國立高雄大學電機工程學系(研究所)
碩士論文
導入電流注入法於矽之金屬輔助化學蝕刻製程
Introducing Current-driving Method to
Metal-assisted Chemical Etching of Silicon
研究生:李玟怜 撰
指導教授:馮瑞陽 博士
導入電流注入法於矽之金屬輔助化學蝕刻製程
指導教授 : 馮瑞陽 博士 國立高雄大學電機工程學系 學生 : 李玟怜 摘要 高深寬比結構於當前半導體元件及光電元件之設計製作,乃極具高應用價值; 然而傳統製備高深寬比結構方式,設備建置較為複雜,成本相對高昂,且反應氣 體大多具危險性。「金屬輔助化學蝕刻技術」為一結合酸與氧化劑,透過貴金屬 催化層定義蝕刻區塊,以其驅動載子交換完成化學反應進行蝕刻,此技術具有簡 易快速的優勢,於製造高深寬比結構上極具潛力。 本研究針對矽之金屬輔助蝕刻技術,設計建置一套鐵氟龍蝕刻載具,以石墨 為電極,透過外部電壓調整樣品之蝕刻電流密度,達調控蝕刻反應速率與蝕刻機 制之目的。實驗上,首先於 P+‒Si 基板上,觀察其微米柱樣品於不同金屬催化層 厚度下(Au = 12、22、32 及 42nm),其「金屬輔助化學蝕刻」之效應;爾後,利 用不同金厚度之 P+−Si (Au = 32nm & 20 nm)及 N−Si 樣品(Au = 15nm),研究其對於不同蝕刻液配比(HF/H2O2)與不同蝕刻時間下,其微米柱與微米洞之蝕刻形貌 與蝕刻深度;另外,本研究於蝕刻時,導入電壓調變機制,透過調整樣品於順偏 蝕刻或逆偏蝕刻,觀察其操作於電洞或電子注入下,對於其樣品之蝕刻影響與作 用機制,並比較有無氧化劑(H2O2)介入之差異。 本研究發現,當蝕刻時間漸長,又週期圖案間距小,因電洞擴散機制造成側 蝕明顯,致蝕刻後,樣品圖案形貌無法維持;另外,本研究比較分次蝕刻與一次 蝕刻之結果,於相同蝕刻時間下,分次蝕刻較能維持樣品圖案表面形貌;並驗證, 經由外部載子調控的方式,確實能調制樣品之蝕刻形貌,更加確認電洞控制為調 控本研究化學蝕刻之關鍵因素。 關鍵字 : 金屬輔助化學蝕刻、高深寬比、氫氟酸
Introducing Current-driving Method to
Metal-assisted Chemical Etching of Silicon
Advisor: Dr. (David) Jui-Yang, Feng
Department of Electrical Engineering in National University of Kaohsiung Student: Wen-Ling Lee
Abstract
High aspect ratio structures have considered as the valuable way of abilities enhancement for semiconductor devices and photonics devices. However, the equipment for fabricating high aspect ratio structure is much more complicated, high-cost and existing risks from processing gases. The so-called Metal-assisted chemical etching method (MaCEtch), only need Acid and Oxidant as the etchant, accompanying with the noble metal as the catalyst meanwhile as patterns to define the etching area. It is much simpler and has high potential in developing high aspect ratio structure.
To introduce MaCEtch for etching Silicon, we designed and built up a Teflon holder assembly as the etch setup – using high density graphite as the electrodes. Overall, we can directly apply voltage into the samples from etchant – to control the etching current density, etching rate and etching mechanism. In experimental, P+-Si samples with micro‒pillar but having different Au film thickness (12, 22, 32 and 42nm) as metal catalyses were firstly introduced into MaCEtch to clarify how the metal thickness influence in MaCEtch. Then, two kinds of samples with different Au thickness, P+‒Si (Au = 32nm & 20nm) and N‒Si (Au = 15nm), were etching under various etchant ratio (HF/H2O2) with different etching time to investigate their etched
hole during etching to study the etching mechanism under bias and current-driving. We also show its results difference when the etchant has no Oxidant (H2O2) involved.
We found as the etching time increased the patterns become not easy to maintain, especially when the patterns pitch is small – it is due to hole-diffusion undercut etching. Furthermore, when comparing the results from one-time etching to multiple etching but operating under the same total etching time, we observe that it is easier to keep the pattern well accompanying with less undercut etching by the way of multiple etching. Also, we demonstrate the pattern shape of samples by MaCEtch can be manipulated by the modulation of electron-hole carriers – it is therefore identified that controlling the electron-hole distribution become the key under operating MaCEtch.
致謝
感謝我的指導教授 馮瑞陽老師。感謝老師在這段時間的悉心指導研究,使 得我對研究能夠充滿興趣,當遇到實驗上的瓶頸時,也給予我研究方向與鼓勵, 使得我能夠充滿前進的動力,並讓此論文能夠順利的完成。 感謝洪勇智老師實驗室提供樣品及量測協助,楊證富老師實驗室提供測量協 助,感謝藍文厚老師對於研究之提點,感謝黃家偉學長提供量測與樣品製程的支 援與協助,感謝實驗室學長及學弟們的協助,使我的實驗室生活更加有趣,感謝 這一路上所有曾經給予協助與鼓勵的同學、朋友和師長們。 最後感謝我的父母與家人,謝謝我的父母無怨無悔的付出和不斷的給予我鼓 勵和支持,才能完成這個學業。目錄
摘要………... I Abstract………… ... II 致謝…………... IV 圖目錄…………... VI 表目錄………... XI 第一章 緒論 ... 1 1.1 前言 ... 1 1.2 相關文獻回顧 ... 6 1.3 研究動機與目的 ... 12 第二章 金屬輔助化學蝕刻及相關文獻 ... 13 2.1 金屬輔助化學蝕刻原理 ... 13 2.2 載子擴散機制 ... 15 2.3 不同催化金屬及其厚度之影響 ... 19 2.4 蝕刻液配比對蝕刻之影響 ... 23 2.5 蝕刻系統之蕭特基能障 ... 26 2.6 金屬輔助陽極蝕刻 ... 28 2.7 偏壓調控金屬輔助化學蝕刻 ... 30 2.8 小結 ... 32 第三章 研究方法及結果與討論 ... 33 3.1 本章架構 ... 33 3.2 研究方法 ... 34 3.2.1 實驗載具與相關架構 ... 35 3.2.2 樣品製備 ... 42 3.3 實驗結果與討論 ... 49 3.3.1 不同樣品於溶液下電壓與電流密度關係 ... 49 3.3.2 GOx granting/P−Si 蝕刻 ... 71 3.3.3 不同金薄膜厚度蝕刻效應 ... 75 3.3.4 Au = 32nm P+−Si 樣品之蝕刻形貌... 77 3.3.5 Au = 20nm P+−Si 樣品之蝕刻形貌... 85 3.3.6 Au = 15nm N−Si 樣品之蝕刻形貌 ... 99 第 四 章 結論 ... 120 參考文獻………... 122 附錄…………... 126圖目錄
