.8 1.2 0.8 1.5 1.1 1.7 2.0 3.6. 9.1 15.2 19.6 27.3 27.8 28.8 37.7 40.1. 0.3 0.8 2.1 0.8 2.5 2.2 2.2 1.3.
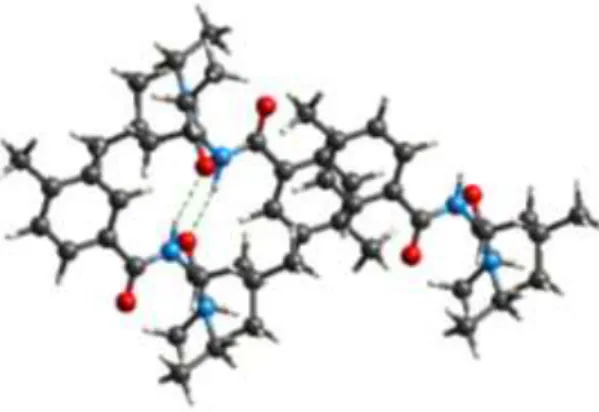
(50) 從表三發現雖然有些%C…C 非零但發現結構並不具有 π-stack,且 二聚體掌性為 RS,根據這些結果我們判斷出%C…C 小於 0.5 的分子晶 體是不具有 π-stack。在此處可下一個結論: %C…C 大於 1.3 具有 π-stack 但%C…C 須接近 2 的分子晶體才是屬於 Homochiral dimer。 從表 3 得知,氟取代數目越少,其%H…H 比例會升高,而%F…F 比例會降低。單看芳香環上三個位置被氟取代的分子( u-2,3,4F、u2,4,5F、u-3,4,5F),發現只有在二號位置沒有氟取代的 u-3,4,5F 具有 Homochiral Dimer,但其 C…H、O…H 都是最低的,接著單看芳香環上 兩個位置被氟取代的分子( u-3,5F、u-2,6F),依然是不在二號位置取代 的屬於 Homochiral Dimer,且其 C…H、O…H 依然都是較低的。 用 Fingerpint Plot 來比較這八個含氟取代 BDA,藉由 Fingerpint Plot 快速比較出他們的相似性及相異性。從圖 8 得知,左下端兩個尖峰 估算出 O-H 鍵長,以 l 2,3,4-F 較下端尖峰最短位置為(de,di)=(1.15,0.8), 將其相加後為 1.95Å 應為氫鍵鍵長對照表 1。若以不同位置含氟取代的 2D Fingerprint Plot 來看,中間也是偏向藍綠色,但也許不一定是 H…H 所造成,因此接下來要拆解 Fingerprint Plot 探討各種相互作用力圖如附 錄 A。 以 u-3,4,5-F 為例(圖 A- 17),F…H 對於中間部分的貢獻也很大(28%) 不只是只有 H…H 的貢獻而已,也發現在有二號位取代的 H…H 有顏色 的部分下端都偏尖,其他沒有二號位取代是較為平坦的。. 38.
(51) u-2,3,4,5,6F. u-2,3,4,5F. u-2,3,4F. u-2,4,5F. u-3,4,5F. u-2,6F. u-3,5F. u-4F. 圖 8 將八個取代 benzoylleucine diethyl amides 所有分子與分子接觸的作 用力用 2D Fingerpint Plot 表示. 39.
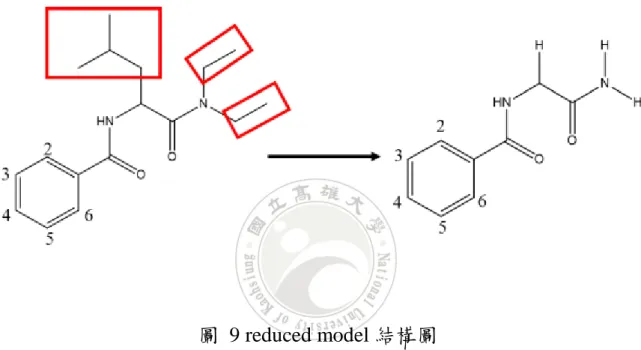
(52) 4.3 Reduced Model 為簡化研究 ππ 相互作用與氫鍵的協同作用,及考慮計算時間成本, 因此本研究將 Benzoylleucine Diethyl Amides 二聚體結構簡化,將異丁 基(isobutyl)以氫原子取代,在氮上面的兩個乙基(ethyl)也分別以氫原子 取代,以此模型(即 reduced moldel)進行計算及探討,其結構如圖 9。. 圖 9 reduced model 結構圖. 取 3-OMe 二聚體的 reduced model 來進行計算,以不同方法(未使 用 counterpoise 計算 BBSE)計算其分子間作用力(假設只包含氫鍵、ππ 相互作用力)。也利用取代的方式,如計算氫鍵時以氫原子取代苯環, 再利用分子間作用力減去氫鍵即得 ππ 相互作用力,以分解出 ππ 相互 作用力及氫鍵作用力,其結果如表 4。. 40.