圖 1-1 向下縮減與向上組裝製作技術示意圖 ... 1 圖 1-2 Si 奈米柱基底之生物感測器工作示意 ... 2 圖 1-3 Si 奈米柱基底之氣體感測器及測量結果 ... 3 圖 1-4 Si 與 Si 陣列結構外觀比較... 3 圖 1-5 Si 基板與 Si 陣列結構之反射率比較... 4 圖 1-6 Si 陣列結構折射率漸變示意圖 ... 4 圖 1-7 3D IC 示意圖 ... 5 圖 1-8 VLS 成長法 ... 7 圖 1-9 以 VLS 生長法生成之矽晶鬚結構 ... 7 圖 1-10 電化學蝕刻架構 ... 8 圖 1-11 HF 介入矽蝕刻之化學反應路徑示意圖 ... 9 圖 1-12 反應性離子蝕刻 ... 11 圖 1-13 蝕刻後側壁形成波浪狀結構 ... 11 圖 2-1 金屬輔助化學蝕刻反應 ... 14 圖 2-2 MaCEtch 電洞注入擴散示意圖及不同間距之實驗結果 ... 16 圖 2-3 式 2-7 模擬圖與實際操作對應 ... 17 圖 2-4 利用空間電位架構於不同電壓下抑制電洞擴散之情形 ... 16 圖 2-5 垂直與側向蝕刻示意圖 ... 17 圖 2-6 電洞注入 Si 之表面蝕刻模式 ... 18 圖 2-7 不同金屬粒子蝕刻形貌 ... 19 圖 2-8 Au = 10nm 金屬輔助化學蝕刻之機制與影響 ... 20 圖 2-9 Au = 20nm 金屬輔助化學蝕刻之機制與影響 ... 20圖 2-10 In−plane 與 Out−of− plane 兩種蝕刻路徑之示意圖 ... 21
圖 2-11 金屬輔助化學蝕刻 : 金屬層厚度與蝕刻速率之關係 ... 22
圖 2-12 不同氧化劑於矽基材下之相對電位,H2O2/H2O 具最大還原電位... 23
圖 2-13 錐狀孔形成機制,蝕刻比例為 0.7 > ρ > 0.2 ... 25
圖 2-16 不同 值的蝕刻液比例對不同摻雜程度之 Si 之影響 ... 27 圖 2-17 SEM, 不同類型 Si 之金屬陽極蝕刻結果 ... 28 圖 2-18 金屬陽極蝕刻架構 ... 29 圖 2-19 金屬陽極蝕刻作用機制及蕭基能障高度關係 ... 29 圖 2-20 偏壓架構比較 ... 30 圖 2-21 偏壓蝕刻結果(V = − 1.75V, 120mins, 100℃) ... 31 圖 3-1 蝕刻裝置架構示意圖 ... 34 圖 3-2 3D 列印製作石墨板夾具 (材質 PLA) ... 36 圖 3-3 PTFE 蝕刻裝置設計示意圖 ... 36 圖 3-4 PTFE 蝕刻裝置 ... 37 圖 3-5 樣品以 PI tape 固定於石墨板上及其結構示意圖 ... 37 圖 3-6 蝕刻架構示意圖 ... 38 圖 3-7 樣品以電流注入方法實際操作圖 ... 38 圖 3-8 升溫電流注入架構示意圖 ... 39 圖 3-9 升溫架構之夾具設計 ... 40 圖 3-10 電流注入法升溫架構實際操作圖 ... 40 圖 3-11 SEM,二維週期性微米圖案柱結構樣品 ... 42
圖 3-12 二維週期 pillar & hole 樣品結構示意圖 ... 43
圖 3-13 樣品蝕刻示意圖 ... 43
圖 3-14 直徑 5m 洞樣品製作流程圖 ... 45
圖 3-15 直徑 10m 柱樣品製作流程圖 ... 47
圖 3-16 樣品進行金薄膜蒸鍍 ... 48
圖 3-17 Au = 32nm pillar & hole 結構樣品實際外觀 ... 48
圖 3-18 以石墨板量測蝕刻液,電壓與電流值關係圖 ... 49 圖 3-19 P+−Si 實驗流程 ... 50 圖 3-20 P+−Si 電流密度與電壓關係圖 ... 52 圖 3-21 P+−Si 半對數電流密度與電壓關係 ... 52 圖 3-22 P+−Si 背面鍍金電流密度與電壓關係圖 ... 55 圖 3-23 P+−Si 背面鍍金半對數電流密度與電壓關係圖 ... 55 圖 3-24 P+−Si 由電流值反推求電阻與電壓關係圖 ... 56 圖 3-25 P+−Si 針對陽極比較電阻與電壓關係圖 ... 56
圖 3-26 樣品於實驗前電阻量測 ... 58 圖 3-27 P+−Si pillar Au = 12nm 之電流密度與電壓關係圖 ... 59 圖 3-28 P+−Si pillar Au = 12nm 之半對數電流密度與電壓關係圖 ... 59 圖 3-29 P+−Si pillar Au = 22nm 之電流密度與電壓關係圖 ... 60 圖 3-30 P+−Si pillar Au = 22nm 之半對數電流密度與電壓關係圖 ... 60 圖 3-31 P+−Si pillar Au = 32nm 之電流密度與電壓關係圖 ... 61 圖 3-32 P+−Si pillar Au = 32nm 之半對數電流密度與電壓關係圖 ... 61 圖 3-33 P+−Si pillar Au = 42nm 之電流密度與電壓關係圖 ... 62 圖 3-34 P+−Si pillar Au = 42nm 之半對數電流密度與電壓關係圖 ... 62 圖 3-35 P+−Si pillar Au = 12nm 電流值反推求電阻與電壓關係圖 ... 63 圖 3-36 P+−Si pillar Au = 12nm 陽極電阻與電壓關係圖 ... 63 圖 3-37 P+−Si pillar Au = 22nm 電流值反推求電阻與電壓關係圖 ... 64 圖 3-38 P+−Si pillar Au =22nm 陽極比較電阻與電壓關係圖 ... 64 圖 3-39 P+−Si pillar Au = 32nm 電流值反推求電阻與電壓關係圖 ... 65 圖 3-40 P+−Si pillar Au = 32nm 陽極電阻與電壓關係圖 ... 65 圖 3-41 P+−Si pillar Au = 42nm 電流值反推求電阻與電壓關係圖 ... 66 圖 3-42 P+−Si pillar Au = 42nm 陽極電阻與電壓關係圖 ... 66 圖 3-43 不同金厚度 P+−Si pillar 之電流密度與電壓關係比較 ... 68 圖 3-44 不同金厚度柱樣品之蕭特基位障高度擬合圖 ... 70 圖 3-45 GOx granting/P−Si 樣品結構示意圖 ... 71
圖 3-46 GOx granting/P−Si SEM, = 0.8, 10mins ... 72
圖 3-47 GOx granting/P−Si SEM, = 0.8, 20mins ... 72
圖 3-48 GOx granting/P−Si SEM,= 0.8, 40mins ... 73
圖 3-49 GOx granting/P−Si SEM, = 0.8, 40˚C, 20mins ... 73
圖 3-50 GOx granting/P−Si SEM, = 0.8, 60˚C, 20mins ... 74
圖 3-51 SEM, Au = 10nm & 20nm 之表面形貌 ... 76
圖 3-52 不同金厚度之 P+−Si pillar SEM, = 0.85, 15mins ... 78
圖 3-53 P+−Si SEM, Au = 32nm, (a) pillar (b) hole, = 0.85, 15mins ... 78
圖 3-54 P+−Si pillar SEM, Au = 32nm, = 0.85, 50℃, 15mins ... 79
圖 3-57 P+−Si pillar SEM, Au = 32nm,
= 0.85, V = 1.5, 50℃, 15mins ... 80
圖 3-58 P+−Si pillar SEM, Au = 32nm, = 0.85, pre-etch, V = 0.5, 1hr ... 81
圖 3-59 P+−Si hole SEM, Au = 32nm, = 0.85, pre-etch, V = 0.5, 50℃,1hr ... 81
圖 3-60 P+−Si SEM, Au = 32nm, = 0.85, 不同蝕刻時間 ... 83
圖 3-61 P+−Si SEM, Au = 20nm, = 0.85, 15mins ... 86
圖 3-62 P+−Si pillar SEM, Au = 20nm, = 0.85, 1hr ... 88
圖 3-63 P+−Si hole SEM, Au = 20nm, = 0.85, 1hr ... 88
圖 3-64 P+−Si pillar SEM, Au = 20nm, = 0.85, 15mins x 4 ... 89
圖 3-65 P+−Si hole SEM, Au = 20nm, = 0.85, 15mins x 4 ... 89
圖 3-66 P+−Si hole SEM, Au = 20nm, = 0.85, pre-etching, V = 0.3 x 3 ... 90
圖 3-67 P+−Si pillar SEM, Au = 20nm, HF]V = 0.9, 15mins x 4 ... 91
圖 3-68 P+−Si pillar SEM, Au = 20nm, HFV = 0.9, 15mins x4 ... 92
圖 3-69 P+−Si hole SEM, Au = 20nm, [HF] = 4(M), V = 0.8, 15mins x 4 ... 92
圖 3-70 P+−Si hole SEM, Au = 20nm, [HF] = 4(M), V = 0.8, 1hr ... 93
圖 3-71 P+−Si Au = 20nm,電壓與電流密度關係 ... 94
圖 3-72 P+−Si pillar, Au = 20nm,V = 0, = 0.85, 15mins, 電流與時間關係 ... 95
圖 3-73 P+−Si pillar, Au = 20nm, I ≦ 0.8mA, = 0.85, 電壓與時間關係 ... 95
圖 3-74 P+−Si pillar SEM, Au = 20nm, = 0.85, 15mins ... 96
圖 3-75 P+−Si pillar SEM, Au = 20nm, = 0.85, V = − 0.5, 15mins ... 97
圖 3-76 P+−Si pillar SEM, Au = 20nm, = 0.85, V = − 0.3, 15mins ... 97
圖 3-77 N−Si SEM, Au = 15nm,= 0.85, 不同蝕刻時間 ... 100
圖 3-78 N−Si pillar SEM, Au = 15nm, = 0.2, 不同蝕刻時間 ... 102
圖 3-79 N−Si pillar SEM, Au = 15nm,= 0.4, 15mins... 103
圖 3-80 N−Si hole SEM, Au = 15nm, = 0.4, 15mins ... 103
圖 3-81 N−Si pillar SEM, Au = 15nm, = 0.5, 15mins... 104
圖 3-82 N−Si hole SEM, Au = 15nm,= 0.5, 15mins ... 104
圖 3-83 N−Si pillar SEM, Au = 15nm, = 0.4, 1hr ... 105
圖 3-84 N−Si hole SEM, Au = 15nm, = 0.4, 1hr ... 105
圖 3-85 樣品於[HF] = 4 (M)之電壓與電流密度關係 ... 106
圖 3-86 N−Si hole SEM, Au = 15nm, [HF] = 4(M), 不同電壓值, 20mins x 3 .. 107
圖 3-88 N−Si hole SEM, Au = 15nm, [HF] = 4(M), V = 5, 50℃, 1hr ... 109
圖 3-89 N−Si Au = 15nm,電壓與電流密度關係 ... 110
圖 3-90 N−Si hole SEM, Au = 15nm,V = − 10, 30mins ... 111
圖 3-91 N−Si hole SEM, Au = 15nm,V = − 10, 30mins ... 111
圖 3-92 N−Si hole SEM, Au = 15nm,= 0.5, V = − 10, 15mins x 4 ... 112
圖 3-93 N−Si hole SEM, Au = 15nm, = 0.5, V = − 10, 15mins x 4 ... 112
圖 3-94 N−Si pillar SEM, Au = 15nm,= 0.5, V = − 5, 1hr x 3 ... 113
圖 3-95 N−Si pillar SEM, Au = 15nm,= 0.5, V = − 5, 1hr x 3 ... 113
圖 3-96 樣品於 之電壓與電流密度關係 ... 114
圖 3-97 N−Si hole SEM, Au = 15nm,= 0.5, V = 5, 15mins x 4 ... 115
圖 3-98 N−Si hole SEM, Au = 15nm,, V = 5, 15mins x 4 ... 115
圖 3-99 N−Si hole SEM, Au = 15nm,= 0.5, V = 20, 15mins x 4 ... 116
圖 3-100 N−Si hole SEM, Au = 15nm,= 0.5, V = 2, 15mins x 4 ... 116
圖 3-101 N−Si hole SEM, Au = 15nm,= 0.5, V = 2, 15mins x 4 ... 117
表目錄
表 3-1 不同金厚度樣品實驗前電阻量測 ... 58 表 3-2 不同金厚度柱樣品之蕭特基位障高度 ... 70 表 3-3 不同 值蝕刻液配比 ... 99
第一章
緒論
1.1 前言
近年來,由於消費性電子產品的崛起,為滿足消費者對於電子產品輕薄短小 及多功能等特性,諸多領域隨著科學技術的進展而有所突破,藉著向下縮減(Top down)與向上組裝(Bottom up)兩個主要製作技術的相輔相成[1],如圖 1-1,向下 縮減(Top down)為光微影技術與蝕刻結合的方式,向上組裝(Bottom up)主要以沉 積合成方式製備結構。
在各式各樣的材料中,以矽優異的半導體性質最受矚目,在地殼中,它是第 二豐富的元素,容易採集成本低廉,常應用在各種積體電路的製程材料,透過不 同的製程如氧化、摻雜與蝕刻等方式,矽材料表現出特別的性質,因此在半導體 產業上扮演十分重要的角色。 矽高深寬比結構除了具有半導體所具有的特殊性質外,同樣顯示出不同於矽 基材的特性,高深寬比結構擁有相當大的表面積/體積比,具容易吸附物質的特 性,如圖 1-2 及圖 1-3,此種結構被當成基底應用於生物之感測元件[2][3]或氣體 感測器[4][5];另外矽高深寬比結構具有場發射、電子傳輸之電學特性,可應用 於電晶體[6],作為源極和汲極的連接;再加上熱傳導、高表面活性與量子限制 效應之特性[7]引起學者們的廣泛討論。矽高深寬比結構作為光電元件的應用也 備受矚目,奈米線具有特殊光學特性,經激發後能發出可見光,且高深寬比結構 能增加發光二極體出光能力,可應用於光學照明。 圖 1-2 Si 奈米柱基底之生物感測器工作示意 [2]
圖 1-3 Si 奈米柱基底之氣體感測器及測量結果 [4] 近年來學者們找尋許多方法來減少光電子元件的光損耗與反射,像是介質干 擾塗料、表面結構建置、奈米陣列圖案製備等[8][9]。如圖 1-4,圖 1-5 及圖 1-6, 若以肉眼觀察矽晶片,會發現其呈現鏡面反光狀,但矽奈米陣列結構卻是呈現黑 色,這是因為奈米柱結構的尺度是小於或近似可見光波長,進入此結構的光不會 產生干涉及繞射現象,而且結構空間中介質的疏密比例也會造成折射率的改變, 降低入射光由於折射率差異而造成的反射,此種特性為抗反射能力,再加上矽高 深寬比結構因具有特殊的光電性質,特別是與矽太陽能電池的結合與應用,藉由 折射率漸變的表面形貌,將入射光留在結構中,藉以達到吸收最大值,表面的結 構可以增加光線進入材料的路徑,更被認為可提升光電效率[10][11][12]。 圖 1-4 Si 與 Si 陣列結構外觀比較 [11]
圖 1-5 Si 基板與 Si 陣列結構之反射率比較 [11]
另一方面,為了因應可攜式電子產品的輕薄短小化的趨勢、光電通訊的產品 的高速化、微系統化的高整合度及環保的需求下,半導體製程技術的開發更快迅 速,需研發更為先進的微連結技術。如圖 1-7,三維(Three Dimension, 3D)整 合系統構裝展現出 3D IC 晶片面積的縮減,並將其性能達到最佳化之規範 [13][14],使得製作成本的下降,其製程大致上而言為一種穿透矽晶圓垂直互連 方式,傳統製作方式為從底部填充入金屬,矽晶圓上以深反應性離子蝕刻或雷射 方式鑽孔,再以導電材料如銅、多晶矽,鎢等材料填滿,但深反性離子蝕刻具無 法精確定義孔洞之疑慮,而金屬輔助蝕刻技術極有潛力取代蝕刻或雷射鑽孔方式, L. Li 等人[15]已將金屬輔助化學蝕刻製程導入矽穿孔技術之發展,深寬比約 11, 期望進一步目標能將此技術導入至次微米與奈米級技術,同時發展三五族化合物 材料之穿孔技術。 圖 1-7 3D IC 示意圖 [14]
1.2 相關文獻回顧