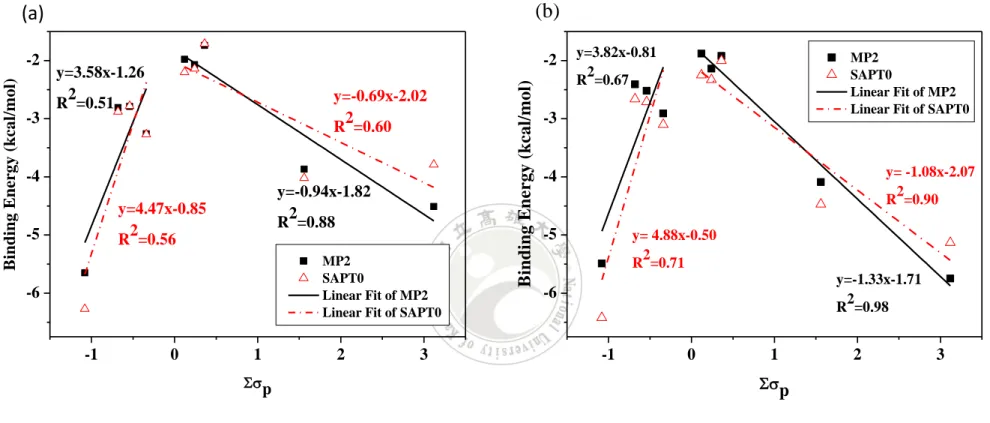
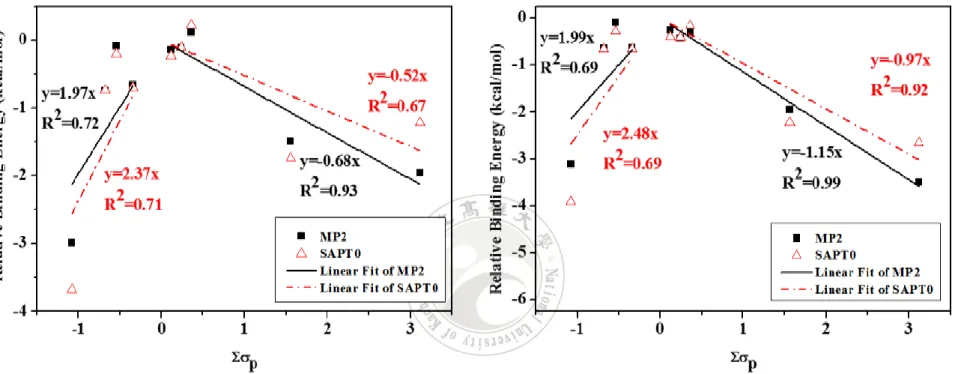
(53) 表 4 r-3OMe 分子間作用力、氫鍵作用力、ππ 相互作用力 a Method. MP2. ΔE ΔE H-bond ΔEππ. -41.07 -19.06 -22.01. B3LYPGD3BJ -39.37 -19.59 -19.78. B3LYP. -20.73 -15.38 -5.35 a Basis set is 6-31G*(0.25) (in kcal/mol). Wb97XD. B2PLYD3. -35.72 -17.97 -17.75. -38.18 -19.14 -19.04. 表 5 表列出 ring model 所計算出來的 ππ 交互作用與利用 DMC 計 算 reduced crystal 的 ππ 交互作用,利用 reduced model 計算出的 π-π 作 用力均比 ring model 來的低,表示氫鍵的協同作用讓 ππ 更加穩定,能 量下降範圍從 0.01~0.65 kcal/mol。3,5OMe 為所有取代基中能量差異最 大,差值為 0.65 kcal/mol,而不同模型所計算 ππ 相互作用差異最小, 其差值為 0.01 kcal/mol,因各個取代基所算出來的能量差異不大,且為 降低計算時間成本,因此取 Benzoylleucine Diethyl Amides 的結構,刪 減到只留下苯環及其取代基的模型(即 Ring Model)來進行計算及研究, 探討取代基效應。 熔點高即可代表結合能越低,二聚體越穩定,對照表 5 單取代部分, 熔點與兩苯環中心距離較無相關,但其與苯環中心到另一苯環垂直距離 有相關,當垂直距離越短熔點越高;雙取代部分,苯環垂直距離最大反 而熔點最高。. 41.