向上組裝與向下縮減是製備矽高深寬比結構的兩大基本架構,向上組裝 (bottom-up)主要以沉積合成的方式製備結構,如氣-液-固(VLS)生長機制、熱蒸鍍 法與電化學沉積等;向下縮減(top-down)為利用微影技術與蝕刻結合的方式,先 於一矽基板上定義奈米結構再進行蝕刻,反應性離子蝕刻屬於此種製備方式。 高溫真空製程 最早有學者提出以固-液-氣相生長法 (Vapor-Liquid-Solid Growth) 製備矽晶 鬚 (whisker) 結構[16],其原理主要為真空環境下通入氣態矽或者四氯矽甲烷 (SiCl4),氣體經擴散進入矽與奈米級觸媒合金液滴,利用欲成長之材料形成共融 點 (eutectic point) 之特性,當矽濃度達到飽和後,即從液滴析出奈米結構,氣態 矽不斷進入腔體進行反應,固態矽也就持續不斷地從液固界面析出形成固態柱狀 結構,如圖 1-8 與圖 1-9,並透過控制溫度壓力等方式來控制矽奈米線的生長, 矽奈米結構直徑與金屬粒子大小成正比關係,因此金屬粒子大小的篩選為控制奈 米結構直徑的關鍵因素;且奈米結構的晶格取向(Orientation)也同樣會影響奈米 結構直徑,再加上由於無法準確控制金屬液滴位置,結構無法均勻一致生長,多 種結構參數的變化對元件特性的變異也有很大的影響。而後人藉由此種技術為基 底衍生其他以化學氣相沉積製備奈米柱結構,以反應氣體通入腔體中,經反應後 生成物擴散而沉積於基板表面上,基於化學氣相沉積法之機制演變出幾種方式 : 氣-液-固成長機制,氣-固成長機制,固-液-固成長機制[17][18][19],主要為改變 氣體來源或催化金屬,其本質原理大致相同,但都需要透過真空與高溫條件下進 行,設備建置複雜,成長耗時且反應氣體具危險性。圖 1-8 VLS 成長法 [17]
電化學蝕刻法
電化學蝕刻法於 1956 年由 Uhlir 所提出[21],以氫氟酸(Hydrofluoric acid, HF) 做為電解液對矽進行電解拋光時,發現矽晶圓表面上覆蓋一層為多孔矽黑色薄膜。 為了有效控制生成長結構通常製作多孔矽的方法為,在 HF 混合溶液中,對矽基 材給予外加偏壓來進行電化學蝕刻,對 P 型半導體而言,給予正偏壓,有電解電 流的產生,進而矽蝕刻,對 N 型而言則是相反,正向偏壓需照光以產生電流, 負偏壓則立即出現電流,因為 N 型半導體多數載子為電子,產生電化學反應需 要照光或者是高電場環境或使用其他方式產生電洞,P 型半導體多數載子為電洞, 無需外加電洞激發即可進行蝕刻反應,影響多孔矽生成型態的因素包括電解液 HF 之濃度、電解溫度、矽晶片摻雜濃度,用以製備不規則之孔洞、樹枝狀或微 孔,而此種方式只能沿著<100>方向進行蝕刻。 圖 1-10 電化學蝕刻架構 [11]
矽於 HF 電解溶液中產生溶解矽的作用,造成矽蝕刻現象,而矽溶解過程由 以下說明: (1) HF 於水中解離出氟離子,因電解作用,矽氫鍵結被氟所取代,氟 與矽形成鍵結。 (2) 由氟−矽鍵結之極性吸引另一個氟離子與矽鍵結,並釋放氫 離子域一個電子,兩個氫原子結合成氫氣釋出。 (3) 因氟與矽鍵結,弱化矽與 矽之間的鍵結能力,容易被蝕刻液解離出的氫離子及氟離子所破壞,使氫與矽分 別與矽表面結合。 (4) 重複 (1) ~ (3) 反應,矽表面形成矽−氫鍵及 SiF4,並與 HF 化合成水溶性 H2SiF6,矽氫鍵繼續重覆以上反應。 圖 1-11 HF 介入矽蝕刻之化學反應路徑示意圖 [21]
深反應性離子蝕刻 (Deep Reactive−Ion Etching) 現今製造高深寬比矽柱結構半導體製程技術主要方式為利用微影技術,結合 物理性的離子轟擊與化學反應蝕刻技術來製備矽高深寬比結構,如圖 1-12 所示, 此種技術,以離子轟擊之方式將表面原子鍵結破壞,加速反應速率,再接著將沉 積產物清除,使蝕刻表面能與蝕刻氣體接觸,沉積在蝕刻圖形表面的沈積物可被離 子清除打掉,故蝕刻可繼續進行,側壁上之沉積物因為受離子轟擊而保留,阻隔蝕刻表 面與蝕刻氣體接觸,成為一非等向性蝕刻[22]。 深反應性離子蝕刻通常會使用含有鹵化物的反應氣體,如四氟化碳 (carbon tetrafluoride, CF4)、六氟化硫 (sulfur hexafluoride, SF6) 以及氯氣 (chlorine, Cl2)
等。此種蝕刻技術,反應氣體同樣具高度危險性,而且設備建置成本費用高昂, 再加上電漿離子蝕刻方式對於材料本身有極大影響,因離子轟擊或是帶電粒子造 成材料損壞缺陷等,有元件特性劣化的可能。深度反應性離子蝕刻雖然可以用來 蝕刻出高深寬比的矽結構,但此蝕刻技術為沉積與蝕刻兩道步驟的循環製程,如 圖 1-13,會造成蝕刻結構側壁產生約 100 nm 左右的波浪結構,無法用於奈米結 構的圖案精確定義。
圖 1-12 反應性離子蝕刻 [22]
1.3 研究動機與目的
傳統製備高深寬比方法需要透過真空與高溫條件下進行,設備建置複雜,成 本高昂,且反應氣體具危險性。2000 年首次有學者提出金屬輔助化學蝕刻法, 使用 HF 與 H2O2配合金屬催化層進行蝕刻,此方式具有簡易快速優勢製備矽基 高深寬比結構。 為提高蝕刻深度,針對金屬輔助化學蝕刻技術,長時間蝕刻可能導致與蝕刻 液接觸之基材因氧化而蝕刻,造成側向蝕刻現象,或是形成緻密孔隙,抑或有結 構倒塌的情況產生。 另外,當元件尺度微小化,容易因為水分影響而沾黏,甚至於損壞,而無論 是氣體風乾或是加溫烘乾即會面臨沾黏問題,為了避免此種情況產生,另一種方 式則是導入超流體乾燥技術,超臨界流體乾燥是一種去除溶劑並且不引起表面張 力效應的方式,在超臨界條件下,不存在表面張力,清除超臨界流體不會影響固 體形狀。 本研究主要工作,是如何將材料蝕刻的更直、更深且不會塌陷與沾黏。為解 決這些問題,如何調控蝕刻穩定朝目標方向蝕刻即是本研究關鍵目標,而解決概 念則是減少蝕刻反應載子向蝕刻反應區周圍擴散,穩定維持蝕刻方向,因此嘗試 利用高密度石墨板建立濕蝕刻架構,以電流導入方式,調控載子移動,並對金屬 薄膜層厚度影響進行探討,並對基材、蝕刻液濃度比例與蝕刻時間等參數進行探 討,比較以上不同因素對於蝕刻形貌之影響。 同時發展超臨界二氧化碳金屬輔助蝕刻技術,以突破目前金屬輔助化學蝕刻 技術濕蝕刻瓶頸,保留金屬輔助蝕刻原具備的技術特性與優點,利用超流體具有 之特性 : 接近零的表面張力,極低的黏滯性與高擴散性,改善結構坍塌與黏滯 現象,期望能克服因濕蝕刻溶液之表面張力所引起的坍塌。第二章
金屬輔助化學蝕刻及相關文獻
2.1 金屬輔助化學蝕刻節原理
金屬輔助化學蝕刻 (Metal − assisted Chemical Etching, MaCEtch )最早發現 於 1997 年,以 HF、HNO3與 H2O2之混合溶液,對鋁覆蓋之矽基板進行蝕刻, 於 2000 年 X. Li 與 P. W. Bohn 兩人首先報導,開啟了利用濕式蝕刻技術製造高 深寬比結構的發展[23][24][25],如圖 2-1,此研究團隊展示於矽基材上沉積一層 極薄貴重金屬層 (如 Au, Pt, Ag),浸泡至含氫氟酸 (HF) 與過氧化氫 (H2O2) 的 混合蝕刻溶液中,能催化矽基材的蝕刻反應,並獲得垂直孔洞或是柱狀結構,金 屬輔助化學蝕刻為一種無須額外熱能或電能的自發性反應,其製程相對簡單,成 本低廉,並能在室溫下進行反應,此種蝕刻機制主要為混合蝕刻溶液催化金屬產 生小區域的氧化還原反應,陽極反應區為貴重金屬層,主要作用為降低氧化劑還 原所需要的活化能,陰極反應區為金屬與半導體界面。 在室溫的環境下經過即可蝕刻形成目標結構,推論金屬與矽接觸位置產生區 域性氧化還原反應的電化學電流傳遞過程,而提出其機制化學反應式如下所示: H2O2 + 2H+ → 2H2O + 2h+ (2-1) 2H+ → H2↑+ 2h+ (2-2) (2H+ + 2e- → H2↑) (2-3) Si + 4h+ + 4HF → SiF4 + 4H+ (2-4) SiF4 + 2HF → H2SiF6 (2-5) 總反應式: Si + H2O2 + 6HF → 2H2O + H2SiF6 + H2↑ (2-6)
2.2 載子擴散機制
P. Liato 等人[26]於 2012 年提出藉由改變長條金屬層間距以及蝕刻液濃度, 觀察表面形成之孔洞,討論電洞擴散距離,如圖 2-2,當 [HF] = 1.73(M),[H2O2] = 1.21(M),蝕刻時間 20 分鐘,A 位於兩金屬之間,寬 1m,B 位金屬層兩邊界 寬 1m 處,當間距越大,AB 區域越平滑,驗證電洞注入為金屬輔助蝕刻技術之 主要機制;並提出二維電洞擴散模型,式 2-7,如圖 2-3,模擬結果與實驗結果 吻合,電洞擴散與時間相關,當蝕刻時間越長,電洞擴散情況越嚴重。 另外,為抑制過量電洞擴散情況,並提出利用一空間電位架構,如圖 2-4, 此結構金屬層間距 20mm,分別施加電位勢 10 及 100(V),樣品金屬層朝陽極平 行置放,以驅動電洞,由其結果發現,有效抑制電洞擴散。 圖 2-2 MaCEtch 電洞注入擴散示意圖及不同間距之實驗結果 [26]𝐶(𝑥, 𝑦, 𝑡) 𝐶0 = 𝑒𝑟𝑓𝑐( 𝑥 2√𝐷𝑡)𝑒𝑟𝑓𝑐 𝑦 2√𝐷𝑡 C0 : Au-Si 介面過量電洞濃度,D : 電洞擴散率,t : 蝕刻時間 (2-7) 圖 2-3 式 2-7 模擬圖與實際操作對應 [26] 圖 2-4 利用空間電位架構於不同電壓下抑制電洞擴散之情形 [26]
L. Li 等人[27]於 2015 年提出電荷的轉移機制報告,如圖 2-5,電洞傳輸牽涉 到兩個過程,垂直蝕刻 Charge transport 1 (CT1) 與側向蝕刻 Charge transport 2 (CT2),電洞傳輸與蝕刻後得到三維結構形貌有相關性。推論整個金屬蝕刻反應 過程,如圖 2-6 : 首先 H2O2接觸金屬表面,經金屬催化電洞生成,H2O2作為氧
化劑提供電洞並注入陰極矽基材表面,產生破壞表面矽價電的矽−氫鍵(Si−H), HF 解離出的氟離子取代矽表面氫鍵位置,因此矽表面含有 Si−H 及生成產物四 氟化矽(SiF4),最後 SiF4與 HF 化合成能溶於水的 H2SiF6,電洞持續地注入,氫
離子不斷的被取代最後形成 H2釋出,而矽表面產生出液態化合物 H2SiF6或氣態
的 SiF6,此反應機制能藉由活化金屬方式持續地進行非等向性蝕刻。
2.3 不同催化金屬及其厚度之影響
不同種類的催化金屬對於蝕刻反應有不同的影響,一般而言,蝕刻結構的形 態隨著貴金屬類型而變化,如圖 2-7 所示,獨立的金或銀粒子能將矽基板蝕刻出 一個直洞,但鉑相對不同,在蝕刻過程中由於顆粒的隨機運動而形成螺旋孔洞, 形成彎曲的孔隙,而且沒有均勻的蝕刻方向[28]。除了蝕刻形貌的有著不同的結 果,不同貴重金屬類型也影響蝕刻速率。銀粒子相較其他金屬,蝕刻速率較慢, 不過整體蝕刻情況穩定,鉑粒子蝕刻速度最為快速,但蝕刻過度迅速無法有效垂 直向下蝕刻,造成蝕刻型態過於雜亂,均勻性不佳,而金薄膜的蝕刻速度適中, 能有效率的垂直蝕刻[29],推論蝕刻速率的差異是由於這些貴金屬的催化活性不 同所致。L. Li 等人[30]於 2013 年提出以多孔奈米級金屬催化層進行垂直均勻溝槽蝕 刻,如圖 2-8 與圖 2-9,金屬輔助化學蝕刻受限於邊界,當金屬催化層太厚,反 應難以抵達中央,造成蝕刻不均勻,顯示催化層穿孔接觸蝕刻液的重要性,並推 論金屬輔助化學蝕刻反應主要分為物質轉移(Mass transport, MT)與電荷的轉移 (charge transport, CT)兩個部分,物質轉移主要是反應物 HF 與過 H2O2的作用及 生成物、水、氟矽酸(H2SiF6),在物質轉移中由催化劑主導反應,電荷轉移為電 洞(electron hole, h+ )生成、移動及消耗之流程。 圖 2-8 Au = 10nm 金屬輔助蝕刻之機制與影響 [30] 圖 2-9 Au = 20nm 金屬輔助蝕刻之機制與影響 [30]
Y. Song 等人[31]於 2014 年提出針對砷化鎵(GaAs)基材進行金屬輔助化學蝕 刻對於金屬層厚度之研究,如圖 2-10,藉由改變不同厚度的金屬催化層,發現貴 重金屬層厚度對於蝕刻反應關係有兩種不同的模式,以金屬催化層厚度為 15nm 為分水嶺,當金屬層厚度大於 15nm 時,物質經催化金屬層與半導體基材介面作 為交換,進行蝕刻交換反應;當金屬層厚度小於 15nm 時,物質直接穿透催化金 屬層作交換反應,由於蝕刻速率隨著金屬催化層的厚度改變而定義出 In−plane 與 out−of−plane 兩種蝕刻模式,如圖 2-11 並量測不同金屬催化層厚度於不同時 間所量測得到的三維矽柱高度,用以計算蝕刻速率。經由此團隊的研究成果做為 參考,因而建立本研究所需的樣品之金屬催化層厚度依序為 12nm、22nm、32nm 及 42nm。 圖 2-10 In−plane 與 Out−of−plane 兩種蝕刻模式之示意圖 [31]
2.4 蝕刻液配比對蝕刻之影響
作為金屬輔助化學蝕刻與 HF 混合的氧化劑有非常多種選擇,如硝酸銀 (AgNO3) 、氯化金鉀 (KAuCl4)、四氯金酸 (HAuCl4)、六氯鉑酸鉀 (K2PtCl6)、
硝酸鐵 (Fe(NO3)3)、硝酸鎳 (Ni(NO3)2)、硝酸鎂 (Mg(NO3)2)、過氧化氫 (H2O2)、
過硫酸鈉 (Na2S2O8)、過錳酸鉀 (KMnO4)、重鉻酸鉀 (K2Cr2O7)、氧氣 (O2 bubble),
或 O2溶解於 H2O。圖 2-12 指出不同氧化劑之標準電位,H2O2電位比矽的電化
學勢高出很多,電洞能注入較深的價帶中,通常做為金屬輔助蝕刻的氧化劑[25]
圖 2-12 不同氧化劑於矽基材下之相對電位,H2O2/H2O 具有最大氧化還原電位
改變蝕刻液比例對於蝕刻樣貌也有不同的結果,C. Chaetier 等人[24]於 2008 年提出,定義莫耳濃度之蝕刻比例 = [HF]+[H[HF] 2O2] (2-8) 當 1 > ρ > 0.7,當金屬催化粒子使用銀,能有效垂直蝕刻,且無明顯側向蝕 刻現象;如圖 2-13,0.7 > ρ > 0.2,蝕刻後,為一錐狀結構,矽表面的直徑大於 金屬粒子直徑,側蝕現象產生;當ρ 值低於 0.3,也就是 0.2 > ρ > 0.09 則會發展 成一個火山口的樣貌,矽的表面蝕刻直徑變得更大,當 0.09 > ρ > 0,無明顯孔 洞或火山口結構產生,傾向生成一個平滑的奈米級孔洞。其蝕刻關係由以下說明, 當 1 > ρ > 0.7,也就是高濃度的 HF,由於具備足夠的 HF 溶解矽或矽氧化物,與 Au−Si 介面生成的電洞有極大的相關性,而電洞生成由 H2O2控制,蝕刻速率主 要由 H2O2濃度主導。當ρ 值低於 0.7 時,蝕刻速率由 HF 濃度所主導,由於電洞 生成的速度遠大於消耗速度,無法被消耗的電洞,由孔洞尖端擴散到側壁,因而 產生細小孔洞。進一步說明,當ρ 值極低或者 H2O2的濃度極高時,暴露於蝕刻 液中的矽基板,電洞可能擴散到整個矽基板,HF 蝕刻整個區域,因此獨立的金 屬粒子會將矽蝕刻出平滑的細小坑洞。 另外,根據 R. A. Lai 等人之研究[32],如圖 2-14 所示,不同 值比例對蝕 刻速率有不同的影響,於各種不同類型的基板,在 = 0.4 ~ 0.6 區間,由於有 足夠的 HF 及 H2O2與樣品進行反應,蝕刻速率為較高,當 值較高或是較低, 蝕刻率下降。
圖 2-13 錐狀孔形成機制,當條件為 0.7 > ρ > 0.2 [24]
2.5 蝕刻系統之蕭特基能障
若以能帶結構來討論金屬輔助蝕刻機制,由於大部分研究多半將金屬僅視為 催化反應半反應 R. A. Lai 等人[32] 於 2016 年提出另一種對金屬層看法,從能 帶結構來看,由於金屬和矽基材之間形成蕭特基接面 (Schottky junction),進而 控制由 H2O2注入矽中的電洞空間分佈,如圖 2-15 與圖 2-16,因電洞遠離 Au–Si 介面或是累積在 Au–Si 介面,造成蝕刻,H2O2注入的電洞分佈與摻雜程度、摻 雜類型、晶體表面方向和蝕刻溶液具有高度相關性,造成不同的蝕刻形貌,定義 = [HF]/[HF]+[H2O2],對於 N−Si 而言,能帶形成一個陷阱,電洞集中矽表面造 成矽氧化進而蝕刻,而 P−Si 因能帶反轉,電洞遠離介面造成蝕刻,但容易因電 洞擴散造成側向蝕刻。2.6 金屬輔助陽極蝕刻
C. Q. Lai 等人[33]於 2016 年提出另一種製造多孔矽的方法,建構金屬輔助 陽極蝕刻架構 (Meatal–assisted anodic etching),如圖 2-17、圖 2-18 及圖 2-19, 以不使用 H2O2 介入方式,利用外部電路直接注入電洞於矽基板,提出電洞對
Au−Si 以及 HF−Si 界面之作用機制及蕭基能障高度關係,不同摻雜程度的基板 有不同的蝕刻結果,使用[HF] = 4 (M), 圖 2-17 (a),P 型(10 − 20 ohm − cm),V = 0.3,J = 7.6 mA/cm2,蝕刻 10 分鐘,圖 2-17 (b),P+
(0.005 − 0.01 ohm − cm),J = 23 mA/cm2,蝕刻 5 分鐘,矽有額外的電洞累積在 Au‒Si 以及 HF‒Si 界面,蝕 刻結果同時具有矽柱結構以及表面多孔矽層,等同於金屬輔助蝕刻與陽極蝕刻之 相加效果,圖 2-17 (c),N 型( 10 − 30 ohm − cm),V = 2.7,1.4 mA/cm2,蝕刻 10
分鐘,矽蝕刻反應發生於 Au−Si 界面,形成矽柱結構,圖 2-17 (d),N+
( < 0.005 ohm − cm),28 mA/cm2,蝕刻 5 分鐘,蝕刻反應 HF/Si 界面,幾乎沒有矽柱結 構形成。
圖 2-17 SEM, 不同類型 Si 之金屬陽極蝕刻結果 (a) P−Si (b) P+−Si (c) N−Si (d) N+−Si [33]
圖 2-18 金屬陽極蝕刻架構 [33]
2.7 偏壓調控金屬輔助化學蝕刻
為了有效控制電洞注入的準直性,L. Li 等人於 2014 年提出[15],將一偏壓 源陰極直接接觸於蝕刻基板,陽極使用鉑金屬,基板 P−Si (1 – 10 ohm−cm),蝕 刻液比例 [HF] = 1.8(M),[H2O2] = 3.06(M),如圖 2-20 與圖 2-21,據其結果得知, 當偏壓越高,蝕刻結果準直性越好,此團隊並將此技術導入矽穿孔發展,其深寬 比約為 11,但其蝕刻溫度達 100℃,操作上可能具有危險性。 圖 2-20 偏壓架構比較 [15]2.8 小結
金屬輔助蝕刻作為一種自發性反應,方法相對簡單,成本低廉,經由以上文 獻探討對反應機制有縱觀的了解,乃本研究之重要參考。 X. Li and P. W. Bohn (2000 年) 金屬輔助化學蝕刻首次報導 C. Cartier 等人 (2008 年) 探討 HF/H2O2之機制及蝕刻形貌 P. Liato 等人 (2008 年) 電洞擴散效應及首次提出空間電位架構抑制電洞擴散 Y. Song 等人 (2014 年) 不同金屬層厚度對蝕刻的影響,提出 In − plane/Out – of − Plane 之機制 L. Li 等人 (2014 年) 偏壓導入架構,抑制電洞擴散,增加蝕刻準直性 C. Q. Lai 等人 (2016 年) 金屬陽極蝕刻,以外部電路注入電洞取代 H2O2, 並探討蝕刻系統中的蕭特基能障關係 R. A. Lai 等人 (2016 年) 探討蝕刻系統中的蕭特基能障,不同蝕刻液濃度 與基板摻雜程度與類型對蝕刻形貌之影響第三章