(54) 表 5 不同模型 π 結合能計算 a 與晶體結構資訊 crystal information de (Å ). Rf (Å ). θg (degree). αh (degree). monosubstitued r-3Me. 3.90. 3.53. 86. 7. r-3OMe r-4F r-4NO2. 3.85 4.51 3.78. 3.52 3.25 3.22. 81 89 77. di-substitued r-3,5Me. 4.16. 3.33. r-3,5OMe r-3,5F r-3,5NO2. 3.78 4.35 3.86. tri-substitued r-3,4,5F cocrystal r-4Me-X r-3,5OMe-Xd. Modelb. binding energy melting point. Reduced modelc. Ring model. 84-85. -. -. 0 9 14. 84-85 132-133 130-131. -3.02 -1.88 -4.13. -2.51 -1.87 -4.07. 89. 0. 107-108. -2.76. -2.39. 3.43 3.38 3.33. 79 91 2. 0 5 87. 143-144 107-108 107-108. -6.12 -2.18 -6.02. -5.47 -2.13 -5.73. 4.41. 3.34. 91. 4. -. -1.99. -1.91. 3.72 3.64. 3.28 3.35. 83 78. 0 2. -. -6.55 -7.47. -6.28 -7.53. a. (˚C). unit in kcal/mol b Basis Set is MP2/6-31g*(0.25) c DMC method d X is r-3,5NO2 e the distance between the centroids of the ring. f the distance of the ring centroid to the plane defined by the opposite ring. g the dihedral angle between carbon1(ring1)centroid(ring1)-centroid(ring2)-carbon2(ring2). h the dihedral angle between tow aromatic planes.. 42.
(55) 4.4 Ring Moldel 以下將分別以不同小節探討 Ring Model ππ 相互作用力的取代基效 應、ππ 之 SAPT 能量分解及其拓撲分析等等。. 4.4.1 ππ 相互作用力 我們以 ring model 進行量子計算,分別使用 G09 及 PSI4 計算 ring model 分子間作用力大小,在 G09 部分計算方法皆為 MP2,而 PSI4 方 法為 SAPT0,基底選擇為 jun-cc-pV(D +d)Z 及 6-31G*(0.25)。在表 11 中,full 代表除了有計算價層電子與內層電子的電子相關能,而 frozen 代表計算電子相關能時不計算內層電子的電子相關能。從表 6 得知, 不管是 SAPT0 或是 MP2,計算內從電子的電子相關能能量下降最大為 0.02kcal/mol。. 43.