研究方法及結果與討論
3.1 本章架構
本章架構主要由研究方法與結果討論所組成,3.2 研究方法分為 : 實驗架構 設置,以及樣品製備;3.3 結果討論。
3.2 研究方法
本研究中,蝕刻架構有三大類別,金屬輔助化學蝕刻,金屬陽極蝕刻以及電 流導入之金屬輔助化學蝕刻,金屬陽極蝕刻以外部電流方式直接注入電洞取代 H2O2,電流注入金屬輔助蝕刻,則是以調控載子方式,進行蝕刻,後兩者的基 底架構相同,差別在於蝕刻液中 H2O2有無;升溫架構以此三種類別為基礎做改 善,再進行隔水加熱與密封蝕刻裝置。 金屬輔助化學蝕刻Metal – assisted chemical etching, MaCEtch Etchant : HF, H2O2
金屬陽極蝕刻
Metal − assisted anodic etching, MAAE Etchant : HF,以外部電路注入載子。 電流注入金屬輔助化學蝕刻
Current driving Metal – assisted chemical etching, CD − MaCEtch Etchant : HF, H2O2,調控載子。
3.2.1 實驗載具與相關架構
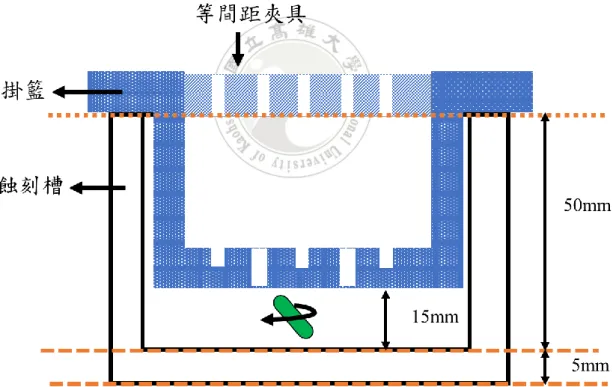
此方法構想為施加電位調控載子至蝕刻液與樣品介面,使矽溶解,建立相對 簡單的蝕刻架構,此實驗設計主要朝向比較陽極與陰極蝕刻差異進行討論,同時 討論 H2O2有無之影響,觀察實驗結果。 實驗架設主要將尺寸 10mm x 10mm 樣品固定於高密度石墨板上,石墨板尺 寸為長 50mm 寬 20mm 厚度 1mm,輔以偏壓,量測電流密度。部分樣品背面鍍 上一層 50nm 鈦及 200nm 金製作歐姆接觸;蝕刻裝置之設計,如圖 3-2,最初使 用 3D 列印技術製作一等間距樣品夾具,夾具僅能固定石墨板上端,底部無法固 定,有位移的可能性;如圖 3-3 及圖 3-4,為了更精準地固定底部石墨板間距, 因此設計基材為聚四氟乙烯(Polytetrafluoroethylene, PTFE)的蝕刻裝置,PTFE 方槽內嵌活動式掛籃,並在掛籃上方再嵌入等間距夾具,掛籃每個面皆具有多處 開口,底部也有凹槽,作為放置石墨板位置,此設計石墨板上下兩端皆有固定處, 改善上下間距不等長的問題。如圖 3-5,樣品固定則是利用 Polyimide(PI) tape 將 樣品固定於石墨板,除反應作用區以外的地方,石墨板接觸蝕刻液之處皆以 PI tape 整個包覆,再置放於夾具中,使兩石墨板有固定距離。 蝕刻條件則有以下情況,如圖 3-6,樣品端分別置放電極其中一端,架構一 為樣品置放於石墨板陽極位置,架構二則是將樣品置固定於石墨板陰極位置,並 連接至電源供應器,接著將整個裝置放入裝有蝕刻液之 PTFE 蝕刻槽中進行實驗, 蝕刻液體積比例分別使用 etchant (I) : [HF] : [H2O2] = 5.7 : 1(M),蝕刻液體積 65ml 與 etchant (II) : [HF] = 7(M),蝕刻液體積 60ml,兩種蝕刻液比例,由電源供應器 設置電壓值,設定蝕刻時間,並量測記錄電流值,觀察樣品於兩種架構下以及 H2O2有無介入之電流密度與樣品表面型態。圖 3-2 3D 列印製作石墨板夾具 (材質 PLA)
圖 3-4 PTFE 蝕刻裝置
圖 3-6 (a) 架構一與 (b) 架構二樣品放置示意圖
升溫電流注入架構 為了增進蝕刻速率,如圖 3-8,建置一電流注入法升溫架構,使用隔水加熱 的方式,讓蝕刻方槽能夠均勻升溫,考慮蝕刻過程中的蝕刻液蒸氣擴散的危險性, 如圖 3-9 與圖 3-10,重新設計石墨板等間距夾具,將整個蝕刻方槽整個封住,為 使電線與石墨板接觸區域避免受 HF 腐蝕,石墨板穿過等間距夾具,使電線與蝕 刻液隔離,因此使用 3D 列印技術製作出新夾具,進行實驗操作。 此架構操作方式為:先將蝕刻液密封進行隔水加熱至目標溫度,接著置換蝕 刻載具,進行蝕刻。 圖 3-8 升溫電流注入架構示意圖
圖 3-9 升溫架構之夾具設計
蝕刻液濃度條配 為調配實驗中不同比例之蝕刻液,以體積莫耳濃度換算所需要的溶液體積, 使用 HF(49 wt%)與 H2O2(35 wt%)調製不同比例,首先,設定蝕刻液總體積,接 著換算 HF、H2O2與 H2O 之個別溶液體積。 𝑉𝐻𝐹 = [𝐻𝐹] × 𝑀𝑟,𝐻𝐹 × 𝑉𝑇𝑜𝑡𝑎𝑙 𝐷𝐻𝐹× 𝑊% (3-1) 𝑉𝐻2𝑂2 = [𝐻2𝑂2] × 𝑀𝑟,𝐻2𝑂2× 𝑉𝑇𝑜𝑡𝑎𝑙 𝐷𝐻2𝑂2× 𝑊% (3-2) 𝑉𝐻2𝑂 = 𝑉𝑇𝑜𝑡𝑎𝑙 − (𝑉𝐻𝐹+ 𝑉𝐻2𝑂2) (3-3) [HF]: HF 體積莫耳濃度(M) [H2O2]: H2O2體積莫耳濃度(M) 𝑉𝐻𝐹: HF 使用體積 (L) 𝑉𝐻2𝑂2: H2O2使用體積 (L) 𝑉𝐻2𝑂: H2O 使用體積 (L) 𝑀𝑟: 分子量 D: 密度 (g/ml) 𝑉𝑇𝑜𝑡𝑎𝑙: 蝕刻液總體積 (L) W%: 重量百分比濃度
3.2.2 樣品製備
本研究所需樣品,如圖 3-11,圖 3-12 及圖 3-13,以光微影製程建置微米級 二維週期圖案,接著以電子束蒸鍍方式成長不同厚度的金薄膜層 12nm、22nm、 32nm 及 42nm 之二維週期性圖案結構,直徑與間距為 1 比 1,並分別製作出兩種 架構,柱及洞結構。 圖 3-11 SEM,二維週期性微米圖案柱結構樣品圖 3-12 二維週期 pillar & hole 樣品結構示意圖
製備週期為直徑 5m hole 結構之樣品 (1) 基板清洗 a. 基板浸泡於丙酮,並使用超音波震盪 15 分鐘。 b. 接著將基板浸泡於異丙醇中,並使用超音波震盪 15 分鐘。 c. 1% HF 浸泡 30 秒清洗矽表面氧化物。 d. 使用去離子水(DI water)將基板清洗乾淨。 e. 使用氮氣將基板吹乾。 (2) 曝光顯影 a. 塗佈抗反射層 ICON7,減少曝光時因駐波效應影響光阻側壁產生波浪結 構,使用轉速 3000rpm,時間 40 秒,塗佈抗反射層後,於加熱板上軟烤, 溫度設置 205˚C,時間 60 秒。 b. 光阻塗佈,使用負光阻 NR7500P,以轉速 3000rpm,40 秒,厚度約為 500nm, 光阻塗佈後於加熱板上軟烤,溫度設置 150˚C,時間 60 秒。 c. 曝光,時間為 5 秒。 d. 曝光後於加熱板進行軟烤,溫度設置 100˚C,時間 60 秒。 e. 顯影,時間為 10 秒,並以去離子水清洗,氮氣吹乾,並以 OM 觀察結構 完整性,以確保光阻經由曝光顯影是否成功將圖案轉移。 (3) 抗反射層清除,以反應式離子蝕刻系統 RIE,使用 O2 流速 20sccm,真空條 件 60mtorr 以下,RF power 100W 條件進行抗反射層清除。
(4) 基板背部電極以電子束蒸鍍系統 E−Gun Evaporation System 依序蒸鍍鎳或鈦 50nm,最後鍍上 200nm 金。
製程流程圖
(1)基板清洗 (2) 抗反射層塗佈 3000rpm, 40sec 軟烤 205˚C, 60sec (3) 負光阻 NR7500P 塗佈 3000rpm, 40sec 軟烤 150℃, 60sec (4)曝光 軟烤 100℃, 60sec (5) 顯影 顯影時間 5sec (6)ARC 清除 (7)背電極製作 (8) Au film 蒸鍍 圖 3-14 直徑 5m 洞樣品製作流程圖製備週期為直徑 10m pillar 之樣品 (1) 基板清洗 a. 基板浸泡於丙酮,並使用超音波震盪 15 分鐘。 b. 接著將基板浸泡於異丙醇中,並使用超音波震盪 15 分鐘。 c. 1% HF 浸泡 30 秒清洗矽表面氧化物。 d. 使用去離子水(DI water)將基板清洗乾淨。 e. 使用氮氣將基板吹乾。 (2) 曝光顯影 a. 塗佈抗反射層 ICON7,減少曝光時因駐波效應影響光阻側壁產生波浪結 構,使用轉速 3000rpm,時間 40 秒,塗佈抗反射層後,於加熱板上軟烤, 溫度設置 205˚C,時間 60 秒。 b. 光阻塗佈,使用負光阻 NR7500P,以轉速 3000rpm,60 秒,厚度約為 500nm, 光阻塗佈後於加熱板上軟烤,溫度設置 150˚C,時間 60 秒。 c. 曝光,時間為 5 秒。 d. 曝光後於加熱板進行軟烤,溫度設置 100˚C,時間 60 秒。 e. 顯影,顯影時間為 5 秒,並以去離子水清洗,氮氣吹乾,並以 OM 觀察 結構完整性,以確保光阻經由曝光顯影是否成功將圖案轉移。 (3) 抗反射層清除,以反應式離子蝕刻系統 RIE,使用 O2 流速 20sccm,真空條 件 60mtorr 以下,RF power 100W 條件進行抗反射層清除。
(4) 基板背部電極生成,以電子束蒸鍍系統 E − Gun Evaporation System 依序蒸 鍍鎳或鈦 50nm,最後鍍上 200nm 金。
(5) 金薄膜,以電子束蒸鍍系統 E − Gun Evaporation System 蒸鍍目標金屬厚 度。
製程流程圖
(1)基板清洗 (2) 抗反射層塗佈 3000rpm, 40sec 軟烤 205˚C, 60sec (3) 負光阻 NR7500P 3000rpm, 60sec 軟烤 150℃, 90sec (4)曝光 軟烤 100℃, 60sec (5) 顯影 顯影時間 5sec (6) ARC 清除 (7)背電極製作 (8)金屬層蒸鍍圖 3-16 樣品進行金薄膜蒸鍍
.