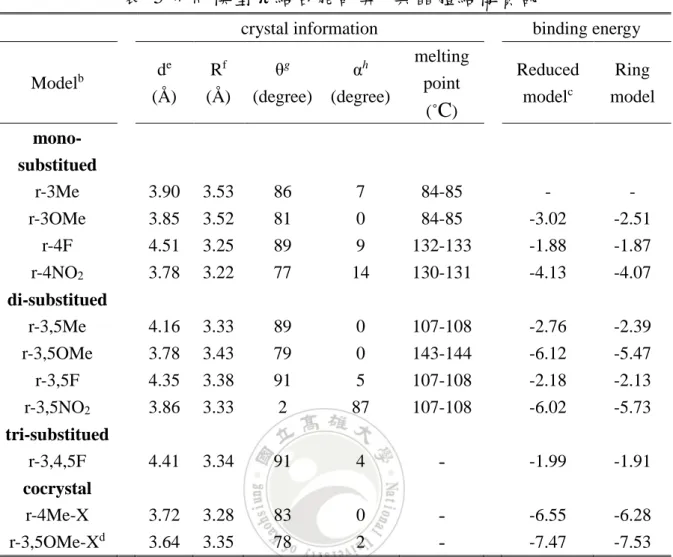
(56) 表 6 ring model 分子間結合能 a SAPT0 Σ|σm|. Σ|σp|. MP2. junb. 025. c. 025c. junb. FCd. Full. FC. full. FC. full. FC. full. mono-substitued 3Me. 0.14. 0.34. -3.25. -3.27. -3.08. -3.10. -3.24. -3.26. -2.89. -2.91. 3OMe. 0.24. 0.54. -2.76. -2.78. -2.70. -2.71. -2.78. -2.79. -2.51. -2.52. 4F. 0.68. 0.12. -2.19. -2.20. -2.25. -2.25. -1.97. -1.98. -1.87. -1.88. 4NO2. 1.42. 1.56. -4.00. -4.02. -4.45. -4.47. -3.85. -3.87. -4.07. -4.09. di-substitued 3,5Me. 0.28. -0.68. -2.86. -2.88. -2.65. -2.66. -2.79. -2.81. -2.39. -2.41. 3,5OMe. 0.48. 1.08. -6.25. -6.27. -6.40. -6.42. -5.63. -5.65. -5.47. -5.49. 3,5F. 1.36. 0.24. -2.13. -2.14. -2.32. -2.33. -2.06. -2.07. -2.13. -2.14. 3,5NO2. 2.84. 3.12. -3.77. -3.79. -5.12. -5.13. -4.49. -4.51. -5.73. -5.75. tri-substitued 3,4,5F. 2.04. 0.36. -1.70. -1.71. -1.99. -2.00. -1.73. -1.74. -1.91. -1.92. cocrystal 4Me-X. 1.49. 1.73. -7.12. -7.14. -7.61. -7.63. -6.17. -6.19. -6.28. -6.30. 1.02. -7.45. -7.47. -8.34. -8.36. -6.96. -6.99. -7.53. -7.55. 3,5OMe-X a. unit in kcal/mol. b. e. 1.66 c. d. e. basis is jun-cc-Pv(D+d) basis is 6-31G*(0.25) Frozen Core X is r-35NO2. 44.
(57) (b). (a). -3. -2. y=3.58x-1.26 2 R =0.51. Binding Energy (kcal/mol). Binding Energy (kcal/mol). -2. y=-0.69x-2.02 2 R =0.60. -4 y=-0.94x-1.82 2 R =0.88. y=4.47x-0.85 2 R =0.56. -5. MP2 SAPT0 Linear Fit of MP2 Linear Fit of SAPT0. -6. -1. 0. 1. 2. 3. y=3.82x-0.81 2 R =0.67. MP2 SAPT0 Linear Fit of MP2 Linear Fit of SAPT0. -3. y= -1.08x-2.07 2 R =0.90. -4. -5. y= 4.88x-0.50 2 R =0.71 y=-1.33x-1.71 2 R =0.98. -6. -1. 0. 1. 2. p. p. 圖 10 MP2 及 SAPT0 之 π 結合能與 Σσp 關係圖,(a)為 jun-cc-pV(D+d)Z (b)為 6-31G*(0.25). 45. 3.
數據


+7

相關文件
核能電廠和原子彈間最大不同處,即在於所 含鈾-235的濃度;鈾-235在核燃料中只佔 3~5%,而在原子彈中高達
– 有些化合物的電子為奇數個,像NO及NO 2 ,其中N 原子 只有7個電子 ( 含共用 ),稱為自由基 (free radical)。由 於具有未成對電子 (unpaired
雖然水是電中性分子,然其具正極區域(氫 原子)和負極區域(氧原子),因此 水是一種極 性溶劑
(1)針對具有中子研究專長者,具備下列要件之 一:①物理、化學、核工系所博士畢業,具 二年以上中子研究經驗;執行中子散射、繞
一般而言,物質的黏度與流體間的凝聚 力和分子間的動量轉移率有關。液體分子與
一般而言,物質的黏度與流體間的凝聚 力和分子間的動量轉移率有關。液體分子與
• 接續之前的例子,若原為 0.288 pF 的液晶 電容 C LC ,再並聯一個亦為 0.288 pF 的電 容C st ,則電位保持的變化值為.
從幾何上看,一個在區間上的每一點都連續的函數,其函數 圖形沒有分斷。直觀上,這樣的連續圖形我們可以一筆劃完