3.3 蝕刻結果與討論
3.3.1 不同樣品於溶液下電壓與電流密度關係
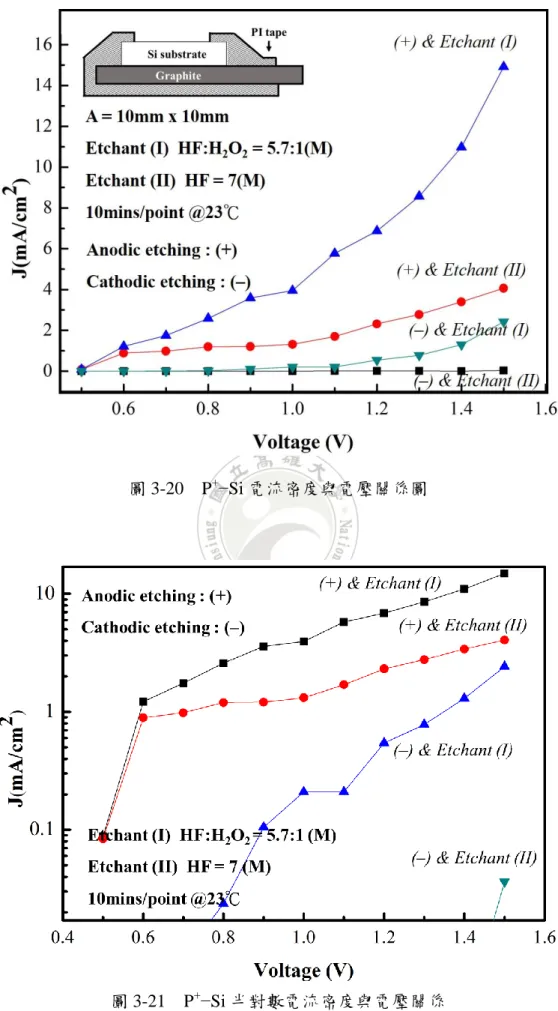
無樣品實驗架構對於不同蝕刻液濃度電壓電流特性 如圖 3-18,蝕刻液比例使用 etchant (I) : [HF] : [H2O2] = 5.7 : 1(M),ρ = 0.85, 蝕刻液體積 65ml,etchant (II) : [HF] = 7(M),蝕刻液體積 60ml,及[HF] = 4(M), 蝕刻液體積 70ml,並以石墨板量測從電壓值 0.5 ~ 5 (V) 所量測電流值,石墨板 尺寸為長 50mm 寬 20mm 厚度 1mm,石墨板之蝕刻液接觸深度約為 40mm,從 etchant (I) : [HF] : [H2O2] = 5.7 : 1 (M)曲線觀察,當電壓值約在 1.5 (V)左右,可觀 察到電流明顯上升趨勢,又以 etchant(I) 含 H2O2蝕刻液整體電流值較大;另一 方面,但當電壓值超過 3.5(V),其電流值已超過儀器負載,因此無實際量測數據, 不同濃度 HF 溶液之結果也有一些不同,濃度較低的蝕刻液在 V = 4(V)後,量測 得到電流值較低。P+−Si (無背鍍金)電流注入法實驗結果與討論 使用 P+−Si (thickness = 430 m, Resistivity < 0.0015 ohm−cm) 尺寸為 10mm x 10mm,一開始僅將清洗後的樣品直接以 PI tape 固定於石墨板上進行蝕刻電流量 測,為降低整體迴路電阻值,之後進一步將 P+−Si 背面依序蒸鍍 200nm 金製作背 部電極,再以 PI tape 固定於石墨板上,並於 4 種條件(樣品分別放置正負極兩端 及 H2O2有無)進行蝕刻,圖中 (+) 表示樣品固定陽極,對應圖 3-6 架構一, (—) 表示樣品固定陰極,對應圖 3-6 架構二,石墨板間距 d 固定為 5mm,同一片樣 品於一條件下連續量測電壓與電流之關係,並繪出電流密度電壓關係圖。 圖 3-19 P+−Si 實驗流程
P+ − Si (無背鍍金) 直接接觸石墨板之製備及量測流程 1. 基板清洗: a. 基板浸泡於丙酮,並使用超音波震盪 15 分鐘。 b. 接著將基板浸泡於異丙醇中,並使用超音波震盪 15 分鐘。 c. 1% HF 浸泡 30 秒清洗矽表面氧化物。 d. 使用去離子水 (DI water) 將基板清洗乾淨。 e. 使用氮氣將基板吹乾。 2. 樣品架設: a. 使用 Polyimide tape 將 10 x 10 mm 矽基板固定基板於石墨板。 b. 量測電阻值,確認基板與石墨板之間接觸。 3. 進行蝕刻,紀錄電流值。
圖 3-20 P+−Si 電流密度與電壓關係圖
P+−Si 直接接觸石墨板以電流注入架構進行實驗,圖 3-20 與圖 3-21 P+−Si 以 PI tape 固定於石墨板上,並以探針量測固定後之基板表面電阻,其值約有 10K ~ 10MΩ,同一樣品連續以電壓 0.5 至 1.5,共取 11 個電壓值,電流值取樣頻率 1Hz, 量測時間為 10 分鐘,再取得電流平均值,並繪製成電流密度與電壓關係圖。 由圖 3-20 可觀察到,在樣品位於陽極上的條件比起陰極位置其電流密度值 較大;另一方面,H2O2之蝕刻液條件之電流密度同樣也比 H2O2排除的條件來的 高,然而,此種結果整體看來與預估的電流密度值還要來的低,觀察陽極含有 H2O2之條件,所得到的電流密度值,最高僅有 16mA/cm2,再加上測量實驗前所 測得的基板表面電阻甚大,考慮可能在固定樣品時,無法有效處理接觸電阻,導 致蝕刻電流因基板與石墨板之間的阻抗而下降,因此嘗試在基板背部製作電極, 蒸鍍一層金屬做改善。
P+−Si (背鍍金)之製備及量測流程 1.基板清洗: a. 基板浸泡於丙酮,並使用超音波震盪 15 分鐘。 b. 接著將基板浸泡於異丙醇中,並使用超音波震盪 15 分鐘。 c. 1% HF 浸泡 30 秒清洗矽表面氧化物。 d. 使用去離子水 (DI water) 將基板清洗乾淨。 e. 使用氮氣將基板吹乾。 2.背電極製作 a. 先於基板背部蒸鍍 50nm 鎳 b. 蒸鍍 200nm 金 3.樣品架設 a. 使用 Polyimide tape 將 10mm x 10mm 矽基板固定基板於石墨板。 b. 量測電阻值,確認基板與石墨板之間接觸。 4.進行蝕刻,紀錄電流值。
圖 3-22 P+−Si 背面鍍金電流密度與電壓關係圖
圖 3-24 P+−Si 由電流值反推求電阻與電壓關係圖
P+−Si 背面鍍金在接觸石墨板方式,以電流注入架構進行實驗,P+−Si 以 PI tape 固定於石墨板上,並以探針量測固定後之基板表面電阻,同一樣品連續以電 壓 0.5 至 1.5,0.1 為一個間距,共取 11 個電壓值,電流值取樣頻率 1Hz,量測 時間為 10 分鐘,再取得電流平均值,並繪製成電流密度與電壓關係圖。 由圖 3-22 與圖 3-23 電流密度與電壓關係圖觀察出,比較架構一,也就是樣 品位於陽極之電流密度,架構一無論是 H2O2的有無,電流值皆遠大於架構二, 電洞直接注入樣品比起電洞遠離的方式有較大的影響, 顯示陽極電洞注入的優 勢。觀察陽極蝕刻之中的兩種條件可發現,蝕刻液中加入 H2O2之電流密度明顯 大於僅有 HF 水溶液之條件,從圖 3-24 與圖 3-25 反推電阻值與電壓關係圖來看, 更容易發現導入 H2O2條件之影響,無論是架構一或架構二,約在電壓值超過 0.8 後,電阻值有下降趨勢,並觀察圖 3-25 陽極與 etchant (II)之曲線趨勢,一開始在 電壓值約在 0.5 ~ 07 之間,電阻值較高,類似一能障,隨者電壓上升,能障被克 服,整體迴路電阻值往下降;而 H2O2介入之條件,與前者相比,電阻值於超過 0.8 後趨近穩定,由曲線趨勢可得知,整體迴路電阻較低,此明顯觀察可得知 H2O2 確實使矽與金之間的能障大幅下降,電流密度得到的結果較大。
不同金厚度之樣品電壓與電流密度關係 針對不同厚度金薄膜分別為 12nm,22nm,32nm 與 42nm 之二維週期圖案 10m 柱結構樣品進行測試,樣品尺寸為 10mm x10mm,基板背面製作電極,蒸 鍍 200nm 金,以電流注入架構進行實驗,樣品以 PI tape 固定於石墨板上,如表 3-1,進行電阻量測,確認電極接觸無虞,圖中 (+) 表示樣品固定陽極,對應圖 3-6 架構一,(−) 樣品固定陰極,對應圖 3-6 架構二,間距 d 為 5mm, 並使用 同一片樣品,連續量測不同電壓所得獲得電流之關係,並繪出電流密度電壓關係 圖。 圖 3-26 樣品於實驗前電阻量測 表 3-1 不同金厚度樣品實驗前電阻量測 Au film thickness (nm) Resistance (Ω) 12 0.7k 22 14 32 10 42 5
圖 3-27 P+−Si pillar Au = 12nm 之電流密度與電壓關係圖
圖 3-29 P+−Si pillar Au = 22nm 之電流密度與電壓關係圖
圖 3-31 P+−Si pillar Au = 32nm 之電流密度與電壓關係圖
圖 3-33 P+−Si pillar Au = 42nm 之電流密度與電壓關係圖
圖 3-35 P+−Si pillar Au = 12nm 電流值反推求電阻與電壓關係圖
圖 3-37 P+−Si pillar Au = 22nm 電流值反推求電阻與電壓關係圖
圖 3-39 P+−Si pillar Au = 32nm 電流值反推求電阻與電壓關係圖
圖 3-41 P+−Si pillar Au = 42nm 電流值反推求電阻與電壓關係圖
觀察圖 3-27 至圖 3-34,比較不同金厚度柱樣品之電流密度,電流密度上升 趨勢接近,就陽極蝕刻針對 H2O2有無之電流密度做比較,陽極與 etchant(I)之測 試,施加電位從電壓值 0.5(V)開始,電流密度即大於其他條件;從半對數電流密 度與電壓關係圖來看,4 種金厚度樣品在 3 種條件,架構一與 etchant(I),架構一 與 etchant(II) (排除 H2O2),架構二與 etchant(I)電流密度有接近的斜率,架構二蝕 刻由於電流密度值較低,大部分情況無法觀察出上升趨勢,僅樣品 Au = 32nm 當 電壓超過 1.2(V)可觀察到上升幅度。 如圖 3-43,在 4 種不同金厚度樣品中,以 Au = 32nm 整體不同蝕刻條件其 電流密度值最大,並決定由此金厚度值 32nm 來繼續測試施加單一不同電壓值之 蝕刻對樣品表面形貌之影響。 將量測得到的電流值紀錄反推,繪製出電壓與電阻關係圖,如圖 3-35 至圖 3-42,可發現於四種不同金厚度之樣品其電阻值趨勢相近,觀察陽極蝕刻兩種蝕 刻液條件,無 H2O2介入之條件,於低電壓之結果,如同有一個位障,當電壓上 升至 0.9 後位障影響減弱;加入 H2O2之蝕刻液由於 Au 與 H2O2之催化反應,位 障降低,電阻值明顯下降,此結果於陰極蝕刻結果相呼應,整體而言陰極蝕刻電 阻值較陽極蝕刻高,但加入 H2O2之條件相較陽極蝕刻較為不穩定,尤其是電壓 值 0.5 ~ 0.8 之間,觀察電流值紀錄,由於電流值是取平均值,一開始可能為負值 到負值趨近 0 的電流,當電壓值越大,電流緩慢往上升,觀察圖 3-37,Au = 22nm 樣品,特別是電壓值 0.6(V),由於量測結果為趨近於 0 的負值電流 (I = − 0.00144mA) ,因此比起其他厚度樣品,電阻值急遽下降。整體觀察,陰極加入 H2O2其下降趨勢同樣較無 H2O2明顯,由此可觀察到 H2O2介入之重要性。
蕭特基位障高度
金屬輔助化學蝕刻主要反應區為 Au − Si 介面,金屬與矽接觸面會形成蕭特 基接面並控制電洞分布,而金屬陽極蝕刻主要為施加電位,克服能障進行蝕刻。 為了獲得每一種樣品接面的蕭特基位障高度(Schottky barrier height),利用實驗數 據電流密度與電壓關係所獲得之數據,以下列關係式,求得:
J = A × 𝑇2𝑒−𝐵𝑘𝑇(𝑒𝑞𝑉𝑘𝑇− 1) (3-4)
A 為理查森常數(Richardson’s constant) 120 A/cm2K2,T 為溫度,k 為波茲曼 常數(Boltzmann’s constant),q 為基本電荷,V 為設定電壓值,B 為蕭基能障高度。 將不同金厚度柱樣品所量測得之電流密度與電壓關係圖,電流密度與電壓值 代入式 3-4,求得 B 值曲線,擬合得到截距值,如圖 3-44 所示,獲得下表 3-2 蕭 基位障高度值,不同金厚度之樣品在同一蝕刻條件下蕭基位障接近,但在不同蝕 刻條件相比,可見差異,在陽極蝕刻導入 H2O2之結果,其位障高度最低,約為 0.3 左右,而無 H2O2介入之條件位障則較高;觀察陰極蝕刻結果則與陽極蝕刻相 反,導入 H2O2之位障高度反而較高。
圖 3-44 不同金厚度柱樣品之蕭特基位障高度擬合圖
表 3-2 不同金厚度柱樣品之蕭特基位障高度 (單位 : eV)
Au film thick. Case1 & H2O2 Case1 Case2 & H2O2 Case2
12nm 0.34 0.46 0.64 0.56
22nm 0.35 0.45 0.61 0.57
32nm 0.33 0.39 0.64 0.58
3.3.2 GO
xgranting/P−Si 蝕刻
從能量角度來看,理論上 H2O2應該可以將電洞直接注入矽基材中,而不受
摻雜類型或摻雜濃度的影響,使用[HF] : [H2O2] = 4 : 1 (M),樣品結構如圖 3-45,
P−Si 上具 Graphene Oxide 條狀週期結構 (GOx granting),厚度約為 20 ~ 30 nm。
如圖 3-46,將 P−Si 浸泡蝕刻液中,蝕刻時間為 10 分鐘,蝕刻深度約為 20 nm, 蝕刻速率 2 nm/min,如圖 3-47,於同一蝕刻液條件下,將蝕刻時間 20 分鐘,蝕 刻深度約為 34 nm,蝕刻速率 1.7 nm/min,圖 3-48,蝕刻時間 40 分鐘,蝕刻深 度約為 95nm,蝕刻速率 2.3 nm/min。 由上述結果可觀察 Graphene Oxide 不會被蝕刻液影響,利用 HF/H2O2進行 矽蝕刻確實發生,但是在 HF/H2O2混合溶液中,矽的蝕刻速率卻是比金屬輔助化 學蝕刻中矽蝕刻速率低得多,即使將蝕刻溫度提高至 40˚C,如圖 3-49,蝕刻時 間 20 分鐘,蝕刻表面形成多孔樣貌,蝕刻深度也僅有 113 nm,蝕刻速率 5.6 nm/min; 如圖 3-50,溫度 60˚C 之蝕刻深度也只有 150 nm 左右,蝕刻速率 7.5 nm/min,因 此為求含有氧化劑之溶液進行快速蝕刻矽基材,金屬催化層的存在是必需的。 圖 3-45 GOx granting/P−Si 樣品結構示意圖
圖 3-46 GOx granting/P−Si SEM, = 0.8, 10mins
3.3.3 不同金薄膜厚度蝕刻效應
圖 3-51 為 10nm 與 20nm 金薄膜表面形貌,當金薄膜越薄,呈現島狀結構, 圖 3-52 為 4 種不同 Au = 12、22、32 與 42nm P+−Si 之樣品,於 = 0.85,[HF] : [H2O2] = 5.7 : 1 (M),蝕刻液體積固定 65ml,蝕刻時間 15 分鐘,於常溫之條件下, 圖 3-52 為 SEM 表面形貌圖,圖 3-52 (a) 金屬層為 12nm 之樣品,相較 22nm, 孔隙更大,由於金薄膜尚未成連續薄膜,金屬層為島狀結構,由於為非均勻連續 薄膜,因此無法有效垂直向下蝕刻;但當金屬厚度越厚,物質交換反應難以到達 中央,造成邊界與中央區域蝕刻速不一致,容易形成錐狀結構,如同圖 3-52 (c) Au = 32nm 之結果,而當金屬層更厚,物質難以做交換,蝕刻速度較慢,如圖 3-52 (d) Au = 42nm 結果所示。 圖 3-51 SEM, Au = 10nm & 20nm 之表面形貌圖 3-52 P+−Si pillar SEM,
3.3.4 Au = 32nm P
+−Si 樣品之蝕刻形貌
P+−Si Au = 32nm 二維週期樣品,根據圖 3-53,以 [HF] : [H2O2] = 5.7 : 1 (M), ρ = 0.85,於室溫下蝕刻 15 分鐘,10 m 柱蝕刻深度約為 2.1 m,蝕刻速率約為 0.14 m;5 m 洞蝕刻深度約 1.79 m/min,蝕刻速率約為 0.12 m/min。 如圖 3-54,為提高蝕刻深度,採用升溫蝕刻,將蝕刻液密封隔水加熱至 50 度,使用 10 m 柱之樣品蝕刻 15 分鐘,側向蝕刻明顯,蝕刻形貌呈現錐狀結構; 如圖 3-55、圖 3-56 與圖 3-57,考慮控制蝕刻準直性,減少側向蝕刻,以 CD − MaCEtch 進行測試,依序設定 V = 0.5、1.0 及 1.5 (V),於 ρ = 0.85,50 度之條件 下,蝕刻 15 分鐘,側向蝕刻仍嚴重,尤其當電壓值達到 1.5(V),電流密度似乎 太大,柱結構明顯倒塌,部分條件可能因為金薄膜為 32nm,已成膜狀結構,於 蝕刻過程中由於氫氣產生造成破裂情況產生;另外,圖 3-58 嘗試於常溫條件下 預蝕刻 10 分鐘,再進行 CD − MaCEtch,電壓值設定 V = 0.5 (V),常溫蝕刻,側 向蝕刻結果仍嚴重,同時大部分柱結構已倒塌,圖 3-59 並以此條件測試 5m 洞 結構並提高溫度,發現無明顯蝕刻,且表面結構混亂,有出現裂痕,這顯示蝕刻 速率可能過於快速,樣品結構不適合進行升溫蝕刻,因此考慮回到常溫條件,進 行蝕刻。
圖 3-54 P+−Si pillar SEM, Au = 32nm, = 0.85, 50℃, 15mins
圖 3-55 P+−Si pillar SEM, Au = 32nm,
圖 3-56 P+−Si pillar SEM, Au = 32nm,
= 0.85, V = 1, 50℃, 15mins
圖 3-57 P+−Si pillar SEM, Au = 32nm,
圖 3-58 P+−Si pillar SEM, Au = 32nm, = 0.85, pre-etch 10mins, V = 0.5, 1hr
圖 3-59 P+−Si hole SEM, Au = 32nm,
由於升溫蝕刻操作結果不如預期,因此改採用常溫增加蝕刻時間進行測試, 根據圖 3-60,使用蝕刻液[HF] : [H2O2] = 5.7 : 1 (M),室溫條件下,進行 1 小時蝕 刻,並以轉速 350rpm 進行磁控攪拌。 如圖 3-60 (a),10m 柱蝕刻 1 小時後,表面無法觀察到任何結構,推測整個 表面可能因為長時間蝕刻,電洞擴散造成表面結構氧化進而蝕刻,結構無法維持; 圖 3-60 (b),而 5m 洞結構側向蝕刻結果導致洞與洞之間蝕刻穿,此兩種結構於 1 小時蝕刻,皆無法維持結構;因此為觀察結構崩塌時機,採用分次蝕刻,每次 蝕刻時間 15 分鐘,重複操作 4 次,共計 1 小時,從 10m 柱之蝕刻結果,觀察 到柱倒塌的情況,而且其表面可觀察到有一層容易分離的結構,呈現破裂同時有 剝離脫落的現象,而當這層不穩定結構整個剝落,就可能產生圖 3-60 (a) 1 小時 蝕刻的結果,導致無法觀察到表面結構;圖 3-60 (d),而 5m 洞結構,從其蝕刻 結果觀察,從表面形貌觀察,其結構較為完整,但切開此樣品進行觀察,結構容 易碎裂,並可以看見分層痕跡,並由上下分層,如圖 3-60 (f) 與圖 3-60 (g),推 測蝕刻深度約有 20m 左右,而造成此結果可能原因為使用 P+ −Si,多數載子為 電洞,基板本身提供過多電洞,再加上 H2O2經金屬層催化而注入的電洞,造成 大量電洞擴散於結構表面,由於電洞無法迅速被消耗,因而形成一矽之多孔隙 層。 分次蝕刻相較於一次性蝕刻 1 小時之結果明顯有些差異,尤其在 5m 洞之 蝕刻結果更為明顯,長時間的一次性蝕刻電洞擴散,可能導致往側壁蝕刻的結果, 而分次蝕刻,相較一次性蝕刻,電洞擴散情況減緩,表面形貌較為完整。
圖 3-60 P+−Si SEM, Au = 32nm,
![圖 1-5 Si 基板與 Si 陣列結構之反射率比較 [11]](https://thumb-ap.123doks.com/thumbv2/9libinfo/8791454.220420/17.892.245.645.132.650/圖15Si基板與Si陣列結構之反射率比較11.webp)
![圖 1-9 以 VLS 生長法生成之矽晶鬚結構 [20]](https://thumb-ap.123doks.com/thumbv2/9libinfo/8791454.220420/20.892.150.747.456.794/圖19以VLS生長法生成之矽晶鬚結構2.webp)
![圖 2-5 垂直與側向蝕刻示意圖 [27]](https://thumb-ap.123doks.com/thumbv2/9libinfo/8791454.220420/30.892.149.747.551.949/圖25垂直與側向蝕刻示意圖27.webp)
![圖 2-6 電洞注入 Si 之表面蝕刻模式 [27]](https://thumb-ap.123doks.com/thumbv2/9libinfo/8791454.220420/31.892.138.815.108.824/圖26電洞注入Si之表面蝕刻模式27.webp)
![圖 2-11 MaCEtch : 金屬層厚度與蝕刻速率之關係 [31]](https://thumb-ap.123doks.com/thumbv2/9libinfo/8791454.220420/35.892.134.753.156.715/圖211MaCEtch金屬層厚度與蝕刻速率之關係31.webp)
![圖 2-14 不同 值與蝕刻速率關係 [32][33]](https://thumb-ap.123doks.com/thumbv2/9libinfo/8791454.220420/38.892.287.623.118.496/圖214不同值與蝕刻速率關係3233.webp)